

Abstract
Background:
Neuromyelitis optica spectrum disease (NMOSD) is a severe autoimmune demyelinating disorder of the central nervous system that throughout epidemiological data, it has not been completely determined. The aim of this study was to assess characteristics of NMOSD patients in Isfahan as one of the most prevalent cities for multiple sclerosis in Iran.
Materials and Methods:
Forty-five patients diagnosed as neuromyelitis optica (NMO) disease through 5 years enrolled in this study. Demographics and characteristics of disease such as Expanded Disability Status Scale (EDSS) score, disease duration, clinical symptoms, laboratory data, and magnetic resonance imaging findings (including T1, T2, and flair protocols) were recorded. NMO-immunoglobulin G serology assay was done in all of the patients by ELISA test.
Results:
Female to male ratio was 5.4:1. The mean age of disease onset was 29.8 ± 11.2 years. NMO antibody was positive in 24.4% of patients. The presenting symptoms were optic neuritis (55.5%), transverse myelitis (40%), and brainstem symptoms (4.5%). The interval between the first and second attack was 19.28 ± 31.27 months (range: 1 month to 17 years). The mean EDSS score of the patients was 2.8 ± 2.25. Frequency of long-extending cervical plaque was higher among men than women (85.7% vs. 57.9%).
Conclusion:
Based on this study, the mean age of NMOSD onset among Isfahan population was considerably lower than other studies, and there was higher frequency of long-extending cervical lesion among male patients which needs more consideration in further studies.
Keywords: Demographics, Devic's syndrome, Iran, neuromyelitis optic
INTRODUCTION
Neuromyelitis optica spectrum disease (NMOSD) is an autoimmune demyelinating disorder involving the central nervous system, with predominance of optic nerve and spinal cord involvement.[1]
Discovery of neuromyelitis optica-immunoglobulin G (NMO-IgG) antibody has increased knowledge in diagnosis of disease.[1]
According to revised criteria of NMOSD, diagnosis can be confirmed by clinical syndromes and/or magnetic resonance imaging (MRI) findings related to the involvement of optic nerve, spinal cord, area postrema, and other parts of brainstem or diencephalon, with/without serum NMO antibody.[2]
Prevalence of NMOSD is varying among different races and is more prevalent in Indian, Asian, and Black populations, where 15–57% of patients with demyelinating diseases suffer from it.[3]
The most common initial symptom of NMO is optic neuritis (ON)[4] that is more severe and less recoverable as compared with multiple sclerosis (MS).[5] Pain, myelitis with painful tonic spasm, vomiting, hiccups, oculomotor dysfunction, pruritus, hearing loss, facial palsy, vertigo, vestibular ataxia, headache, and paresthesia are other reported symptoms of NMOSD.[4,6,7]
Notably, NMOSD has female predominance in different populations.[8] In one study performed in the United States, the age of onset was 41.1 years on an average,[8] but Collongues et al. reported the mean age of 34.5 years for onset of the disease in French population.[9]
Course of NMOSD disease can be monophasic or relapsing, but relapsing course was reported in 80%–90% of patients.[10] Monophasic course of NMOSD occurs equally among men and women, but relapsing course is significantly more frequent among females.[11] Relapsing course during first 2 years of disease onset is in association with poor prognosis, severe disability, and even death.[12]
Based on lacking enough data about NMOSD in different populations and especially few available studies about NMOSD in Isfahan, Iran, which is one of the most prevalent cities of MS in Iran and also lack of enough study based on 2014 criteria of Wingerchuk et al.,[2] this study was designed to assess demographics of NMOSD patients and increasing knowledge for better diagnosis and management of patients.
MATERIALS AND METHODS
This is a cross-sectional study conducted in Kashani Hospital as an MS referral center in Isfahan, Iran. Among 2300 patients registered in Kashani MS Clinic during 2009–2014, 45 patients were diagnosed with NMO patients according to the revised NMOSD diagnostic criteria 2014.[2] The study was approved by the Isfahan University of Medical Sciences.
Inclusion criterion was diagnosis of NMOSD based on Wingerchuk et al., 2014 criteria[2] and patients who did not match criteria completely were excluded.
All patients were informed about the study and consent form was signed by all of them. Demographic data and characteristics of disease such as Expanded Disability Status Scale (EDSS) score, disease duration, clinical symptoms, laboratory data, and brain and spine MRI findings were recorded by a trained nurse.
MRI device was Philips Ingenia 1.5 Tesla and all patients had T1, T2, and flair MRI.
Patients whose brain MRI was not matched with McDonald criteria were followed in order to be assessed for criteria of NMOSD.[13]
Long-extending transverse myelitis (LETM) was defined as ≥2 segment involvement in the spine MRI.
All of the patients were examined and EDSS score was assessed by an expert neurologist. Semiquantitative NMO-IgG serology assay with ELISA method was done for all patients. 2 ml serum of each patient was needed for this test. Cutoff value for positive results was 1.6 U/ml.
Data were analyzed by using IBM SPSS version 20, United States software. Because the distribution of much of the quantitative data was skewed, medians and ranges are presented. Mann–Whitney test was applied to assess the differences. Chi-square or Fisher's exact test was used for dichotomous variables. Values of P < 0.05 were considered statistically significant.
RESULTS
Forty-five patients with diagnosis of NMOSD including 7 male and 38 female enrolled in this study (female to male ratio was 5.4:1). The mean age of patients, mean age of disease onset, and mean duration of disease were 37.2 ± 10.8 years (mean ± standard deviation [SD]; range: 19–62), 29.8 ± 11.2 years (mean ± SD; range: 11–58), and 7.43 ± 2.6 years (mean ± SD; range: 1–14), respectively [Table 1].
Table 1.
Demographic data of the patients with neuromyelitis optica

The first presenting symptoms were ON in 25 (55.6%), transverse myelitis (TM) in 18 (40%), and brainstem symptom (including manifestations such as intractable vomiting, nausea, or vertigo) in 2 (4.45%) patients. The presentation of second attacks was ON in 23 (51.1%), TM in 18 (40%), and brainstem deficit in 2 (4.45%) and 2 (4.45%) with no new presentations.
The interval between the first and the second attack was 19.28 ± 31.27 months (range: 1 month to 17 years).
The mean EDSS score in patients at the time of the study was 2.8 ± 2.25 (mean ± SD; range: 1–8). EDSS score <3.5 in 25 patients (55%), between 4 and 5.5 in 11 patients (24.4%), and ≥6 in nine patients 9 (20%) was detected.
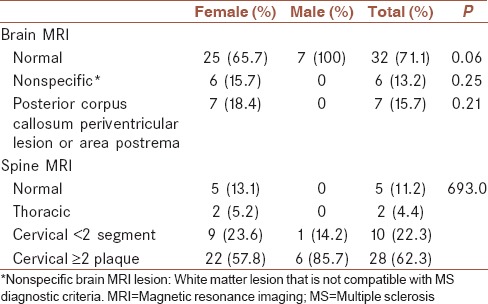
Brain MRI was normal in 32 (71.1%) patients and nonspecific abnormality in white matter of hemispheres and brainstem was detected in six patients (13.3%). Seven (15.7%) patients had periventricular lesions mostly in the posterior of corpus callosum.
Spinal MRI showed thoracic cord lesion in two patients and cervical cord lesion in 38 and no lesion in 5 (11.2%) patients. Cervical MRI findings included ten patients (22.3%) with short-segment T2 hyperintense lesions. Long-extending lesion of two vertebral segments preferentially in central gray matter was detected in three patients (6.7%) and of three vertebral segments in 25 patients (55.6%). Higher rate of LETM was found among male (85.7%) in comparison with female (59.7%) that was not statistically significant.
Patients were treated by azathioprine (80%), mycophenolate mofetil (11.9%), methotrexate (4.45%), or cyclophosphamide (4.45%). Eight patients (17.7%) were treated by low-dose corticosteroids as added on therapy as well [Tables 1–3].
Table 3.
First and second symptoms of patients with neuromyelitis optica and frequencies

Table 2.
Magnetic resonance imaging findings of the patients with neuromyelitis optica and frequencies
DISCUSSION
As NMOSD is a new disorder, more considerations can lead to better diagnosis and thus more effective treatment. The aim of this study was assessing demographics, clinical course, and type of treatment in Isfahan NMOSD patients. In the current study, female/male ratio was 5.4:1 and was consistent with previous studies that showed various female/male ratio ranged 3–9.[11]
The mean age of onset in this study was 29.8 (range 11–58) years that was considerably lower than existing data.[11]
ON was the most common first presenting sign at onset of disease (55.6%) such as Wingerchuk et al. that reported ON in 52% and myelitis in 35% of patients.[5] Sahraian et al. reported vertigo and ataxia as other symptoms of their patients like 5.45% of our patients who mentioned these symptoms.[14]
NMO has a monophasic or relapsing course. In monophasic type, ON and myelitis occur closely or simultaneously in <30 days with no other relapse,[10] but in relapsing course, 55% of patients may experience second attacks in a year after clinical initiation, 78% within 3 years and 90% within 5 years.[15] Most of our patients had relapsing course with a mean period of relapses interval of 19.28 ± 31.27 months (with the range of 1 month to 17 years).
The mean EDSS score at the time of participation was 2.8 (range 1–8) and 55% of patients had mild disability. Severe disability (EDSS score ≥ 6) was noticeably lower than many other studies.[16,17]
In this presentation, 71.1% of patients had normal brain MRI that was consistence with most of previous studies.[18] In 15.7% of the patients, periventricular lesions in the posterior of corpus callosum or area postrema were detected.
Long-extending lesion in central gray matter of the cervical MRI was detected in 28 patients (62.2%) that had two vertebral segments length in 6.7% and three vertebral segments length in 55.6%. These findings are suggestive of NMO disease.[10,19] 22.3% showed short segment lesion in their cervical MRI that is similar to MS neuroimaging findings and should be considered by other neurologists.
We found higher frequency of LETM in male (85.7%) in comparison with female (59.7%) though it was not statistically significant.
Introduction of NMO-IgG by Lennon et al., in 2005 had significant effect on differentiation of MS from NMOSD. This antibody which binds to aquaporin-4 is 73% sensitive and 91% specific for NMO diagnosis.[20] In our study, positive result of NMO antibody was low that may be due to methodological problem.
Treatment and prevention of relapses are really important because of poorer prognosis of NMO in comparison to MS. Late initiation of treatment and relapse occurrence may lead to permanent disability.[10] Most of our patients were treated with azathioprine (80%) that resulted in better response to treatment in our patients.
The limitation of this study was measurement of NMO by ELISA instead of cell-based assay.
CONCLUSION
According to our study, the mean age of NMO onset was considerably lower than other populations. Frequency of LETM in the spinal MRI was higher in male patients; thus, if a patient presents signs and symptoms of ON, cervical spine should be evaluated. Low EDSS score of patients and good response to azathioprine would suggest better prognosis even though further studies is necessary to confirm it.
Limitation
Although gold standard method of NMO antibody measurement is cell-based assay; however, as we do not have reliable center which uses this method in Iran, Eliza is used in this study
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
AUTHORS’ CONTRIBUTION
FA contributed in the conception of the work, conducting the study, revising the draft, approval of the final version of the manuscript, and agreed for all aspects of the work AS contributed in the conception of the work, revising the draft, the acquisition and interpretation of data for the work
VSh contributed in the conception of the work, conducting the study, revising the draft, approval of the final version of the manuscript, and agreed for all aspects of the work
MAN contributed in the conception of the work, revising the draft, the acquisition and interpretation of data for the work
SV Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work
REFERENCES
- 1.Jurynczyk M, Craner M, Palace J. Overlapping CNS inflammatory diseases: Differentiating features of NMO and MS. J Neurol Neurosurg Psychiatry. 2015;86:20–5. doi: 10.1136/jnnp-2014-308984. [DOI] [PubMed] [Google Scholar]
- 2.Wingerchuk D, Banwell B, Bennett J, Cabre P, Carroll W, Chitnis T, et al. Revised diagnostic criteria for neuromyelitis optica spectrum disorders (S63. 001) Neurology. 2014;82(10 Supplement):S63–001. doi: 10.1212/WNL.0000000000001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Etemadifar M, Nasr Z, Khalili B, Taherioun M, Vosoughi R. Epidemiology of neuromyelitis optica in the world: A systematic review and meta-analysis. Mult Scler Int 2015. 2015:174720. doi: 10.1155/2015/174720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakajima H, Hosokawa T, Sugino M, Kimura F, Sugasawa J, Hanafusa T, et al. Visual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosis. BMC Neurol. 2010;10:45. doi: 10.1186/1471-2377-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome) Neurology. 1999;53:1107–14. doi: 10.1212/wnl.53.5.1107. [DOI] [PubMed] [Google Scholar]
- 6.Bradl M, Kanamori Y, Nakashima I, Misu T, Fujihara K, Lassmann H, et al. Pain in neuromyelitis optica – Prevalence, pathogenesis and therapy. Nat Rev Neurol. 2014;10:529–36. doi: 10.1038/nrneurol.2014.129. [DOI] [PubMed] [Google Scholar]
- 7.Kremer L, Mealy M, Jacob A, Nakashima I, Cabre P, Bigi S, et al. Brainstem manifestations in neuromyelitis optica: A multicenter study of 258 patients. Mult Scler. 2014;20:843–7. doi: 10.1177/1352458513507822. [DOI] [PubMed] [Google Scholar]
- 8.Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: A multicenter analysis. Arch Neurol. 2012;69:1176–80. doi: 10.1001/archneurol.2012.314. [DOI] [PubMed] [Google Scholar]
- 9.Collongues N, Marignier R, Zéphir H, Papeix C, Blanc F, Ritleng C, et al. Neuromyelitis optica in France: A multicenter study of 125 patients. Neurology. 2010;74:736–42. doi: 10.1212/WNL.0b013e3181d31e35. [DOI] [PubMed] [Google Scholar]
- 10.Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019–32. doi: 10.1111/j.1468-1331.2010.03066.x. [DOI] [PubMed] [Google Scholar]
- 11.Sahraian MA, Radue EW, Minagar A. Neuromyelitis optica: Clinical manifestations and neuroimaging features. Neurol Clin. 2013;31:139–52. doi: 10.1016/j.ncl.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Bergamaschi R, Ghezzi A. Devic's neuromyelitis optica: Clinical features and prognostic factors. Neurol Sci. 2004;25(Suppl 4):S364–7. doi: 10.1007/s10072-004-0342-0. [DOI] [PubMed] [Google Scholar]
- 13.Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahraian MA, Moinfar Z, Khorramnia S, Ebrahim MM. Relapsing neuromyelitis optica: Demographic and clinical features in Iranian patients. Eur J Neurol. 2010;17:794–9. doi: 10.1111/j.1468-1331.2009.02928.x. [DOI] [PubMed] [Google Scholar]
- 15.Mandler RN. Neuromyelitis optica – Devic's syndrome, update. Autoimmun Rev. 2006;5:537–43. doi: 10.1016/j.autrev.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Papais-Alvarenga RM, Miranda-Santos CM, Puccioni-Sohler M, de Almeida AM, Oliveira S, Basilio De Oliveira CA, et al. Optic neuromyelitis syndrome in Brazilian patients. J Neurol Neurosurg Psychiatry. 2002;73:429–35. doi: 10.1136/jnnp.73.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Riordan JI, Gallagher HL, Thompson AJ, Howard RS, Kingsley DP, Thompson EJ, et al. Clinical, CSF, and MRI findings in Devic's neuromyelitis optica. J Neurol Neurosurg Psychiatry. 1996;60:382–7. doi: 10.1136/jnnp.60.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller DH, Weinshenker BG, Filippi M, Banwell BL, Cohen JA, Freedman MS, et al. Differential diagnosis of suspected multiple sclerosis: A consensus approach. Mult Scler. 2008;14:1157–74. doi: 10.1177/1352458508096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–9. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 20.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]