

Abstract
Circular RNAs (circRNAs) are highly stable, covalently closed RNAs that are regulated in a spatiotemporal manner and whose functions are largely unknown. These molecules have the potential to be incorporated into engineered systems with broad technological implications. Here we describe a switch for inducing back-splicing of an engineered circRNA that relies on the CRISPR endoribonuclease, Csy4, as an activator of circularization. The endoribonuclease activity and 3′ end-stabilizing properties of Csy4 are particularly suited for this task. Coexpression of Csy4 and the circRNA switch allows for the removal of downstream competitive splice sites and stabilization of the 5′ cleavage product. This subsequently results in back-splicing of the 5′ cleavage product into a circRNA that can translate a reporter protein from an internal ribosomal entry site (IRES). Our platform outlines a straightforward approach toward regulating splicing and could find potential applications in synthetic biology as well as in studying the properties of different circRNAs.
Keywords: CRISPR, Csy4, circular RNA, splicing regulation, gene regulation
INTRODUCTION
Circular RNAs (circRNAs) comprise an emerging large class of noncoding, covalently closed RNAs present in a wide variety of eukaryotic organisms ranging from protists to humans (Wang et al. 2014). In human cells, they are typically generated through the covalent joining of a downstream splicing donor site with an upstream splicing acceptor site through a process called direct back-splicing (Salzman et al. 2012; Jeck et al. 2013). Due to their circular nature, circRNAs exhibit marked stability relative to their corresponding linear isoforms (Jeck et al. 2013; Lasda and Parker 2014; Liang and Wilusz 2014; Enuka et al. 2015). The median half-life of circRNAs is at least 2.5× longer than their linear counterparts (Enuka et al. 2015). Furthermore, for some genes, the abundance of the circRNA exceeds that of the associated linear mRNA by a factor of 10 (Salzman et al. 2012; Jeck et al. 2013).
Despite their prevalence, the function, if any, of most circRNAs remains to be determined (Ebbesen et al. 2015). Some circRNAs such as ciRS-7 (Cdr1as) and the Sry circRNA act as microRNA sponges (Hansen et al. 2013; Memczak et al. 2013), while circMbl has been proposed to act as a sponge for MBL (Muscleblind) protein (Ashwal-Fluss et al. 2014). Though most circRNAs arise from protein-coding exons, endogenously encoded circRNAs do not appear to be associated with ribosomes or translated (Capel et al. 1993; Jeck et al. 2013; Guo et al. 2014). Engineered circRNAs can be translated, however, if an IRES site is included in the construct to drive translation (Chen and Sarnow 1995; Wang and Wang 2015). This property allows for expression of proteins from stable, persistent RNA molecules. Accordingly, the development of molecular tools to exogenously control the circularization of a particular RNA of interest could prove useful.
In this regard, an interesting RNA targeting enzyme that has garnered recent interest from the synthetic biology community is Csy4 (Cas6f), from the bacteria Pseudomonas aeruginosa. Csy4 is an endoribonuclease belonging to the clustered regularly interspaced short palindromic repeats (CRISPR) family. CRISPR systems utilize small RNAs (crRNAs) to guide CRISPR nucleases to nucleic acids, mediating sequence-specific cleavage (Marraffini 2015). Csy4 recognizes a 16 ribonucleotide hairpin, which repeats throughout a precursor CRISPR RNA, cleaving at the 3′ base of the hairpin stem to generate crRNAs (Haurwitz et al. 2010; Cady and O'Toole 2011). Importantly, Csy4 remains associated with the 5′ cleavage product (Haurwitz et al. 2010). These properties of Csy4 have been adapted recently to process operon-encoding RNAs for gene expression without interference from cis sequences in E. coli, B. subtilis, and S. cerevisiae (Qi et al. 2012). Further, Csy4 was used for processing RNA encoding multiple guide-RNA (gRNA) to single gRNAs for Cas9-based applications (Nissim et al. 2014; Tsai et al. 2014). Csy4 has also been used to isolate and identify proteins associated with a specific RNA of interest (Lee et al. 2013). In addition, we recently demonstrated that Csy4 cleavage within the 3′ UTR of an mRNA results in stabilization of the mRNA, and supports translation of the nonpolyadenylated message in mammalian cells (Borchardt et al. 2015).
The specific cleavage activity of Csy4 can be applied to remove RNA sequences downstream from the targeted hairpin. Here we take advantage of Csy4 to activate circularization of RNA. By directing Csy4 to cleave within an intron, we remove downstream competing splicing signals while stabilizing the cleaved upstream product. This cleaved product is back-spliced into a circRNA through use of the remaining splicing signals. We demonstrate that Csy4 cleavage results in circularization of RNA as efficiently, if not more efficiently, than from a gene expressing the same circRNA constitutively, with no competing introns. We measure circRNA biogenesis using a Csy4-dependent fluorescent reporter system (circGFP-Csy4-dependent, or circGFP-CD), Northern blotting, and qRT-PCR. This reporter may serve as a switch-based platform for the regulation of a variety of natural and engineered circRNA-mediated processes while also providing a new tool for shifting expression from one RNA isoform or protein to another. Additionally, the Csy4-activated circularization system has the potential for application in the study of both cis- and trans-factors involved in circRNA biogenesis.
RESULTS AND DISCUSSION
The reporter design was based on a circGFP construct reported earlier (Wang and Wang 2015), which encodes a polyadenylated pre-mRNA whose back-splicing is directed by inverted repeat sequences flanking the exon. The circGFP cassette consists of a split GFP in which the N-terminal fragment of GFP lies downstream from the C-terminal fragment. The C-terminal fragment is preceded by a canonical splice acceptor sequence while the N-terminal fragment is immediately followed by a canonical splice donor sequence. The two fragments are separated by an IRES sequence such that, upon back-splicing, full-length GFP is reconstituted and can be expressed as protein (Fig. 1A). In the current study, we added a competing splice acceptor site downstream from the split-GFP cassette. This competing splice acceptor is followed by a P2A-dsRed cassette, where the P2A peptide signal disrupts upstream translation, while simultaneously allowing translation of the dsRed ORF without being directly fused to GFP (Szymczak and Vignali 2005). To make circRNA formation from the circGFP cassette responsive to Csy4, the split GFP and P2A-dsRed cassettes were separated by a Csy4 substrate hairpin. The circGFP-CD (circGFP-Csy4-dependent, depicted in Fig. 1B,C) reporter expresses dsRed upon forward splicing (linear RNA) or GFP upon back-splicing (circRNA). This allows us to test whether circularization can compete with forward splicing, and whether expressing Csy4 alters the amounts of circular and linear products. Expression of circGFP-CD in the absence of Csy4 results in a competition between forward splicing and circularization (Fig. 1B). Conversely, when circGFP-CD is coexpressed with Csy4, the competing splice acceptor and P2A-dsRed encoding sequence may be cleaved away before splicing occurs, leaving behind the 5′ splice acceptor site (Fig. 1C). We hypothesized that the stabilization of the 5′ cleavage product by Csy4 could permit the cleaved split-GFP RNA to persist long enough for back-splicing to occur and support increased GFP expression (Fig. 1C). Note that should forward splicing occur prior to Csy4 cleavage, the dsRed signal will persist.
FIGURE 1.

Schematic of splicing patterns and reporter outputs for circGFP and circGFP-CD. The RNAs derived from the circGFP (A) and circGFP-CD (B,C) plasmids are expressed from the CMV promoter and are capped in their linear isoforms. Donor and acceptor splice sites are represented by gray triangles, and the dotted lines indicate the expected predominant splicing patterns. (A) The precursor circGFP transcript contains a split-GFP cassette flanked by intron sequences that have been engineered to contain inverted repeat sequences (blue boxes). The GFP fragments are separated by an EMCV IRES such that upon RNA circularization, full-length GFP is expressed. (B) The circGFP-CD precursor RNA contains the full circGFP sequence followed by a Csy4 targeted hairpin and a P2A-dsRed cassette. In the absence of Csy4 (B), forward splicing is favored and dsRed is translated. A nonfluorescent N-terminal GFP fragment by-product is released via the P2A sequence. In the presence of Csy4 (C), Csy4 can cleave the RNA at the base of the hairpin stem, releasing the forward splice acceptor and P2A-dsRed cassette. Csy4 remains associated with the cleaved hairpin, stabilizing the RNA and allowing back-splicing to occur. The IRES subsequently drives full-length GFP expression from the circular RNA.
We first tested the circGFP-CD construct by transfecting this plasmid into mammalian cells and observing reporter expression via fluorescence microscopy. CircGFP, which is constitutively back-spliced for GFP expression, is included for comparison (Fig. 2). When circGFP-CD is expressed alone, the cells strongly express dsRed but lack GFP fluorescence (Fig. 2A). This expression pattern is consistent with predominant forward splicing and minimal back-splicing. It is important to note that the forward splice acceptor and preceding intron sequence is identical to the back splice acceptor and intron sequence. Therefore, the abundance of the dsRed signal indicates that forward splicing is strongly favored over back-splicing in this intron/exon configuration and host cell type. In contrast, when Csy4 is coexpressed with circGFP-CD, the dsRed signal decreases and a pronounced GFP signal appears, consistent with activation of circRNA biogenesis (Fig. 2A). Fluorescence was quantified using FIJI image processing software (Schindelin et al. 2012), and the dsRed signal decreased 5.6-fold upon coexpression with Csy4. The corresponding increase in the GFP signal was measured at 115.8-fold relative to Csy4 absent conditions (Fig. 2B,C). The appearance of the GFP signal concurrent with diminishing dsRed signal is consistent with Csy4 cleaving away the P2A-dsRed element and stabilizing the 5′ cleavage product, allowing back-splicing and subsequent GFP expression to occur.
FIGURE 2.

Csy4 activates circGFP-CD reporter expression to switch from dsRed to GFP. (A) Representative images of dsRed (top) and GFP (bottom) fluorescence from HEK293 cells expressing circGFP (far left), or circGFP-CD in the absence of Csy4 (middle left), presence of native Csy4 (middle right), or presence of Csy4-H29A (far right) 72 h post-transfection. Schematics of major pre-mRNA species present before splicing are depicted above each column, and schematics of the major RNA species post-splicing are depicted on the left of each row. The dsRed (B) and GFP (C) fluorescence were quantified from images taken 72 h post-transfection of HEK293 cells using the FIJI image processing package (Schindelin et al. 2012). Error bars indicate standard deviation of integrated density calculated from nine images from three biological replicates (three images per replicate). Statistical significance was calculated using a two-tailed Student's t-test ([****] P ≤ 0.0001, [*] P ≤ 0.05). (D) Western blot detecting GFP or loading control Actin for lysates of HEK293 cells transfected as indicated.
To confirm that RNA cleavage and removal of the forward splicing acceptor is essential for favoring back-splicing in our system, we coexpressed circGFP-CD with a catalytically dead Csy4 mutant, Csy4-H29A (H29A). This mutant is capable of binding to the target hairpin, but is unable to cleave the RNA (Haurwitz et al. 2010). In the presence of H29A, the GFP signal showed only a slight increase (1.8-fold), consistent with Csy4 cleavage being required for predominant back-splicing to occur (Fig. 2C). H29A coexpression also resulted in a 2.4-fold increase in the dsRed signal (Fig. 2A,B). This modest effect may result from H29A-mediated stabilization of the precursor RNA. We performed Western blot analysis, which demonstrated that detectable amounts of GFP protein were only observed when circGFP-CD RNA was coexpressed with native Csy4 (Fig. 2D). These results corroborate the requirement of Csy4 for circularization and consequent GFP expression in our reporter system.
We characterized the different RNA species generated from circGFP-CD using RT-PCR, Northern blotting, RNase R treatment, and qRT-PCR. PCR primers were designed for circRNA detection such that a PCR product will only be generated when the GFP fragments have been spliced in the appropriate orientation, for example, via RNA circularization (Fig. 3A). In the absence of Csy4, a faint PCR product was detected using the circular RNA detection primers, suggesting that back-splicing from circGFP-CD RNA may occur at a low basal rate (Fig. 3A). However, when circGFP-CD RNA is coexpressed with Csy4, a robust signal is observed, consistent with a substantial increase in circRNA levels (Fig. 3A). It is important to note that circRNA levels expressed from circGFP-CD remain 25- to 40-fold higher in the presence of Csy4 relative to basal expression in the absence of Csy4 when assayed by qRT-PCR (see Fig. 4A). A modest increase in circRNA levels was observed upon coexpression with H29A (Fig. 3A), consistent with quantified fluorescence data shown earlier (Fig. 2C).
FIGURE 3.
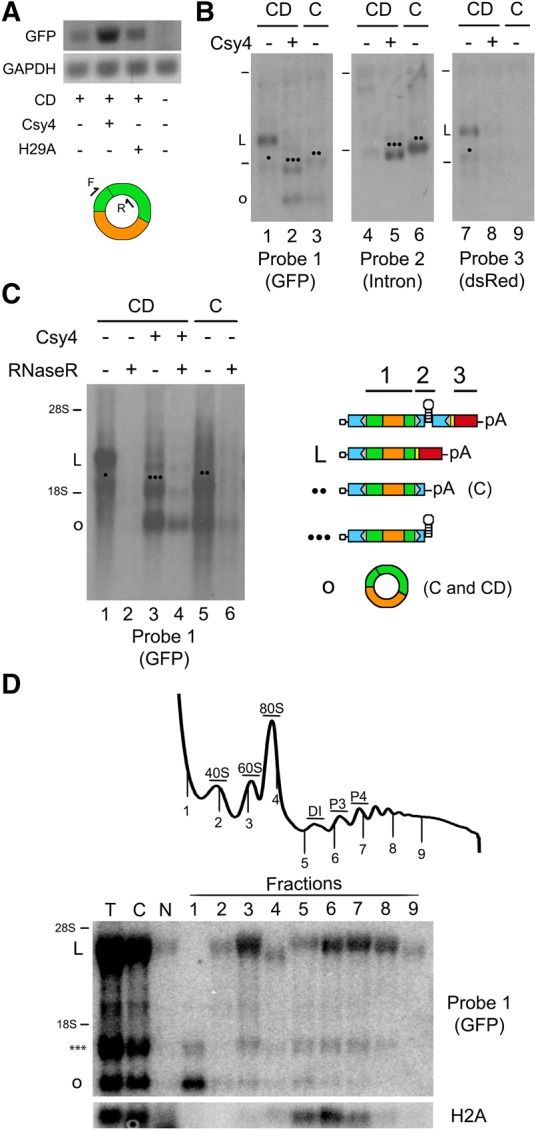
Detection of circGFP-CD expression products. (A) PCR products obtained using cDNA template generated from randomly primed RNA. RNA was isolated from HEK293 cells transfected with circGFP-CD (CD), Csy4, or H29A as indicated. Primers were designed to detect reconstituted, full-length GFP, resulting from back-splicing. GAPDH PCR products were amplified as loading controls from the same templates. (B) Northern blotting of total cellular RNA probed against GFP, intron sequences, or dsRed to characterize various RNA species expressed from the circGFP-CD (CD) and circGFP (C) plasmids in the presence and absence of Csy4. Tick marks denote the position of 28S and 18S rRNAs. Schematics of expected RNA species are depicted with a corresponding symbol for band identification on the Northern blot images. The (•) symbol refers to the unknown product. Representations of probes utilized for detecting GFP (1), intron (2), and dsRed (3) sequence are depicted above a schematic of the circGFP-CD pre-mRNA. (C) Total RNA from the samples in B was analyzed by Northern blotting with or without RNaseR digestion and probed against GFP. Bands for each species were quantified on a PhosphorImager. (D) Polyribosome analysis of RNA from cells cotransfected with circGFP-CD and Csy4. The absorbance trace of the gradient fractionation is shown, and lines denote where each collected fraction begins. (DI) disome, (P3) three bound ribosomes, (P4) four bound ribosomes. Equal proportions of each fraction were analyzed by Northern blotting and probed for GFP and histone H2A, scanned on a PhosphorImager for visualization. (T) Total RNA, (C) cytoplasmic RNA, (N) nuclear RNA.
FIGURE 4.
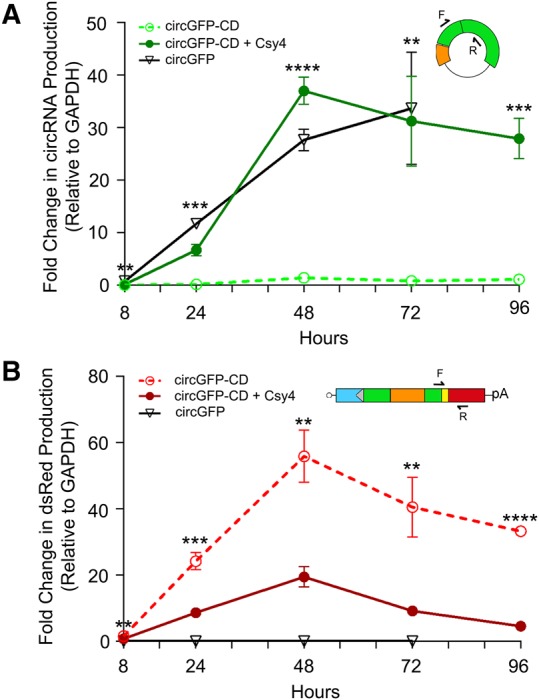
qRT-PCR analysis of the accumulation of circRNA and linear splicing products after transfection. qRT-PCR analysis of cDNA generated from RNA isolated at 8, 24, 48, 72, and 96 h for circRNA (A) or linear RNA (B) was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Cells were transfected with circGFP-CD (open circles, dotted line), circGFP-CD and Csy4 (closed circles, solid line), or with a circGFP plasmid (open triangles, solid line). qRT-PCR was carried out using primers specific for either circular (A) or linear (B) splicing products as depicted above each graph. Error bars indicate standard deviation of three biological replicates. Statistical significance was calculated using a two-tailed Student's t-test ([****] P ≤ 0.0001, [***] P ≤ 0.001, [**] P ≤ 0.01, [*] P ≤ 0.05).
We also performed Northern blots to directly visualize and quantify the abundance of the various RNA species expressed from the circGFP-CD plasmid in the presence and absence of Csy4 (Fig. 3B). We analyzed total RNA expressed from cells transfected with circGFP or circGFP-CD with and without Csy4. Blotting with a probe against the GFP ORF revealed all the RNA isoforms expressed from the circGFP-CD construct. In the absence of Csy4, the most prominent band corresponded to the expected size for the forward-spliced RNA species (Fig. 3B, lane 1). We also detected a second, less-abundant, lower molecular weight product of unknown origin under these conditions (denoted by [•] in the Northern blots). In the presence of Csy4, the amount of forward-spliced product detected is decreased. Instead, the predominant bands are found at sizes corresponding to the linear Csy4-cleaved RNA species and the circular RNA (Fig. 3B, lane 2). The band representing the circRNA is also present in the RNA isolated from cells expressing the circGFP plasmid. A polyadenylated, unspliced precursor RNA was also expressed from the circGFP plasmid (Fig. 3B, lane 3). The identity of each RNA species was confirmed by blotting with additional probes. An intron-specific probe detected only the Csy4-cleaved species from circGFP-CD (lane 5) and the polyadenylated, unspliced precursor from circGFP (lane 6), confirming their identity. A probe against the dsRed ORF detected only the circGFP-CD forward splice product and unknown product (lane 7).
We can quantify the apparent efficiency of circularization by comparing the amount of radiolabel signal detected for the circular product to that from the other major RNA products. It should be noted that the measurements described here represent steady-state levels of each product, and are a balance of RNA biogenesis and degradation. The signal from each blot described in Figure 3B was quantified using a PhosphorImager. Circularization efficiency is expressed as a percentage of the sum of values for the circRNA and its direct precursor. For circGFP-CD, the precursor is the Csy4-cleaved species, and for circGFP it is the polyadenylated unspliced transcript. Also, it is important to note that the band representing the cleaved precursor RNA (denoted by [•••] in the Northern blots) overlaps with that of the slightly higher molecular weight unknown product (•). Because both the unknown product and cleaved precursor RNA are produced when circGFP-CD is coexpressed with Csy4, we estimated the values representing each product using values obtained from the blot probed for dsRed (Probe 3) (see Materials and Methods). When RNA expressed from the circGFP plasmid is analyzed, the circularization efficiency is 49.0% (±9.3%; percentages expressed as mean ± SE, n = 3), whereas when circGFP-CD is coexpressed with Csy4, circularization efficiency is 62.8% (±10.1). Note that if the circGFP precursor RNA or Csy4-cleaved RNA is transported from the nucleus, then splicing will be prevented. Similarly, if Csy4 cleavage does not occur prior to forward splicing, then the linear polyadenylated transcript will be formed. We can estimate the efficiency of Csy4 cleavage by comparing the amount of forward-spliced species (not cleaved) to cleaved products (cleaved precursor + circRNA). When Csy4 is present, forward-spliced products represent 20.7% of the signal, while the cleaved precursor and circRNA combined represent the remaining radiolabel signal. Thus we conclude that the large majority of the transcripts (∼80%) are cleaved prior to forward splicing. Since the cleaved product is primarily cytoplasmic (see Fig. 3D), failure to back-splice the cleaved product completely is likely due to export from the nucleus. This indicates that Csy4 is able to access and cleave the circGFP-CD RNA rapidly, in most cases, before splicing can remove the target hairpin.
We further confirmed circRNA production by RNase R digestion and subsequent visualization of products by Northern blotting with the GFP probe. RNase R is a 3′- to 5′-exonuclease to which circRNAs are inherently resistant due to their lack of a 3′ end (Suzuki et al. 2006). When circGFP-CD RNA expressed in the absence of Csy4 was treated with RNaseR, no resistant products were detected (Fig. 3C, lanes 1,2). This is consistent with predominant forward splicing to produce linear species in the absence of Csy4. In contrast, when circGFP-CD is coexpressed with Csy4, an RNase R resistant product was observed, representing circular RNA (Fig. 3C, lanes 3,4; 45.1% of the undigested). These results are consistent with Csy4-activated circRNA production. In addition, the cleaved, hairpin-containing linear RNA demonstrated some resistance to RNaseR (Fig. 3C, lanes 3,4; 8.3% of the undigested). This may be due to the stem–loop having a very short 3′ overhang, interfering with efficient RNaseR loading after Csy4 cleavage. Total RNA derived from cells transfected with circGFP plasmid was included as a control for circRNA resistance to RNaseR. The linear precursor RNA expressed from the circGFP plasmid was susceptible to RNaseR, while the circRNA was resistant (Fig. 3C, lanes 5,6; 22.9% of the undigested). Taken together, these data confirm that the predominant splicing product of circGFP-CD switches from linear to circular when coexpressed with Csy4.
To determine how or if the circRNA produced by our system is actively translated, we measured the association of circRNA with polyribosomes by sucrose gradient fractionation in cells expressing circGFP-CD in the presence of Csy4. The presence of specific RNAs in each fraction was analyzed by Northern blotting, probing separately for GFP and histone H2A mRNA, an mRNA with a 125 aa ORF. All of the histone mRNA was present on small polyribosomes (2–4 ribosomes), in agreement with previous reports (Fig. 3D, fractions 5–7; Slevin et al. 2014). The linear dsRed RNA was present on larger polyribosomes (3–7 ribosomes). The two major GFP-containing RNA species observed in earlier blots (Fig. 3B) were found in both the total and cytoplasmic RNA fractions. Note that the cleaved precursor and circRNA were predominantly cytoplasmic (Fig. 3D, lane C). A substantial amount of the circRNA and a smaller fraction of the cleaved RNA were found in the portion of the gradient that did not contain ribosomes. CircRNA was also detected in the monosome (fraction 3) and disome (fraction 5) regions, with lesser amounts in fractions 6 and 7 (3–4 bound ribosomes; Fig. 3D). The Csy4-cleaved RNA was also present on polyribosomes of similar size. Note that this cleaved RNA contains a predicted 180 nt ORF, which likely accounts for its being bound by ribosomes, since Csy4 bound at the 3′ end of an mRNA has been shown to support translation (Borchardt et al. 2015). Therefore, these data demonstrate for the first time that a circRNA is actively translated on polyribosomes, although it is inefficiently translated compared to the linear dsRed RNA.
A potential question is whether there might be transcripts in which the GFP ORF has been restored that are not circular. This might occur due to production of concatameric transcripts from run-on transcription that could then be forward-spliced or generated by trans-splicing of two separate RNA transcripts. However, the observation that GFP expression is dependent on active Csy4 cleavage strongly argues against concatamerization (which would have been observed in the absence of Csy4 and resolved by Csy4 cleavage). Another possibility is that in the presence of Csy4, the cleaved transcript bound to Csy4 could be trans-spliced either to a second linear transcript or with a nascent pre-mRNA. In all these cases, the predicted product would contain intron sequences. Specifically, a transpliced RNA of two cleaved species would be 2.5 kb long, compared to the 1.8 kb cleaved product. However, when we probed the RNA from circGFP-CD in the presence of Csy4 with the intron probe (Fig. 3B, lanes 4–6), we did not observe any transcripts larger than 1.8 kb. Thus, we are confident that translation occurs from the circRNA transcript.
To observe the kinetics of circRNA formation from the circGFP-CD plasmid, we measured RNA levels for linear forward-spliced and circular RNAs over time by qRT-PCR. CircRNA and forward splicing products were differentiated by primers designed against circular and linear splicing products, as depicted in Figure 4. Consistent with earlier studies, we observed circGFP-expressed circular RNA levels rising over the course of 72 h (Fig. 4A; Wang and Wang 2015). Accumulation of circRNA from cells cotransfected with circGFP-CD and Csy4 also increased over time in a manner similar to circGFP (Fig. 4A). This observation suggests that the efficiency of circRNA formation from circGFP-CD is not negatively affected by the presence of Csy4, consistent with the Northern blotting data described earlier (Fig. 3B). Importantly, in the absence of Csy4, circRNA levels derived from the circGFP-CD cassette remained 25- to 40-fold lower relative to those generated in the presence of Csy4 throughout the duration of the timecourse (Fig. 4A). We also performed qRT-PCR on the same samples with primers detecting the forward splicing product (Fig. 4B). This product increased initially, reaching a peak at 48 h before declining. Higher levels were detected when circGFP-CD was expressed in the absence of Csy4 (Fig. 4B), compared to cotransfection of circGFP-CD with Csy4, which resulted in consistently lowered expression levels of the forward splice product (Fig. 4B), in agreement with fluorescence imaging (Fig. 2). These results confirm that a fraction of the RNA expressed from circGFP-CD is forward spliced before Csy4 cleaves away the forward splice acceptor.
In conclusion, we have successfully developed a CRISPR-activated system that results in the preferential circularization of a specific RNA target in mammalian cells. Csy4 acts as a reliable regulatory part for controlling the circGFP-CD switch, inducing circRNA biogenesis and protein expression. Thus, circGFP-CD is an adaptable device for the expression of a variety of proteins or functional noncoding RNA sequences for use in genetic circuits. A wide range of RNA-based tools has been engineered for manipulating gene expression (Liang et al. 2011). For example, riboswitches and ribozymes are popular ligand responsive tools for controlling gene expression at the RNA level (Groher and Suess 2014). Other tools for controlling RNA splicing include engineered splicing factors (ESFs) (Wang et al. 2009). ESFs may serve as alternative options for regulating back-splicing efficiency. Here, we expand the RNA toolkit by developing a Csy4-dependent circRNA platform that provides a unique approach to regulate splicing, RNA stability, and protein translation. The functional capability of this Csy4-inducible circRNA platform could potentially be expanded by replacing the fluorescent reporter ORFs with other components, including miRNA or protein sponge sequences as well as ORFs of proteins of interest.
While the current system enables toggling from linear to circular RNA using Csy4, it might be possible to further optimize this system with the addition of cis-acting sequences and target sequences for trans-acting factors that have been shown to favor circRNA formation (Jeck et al. 2013; Ashwal-Fluss et al. 2014; Liang and Wilusz 2014; Zhang et al. 2014; Conn et al. 2015; Ivanov et al. 2015; Kramer et al. 2015). As a circRNA expression tool, the addition of a variety of intron sequences with inverted repeats might improve the back-splicing efficiency of the reporter circRNA. In addition to temporal regulation, it might be possible to regulate circRNA expression in a cell/tissue-specific manner by changing promoter elements. Choice of promoter element could also influence the efficiency of circRNA formation. A recent study indicated that transcription elongation rate positively correlates with RNA circularization efficiency (Zhang et al. 2016). Therefore, it may be worthwhile to investigate any potential interplay between Csy4 cleavage and transcription elongation rate in relation to circRNA biogenesis in the future, as Csy4 must cleave the RNA prior to the cotranscriptional forward splicing event. It may also be worthwhile to compare Csy4 activation of circRNA biogenesis to circRNA expression from an inducible promoter. We would expect that these two mechanisms may differ in their kinetics of circRNA expression as Csy4-mediated activation occurs post-transcriptionally, while inducible promoters are regulated at the transcriptional level. Importantly, we show here that the efficiency of processing for circRNA activation from circGFP-CD is at least as high, if not higher than from circGFP (which is not processed by Csy4). Further, the circGFP-CD system may provide certain advantages for studying circRNA expression. In particular, circGFP-CD more accurately recapitulates the endogenous context for circRNA production by providing a competitive forward splicing alternative. Additionally, the circGFP-CD cassette can be customized for expression of different proteins, as well as used as a switch for the expression of different isoforms. Ultimately, Csy4-activated circRNA formation could serve as a versatile platform for a range of potential applications in synthetic biology.
MATERIALS AND METHODS
Plasmids
The Csy4 gene was amplified from the Pseudomonas aeruginosa UCBP-PA14 genome using primers 5′-ATCGTCTAGAATGGACCACTACCTCGACATTCGCTTGC-3′ and 5′-CGATGCGGCCGCTCAGAACCAGGGAACGAAACCTCCTTTGC-3′. The PCR product was cloned under the control of the CBA promoter in a plasmid backbone. Csy4-H29A was amplified from pHMGWA-Pa14Csy4H29A (Addgene plasmid #41092), which was a gift from Dr. Jennifer Doudna (Haurwitz et al. 2010). Csy4-H29A was cloned under the control of the CBA promoter in a plasmid backbone. The circGFP plasmid was a gift from Dr. Zefeng Wang (Wang and Wang 2015). To construct circGFP-CD, a gBlock (IDT DNA Technologies) was synthesized consisting of the Csy4 targeted hairpin followed by the canonical intron 12 of IGF2BP1 and the P2A sequence in-frame with a dsRed coding sequence. This gBlock was then cloned downstream from the split-GFP cassette such that the entire transcript is driven by the CMV promoter and terminated with a SV40 poly(A) signal.
Cell culture
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (GIBCO/Life Technologies) supplemented with 10% FBS (Atlanta Biologicals), 1% penicillin/streptomycin, and 2.5 µg/mL amphotericin B (Sigma-Aldrich) and maintained at 37°C and 5% CO2.
Fluorescence microscopy and quantification of fluorescence
5 × 104 HEK293 cells were seeded overnight into 24-well plates and transfected with the indicated plasmids at equimolar amounts (totaling 500 ng). Cells were imaged at 72 h post-transfection using an EVOS FL epifluorescence cell imaging system (AMC/Life Technologies) with the GFP light cube (excitation 470 nm, emission 510 nm), or RFP light cube (excitation 530, emission 590). Cells were imaged using the same exposure settings across each set of plasmid conditions. Three images were analyzed for each of three biological replicates using the FIJI image processing package to measure integrated density. Values for all nine images were averaged. Error bars indicate standard deviation of integrated density calculated from these nine images. Statistical significance was calculated using a two-tailed Student's t-test ([****] P ≤ 0.0001, [*] P ≤ 0.05).
RT-PCR
3 × 105 HEK293 cells were seeded overnight into wells of a six-well plate and transfected with the indicated plasmids at equimolar quantities (totaling 2500 ng). RNA was harvested from these cells 48 h post-transfection using the Total RNA Purification Kit (Norgen Biotek) and DNase treated using the Turbo DNA-free kit (Ambion/Life Technologies). Equal nanogram amounts of DNase treated RNA was converted to cDNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems/Life Technologies). Products of this reverse transcription reaction were utilized as template for PCR amplification using gene-specific primers for GFP (5′-AGTGCTTCAGCCGCTACCC-3′, 5′-GTTGTACTCCAGCTTGTGCC-3′) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (5′-CCACTCCTCCACCTTTGAC-3′, 5′-ACCCTGTTGCTGTAGCC-3′). PCR products were visualized on an agarose gel.
Western blot
HEK293 cells seeded overnight in six-well plates at a density of 3 × 105 cells per well were transfected with a total of 2 µg DNA as indicated. Lysates were recovered 48 h post-transfection using 1× Passive Lysis Buffer (Promega) and diluted 1 to 10. Samples were heated to 100°C before separation on a 10% Bis–Tris gel and transfer to a nitrocellulose membrane. Membranes were blocked overnight in 2% nonfat milk in TBST. After overnight incubation, membranes were blotted with primary antibody against either GFP (1:1000 Santa Cruz, SC9996) or Actin (1:2000, Abcam, Ab3280). Stabilized peroxidase-conjugated goat anti-mouse antibody was used as secondary antibody (1:20,000, Jackson Immunologicals, 31430). Blots were developed using SuperSignal West Femto substrate (Thermo Scientific/Life Technologies).
RNaseR digestion
2.2 × 106 HEK293 cells were seeded overnight into 10 cm plates and transfected with the indicated plasmids at equimolar quantities (totaling 16 μg). RNA was harvested at 72 h post-transfection via TRIzol Reagent according to the manufacturer's protocol. Five micrograms of RNA were treated with five units of RNaseR (Epicentre) at 37°C for 10 min. Enzyme was inactivated at 95°C for 5 min. The resulting RNA was visualized by Northern blotting.
Northern blot
2.2 × 106 HEK293 cells were seeded overnight into 10 cm plates and were transfected using PEI Max with the indicated plasmids for a total of 16 µg. RNA was harvested at 72 h post-transfection via TRIzol Reagent according to the manufacturer's protocol. Five micrograms of RNA were resuspended in denaturing buffer (67% deionized formamide, 6.7% formaldehyde, 1× MOPS running buffer) incubated at 60°C for 10 min, and cooled on ice. Samples were separated on a 1.2% denaturing agarose gel and subsequently transferred to Hybond N+ membrane (GE Healthcare). The radiolabeled probe was generated using the Prime-It II Random Primer Labeling Kit (Agilent Technologies) according to manufacturer's instructions. DNA templates for probe labeling were generated by PCR with the following primers amplifying either GFP (5′-GCATGCTCTTCTCAGGAGCGCACCATCTTCTTCAAGGACGACGG-3′, 5′-GCATGCTCTTCTTACCTGGACGTAGCCTTCGGGCATGGC-3′) dsRed (5′-TCGGCCTCCTCCGAGGAC-3′, 5′-CTACAGGAACAGGTGGTGGCG-3′), or an intronic region of circGFP-CD (5′-GTAAGTCTCGACGGATCCCAAATAAGATGCC-3′, 5′-CCGGTAGCGCTAGCGTTTCTTAGC-3′). The radiolabeled probe was purified using illustra MicroSpin G-50 columns (GE Healthcare) following the manufacturer's protocol. Probe was then hybridized to the membrane in Rapid-Hyb buffer (GE Healthcare). Blots were visualized by exposure to film and radiolabel signal was quantified by exposure to a PhosphorImager screen.
Quantification of Northern blot signal
Blots were exposed to a PhosphorImager screen and scanned using a Typhoon imager. ImageQuant software was used to quantify total counts for each RNA species and values were normalized to blot background. Circularization efficiency (%) was derived from the ratio of circRNA to the sum of circRNA and precursor species using the values obtained from the GFP blot (probe 1) (Fig. 3C). Since the cleaved hairpin-containing precursor species (•••) and the unknown species (•) in the CD(+Csy4) lane of the GFP blot were too close in size to distinguish separate values, we utilized the Northern blot for dsRed (probe 3) to obtain approximate values of each product. To achieve this, we took the ratio of the signal for the unknown product (•) in the CD(+Csy4) lane to the forward splice product from the dsRed blot (probe 3). This ratio was then multiplied by the value of the signal for the forward splicing product in the CD(+Csy4) lane on the blot for GFP (probe 1) to estimate the amount of signal in the GFP blot corresponding to the unknown product. The approximate amount of cleaved precursor (•••) signal in the CD(+Csy4) lane of the GFP blot (probe 1) was then calculated by subtracting the estimated unknown product signal from the raw value detected for the band.
Polyribosome isolation
2.2 × 106 HEK293 cells were seeded overnight into 10 cm plates and were transfected using PEI Max with the indicated plasmids for a total of 16 µg. Twenty-four hours post-transfection, cells were split and seeded at 500,000 cells into 10 cm plates. Cells were grown to 60%–70% confluency, then incubated in media containing cycloheximide (CHX, 100 µg/mL) for 10 min at 37°C, followed by two washes with ice-cold PBS containing CHX. Cells were lysed (20 mM Tris–HCl, pH 7.4, 140 mM KCl, 5 mM MgCl2, 1 mM DTT, 1% Triton X-100) and passed through a 27 1/2-gauge needle five times to disrupt cell membranes. Lysates were spun to remove nuclei, and spun again to remove remaining cellular debris. Clarified lysate was loaded on top of a linear 10%–50% sucrose gradient prepared in polysome gradient buffer (20 mM Tris–HCl, pH 7.4, 140 mM KCl, 5 mM MgCl2), and spun for 2 h at 32,000 rpm in a SW41 swinging bucket rotor with no break. Gradients were fractionated into 750 µL fractions with continuous monitoring of absorbance at 254 nM using a Brandel gradient fractionator system. RNA was extracted from each gradient fraction using TRIzol reagent according to the manufacturer's directions, and visualized by Northern blot as described above.
qRT-PCR
3 × 105 HEK293 cells were seeded overnight into wells of a six-well plate. Before transfection, t = 0 control RNA was harvested from cells using the Total RNA Purification Kit (#17200, Norgen Biotek). Remaining cells in separate wells were transfected with equimolar amounts (totaling 6 µg) of the indicated plasmids using PEI Max. RNA from each condition was harvested at time intervals of 8, 24, 48, 72, and 96 h post-transfection using the Total RNA Purification Kit (Norgen Biotek) and treated with DNase using the Turbo DNA-free kit (Ambion/Life Technologies). Two hundred and fifty nanograms of DNase treated RNA was converted to cDNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems/Life Technologies). Products of this reaction were diluted 1:100 and used as template for qPCR. qPCR was carried out using the Faststart Essential DNA Green Master Mix (Roche) and primers for detecting GFP (5′-AGTGCTTCAGCCGCTACCC-3′, 5′-GTTGTACTCCAGCTTGTGCC-3′), dsRed (5′-GCTACGTCCAGGGATCCGGCG-3′, 5′-CTTGGTCACCTTCAGCTTGGCGGTCTG-3′), or GAPDH (5′-CCACTCCTCCACCTTTGAC-3′, 5′-ACCCTGTTGCTGTAGCC-3′). Data were analyzed using the ΔCt method and GFP and dsRed values are expressed relative to GAPDH values. Error bars represent standard deviation of three biological replicates. Statistical significance was calculated using a two-tailed Student's t-test. ([****] P ≤ 0.0001, [***] P ≤ 0.001, [**] P ≤ 0.01, [*] P ≤ 0.05).
ACKNOWLEDGMENTS
We thank Dr. Zefeng Wang for providing the circGFP plasmid. This study was supported by National Institutes of Health grants awarded to A.A. (R01HL089221; P01HL112761) and W.F.M. (GM29832). E.K.B. and R.M.M. were partially supported by a National Institutes of Health training grant awarded to the Curriculum in Genetics and Molecular Biology (5T32GM007092).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.056838.116.
REFERENCES
- Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. 2014. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56: 55–66. [DOI] [PubMed] [Google Scholar]
- Borchardt EK, Vandoros LA, Huang M, Lackey PE, Marzluff WF, Asokan A. 2015. Controlling mRNA stability and translation with the CRISPR endoribonuclease Csy4. RNA 21: 1921–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady KC, O'Toole GA. 2011. Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J Bacteriol 193: 3433–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell-Badge R. 1993. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 73: 1019–1030. [DOI] [PubMed] [Google Scholar]
- Chen CY, Sarnow P. 1995. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268: 415–417. [DOI] [PubMed] [Google Scholar]
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. 2015. The RNA binding protein quaking regulates formation of circRNAs. Cell 160: 1125–1134. [DOI] [PubMed] [Google Scholar]
- Ebbesen KK, Kjems J, Hansen TB. 2015. Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta 1859: 163–168. [DOI] [PubMed] [Google Scholar]
- Enuka Y, Lauriola M, Feldman ME, Sas-Chen A, Ulitsky I, Yarden Y. 2015. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res 44: 1370–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groher F, Suess B. 2014. Synthetic riboswitches—a tool comes of age. Biochim Biophys Acta 1839: 964–973. [DOI] [PubMed] [Google Scholar]
- Guo JU, Agarwal V, Guo H, Bartel DP. 2014. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 15: 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. 2013. Natural RNA circles function as efficient microRNA sponges. Nature 495: 384–388. [DOI] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. 2010. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329: 1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, et al. 2015. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 10: 170–177. [DOI] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. 2013. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19: 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S, Wilusz JE. 2015. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev 29: 2168–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasda E, Parker R. 2014. Circular RNAs: diversity of form and function. RNA 20: 1829–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Haurwitz RE, Apffel A, Zhou K, Smart B, Wenger CD, Laderman S, Bruhn L, Doudna JA. 2013. RNA-protein analysis using a conditional CRISPR nuclease. Proc Natl Acad Sci 110: 5416–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Wilusz JE. 2014. Short intronic repeat sequences facilitate circular RNA production. Genes Dev 28: 2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JC, Bloom RJ, Smolke CD. 2011. Engineering biological systems with synthetic RNA molecules. Mol Cell 43: 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA. 2015. CRISPR-Cas immunity in prokaryotes. Nature 526: 55–61. [DOI] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. 2013. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333–338. [DOI] [PubMed] [Google Scholar]
- Nissim L, Perli SD, Fridkin A, Perez-Pinera P, Lu TK. 2014. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell 54: 698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. 2012. RNA processing enables predictable programming of gene expression. Nat Biotechnol 30: 1002–1006. [DOI] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. 2012. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7: e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slevin MK, Meaux S, Welch JD, Bigler R, Miliani de Marval P, Su W, Rhoads RE, Prins JF, Marzluff WF. 2014. Deep sequencing shows multiple oligouridylations are required for 3′ to 5′ degradation of histone mRNAs on polyribosomes. Mol Cell 53: 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A. 2006. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 34: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak AL, Vignali DA. 2005. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin Biol Ther 5: 627–638. [DOI] [PubMed] [Google Scholar]
- Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK. 2014. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang Z. 2015. Efficient backsplicing produces translatable circular mRNAs. RNA 21: 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cheong CG, Hall TM, Wang Z. 2009. Engineering splicing factors with designed specificities. Nat Methods 6: 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PL, Bao Y, Yee MC, Barrett SP, Hogan GJ, Olsen MN, Dinneny JR, Brown PO, Salzman J. 2014. Circular RNA is expressed across the eukaryotic tree of life. PLoS One 9: e90859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. 2014. Complementary sequence-mediated exon circularization. Cell 159: 134–147. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, Yang L, Chen LL. 2016. The biogenesis of nascent circular RNAs. Cell Rep 15: 611–624. [DOI] [PubMed] [Google Scholar]