

Abstract
Nonsense-mediated mRNA decay (NMD) plays an important role in eukaryotic gene expression, yet the scope and the defining features of NMD-targeted transcripts remain elusive. To address these issues, we reevaluated the genome-wide expression of annotated transcripts in yeast cells harboring deletions of the UPF1, UPF2, or UPF3 genes. Our new RNA-seq analyses confirm previous results of microarray studies, but also uncover hundreds of new NMD-regulated transcripts that had escaped previous detection, including many intron-containing pre-mRNAs and several noncoding RNAs. The vast majority of NMD-regulated transcripts are normal-looking protein-coding mRNAs. Our bioinformatics analyses reveal that this set of NMD-regulated transcripts generally have lower translational efficiency and higher ratios of out-of-frame translation. NMD-regulated transcripts also have lower average codon optimality scores and higher transition probability to nonoptimal codons. Collectively, our results generate a comprehensive catalog of yeast NMD substrates and yield new insights into the mechanisms by which these transcripts are targeted by NMD.
Keywords: NMD substrates, translational fidelity and efficiency, codon optimality
INTRODUCTION
Nonsense-mediated mRNA decay (NMD) is a eukaryotic surveillance mechanism that targets mRNAs undergoing premature translation termination for rapid degradation (Kervestin and Jacobson 2012; Lykke-Andersen and Bennett 2014; He and Jacobson 2015b). The pathway was initially uncovered in Saccharomyces cerevisiae and Caenorhabditis elegans (Leeds et al. 1991; Peltz et al. 1993; Pulak and Anderson 1993) and later shown to be conserved from yeast to humans (Behm-Ansmant et al. 2007; Schoenberg and Maquat 2012). NMD's function was originally thought to be limited to quality control, i.e., the elimination of mRNAs derived from genes harboring nonsense mutations to prevent the accumulation of potentially deleterious truncated polypeptides (He et al. 1993; Pulak and Anderson 1993). However, NMD also targets a significant fraction of apparently normal and physiologically functional wild-type mRNAs (Schweingruber et al. 2013), indicating that it also serves as a fundamental post-transcriptional regulatory mechanism for eukaryotic gene expression. Consistent with these important roles, NMD function is linked to diverse cellular processes, including cell growth and proliferation (Weischenfeldt et al. 2008; Avery et al. 2011; Lou et al. 2014), development and differentiation (Medghalchi et al. 2001; Metzstein and Krasnow 2006; Gong et al. 2009; Wittkopp et al. 2009), innate immunity (Gloggnitzer et al. 2014), antiviral or stress responses (Sakaki et al. 2012; Balistreri et al. 2014), and neuronal activity or behavior (Giorgi et al. 2007; Colak et al. 2013).
In all organisms examined, the activation of NMD requires a set of conserved core regulatory factors, Upf1, Upf2, and Upf3 (Kervestin and Jacobson 2012; He and Jacobson 2015b). These three proteins interact with each other, the ribosome, and multiple translation and mRNA decay factors (Kervestin and Jacobson 2012). Based on these molecular interactions, several potential functions have been proposed for the Upf factors, including remodeling terminating mRNPs (Franks et al. 2010), releasing and recycling ribosomal subunits (Ghosh et al. 2010), and recruiting mRNA decay factors (Okada-Katsuhata et al. 2012; Nicholson et al. 2014; He and Jacobson 2015a). However, the exact roles for the Upfs, and their modes of action in NMD, remain largely unknown.
Despite the conservation of the core Upf proteins, NMD-targeted mRNAs appear to be degraded by different mechanisms in different eukaryotic cells. In yeast, NMD-targeted mRNAs are degraded predominantly through a deadenylation-independent mechanism involving decapping by the Dcp1/Dcp2 decapping enzyme and 5′–3′ exonucleolytic digestion by Xrn1 (Muhlrad and Parker 1994; He and Jacobson 2001). In human cells, NMD-targeted mRNAs are degraded through multiple mechanisms including endonucleolytic cleavage (Huntzinger et al. 2008; Eberle et al. 2009; Lykke-Andersen et al. 2014), deadenylation-dependent decapping (Unterholzner and Izaurralde 2004; Yamashita et al. 2005; Loh et al. 2013), and exosome-mediated 3′–5′ decay (Lejeune et al. 2003), with endonucleolytic decay appearing to be the predominant initiating mechanism in human cells (Boehm et al. 2014). In the latter decay pathway, Smg6 cleaves its substrate mRNAs in the vicinity of PTCs and the resulting 5′ and 3′ fragments are degraded by the exosome and Xrn1, respectively (Huntzinger et al. 2008; Eberle et al. 2009; Boehm et al. 2014).
Depending on the organism or cell type, ∼5%–20% of the transcripts in a typical transcriptome are substrates of NMD (Lelivelt and Culbertson 1999; He et al. 2003; Mendell et al. 2004; Rehwinkel et al. 2005; Weischenfeldt et al. 2008; Ramani et al. 2009) and these transcripts can be classified into several general categories. One category, exemplifying typical NMD substrates, includes mRNAs with a destabilizing premature termination codon (PTC) in their coding region. These transcripts are generated from endogenous genes harboring nonsense or frameshift mutations (He et al. 2003), pseudogenes (He et al. 2003; McGlincy and Smith 2008), nonproductively rearranged genetic loci (Li and Wilkinson 1998), or from alternative splicing events that lead to intron retention or inclusion of a PTC-containing exon (Lareau et al. 2007; Ni et al. 2007; Jaillon et al. 2008; Lykke-Andersen et al. 2014). A second category contains mRNA-like transcripts with limited or no apparent coding potential, such as long noncoding RNAs (Kurihara et al. 2009; Tani et al. 2013; Lykke-Andersen et al. 2014), small RNAs derived from intragenic regions (Thompson and Parker 2007; Smith et al. 2014), or transcripts of inactivated transposable elements (He et al. 2003). A third category contains a subset of physiologically relevant transcripts that appear to be “normal,” such as mRNAs with upstream open reading frames (uORFs) (He et al. 2003; Gaba et al. 2005; Arribere and Gilbert 2013), or with atypically long 3′ UTRs (Singh et al. 2008; Kebaara and Atkin 2009), or normal-looking wild-type mRNAs with no atypical features (He et al. 2003).
To generate a comprehensive and high resolution catalog of NMD-regulated transcripts, and to delineate the defining features of these transcripts in NMD targeting, we utilized RNA-seq to reevaluate the effects of deleting the UPF1, UPF2, or UPF3 genes on the transcriptome-wide expression of annotated yeast genes. Our new analyses confirm previous results of microarray studies, but also uncover hundreds of new NMD-regulated transcripts that had escaped previous detection, including many intron-containing pre-mRNAs. Our bioinformatics analyses reveal several intrinsic features of NMD-regulated transcripts, yield new insights into the mechanisms by which translation of these transcripts targets them for NMD, and provide strong support for the notion that transcripts can become NMD substrates at any time during their translational life cycle.
RESULTS
Upf1, Upf2, and Upf3 regulate a common set of transcripts in yeast cells
To obtain a high-resolution catalog of yeast NMD substrates, we analyzed expression profiles of wild-type (WT), upf1Δ, upf2Δ, and upf3Δ strains. RNA-seq libraries were prepared from these strains in three biological replicates and the sequence reads from each library were aligned to: (i) a yeast transcriptome comprised of 7473 transcripts, including all annotated protein-coding sequences, functional and noncoding RNAs, and the unspliced isoforms of all intron-containing mRNAs and (ii) a separate transcriptome comprising all 3569 CUT, SUT, and XUT transcripts annotated previously (Wery et al. 2016). We opted to use two separate transcriptomes because combining them resulted in a substantial loss of power to detect differential expression. Further investigation revealed that overlapping annotations were the principal cause: 1655 out of 3569 genomic coordinates of CUT, SUT, and XUT sequences had some extent of overlap, sometimes with multiple annotations (2024 total overlaps). Less than 1% of reads mapping to CUTs, SUTs, and XUTs were unique, compared to >55% for other transcripts (Supplemental Table 1). Based on posterior probabilities calculated by the RSEM software tool (Li and Dewey 2011), including CUTs, SUTs, and XUTs in a combined analysis would have resulted in a substantial increase in read quantification error for all transcripts, compromising dispersion estimates used for subsequent differential expression calculations (Anders and Huber 2010; Robinson et al. 2010). Similarly, because of their repetitive nature, we also excluded autonomous replicating sequences and long terminal repeats of transposable elements from our analysis.
We used RSEM (Li and Dewey 2011) for transcript quantification and the DESeq R package (Anders and Huber 2010) for differential expression analysis. To account for replicate variability, we used a false discovery rate threshold of 0.01 instead of arbitrary fold-change as a criterion for differential expression. All libraries exhibited similar distributions of read counts with few outliers (Fig. 1A) and biological replicates were extremely consistent, with Pearson correlation coefficients ranging from 0.84 to 0.99 (Supplemental Fig. 1). We refer to the set of sequences that contains all annotated transcripts except for CUTs, SUTs, and XUTs as transcriptome 1 (T1) and the set with the sequences for CUTs, SUTs, XUTs as transcriptome 2 (T2).
FIGURE 1.

Upf1, Upf2, and Upf3 regulate the same set of transcripts in yeast. (A) RNA-seq libraries from WT, upf1Δ, upf2Δ, and upf3Δ strains display comparable overall read count distributions for both transcriptome 1 (T1; left panel) and transcriptome 2 (T2; right panel). Violin and box-plots were used to visualize the average sequence reads distribution of the transcriptomes of the indicated strains from three independent experiments. (B,C) Transcripts up- and down-regulated in upf1Δ, upf2Δ, and upf3Δ strains show significant overlap. Transcripts up- or down-regulated in each UPF deletion strain were identified by comparisons to the WT strain. Venn diagrams were used to display the relationships among the sets of transcripts that are up-regulated (B) and down-regulated (C) in T1 or T2 of upf1Δ, upf2Δ, and upf3Δ strains. (D) All three UPF deletion strains display similar genome-wide expression patterns. Scatterplots were used to compare the read count values of differentially expressed transcripts between WT and upf1Δ, upf2Δ, or upf3Δ strains. The vast majority of differentially expressed transcripts in UPF deletion strains showed up-regulation and a small number of transcripts showed down-regulation. The y = x line is shown in red. (Top panel) Pairwise comparisons of the expression levels between WT and each UPF deletion strain for 936 differentially expressed transcripts from transcriptome 1. (Bottom panel) Pairwise comparisons of the expression levels between WT and each UPF deletion strain for 456 differentially expressed transcripts from transcriptome 2. (E) Transcripts commonly regulated by NMD each have virtually identical expression values in upf1Δ, upf2Δ, or upf3Δ strains. As in D, scatterplots were used to compare the read count values of NMD-regulated transcripts between upf1Δ and upf2Δ, upf1Δ and upf3Δ, and upf2Δ or upf3Δ strains. (Top panel) Differentially expressed transcripts from transcriptome 1. (Bottom panel) Differentially expressed transcripts from transcriptome 2.
Deletion of UPF1, UPF2, or UPF3 led to differential expression of a subset of transcripts in yeast cells. In each of the UPF deletion strains, the vast majority of differentially expressed transcripts were up-regulated and only a small number of transcripts were down-regulated (Fig. 1B–D). The number of up- or down-regulated transcripts in individual UPF deletion strains was comparable and exhibited substantial overlap (Fig. 1B,C). These results indicate that the three Upf factors control the expression of a common set of transcripts in yeast cells. In transcriptome 1, we identified 907 transcripts that were up-regulated and 29 that were down-regulated upon UPF deletion; under the same circumstances transcriptome 2 had 332 up-regulated transcripts (including eight CUTs, 114 SUTs, and 210 XUTs; Supplemental Table 2) and 124 down-regulated transcripts (Fig. 1B,C; Supplemental Fig. 2). Pairwise comparisons of read counts for each of the 1392 differentially expressed transcripts in upf1Δ, upf2Δ, and upf3Δ cells manifested almost equivalent expression values in each case (Fig. 1E), indicating that deletion of UPF1, UPF2, or UPF3 has almost identical quantitative effects on NMD-regulated transcripts. Similarly, when pairwise comparisons were conducted for all transcripts the three UPF deletion strains exhibited almost identical expression patterns (Supplemental Fig. 2). These results strengthen the idea that, at least in yeast, Upf1, Upf2, and Upf3 have equivalent effects on the execution of a single NMD pathway (He et al. 1997).
Transcripts regulated by NMD have also been identified in previous analyses (He et al. 2003; Johansson et al. 2007; Sayani et al. 2008; Smith et al. 2014; Malabat et al. 2015) and we sought to compare our new transcript lists to those generated earlier. However, naming conventions for Affymetrix microarray probes are not completely consistent with annotated gene names, and a significant fraction (∼20%) of transcripts represented by microarray probes are not in our new reference transcriptomes. Therefore, we restricted our comparisons to microarray probes that have definitively matched transcripts in our transcriptomes. As shown in Table 1, there is substantial overlap (>60%) with our results and previously published data sets. These results indicate that our RNA-seq analyses yielded comprehensive sets of NMD-regulated transcripts in yeast cells that include the majority of transcripts identified by previous analyses and numerous transcripts that were not detected in previous studies. Transcripts commonly up-regulated in all three UPF deletion strains did not show any significant enrichment for those encoding factors involved in signal transduction or transcriptional regulation and, although a handful of up-regulated transcripts encode transcription factors, the annotated targets for each of these transcription factors were not particularly enriched in our NMD-regulated transcript list. Although we cannot definitively rule out indirect effects that alter mRNA abundance, these observations and the known roles of the three Upf factors as positive regulators of NMD (He and Jacobson 2001) suggest that the 1239 transcripts commonly up-regulated in the two transcriptomes analyzed here likely constitute direct substrates of NMD. Because of the ambiguity of read alignments and lack of information about the biological role of CUT, SUT, and XUT sequences in the literature, most of our subsequent analyses were focused on the 907 up-regulated transcripts present in transcriptome 1.
TABLE 1.
Overlap of NMD-regulated transcripts identified in this study and in several previous studies
Structural and functional classes of NMD-regulated RNAs in transcriptome 1
To gain insight into the mechanism targeting well-annotated NMD substrates, we classified the NMD-regulated transcripts of transcriptome 1 into their respective structural and functional categories. Among the 907 up-regulated transcripts of transcriptome 1, 902 were from annotated protein-coding genes and five were from annotated “noncoding” RNA genes (Supplemental Table 2). Since NMD requires ongoing translation (Zhang et al. 1997; Hu et al. 2010), our observation that some “noncoding” RNAs of transcriptome 1 are substrates of NMD suggests that these transcripts are actually translated and may encode bona fide polypeptides. Among the 902 NMD-regulated transcripts coming from protein coding genes, 88% appear to be “normal” mRNAs and to lack any structural features indicative of substrates of NMD (see below). The remaining 12% can be classified into five known structural classes of NMD substrates (He et al. 2003), namely: (i) mRNAs encoded by genes harboring nonsense mutations in their coding regions (e.g., CAN1, LEU2), (ii) mRNAs utilizing frameshifting in their translation (e.g., YGR109W-B, YIL082W-A, and YIL009C-A), (iii) transcripts originated from pseudogenes (e.g., YAR061W, YFL056C, YOL153C), (iv) mRNAs that contain annotated and putative upstream open reading frames (Ingolia et al. 2009) (uORFs; e.g., CPA1), and (v) pre-mRNAs that retain their introns and enter the cytoplasm as a consequence of inefficient or regulated splicing (e.g., RPL22B, RPL24B, HRB1). The latter two classes (uORF-containing mRNAs and intron-containing pre-mRNAs) were enriched significantly in NMD substrates: 89 out of 356 putative uORF-containing yeast transcripts (Ingolia et al. 2009) and 57 out of 351 potential intron-containing pre-mRNAs were present at a higher ratio than would be expected by chance (χ2 P = 4.8 × 10−9 and 0.0006, respectively). Our observation that NMD targets a large number of intron-containing pre-mRNAs indicates that there is a widespread entry of intron-containing transcripts into the yeast cytoplasm and that, in yeast, NMD plays a general role in the degradation of a subset of intron-containing pre-mRNAs.
Validation of newly identified NMD substrates
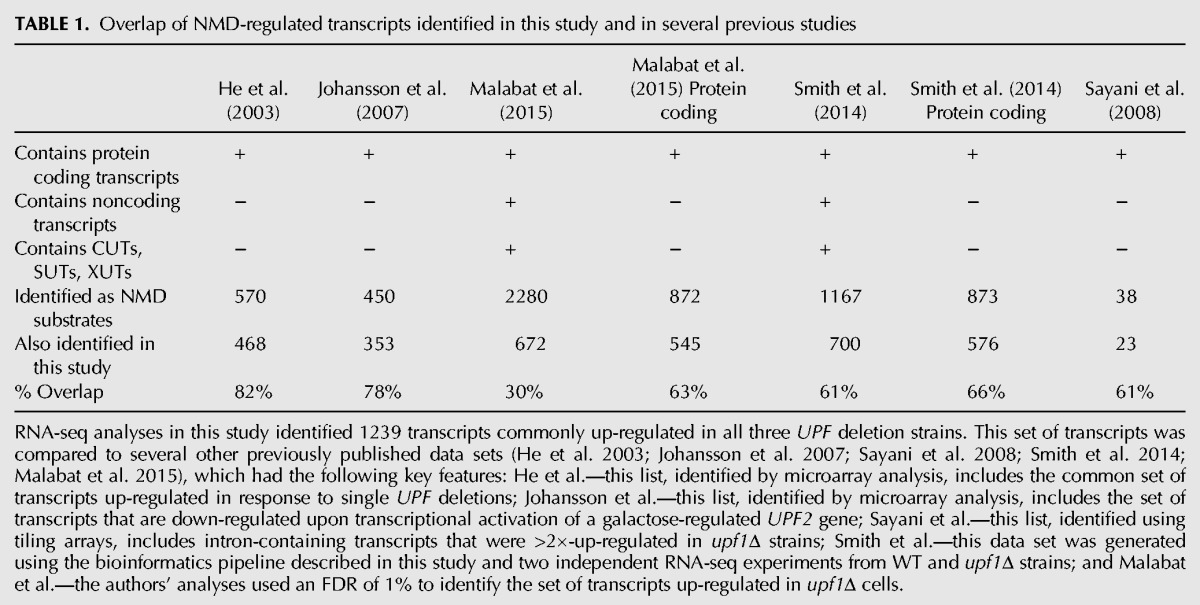
To validate our RNA-seq and bioinformatics results, we assessed the levels of expression of seven newly identified NMD-regulated transcripts in wild-type, upf1Δ, upf2Δ, and upf3Δ strains by Northern blotting. To ascertain NMD substrate specificity, we also analyzed yeast strains harboring single deletions of genes encoding the major cytoplasmic 5′–3′ exonuclease (Xrn1), the Dcp1/Dcp2 decapping enzyme, and several decapping activators (Edc3, Pat1, Lsm1, Dhh1, and Scd6). Among the seven selected NMD-regulated transcripts analyzed, four (HRB1, RPL22B, NHP6B, MTR2) are intron-containing pre-mRNAs, two are annotated as “noncoding” RNAs, and one utilizes frameshifting during translation (Ty4 transposon). Of the intron-containing pre-mRNAs, two (HRB1, RPL22B) contain an intron in their coding regions and two (NHP6B, MTR2) contain an intron in their 5′ UTRs. As a negative control, we analyzed the intron-containing HAC1 pre-mRNA in these strains. All seven NMD-regulated transcripts showed the expected expression patterns in these yeast strains (Fig. 2A–C). Compared to their expression in wild-type cells, each of these transcripts showed increased levels in upf1Δ, upf2Δ, and upf3Δ strains, and also in xrn1Δ, dcp1Δ, and dcp2Δ strains. However, the levels of expression of each of these transcripts were unchanged in edc3Δ, pat1Δ, lsm1Δ, dhh1Δ, and scd6Δ strains. The levels of the HAC1 pre-mRNA were essentially unchanged in all the deletion strains. Collectively, these observations confirm that all seven transcripts are NMD-regulated and degraded through decapping and 5′–3′ exonucleolytic decay. The latter results mirror our analyses of RNA-seq data (see below).
FIGURE 2.
Validation of several different classes of NMD substrates by Northern blotting. Northern blotting analyses of (A) intron-containing transcripts (HRB1, RPL22B, NHP6B, and MTR2), (B) transcripts using frameshifting during translation (Ty-4 transposons), (C) “noncoding” RNAs (ICR1 and IRT1), and (D) negative control transcripts (HAC1 pre-mRNA). Total RNA was isolated from the indicated strains, and the steady-state levels of individual transcripts in these strains were analyzed by Northern blotting. In each case, a random-primed probe was hybridized to the blot and SCR1 served as the loading control.
NMD substrates are principally degraded by decapping and 5′–3′ exonucleolytic decay
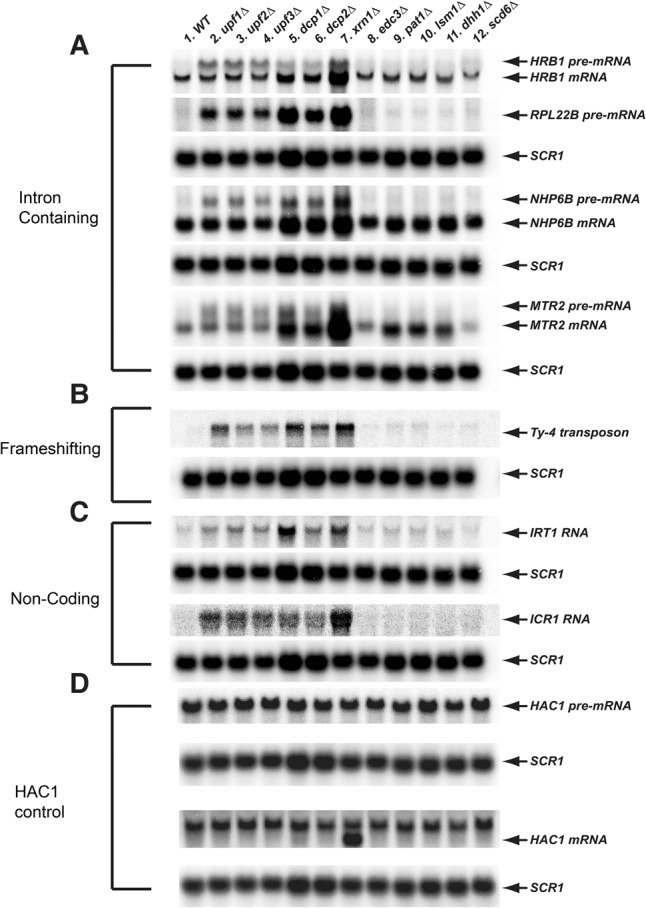
Several NMD substrates have previously been shown to be degraded by decapping and 5′–3′ exonucleolytic degradation (Muhlrad and Parker 1994; He and Jacobson 2001), including those analyzed in Figure 2. To evaluate the prevalence of this mechanism in NMD at a transcriptome-wide level, we analyzed the expression patterns of the 907 NMD regulated transcripts of transcriptome 1 in dcp1Δ, dcp2Δ, and xrn1Δ strains. We prepared RNA-seq libraries from dcp1Δ, dcp2Δ, and xrn1Δ strains and subjected them to the same analysis pipeline as described above for libraries prepared from upf strains. These libraries showed similar consistencies and read count distributions as single WT and UPF deletion libraries (Fig. 3A; Supplemental Fig. 1). Consistent with current concepts of the roles of Dcp1, Dcp2, and Xrn1 in NMD and general 5′–3′ decay (Parker 2012), deletion of DCP1, DCP2, and XRN1 each caused up-regulation of >1000 transcripts (Fig. 3B). As expected, the up-regulated transcripts in dcp1Δ, dcp2Δ, and xrn1Δ strains had significant overlap, with overlapping fractions ranging from 52% to 88% (Fig. 3B). The 907 NMD-regulated transcripts had significant overlap with transcripts that were up-regulated in both dcp1Δ and dcp2Δ cells or xrn1Δ cells (Fig. 3C). Overall, ∼70% of NMD-regulated transcripts had increased levels of expression in dcp1Δ, dcp2Δ, and xrn1Δ strains. Consistent with our earlier microarray analyses (He et al. 2003), these results indicate that yeast NMD substrates are largely but probably not exclusively degraded by decapping and 5′–3′ exonucleolytic decay.
FIGURE 3.
NMD substrates are principally degraded by decapping and 5′–3′ exonucleolytic decay. (A) RNA-seq libraries from WT, dcp1Δ, dcp2Δ, and xrn1Δ strains display normal and comparable overall read count distributions. As in Figure 1A, violin and box-plots were used to visualize the average sequence reads distribution of the transcriptomes of the indicated strains from three independent experiments. (B) Transcripts up- or down-regulated in dcp1Δ, dcp2Δ, and xrn1Δ strains show significant overlap. Transcripts up- or down-regulated in dcp1Δ, dcp2Δ, and xrn1Δ strains were identified by comparisons to the WT strain. (C) Transcripts commonly up- or down-regulated from transcriptome 1 in all three UPF deletion strains show significant overlap with transcripts up-regulated in both dcp1Δ and dcp2Δ strains or an xrn1Δ strain. Venn diagrams were used to display the relationships among the up- or down-regulated transcripts from the indicated strains.
Intron-containing pre-mRNAs targeted by NMD are engaged in translation
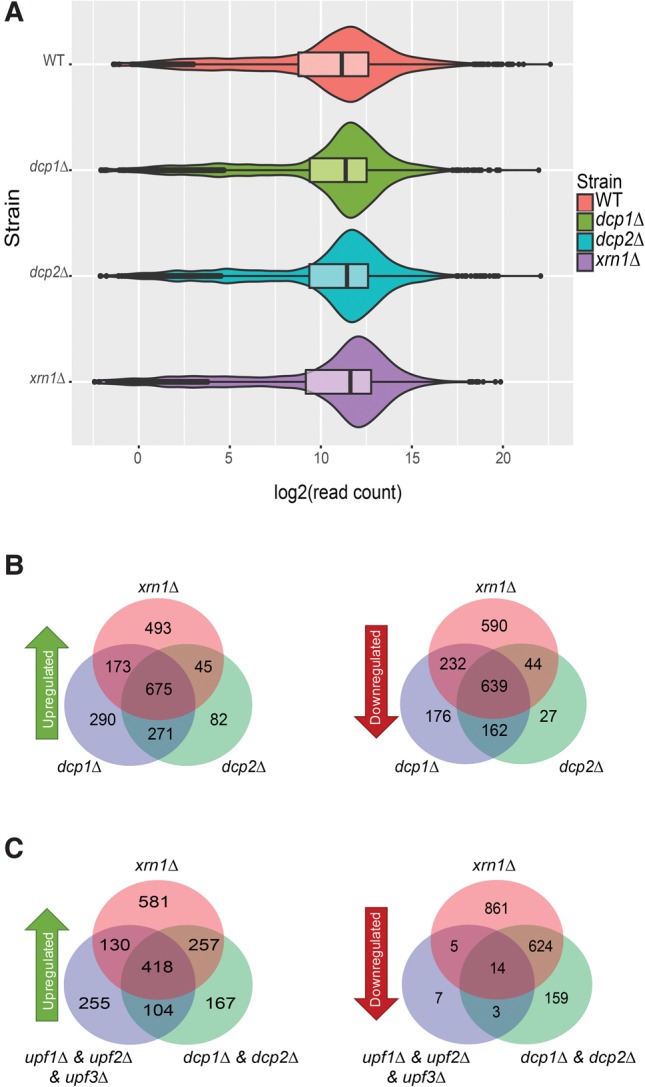
To further elucidate the role of NMD in the degradation of intron-containing pre-mRNAs, we analyzed ribosome occupancy within the intronic regions of pre-mRNAs targeted by NMD (n = 57) and those that are not (n = 244). Using published ribosome profiling data (Young et al. 2015), we measured ribosome densities (coverageprofiling/coverageRNA-Seq) within the intronic regions for these two groups of intron-containing transcripts. We found that introns from the pre-mRNAs targeted by NMD do in fact show a subtle, but statistically significant, higher ribosome occupancy than introns from the pre-mRNAs not targeted by NMD (two-sample KS test P = 0.038; Fig. 4A,B). These results indicate that intron-containing pre-mRNAs targeted by NMD are generally engaged in translation. In support of this conclusion, several of these NMD-targeted pre-mRNAs were previously shown to be associated with polyribosomes (He et al. 1993).
FIGURE 4.
NMD targeted intron-containing pre-mRNAs are engaged in translation. (A) Cumulative density plot of ribosome density of the intronic regions for pre-mRNAs targeted (blue, n = 57) or not targeted (red, n = 244) by NMD. This plot illustrates the fraction (on the y-axis) of transcripts having the indicated ribosome densities (on the x-axis). (B) Distribution of mean ribosome densities over normalized intronic regions for the same two sets of intron-containing transcripts as in A. Plots in A and B were derived from the ribosome profiling data of WT cells by Young et al. (2015). Ribosome densities were calculated as profilingcoverage/RNA-seqcoverage for each intron. Introns of NMD-targeted pre-mRNAs show higher ribosome densities than introns of the pre-mRNAs that are not targeted by NMD (two-sample KS test P = 0.038).
NMD substrates similar to normal mRNAs are poorly translated regardless of the NMD status of the cell
The compilation of a comprehensive list of NMD substrates raises the general question of what dictates NMD specificity for these transcripts. While the NMD targeting of PTC-, uORF-, and intron-containing transcripts, and “noncoding” RNAs, can all be attributed to premature translation termination, the vast majority (almost 90%) of NMD substrates in transcriptome 1 are protein-coding transcripts that look like normal, wild-type mRNAs. To identify potential features associated with this “normal-looking” group of NMD substrates, we evaluated several parameters, including 5′ UTR, ORF, and 3′ UTR lengths, and ribosome densities, for this group of NMD substrates and compared these parameters to those generated from protein-coding mRNAs not subject to NMD regulation. Using previously published annotations (Nagalakshmi et al. 2008; Xu et al. 2009; Pelechano et al. 2013), we observed conflicting results for the potential role of UTR lengths in substrate selection. Using the annotations of Nagalakshmi and colleagues, we found no discernible difference between 5′- and 3′ UTR lengths of normal-looking mRNAs targeted versus not targeted by NMD, while the annotations of Pelechano and colleagues suggested that both UTRs are longer for NMD substrates and the annotations of Xu and colleagues suggested that 5′ UTRs are shorter for NMD substrates (data not shown). These conflicting annotations precluded any conclusions about the role of UTR lengths in the determination of NMD substrate status. However, by comparing the published (Young et al. 2015) ribosome occupancies of these subsets of transcripts we observed a striking difference in normalized ribosome occupancy in wild-type cells. The normal-looking NMD substrates had significantly lower ribosomal density throughout their open reading frames than the non-NMD substrates (two-sample KS test P < 2.2 × 10−16) (Fig. 5A,B, blue and red lines, respectively). Based on this observation, we also analyzed the normalized ribosome occupancy of all putative uORF-containing transcripts. We separated the transcripts into two different groups: those regulated by NMD and those not regulated by NMD. Much like the normal-looking NMD substrates, the NMD-regulated uORF-containing transcripts also exhibited lower ribosome densities than those not subject to NMD regulation (Fig. 5C; two-sample KS test P < 2.2 × 10−16 and P = 3.57 × 10−10 for transcripts without and with uORFs, respectively). Together, these results indicated that NMD substrates are probably translated less efficiently than non-NMD substrates.
FIGURE 5.
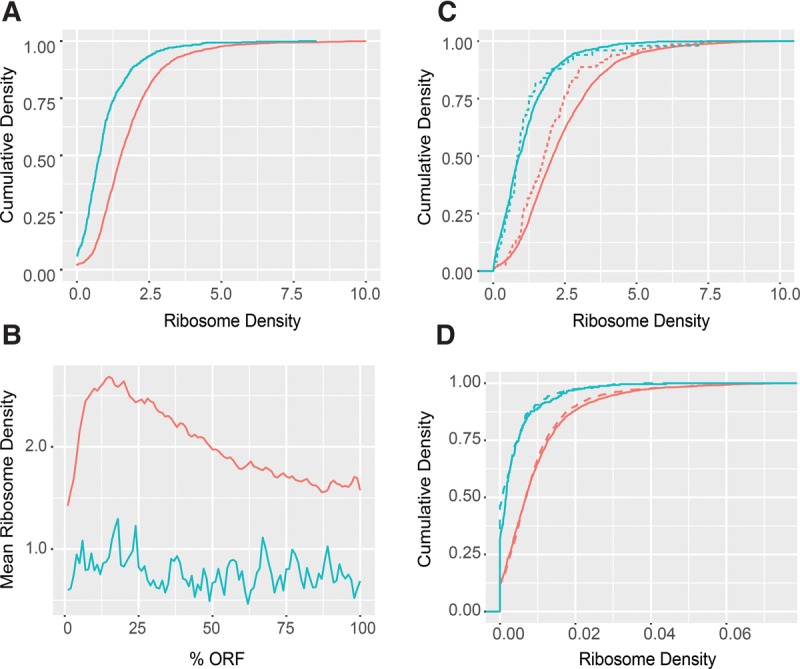
NMD substrates are less efficiently translated than nonsubstrates independent of the NMD machinery. (A) Cumulative density plots of ribosome densities derived from ribosome profiling data of WT cells for normal-looking NMD substrates (blue n = 746) and non-NMD substrates (red = 4633). (B) Mean ribosome densities over normalized ORFs derived from the same data and for the same two sets of transcripts shown in A. (C) Cumulative density plots of ribosome densities derived from the same data in A for uORF-containing transcripts targeted (dashed, blue n = 42) or not targeted (dashed, red n = 199) by NMD and for uORF-lacking transcripts targeted (solid, blue n = 704) or not targeted (solid, red n = 4434) by NMD. (D) Cumulative density plots of ribosome densities derived from other ribosome profiling data sets of WT (solid) and upf1Δ (dashed) cells for normal-looking NMD substrates (blue) and non-NMD substrates (red) shown in A. Plots in A, B, and C were derived from the ribosome profiling data of Young et al. (2015), and plots in D were derived from the ribosome profiling data of Smith et al. (2014). Ribosome densities were calculated as profilingcoverage/RNA-seqcoverage for each transcript. Two-sample KS test P-values are described in the Results section.
In addition to triggering rapid transcript degradation, recognition of an mRNA as an NMD substrate has been suggested to lead to concomitant Upf1-dependent translational repression (Muhlrad and Parker 1999). To assess whether the observed lower ribosome density of normal-looking NMD substrates in wild-type cells reflected this phenomenon, we utilized published ribosome footprinting libraries (Smith et al. 2014) to compare the normalized ribosome densities of NMD substrates in wild-type and upf1Δ cells. These analyses demonstrated that the ribosome density profiles for both normal-looking NMD substrates and non-NMD substrates showed similar differences in ribosome density regardless of the strain (Fig. 5D; two-sample KS test P < 2.2 × 10−16 for both WT and upf1Δ, between NMD substrates and nonsubstrates), i.e., the NMD substrates were also translated less efficiently in upf1Δ cells. Collectively, our bioinformatics analyses indicate that low ribosome density is an intrinsic property of NMD-targeted transcripts.
Normal-looking NMD substrates have a higher rate of out-of-frame translation
The comparatively reduced ribosome densities of NMD substrates observed in Figure 5 suggested that these mRNAs may share a common impairment. We thus tested whether normal-looking NMD substrates and non-NMD substrates may have different amounts of out-of-frame translation in their coding regions. For this analysis, we analyzed published (Young et al. 2015) ribosome profiling data and only used read lengths that displayed a strong preference (>80% of reads) for one frame, mapping these sequence reads to NMD and non-NMD-regulated transcripts. We calculated the ratio of out-of-frame reads to total mapped reads for each of these transcripts and compared the out-of-frame ratio distributions between the normal-looking NMD and non-NMD populations. This analysis indicated that the NMD-regulated transcripts showed a significantly higher ratio of out-of-frame reads (Fig. 6A; two-sample KS test P < 2.2 × 10−16). These results indicate that in addition to lower ribosome density, NMD substrates also exhibit higher out-of-frame read ratios and thus a higher rate of out-of-frame translation. One potential explanation for increased out-of-frame translation could be that these genes have internal transcription start sites (iTSSs) and the subsequent isoforms are the main substrates for NMD. To test this hypothesis, we used iTSS-containing gene lists published by two independent groups (Pelechano et al. 2013; Malabat et al. 2015). We compared the overlaps between transcripts with iTSSs and our list of NMD substrates. Interestingly, we found little overlap between these three groups of transcripts. In addition, when we compared the ribosome densities and out-of-frame read ratios between transcripts that have been suggested to have iTSSs by Malabat and colleagues and those that do not we observed similar differences between NMD substrates and nonsubstrates. That is, regardless of iTSS status, transcripts that we have identified as NMD substrates show higher rates of out-of-frame translation and lower ribosome densities (Supplemental Fig. 3).
FIGURE 6.

NMD substrates have lower translation fidelity and lower codon optimality. (A) Cumulative density plots of in-frame read ratios over total reads derived from ribosome profiling data of WT cells for intron-lacking NMD substrates (n = 746, blue) and non-NMD substrates (n = 4633, red). (B) Cumulative density plots of mean codon optimality scores for two sets of transcripts shown in A. (C) Mean transition probabilities of a two-state discrete time Markov chain between optimal (O) and nonoptimal (N) codons for intron-lacking NMD substrates (blue) and non-NMD substrates (red). (D) Distributions of Markov chain codon transition probabilities for intron-lacking NMD substrates (blue) and non-NMD substrates (red). Plots in A were derived from the ribosome profiling data of Young et al. (2015), and plots in B, C, and D were based on codon optimality assignments and scores published by Pechmann and Frydman (2013). Two-sample KS test P-values are described in the Results section.
Normal looking NMD substrates have lower average codon optimality and a biased distribution pattern of nonoptimal codons
To further understand the basis of lower ribosome density and increased out-of-frame translation of NMD substrates, we explored potential differences in codon usage for normal looking NMD and non-NMD-regulated transcripts. We used published codon optimality data (Pechmann and Frydman 2013), to calculate average codon optimality scores for both NMD and non-NMD-regulated transcripts, and compared the score distributions of these two populations. We found that the NMD-regulated transcripts had a subtle, but statistically significant lower average codon optimality score (Fig. 6B; two-sample KS test P = 3.9 × 10−7). Based on this finding, we then recoded the codon sequences of each NMD- and non-NMD-regulated transcript as a binary series of optimal (O) or nonoptimal (N) codons, treated each recoded transcript as a discrete time Markov chain, and calculated the transition probabilities from one state to another for each transcript (i.e., O to O, O to N, N to N, and N to O). We then compared the distributions of transition probabilities of the NMD and non-NMD-regulated transcripts. We found that NMD-regulated transcripts again showed a subtle but statistically significant preference toward nonoptimal codons (i.e., having higher N to N and O to N, but lower N to O and O to O transition probabilities; two sample KS test P = 2.4 × 10−7, 1.2 × 10−7, 5.4 × 10−14, 5.3 × 10−14, respectively; Fig. 6C,D). Our analyses thus indicated that, as individual metrics, average codon optimality and N to N and O to O transition probabilities all seem to contribute to NMD susceptibility. Because average codon optimality is highly correlated with transition probabilities we were unable to conclude whether NMD substrates still had a higher tendency to exhibit longer stretches of nonoptimal codons when controlled for overall codon optimality.
DISCUSSION
A comprehensive catalog of annotated yeast NMD substrates
Using RNA-seq analyses, we have redefined the set of transcripts regulated by NMD in Saccharomyces cerevisiae. Our new comprehensive list of yeast NMD substrates originates from the well-annotated genes of transcriptome 1 and includes the vast majority of NMD-regulated transcripts identified by previous analyses (He et al. 2003; Johansson et al. 2007; Sayani et al. 2008; Smith et al. 2014; Malabat et al. 2015), as well as hundreds of new transcripts that escaped prior detection. While many CUTs, SUTs, and XUTs of transcriptome 2 manifest changes in abundance in response to inactivation of each Upf factor the relevance of these transcripts to the conventional understanding of NMD remains obscure, particularly in light of the lack of precise mapping information for these transcripts. Accordingly, our attention has largely been drawn to the components of transcriptome 1. Consistent with the positive roles of Upf1, Upf2, and Upf3 in NMD activation (Kervestin and Jacobson 2012; He and Jacobson 2015b), almost all NMD substrates in the transcriptome 1 list are up-regulated by NMD inactivation, with only a handful of transcripts showing down-regulation under the same conditions. Further, the strict requirement for translation in NMD activation (Zhang et al. 1997; Hu et al. 2010) was reflected in the observation that 99% of the NMD substrates in transcriptome 1 were annotated as protein coding transcripts.
Each of the up-regulated transcripts shares a nearly identical quantitative response to deletion of the UPF1, UPF2, or UPF3 genes and exhibits comparable expression levels in the three UPF deletion strains (Fig. 1E; Supplemental Fig. 2). In addition, most transcripts in the up-regulated group also exhibit increased accumulation upon inactivation of the Dcp1/Dcp2 decapping enzyme and the 5′–3′ Xrn1 exonuclease (Fig. 3C), two critical components that function downstream from the yeast NMD pathway (Parker 2012). The set of up-regulated transcripts also includes several known structural classes of NMD substrates including mRNAs encoded by genes harboring nonsense mutations in their coding regions, mRNAs utilizing frameshifting in their translation, pseudogene transcripts, mRNAs that contain uORFs, and pre-mRNAs that retain their introns and enter the cytoplasm as a consequence of inefficient or regulated splicing (Fig. 7A). Collectively, these observations lead us to conclude that the bulk of the up-regulated transcripts identified here are likely to be bona fide substrates of the NMD pathway in yeast cells.
FIGURE 7.
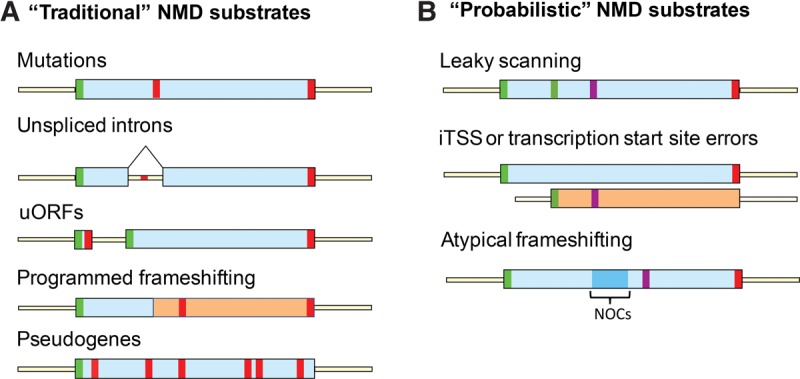
Different classes of NMD substrates. (A) “Traditional” NMD substrates. Translation of these NMD substrates commences at initiation codons located at ORF (or uORF) 5′ ends, proceeds 3′, and leads to an in-frame encounter with a coding region premature termination codon. Transcripts in this class include mRNAs derived from nonsense alleles, pre-mRNAs that enter the cytoplasm with unspliced introns, uORF-containing mRNAs, mRNAs in which programmed frameshifting allows a fraction of ribosomes to avoid premature termination, and mRNAs transcribed from pseudogenes. (B) “Probabilistic” NMD substrates. These NMD substrates lack in-frame premature termination codons in their coding regions, but contain mRNA features that promote either downstream out-of-frame translational initiation or frameshifting and thus trigger premature termination in a new reading frame. Transcripts in this category include mRNAs with poor sequence context around the normal initiation codon, mRNAs whose transcription start site is internal to the principal ORF, and mRNAs with lower overall codon optimality or a long stretch of nonoptimal codons (NOCs). In each of these cases, a subset of ribosomes translates the mRNA in a frame different from that of the annotated ORF. (Green) Initiation codon, (red) stop codon, (yellow) UTR, (purple) stop codon encountered in the +1 or +2 reading frame; (blue) cluster of nonoptimal codons.
A significant fraction of yeast intron-containing mRNAs are targeted by cytoplasmic NMD
In addition to generating a comprehensive catalog of yeast NMD substrates, our RNA-seq analyses also revealed that a significant fraction (∼16%) of yeast intron-containing genes produce intron-containing pre-mRNA isoforms that are engaged with translating ribosomes and subject to NMD regulation (Fig. 4; Supplemental Table 2). These observations indicate that, even under normal growth conditions, a significant fraction of intron-containing pre-mRNAs are exported from the nucleus to the cytoplasm, where they are degraded by NMD. The large number of yeast intron-containing pre-mRNAs subject to NMD regulation suggests that NMD plays a much more significant than anticipated role in intron-containing pre-mRNA degradation in yeast cells. Consistent with this conclusion, previous tiling microarray analyses also revealed a large overlapping set of intron-containing pre-mRNAs subject to NMD (Sayani et al. 2008).
Potential targeting mechanisms of the normal-looking NMD substrates
The largest fraction of NMD substrates identified here is comprised of normal-looking mRNAs that appear to lack defining features of premature termination. Our bioinformatics analyses reveal several intrinsic properties of these normal-looking NMD substrates that suggest a potential NMD-targeting mechanism. Compared to non-NMD substrates, this group of mRNAs has lower translation efficiency, a higher rate of out-of-frame translation, lower average codon optimality, and a propensity to have stretches of nonoptimal codons. Further, in contrast to an earlier proposition (Muhlrad and Parker 1999; Isken et al. 2008), the lower translation efficiency for this group of substrates appears to be independent of the NMD machinery.
The intrinsic properties that we uncovered for the normal-looking NMD substrates could reflect direct causes or indirect consequences of NMD targeting for these mRNAs, i.e., some of these properties may function independently or synergistically in NMD targeting. One possible mechanism of NMD targeting may be attributable to translational elongation through a stretch of nonoptimal codons. The lower average codon optimality or longer stretches of nonoptimal codons of the normal-looking NMD substrates might lead to less efficient translation for this group of transcripts, an increased probability that an error will be made during translation elongation, and the observed higher rate of out-of-frame translation. Clearly, the latter offers a greater likelihood for premature translation termination.
The less efficient translation and the higher rate of out-of-frame translation that we observed for normal-looking NMD substrates could also be caused by events independent of the translation of a stretch of nonoptimal codons. One possibility could be heterogeneity in the primary structure of this group of NMD substrates, i.e., the normal-looking NMD substrates may each have multiple transcript isoforms. Some of the isoforms may have very short 5′ UTRs (Arribere and Gilbert 2013) and some of the isoforms may result from internal transcriptional initiation in protein coding regions (Malabat et al. 2015). These unusual isoforms could have lower efficiencies of translation initiation at conventional ORF start sites and higher rates of out-of-frame downstream translation initiation. However, the detection of such isoforms as NMD substrates in a transcriptome-wide study would require that they constitute a significant fraction of the mRNA isoform population for a particular gene. Significantly, the less efficient translation and the higher rate of out-of-frame translation that we observed for normal-looking NMD substrates are largely independent of iTSS status (Supplemental Fig. 3). Similarly, alternative splicing events could also produce a subset of transcripts targeted by NMD, but previously reported alternative splicing events in yeast appear to generate only minor mRNA isoforms (Kawashima et al. 2014) and are thus unlikely to be detected as NMD substrates in our study. Therefore, the less efficient translation and higher rates of out-of-frame translation of the normal-looking NMD substrates are unlikely to be caused by transcript isoform heterogeneities, and most likely originate from the intrinsic translation properties of these mRNAs. Further, their propensity for frameshifting is most likely the cause of subsequent premature termination and NMD substrate status. In short, NMD can serve as a probabilistic quality control mechanism that allows for detection of errors during translation elongation. Collectively, atypical transcription or translation initiation, or unexpected frameshifting events, could all be targeted by NMD. In each of these cases, NMD activation is linked to premature or premature-like translation termination, as observed for several previously characterized classes of transcripts targeted by NMD (Fig. 7). This mode of action for NMD is consistent with previously published results in which NMD could target transcripts even after their first round of translation (Maderazo et al. 2003; Gaba et al. 2005).
MATERIALS AND METHODS
Yeast strains
All strains used in this study are in the W303 background. The wild-type strain (HFY114) and its isogenic derivatives harboring deletions of UPF1 (HFY871), UPF2 (HFY116), UPF3 (HFY861), DCP1 (HFY1067), or XRN1 (HFY1080) were described in He et al. (2003). Isogenic strains harboring deletion of DCP2 (CFY1016), EDC3 (CFY25), PAT1 (SYY2674), LSM1 (SYY2680), and DHH1 (SYY2686) were described in He and Jacobson (2015a). A strain harboring a deletion of SCD6 (SSY2352) was constructed by gene replacement (Guthrie and Fink 1991) using a DNA fragment harboring the scd6::KanMX6 null allele.
Cell growth and RNA isolation
Cells were all grown in YEPD media at 30°C. In each case, cells (15 mL) were grown to an OD600 of 0.7 and harvested by centrifugation. Cell pellets were frozen on dry ice and then stored at −80°C until RNA isolation. The procedures for RNA isolation were as previously described (He and Jacobson 1995).
RNA-seq library preparation
Total RNA was treated with Baseline-Zero DNase (Epicenter) to remove any genomic DNA contamination. Five micrograms of DNase-treated total RNA was then depleted of rRNA using the Illumina yeast RiboZero Removal Kit and the resulting RNA was used for RNA-seq library preparation. Multiplex strand-specific cDNA libraries were constructed using the Illumina TruSeq Stranded mRNA LT Sample Prep Kit. Three independent cDNA libraries were prepared for each yeast strain analyzed.
RNA sequencing
Total RNA cDNA libraries were sequenced on the Illumina HiSeq4000 platform at Beijing Genomics Institute. Four independent libraries were pooled into a single lane and single-end 50-cycle sequencing was carried out for all cDNA libraries.
Northern analysis
Procedures for Northern blotting were as previously described (He and Jacobson 1995). In each case, the blot was hybridized to a random primed probe for a specific transcript, with SCR1 serving as a loading control. Transcript-specific signals on Northern blots were determined with a FUJI BAS-2500 analyzer. Specific PCR fragments from the following genes were used as probes for Northern blotting analyses presented in Figure 4: RPL22B, entire 321-nt intron; HAC1, entire 252-nt intron, HRB1, exon 2 nt 784-1278; NHP6B, CDS nt 1-300; MTR2, CDS nt 1-555; ICR1, nt 2461-3040; IRT1, nt 811-1340; and TY4, CDS nt 4801-5410.
Bioinformatics methods
General computational methods
All statistical analyses were carried out using the R statistical programming environment, versions 3.2.4 and 3.2.5. R packages ggplot2, gplots, plyr, reshape2, and gridExtra were used for data preprocessing and visualization and foreach, doSNOW, and doParallel were used for parallel processing.
Differential expression analysis
The R64-2-1 S288C reference genome assembly (sacCer3) (Saccharomyces Genome Database Project) was used for sequencing reads mapping and transcriptome construction. We generated a yeast transcriptome comprised of 7473 transcripts that includes all annotated protein-coding sequences, functional and noncoding RNAs, and the unspliced isoforms of all intron-containing genes. Because of their repetitive nature, autonomous replicating sequences and long terminal repeats of transposable elements were excluded from the transcriptome. RSEM program (Li and Dewey 2011) was used to map the sequence reads to the transcriptome and to quantify individual transcript levels with settings --bowtie-m 30 --no-bam-output --forward-prob 0. The expected read counts for individual transcripts from RSEM were considered as the number of reads mapped to each transcript and were then imported into the Bioconductor DESeq package (Anders and Huber 2010) for differential expression analysis.
The Benjamini–Hochberg procedure was used for multiple testing corrections. To account for replicate variability, we used a false discovery threshold of 0.01 (1%) instead of an arbitrary fold change cutoff as the criterion for differential expression. We repeated the same pipeline for a separate transcriptome that contained CUT, SUT, and XUT sequences. These sequences are extracted from the yeast genome based on the previous annotations (Wery et al. 2016).
Ribosome footprint profiling analysis
We generated a second transcriptome for ribosome profiling analysis that only included mRNAs and unspliced isoforms from verified protein-coding genes. Because there are no formal 5′- and 3′ UTR annotations for most yeast transcripts, we used sequences 300 nt upstream of start codons or downstream from stop codons as the 5′- and 3′ UTR sequences. For transcripts that have annotated 5′ UTR introns, 300 nt immediately upstream of the annotated introns were considered as their 5′ UTRs. We used raw data from previously published ribosomal profiling experiments (Smith et al. 2014; Young et al. 2015). Raw fastq files and sequence reads were trimmed for adapter sequences with cutadapt with settings -a CTGTAGGCA -q 10 --trim-n -m 10. After adapter trimming, sequence reads were mapped to the transcriptome with bowtie (Langmead et al. 2009) with settings -m 4 -n 2 -l 15 --suppress 1,6,7,8 --best –strata. Because our transcriptome contains both spliced and unspliced isoforms from hundreds of the intron-containing genes, we allowed as many as four multiple mappings. After bowtie alignment, the riboSeqR package (Chung et al. 2015) was used for initial visualizations and frame calling. All other analyses were carried out by R scripts written in-house.
Ribosome density calculations
The ratio of profilingcoverage/RNA-seqcoverage for each transcript along the entirety of either intronic or coding regions (Figs. 3A, 5A,C) or over 100 bins (percentages) of the entire intron or coding regions was calculated using in-house scripts. For this analysis, we only used ribosome footprint read lengths that showed a strong preference (>80%) to a specific reading frame.
Calculation of in- and out-of-frame read ratios
We used only ribosome footprint read lengths that showed a strong preference (>80%) to a specific reading frame. Accounting for A-site occupancy, we mapped the reads from each length to our transcriptome and calculated the number of reads mapped to each transcript and the number of out-of-frame reads for each transcript for each read length. We then pooled the total and out-of-frame reads together and calculated the ratio of out-of-frame over total reads for each transcript.
Codon optimality calculations
We used previously published codon optimality assignments and scores (Pechmann and Frydman 2013) in our analyses. The average codon optimality score for each transcript in our ribosome profiling transcriptome was calculated using the Biostrings R package and in-house scripts. We took the sum of optimality scores for all codons in a transcript and then divided the sum by the total number of codons in the corresponding transcript. For discrete time Markov chain analysis, we labeled each codon as optimal (O) or nonoptimal (N) and then calculated the transition probabilities using maximum likelihood estimates as an unbiased measure for each transcript using the markovchain R package.
Statistical tests
We used χ2 tests with Yates continuity correction to assess different subsets of transcripts for either enrichment or depletion of a particular group of transcripts. Because the data for ribosome densities, transcript in-frame reads ratios, average codon optimality, and codon transition probabilities did not show normal (Gaussian) distributions, we used nonparametric two-sample Kolmogrov–Smirnov (KS) tests to assess the significance between different groups of transcripts. As KS tests compare the empirical distributions of two population samples, for consistency we used cumulative density plots in Figures 4–6.
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE86428.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant to A.J. (5R01 GM27757-37) from the National Institute of General Medical Sciences, National Institutes of Health.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.060541.116.
Freely available online through the RNA Open Access option.
REFERENCES
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Gilbert WV. 2013. Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res 23: 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery P, Vicente-Crespo M, Francis D, Nashchekina O, Alonso CR, Palacios IM. 2011. Drosophila Upf1 and Upf2 loss of function inhibits cell growth and causes animal death in a Upf3-independent manner. RNA 17: 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balistreri G, Horvath P, Schweingruber C, Zünd D, McInerney G, Merits A, Mühlemann O, Azzalin C, Helenius A. 2014. The host nonsense-mediated mRNA decay pathway restricts mammalian RNA virus replication. Cell Host Microbe 16: 403–411. [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I, Kashima I, Rehwinkel J, Sauliere J, Wittkopp N, Izaurralde E. 2007. mRNA quality control: an ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett 581: 2845–2853. [DOI] [PubMed] [Google Scholar]
- Boehm V, Haberman N, Ottens F, Ule J, Gehring NH. 2014. 3′ UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep 9: 555–568. [DOI] [PubMed] [Google Scholar]
- Chung BY, Hardcastle TJ, Jones JD, Irigoyen N, Firth AE, Baulcombe DC, Brierley I. 2015. The use of duplex-specific nuclease in ribosome profiling and a user-friendly software package for Ribo-seq data analysis. RNA 21: 1731–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Ji SJ, Porse BT, Jaffrey SR. 2013. Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell 153: 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH. 2009. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16: 49–55. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TM, Singh G, Lykke-Andersen J. 2010. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense- mediated mRNA decay. Cell 143: 938–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaba A, Jacobson A, Sachs MS. 2005. Ribosome occupancy of the yeast CPA1 upstream open reading frame termination codon modulates nonsense-mediated mRNA decay. Mol Cell 20: 449–460. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Ganesan R, Amrani N, Jacobson A. 2010. Translational competence of ribosomes released from a premature termination codon is modulated by NMD factors. RNA 16: 1832–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Yeo GW, Stone ME, Katz DB, Burge C, Turrigiano G, Moore MJ. 2007. The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell 130: 179–191. [DOI] [PubMed] [Google Scholar]
- Gloggnitzer J, Akimcheva S, Srinivasan A, Kusenda B, Riehs N, Stampfl H, Bautor J, Dekrout B, Jonak C, Jiménez-Gómez JM, et al. 2014. Nonsense-mediated mRNA decay modulates immune receptor levels to regulate plant antibacterial defense. Cell Host Microbe 16: 376–390. [DOI] [PubMed] [Google Scholar]
- Gong C, Kim YK, Woeller CF, Tang Y, Maquat LE. 2009. SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev 23: 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. 1991. Methods in enzymology: molecular biology of Saccharomyces cerevisiae. Academic Press, NY. [Google Scholar]
- He F, Jacobson A. 1995. Identification of a novel component of the nonsense-mediated mRNA decay pathway by use of an interacting protein screen. Genes Dev 9: 437–454. [DOI] [PubMed] [Google Scholar]
- He F, Jacobson A. 2001. Upf1p, Nmd2p, and Upf3p regulate the decapping and exonucleolytic degradation of both nonsense-containing mRNAs and wild-type mRNAs. Mol Cell Biol 21: 1515–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A. 2015a. Control of mRNA decapping by positive and negative regulatory elements in the Dcp2 C-terminal domain. RNA 21: 1633–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A. 2015b. Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu Rev Genet 49: 339–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Peltz SW, Donahue JL, Rosbash M, Jacobson A. 1993. Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1- mutant. Proc Natl Acad Sci 90: 7034–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Brown AH, Jacobson A. 1997. Upf1p, Nmd2p, and Upf3p are interacting components of the yeast nonsense-mediated mRNA decay pathway. Mol Cell Biol 17: 1580–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Li X, Spatrick P, Casillo R, Dong S, Jacobson A. 2003. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol Cell 12: 1439–1452. [DOI] [PubMed] [Google Scholar]
- Hu W, Petzold C, Coller J, Baker KE. 2010. Nonsense-mediated mRNA decapping occurs on polyribosomes in Saccharomyces cerevisiae. Nat Struct Mol Biol 17: 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E. 2008. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14: 2609–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Kim YK, Hosoda N, Mayeur GL, Hershey JW, Maquat LE. 2008. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 133: 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Bouhouche K, Gout JF, Aury JM, Noel B, Saudemont B, Nowacki M, Serrano V, Porcel BM, Segurens B, et al. 2008. Translational control of intron splicing in eukaryotes. Nature 451: 359–362. [DOI] [PubMed] [Google Scholar]
- Johansson MJ, He F, Spatrick P, Li C, Jacobson A. 2007. Association of yeast Upf1p with direct substrates of the NMD pathway. Proc Natl Acad Sci 104: 20872–20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Douglass S, Gabunilas J, Pellegrini M, Chanfreau GF. 2014. Widespread use of non-productive alternative splice sites in Saccharomyces cerevisiae. PLoS Genet 10: e1004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebaara BW, Atkin AL. 2009. Long 3′-UTRs target wild-type mRNAs for nonsense-mediated mRNA decay in Saccharomyces cerevisiae. Nucleic Acids Res 37: 2771–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervestin S, Jacobson A. 2012. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 13: 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Matsui A, Hanada K, Kawashima M, Ishida J, Morosawa T, Tanaka M, Kaminuma E, Mochizuki Y, Matsushima A, et al. 2009. Genome-wide suppression of aberrant mRNA-like noncoding RNAs by NMD in Arabidopsis. Proc Natl Acad Sci 106: 2453–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. 2007. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446: 926–929. [DOI] [PubMed] [Google Scholar]
- Leeds P, Peltz SW, Jacobson A, Culbertson MR. 1991. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev 5: 2303–2314. [DOI] [PubMed] [Google Scholar]
- Lejeune F, Li X, Maquat LE. 2003. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell 12: 675–687. [DOI] [PubMed] [Google Scholar]
- Lelivelt MJ, Culbertson MR. 1999. Yeast Upf proteins required for RNA surveillance affect global expression of the yeast transcriptome. Mol Cell Biol 19: 6710–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wilkinson MF. 1998. Nonsense surveillance in lymphocytes? Immunity 8: 135–141. [DOI] [PubMed] [Google Scholar]
- Loh B, Jonas S, Izaurralde E. 2013. The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev 27: 2125–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou CH, Shao A, Shum EY, Espinoza JL, Huang L, Karam R, Wilkinson MF. 2014. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep 6: 748–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J, Bennett EJ. 2014. Protecting the proteome: eukaryotic cotranslational quality control pathways. J Cell Biol 204: 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Chen Y, Ardal BR, Lilje B, Waage J, Sandelin A, Jensen TH. 2014. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev 28: 2498–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maderazo AB, Belk JP, He F, Jacobson A. 2003. Nonsense-containing mRNAs that accumulate in the absence of a functional nonsense-mediated mRNA decay pathway are destabilized rapidly upon its restitution. Mol Cell Biol 23: 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabat C, Feuerbach F, Ma L, Saveanu C, Jacquier A. 2015. Quality control of transcription start site selection by nonsense-mediated-mRNA decay. Elife 4: 06722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, Smith CW. 2008. Alternative splicing resulting in nonsense-mediated mRNA decay: what is the meaning of nonsense? Trends Biochem Sci 33: 385–393. [DOI] [PubMed] [Google Scholar]
- Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, Dietz HC. 2001. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet 10: 99–105. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. 2004. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36: 1073–1078. [DOI] [PubMed] [Google Scholar]
- Metzstein MM, Krasnow MA. 2006. Functions of the nonsense-mediated mRNA decay pathway in Drosophila development. PLoS Genet 2: e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Parker R. 1994. Premature translational termination triggers mRNA decapping. Nature 370: 578–581. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Parker R. 1999. Recognition of yeast mRNAs as “nonsense containing” leads to both inhibition of mRNA translation and mRNA degradation: implications for the control of mRNA decapping. Mol Biol Cell 10: 3971–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Grate L, Donohue J, Preston C, Nobida N, O'Brien G, Shiue L, Clark T, Blume J, Ares M. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P, Josi C, Kurosawa H, Yamashita A, Muhlemann O. 2014. A novel phosphorylation-independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res 42: 9217–9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada-Katsuhata Y, Yamashita A, Kutsuzawa K, Izumi N, Hirahara F, Ohno S. 2012. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res 40: 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R. 2012. RNA degradation in Saccharomyces cerevisae. Genetics 191: 671–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Frydman J. 2013. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol 20: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM. 2013. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497: 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltz SW, Brown AH, Jacobson A. 1993. mRNA destabilization triggered by premature translational termination depends on at least three cis-acting sequence elements and one trans-acting factor. Genes Dev 7: 1737–1754. [DOI] [PubMed] [Google Scholar]
- Pulak R, Anderson P. 1993. mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev 7: 1885–1897. [DOI] [PubMed] [Google Scholar]
- Ramani AK, Nelson AC, Kapranov P, Bell I, Gingeras TR, Fraser AG. 2009. High resolution transcriptome maps for wild-type and nonsense-mediated decay-defective Caenorhabditis elegans. Genome Biol 10: R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Letunic I, Raes J, Bork P, Izaurralde E. 2005. Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA 11: 1530–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki K, Yoshina S, Shen X, Han J, DeSantis MR, Xiong M, Mitani S, Kaufman RJ. 2012. RNA surveillance is required for endoplasmic reticulum homeostasis. Proc Natl Acad Sci 109: 8079–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayani S, Janis M, Lee C, Toesca I, Chanfreau G. 2008. Widespread impact of nonsense-mediated mRNA decay on the yeast intronome. Mol Cell 31: 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg DR, Maquat LE. 2012. Regulation of cytoplasmic mRNA decay. Nature Rev Genet 13: 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweingruber C, Rufener SC, Zund D, Yamashita A, Muhlemann O. 2013. Nonsense-mediated mRNA decay—mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta 1829: 612–623. [DOI] [PubMed] [Google Scholar]
- Singh G, Rebbapragada I, Lykke-Andersen J. 2008. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol 6: e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JE, Alvarez-Dominguez JR, Kline N, Huynh NJ, Geisler S, Hu W, Coller J, Baker KE. 2014. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep 7: 1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Torimura M, Akimitsu N. 2013. The RNA degradation pathway regulates the function of GAS5 a non-coding RNA in mammalian cells. PLoS One 8: e55684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DM, Parker R. 2007. Cytoplasmic decay of intergenic transcripts in Saccharomyces cerevisiae. Mol Cell Biol 27: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Izaurralde E. 2004. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol Cell 16: 587–596. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, Jacobsen SE, Nerlov C, Porse BT. 2008. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev 22: 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wery M, Descrimes M, Vogt N, Dallongeville AS, Gautheret D, Morillon A. 2016. Nonsense-mediated decay restricts LncRNA levels in yeast unless blocked by double-stranded RNA structure. Mol Cell 61: 379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp N, Huntzinger E, Weiler C, Sauliere J, Schmidt S, Sonawane M, Izaurralde E. 2009. Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol Cell Biol 29: 3517–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. 2009. Bidirectional promoters generate pervasive transcription in yeast. Nature 457: 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Chang TC, Yamashita Y, Zhu W, Zhong Z, Chen CY, Shyu AB. 2005. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat Struct Mol Biol 12: 1054–1063. [DOI] [PubMed] [Google Scholar]
- Young DJ, Guydosh NR, Zhang F, Hinnebusch AG, Green R. 2015. Rli1/ABCE1 recycles terminating ribosomes and controls translation reinitiation in 3′UTRs in vivo. Cell 162: 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Welch EM, Hogan K, Brown AH, Peltz SW, Jacobson A. 1997. Polysome-associated mRNAs are substrates for the nonsense-mediated mRNA decay pathway in Saccharomyces cerevisiae. RNA 3: 234–244. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.