

Abstract
Translation of poly(A) tails leads to mRNA cleavage but the mechanism and global pervasiveness of this “nonstop/no-go” decay process is not understood. Here we performed ribosome profiling (in a yeast strain lacking exosome function) of short 15–18 nucleotides mRNA footprints to identify ribosomes stalled at 3′ ends of mRNA decay intermediates. In this background, we found mRNA cleavage extending hundreds of nucleotides upstream of ribosome stalling in poly(A) and predominantly in one reading frame. These observations suggest that decay-triggering endonucleolytic cleavage is closely associated with the ribosome. Surprisingly, ribosomes appeared to accumulate (i.e., stall) in the transcriptome when as few as three consecutive ORF-internal lysine codons were positioned in the A, P, and E sites though significant mRNA degradation was not observed. Endonucleolytic cleavage was found, however, at sites of premature polyadenylation (encoding polylysine) and rescue of the ribosomes stalled at these sites was dependent on Dom34. These results suggest this process may be critical when changes in the polyadenylation site occur during development, tumorigenesis, or when translation termination/recycling is impaired.
Keywords: Hbs1, PELO, ribosome stalling, cryptic polyadenylation, ski complex
INTRODUCTION
The poly(A) tail is a fundamental feature of many cytoplasmic mRNAs and is thought to promote translation in some cases and prevent degradation of the message (Moore and Proudfoot 2009; Chang et al. 2014; Subtelny et al. 2014). In general, poly(A) tails are not translated because most mRNAs encode a stop codon that terminates translation and prevents the ribosome from reaching the 3′ end of the message. However, it is known that some number of transcripts are synthesized without a stop codon due to premature cleavage and polyadenylation at “cryptic” poly(A) sites (Frischmeyer et al. 2002; Ozsolak et al. 2010; Pelechano et al. 2013).
In such cases, translation of the poly(A) tail invokes an mRNA degradation pathway known as “nonstop decay” (NSD) and results in rapid loss of the message via a mechanism that is not fully understood (Frischmeyer et al. 2002; van Hoof et al. 2002) though the process is thought to begin with a ribosome stalling event in poly(A). Subsequent experiments revealed that ribosome stalling generally results in endonucleolytic cleavage of mRNA via a process called “no-go decay” (NGD) (Doma and Parker 2006). The combined “nonstop/no-go decay” (NSD/NGD) pathway is thought to be self-perpetuating because any upstream ribosome that reaches a cleaved 3′ end will arrest and can trigger additional mRNA cleavage (Tsuboi et al. 2012). The ribosomes that arrest on the truncated ends are known to be “rescued” by the factors Dom34/Hbs1/Rli1 (Shoemaker et al. 2010; Pisareva et al. 2011; Shoemaker and Green 2011; Guydosh and Green 2014), (i) permitting them to be recycled for new rounds of translation and (ii) deprotecting the 3′ end of the mRNA for 3′–5′ degradation by the ski complex-associated exosome (Halbach et al. 2013; Kowalinski et al. 2016). While cleavage has been observed in the vicinity of putative stalls (Chen et al. 2010; Tsuboi et al. 2012), the relative roles of endonucleases and exonucleases in the full degradation of the message is not clear. Moreover, the prevalence of endonucleolytic cleavage and no-go decay is unknown.
In principle, there are many potential mechanisms by which the ribosome could detect an encounter with the poly(A) tail and initiate the nonstop/no-go decay process. The ribosome could detect the presence of poly(A) in the mRNA entry channel or interact with poly(A)-tail binding proteins and undergo a conformational change that would recruit nonstop/no-go decay factors. Another reasonable possibility is that translation of AAA codon repeats into polylysine could stall the ribosome due to favorable interactions between positively charged nascent peptide and negatively charged rRNA (Lu and Deutsch 2008). In reconstituted systems, peptide bond formation between consecutive lysines is known to be slow (Koutmou et al. 2015). Consistent with this view that the ribosome arrests on messages encoding polylysine, gel-shift assays on reporters that encode runs of polylysine (Bengtson and Joazeiro 2010; Shao et al. 2013) or allow translation of the poly(A) tail (due to the absence of a stop codon) (Chiabudini et al. 2012; Shao et al. 2013) suggest that the addition of multiple lysine codons to a mRNA ORF does not result in the anticipated increase in the size of the translated protein products; these observations suggest that translation of lysines is minimal. It has also been shown that other factors, including Asc1 and the ubiquitin ligase Hel2, are involved in ribosome complex recognition and ribosomal ubiquitylation for quality control when long tracts of polybasic residues (arginine or lysine) are encountered (Kuroha et al. 2010; Brandman et al. 2012; Ikeuchi and Inada 2016; Juszkiewicz and Hegde 2016; Sundaramoorthy et al. 2017). These factors were also shown to be important during decoding of the I–A wobble pair at CGA codons (Letzring et al. 2013; Wolf and Grayhack 2015). Whether polybasic tracts are recognized in the same way as poly(A) translation is not clear. Moreover, whether ribosome-profiling data covering sequences encoding peptides with a net positive charge support a general stalling model has been extensively debated (Brandman et al. 2012; Charneski and Hurst 2013; Artieri and Fraser 2014; Sabi and Tuller 2015; Weinberg et al. 2016). A recent comprehensive reexamination of many data sets has shown the extent of apparent pausing when polybasic sequences are positioned in the exit tunnel to be highly variable, depending, at least partly, on whether cells were treated with cycloheximide (CHX) in culture (Hussmann et al. 2015; Sabi and Tuller 2015; Requiao et al. 2016; Weinberg et al. 2016).
Despite the lack of consensus on how sequences encoding polybasic peptides are detected, it is clear that the peptides from such translation events are ultimately degraded following ubiquitination by the E3 ligase Ltn1 (Bengtson and Joazeiro 2010). More recently, it was shown that Ltn1 is part of a “ribosome quality control complex” (RQC) that associates with the 60S ribosome and includes the ATPase Cdc48, the RNA binding protein Tae2, and another component, Rqc1 (Brandman et al. 2012; Shen et al. 2015). This RQC was shown to tag nascent chains with C-terminal Ala-Thr extensions or “CAT tails” (Shen et al. 2015), whose function is still being determined (Shen et al. 2015; Yonashiro et al. 2016). There is clear substrate overlap between the NSD/NGD pathway and the RQC pathway. Both processes have been observed on ORF-internal polylysine reporters as well as reporters where the ribosome translates authentic poly(A) tail (Tsuboi et al. 2012; Shao et al. 2013; Shcherbik et al. 2016). More generally, runs of polybasic residues in the genome have been reported to increase mRNA cleavage, possibly due to the NSD/NGD pathway (Peach et al. 2015). However, it remains unclear whether these pathways operate independently or specialize for particular substrates. Arguing that there may be some differences, several studies have suggested that the C-terminal domain of the ski-complex member Ski7 (Marshall et al. 2013; Kowalinski et al. 2016) is required for nonstop decay [i.e., translation of the poly(A) tail], but not for more general ribosome stalling, such as for cleaved RNAs (Meaux and Van Hoof 2006; Tsuboi et al. 2012).
Here we applied the short-read ribosome profiling methodology that we previously introduced (Guydosh and Green 2014) to examine the mechanism and extent of mRNA-cleavage and ribosome rescue at sites of poly(A) translation across the yeast transcriptome. Our method revealed a number of new properties of nonstop/no-go decay, including the existence of a frame for the endonucleolytic cleavage of mRNA and the requirement that ribosomes be arrested due to an actual failed translation event. In addition, we show that polylysine stalling is initially observed after just a few consecutive lysines are encountered, in the active site of the ribosome. Finally, we reveal the widespread role of Dom34 in rescuing ribosomes that stall in the vicinity of prematurely polyadenylated sequences throughout the genome. Because premature polyadenylation is abundant and often occurs in the middle of coding sequence (Ozsolak et al. 2010; Pelechano et al. 2013), the NSD/NGD quality-control pathway is likely active in regulating the levels of many gene products with important implications for human health (Klauer and van Hoof 2012).
RESULTS
Translation of poly(A) tails via nonsense suppression triggers widespread mRNA cleavage
We previously showed that short (15–18 nucleotides [nt]) ribosome footprints are generated by ribosomes that stall upon reaching the truncated 3′ end of the HAC1 mRNA after cleavage by the endonuclease Ire1 (Guydosh and Green 2014). Since these ribosomes are targeted by the factor Dom34 for recycling (Shoemaker et al. 2010), these footprints are only detectable in a Saccharomyces cerevisiae strain where DOM34 is deleted. Because stalled ribosomes are thought to trigger endonucleolytic cleavage of the mRNA during nonstop/no-go decay (Tsuboi et al. 2012), we reasoned that ribosome profiling of short footprints should offer new mechanistic insights into this phenomenon. To trigger this process, we transformed a dom34Δ yeast strain with the ochre suppressor tRNA, SUP4-o, to allow low-level ribosome readthrough at UAA codons and then performed enhanced ribosome profiling of footprints 15–34 nt in size. Because cleaved mRNAs can be degraded by the exosome, we performed many of our experiments in a ski2Δ background (indicated in some cases by [*]) to allow visualization of intermediates (Tsuboi et al. 2012). In initial data sets, we found that short footprints were heavily contaminated with fragments of tRNA and rRNA. For the libraries prepared here, we used the Ribo-Zero kit to eliminate the rRNA oligonucleotide reads by subtracting them from our purified RNA sample. However, the kit appeared to introduce short oligos of unknown origin that stochastically mapped across coding and noncoding regions of the genome. Because these oligos typically generated very sharp (1–2 nt wide) peaks in the data, we were able to eliminate them from our analysis with filtering algorithms (described in Materials and Methods).
We first analyzed the size distribution of read lengths that mapped near the 3′ ends of 315 mRNAs with UAA stop codons (where readthrough was anticipated) and no in-frame stop codons in their 3′UTRs [so ribosomes that read past the main stop codon would generally read into the poly(A) tail (Fig. 1A)]. In the dom34Δ* SUP4-o strain, the distribution of short reads was proportionately enriched for reads 15–18 nt in length. In contrast, the 15–18 nt footprints were less abundant in strains where Dom34 was present (and therefore able to recycle ribosomes stalled on truncated ends) or where SUP4-o was absent [and thus where readthrough into poly(A) tail is minimized]. When we expand our analysis to compare the abundance of short (15–18 nt) footprints to long (25–34 nt) footprints on all genes in many data sets, we generally find that short footprints represent ∼10% of the total reads and that strains lacking both DOM34 and SKI2 usually have more. Getting a more quantitative estimate is hampered by the occurrence of contaminating oligos introduced during Ribo-Zero subtraction, as noted above.
FIGURE 1.
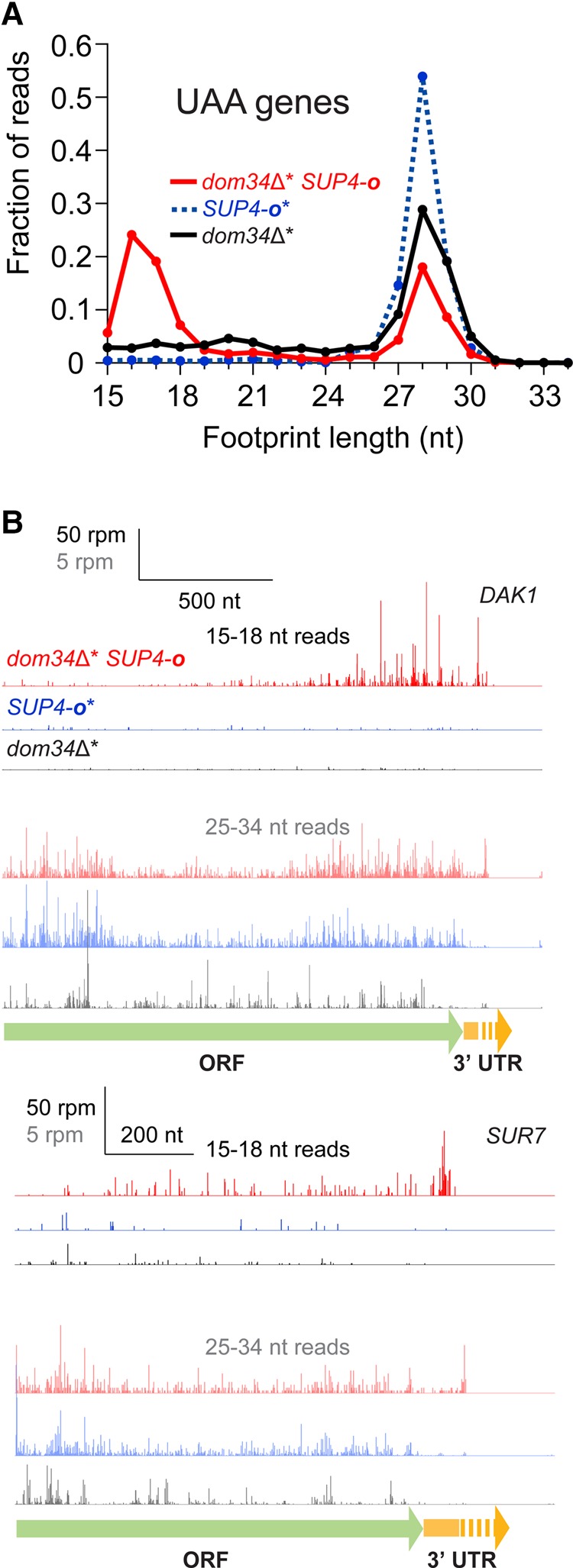
Translation of the poly(A) tail leads to upstream mRNA cleavage and ribosome stalling at the 3′ ends of the resulting truncated fragments. (A) Size distributions of short (15–34 nt) ribosome footprints that mapped without mismatches to sequences comprised of the 3′UTR of a gene and an equal length of ORF sequence immediately upstream of the 3′UTR. Footprints mapping to any part of the poly(A) tail were not included. (B) Examples of mapped footprints on genes (DAK1 and SUR7) both containing UAA stop codons and no in-frame stop codons in the 3′UTR. Data reveal enrichment of long reads near poly(A) and short reads over a more extensive region upstream when DOM34 is knocked out and the suppressor tRNA is present compared to strains where only one of these mutations is present. In the case of DAK1, long reads are already enriched in the absence of the suppressor tRNA (as shown previously [Guydosh and Green 2014]) due to the accumulation of likely nontranslating ribosomes. However, short reads are not enriched upstream in this case. Approximate region where polyadenylation takes place is indicated with a dotted arrow.
We next examined ribosome distributions in the dom34Δ* SUP4-o strains for genes that encode a UAA stop codon and no subsequent stop codons before the poly(A) tail. While the majority of long footprints (25–34 nt) mapped to ORFs, we also found long footprints that mapped to 3′UTRs, typically enriched at the very 3′ end of the UTR and partially overlapping the poly(A) tail (Fig. 1B; Supplemental Fig. S1A). To allow alignment of such chimeric reads, we trimmed the terminal poly(A)s from those reads that failed to map to the transcriptome in our initial alignment and then remapped them. The short reads (15–18 nt), in contrast, only sparsely covered ORFs but were strongly enriched in 3′UTRs (see examples in Fig. 1B). To examine this trend globally, we averaged ribosome profiling data across all such genes with a UAA stop codon and no further in-frame stop codons in the 3′UTR. With this analysis, it was clear that short read enrichment extended upstream of the poly(A) tail for hundreds of nucleotides (Fig. 2A), consistent with a chain-reaction type mechanism whereby an initial stall at the end of the 3′UTR triggers successive rounds of upstream cleavage and ribosome stalling, in this dom34Δ* yeast background. When we performed a similar analysis with SKI2 present (Fig. 2A, yellow trace), we found that accumulation of long footprints in the 3′UTR was essentially unchanged but that short footprints were diminished to a level somewhat above the background. In this case, we define the background as UAA genes with DOM34 present or non-UAA genes (all genes with a UAG or UGA stop codon). We included both of these controls to ensure that any biases associated subsets of genes under study did not affect our conclusions. These results suggest that the ribosomes protecting full-length footprints stall independently of the generation of short footprints. When SKI2 is present, the mRNAs are typically degraded rapidly before ribosomes can reach the endonucleolytically cleaved ends. These trends were also reflected in a similar analysis of individual genes (Supplemental Fig. S1B, red dots show higher short-read ratios than dark yellow dots). These striking genome-wide observations of ribosome distribution are broadly consistent with models previously proposed based on observations of mRNA decay for reporter mRNAs lacking stop codons (Tsuboi et al. 2012). We note that the involvement of iterated endonucleolytic cleavage is likely exaggerated by the particular yeast background used (dom34Δ*) as under normal conditions the exosome would be more actively involved once the first endonucleolytic cleavage event has occurred (again, compare heavy yellow and red traces in Fig. 2A).
FIGURE 2.
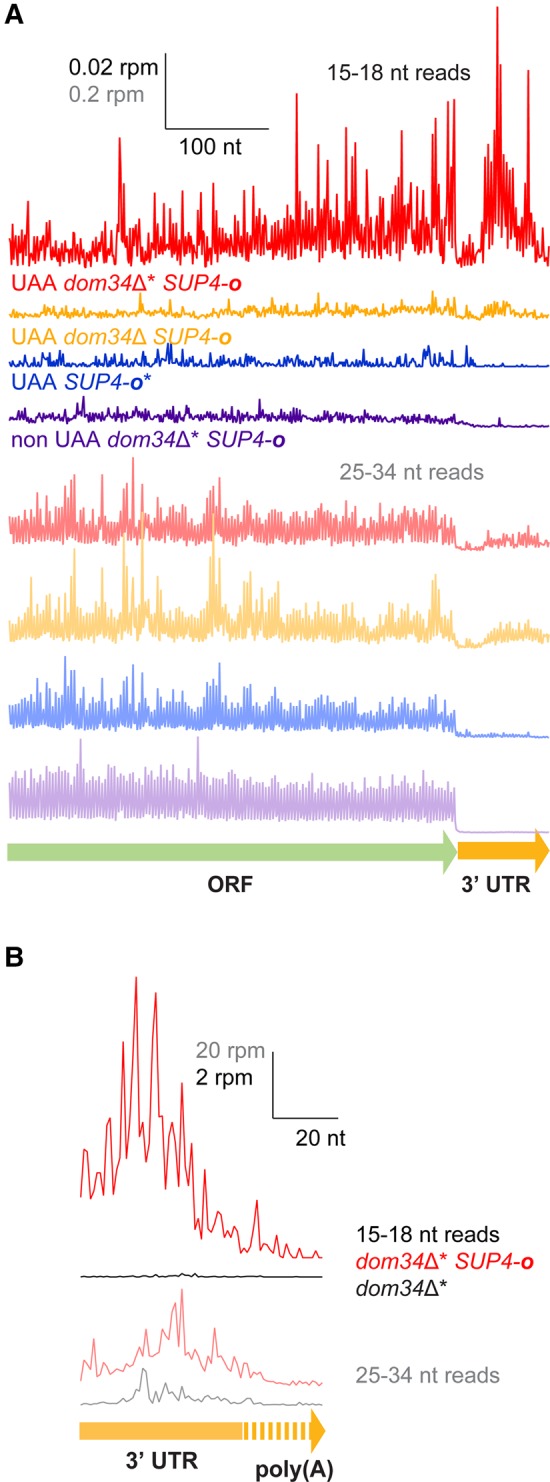
Observation of stall-induced cleavage requires active translation and the absence of Dom34. (A) Average ribosome occupancy for short reads (top dark traces) and long reads (bottom light traces) from genes with UAA stop codons and no in-frame stop codons in the 3′UTR. Gene sequences are aligned at their stop codons and reads are mapped by their 5′ ends. ORF annotation drawn to coincide with ribosome A sites. (B) Average ribosome occupancy from genes with UAA stop codons and no in-frame stop codons in the 3′UTR. Gene sequences are aligned at their primary polyadenylation sites and reads are mapped by their 5′ ends and unshifted. Only genes translating 3′UTRs after nonsense suppression generate mRNA cleavage and resultant short-read enrichment in the dom34Δ strain.
Active translation is critical for cleavage induction
We noticed in our analysis of individual genes (e.g., DAK1 in Fig. 1B or SMX2 in Supplemental Fig. S1A, and many examples in Supplemental Fig. S1B) that ribosomes protecting long (25–34 nt) footprints were strongly (>10×) enriched in the 3′UTRs in the dom34Δ* strain (black reads), even in the absence of nonsense suppression. We previously observed this phenomenon on a subset (∼11%) of genes in our previous study of dom34Δ yeast (Guydosh and Green 2014) and attributed this density to ribosomes that primarily were not engaged in active translation. To reconcile our new data with our old observations, we examined average ribosome density after alignment of genes at the ends of their 3′UTRs for the dom34Δ* strain in the presence or absence of SUP4-o (Fig. 2B); importantly, in this analysis, we only included genes with UAA stop codons and no subsequent in-frame stop codons. Long footprints were indeed enriched in the dom34Δ* background regardless of whether the suppressor tRNA was present. However, short footprints were only present when SUP4-o was present and ribosomes were translating through the 3′UTR. These observations provide striking evidence for the idea that ribosomes must be actively translating to trigger endonucleolytic mRNA cleavage.
Trimming by RNase 1 at 5′ ends is imprecise
To better understand the mechanism of mRNA cleavage during nonstop/no-go decay, we performed a careful analysis of the reading frame to which footprints aligned. To first establish the positioning of the ribosome in our dom34Δ* SUP4-o strain, we plotted ribosome density averaged across all genes aligned at their start codons (Fig. 3A). Reads were assigned by either their 5′ or 3′ ends and the separation in footprint peaks by these two methods was 28 nt apart, as anticipated for the footprint of a ribosome protecting the mRNA (Wolin and Walter 1988). The distance from the 5′ end of the footprint to the first nucleotide of the P site was 12 nt, again similar to what has been reported in other studies (Ingolia et al. 2009; Guydosh and Green 2014).
FIGURE 3.
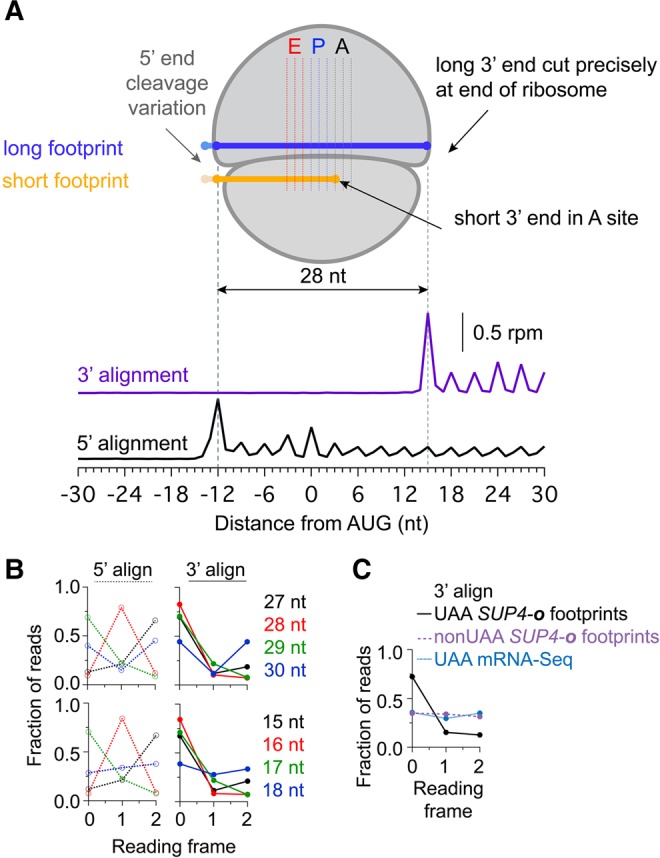
Analysis of mapped reading frame in 3′UTRs. (A) Schematic diagram of ribosome associated with long or short footprint reads. Short reads are always observed such that the 3′ end of the mRNA is positioned at the first nucleotide position of the A site. For reference, data underneath is average ribosome occupancy of long reads aligned by either 5′ or 3′ ends at start codons. Data are taken from the dom34Δ* SUP4-o strain on genes with non-UAA stop codons. This analysis also shows that the 3′ end of footprints maps more precisely to a single reading frame than the 5′ end (leading to reduced blurring of peaks at each codon). (B) Fraction of 5′ (left, dotted) or 3′ (right, solid) read ends mapping to each reading frame for long (top) and short (bottom) reads. Data derived from the dom34Δ* SUP4-o strain for 3′UTRs preceded by a UAA stop codon and no subsequent in-frame stop codons. (C) Fraction of 3′ read ends mapping to each reading frame for pooled short (15–18 nt) reads. Footprint reads from the dom34Δ* SUP4-o strain in cases where 3′UTRs are preceded by a UAA stop codon at the end of the ORF and include no subsequent in-frame stop codons are shown (black line) to preferentially occur in frame 0. In contrast, footprint reads from the same strain in cases with a non-UAA stop codon (purple line) are found equally well in all three reading frames. Similarly, mRNA-seq reads derived from the dom34Δ* strain for 3′UTRs preceded by a UAA stop codon at the end of the ORF and no subsequent in-frame stop codons in the 3′UTR (blue line) are equally distributed in all three frames.
We then checked the reading frame to which the 5′ and 3′ ends of a subset of long footprints (limited here to 27–30 nt) aligned (Fig. 3B, top). As has been observed previously (Ji et al. 2015), the 5′ ends for a given read length generally mapped to a single frame, but the frame to which they mapped varied according to read length; in contrast, the 3′ ends of all footprint lengths generally aligned to a single frame (frame 0). These results suggest that the majority of variation in footprint length occurs on the 5′ end of the read (these heterogeneities could reflect ribosome-based events or technical issues associated with library preparation). These results imply that 3′ alignment is superior in applications where reading frame analysis is critical, a finding that has been clearly established for profiling of bacterial ribosomes (Woolstenhulme et al. 2015). Increased resolution of average ribosome density peaks (Fig. 3A) in the 3′ aligned reads supports this conclusion.
Endonucleolytic cleavage of mRNA during nonstop/no-go decay occurs primarily in a single reading frame
We next turned to alignment of short ribosome footprints. Much like 5′ alignment of long footprints, we found that the reading frame to which the 5′ ends of short-reads aligned was uniform for a given fragment size but varied with length (Fig. 3B, bottom). This finding suggests that RNase 1 cleaves the mRNA upstream of ribosomes stalled on truncated mRNAs in much the same way as it does for all ribosomes. We then examined the frame to which 3′ ends mapped with no expectation that the reading frame would be the same for all read lengths since the 3′ end of the read is set by endonucleolytic cleavage that takes place during no-go decay and not by RNase 1. Surprisingly, we found that the reading frame for the 3′ ends of all footprint lengths consistently mapped to a single reading frame (72% in frame 0). Thus, for these short reads, the ribosome stalls on truncated footprints in a single frame where the end of the mRNA aligns to the first nucleotide position of the A site (and thus there is an incomplete codon that cannot be deciphered). Similar analysis for short reads mapping to genes without a UAA stop codon or for mRNA-seq reads (also cut to lengths of 15–18 nt) revealed no bias in the reading frame to which reads mapped on their 3′ ends (all three frames similar in blue/purple traces versus frame 0 preferred in black traces, Fig. 3C). These latter observations rule out artifactual causes from library cloning or computational alignment for the strong frame exhibited by the 3′ end of short reads. This unexpected finding implies that the endonuclease that cleaves the mRNA during nonstop/no-go decay is intimately associated with the triggering stalled ribosome (presumably on its upstream end).
Only three lysines are necessary to stall the ribosome
Our results point to a model where ribosomes that translate into the poly(A) tail eventually stall and trigger a precise endonucleolytic cleavage upstream. We wondered what molecular features can induce a sufficient initial stalling event to trigger efficient mRNA cleavage. We therefore tested the idea that ribosomes stall because of iterated lysine incorporations (since AAA encodes lysine). We tested this idea by averaging ribosome profiling data from coding regions that include tracts of 1–4 consecutive lysines (Fig. 4). Long (25–34 nt) footprints provided clear evidence for ribosome pausing when the ribosome was positioned with three sequential lysine codons in the A, P, and E sites (light traces, highlighted with red arrow and shown for replicate samples). Separate analysis of AAA and AAG codon stretches also revealed that the stalling evident in the long-read data was readily observable for both types of codon, implying that stalling induced by three consecutive lysines is triggered by the chemical properties of lysine rather than the sequence specificities of the mRNA:tRNA interaction (light gray traces, Supplemental Fig. S2A). Importantly, no accumulation of reads was observed at these locations in mRNA-seq data (light blue traces, Fig. 4 or Supplemental Fig. S2A), ruling out the possibility that these signals result from artifacts of preparing the deep-sequencing library [or sequencing issues related to poly(A) tracts]. We were even careful to cut mRNA-seq fragments of the same size range (from 25–34 nt) to eliminate the possibility that longer fragments typically used for mRNA-seq may not accurately reproduce cloning biases present in the 25–34 nt footprint library. We have observed this three lysine pause trigger in every data set that we have analyzed, including those with DOM34 present (light gray traces, Supplemental Fig. S2B), strongly supporting the generality of this finding. In addition, we asked whether runs of multiple arginines (also positively charged) exhibited a similar phenotype (light gray traces, Supplemental Fig. S2C) and found that arginine codons stall ribosomes to a very small extent with a single codon in the A site, and the signal is modestly increased as additional arginine codons are added to the sequence. We note that these average gene analyses become increasingly bouncy as the number of lysines or arginines increase since there are diminishing numbers of such sequence motifs in the genome.
FIGURE 4.
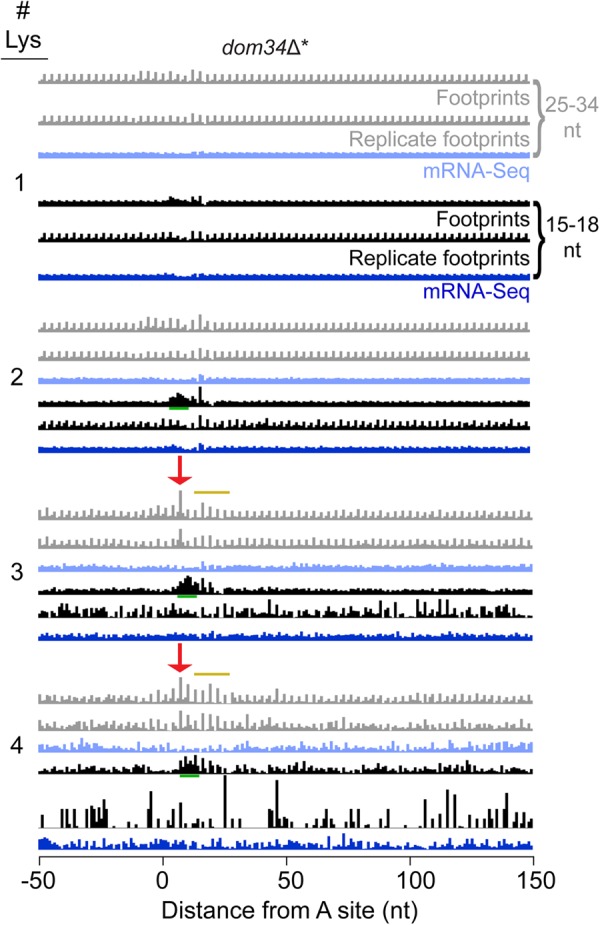
Ribosome profiling data reveals minor pausing on polylysine sequences but no mRNA cleavage upstream. Average fraction of ribosome occupancy from the dom34Δ* strain over a window around one or more consecutive Lys codons found within open reading frames. Only the first instance of multiple repeats was included when multiple repeats were consecutively encoded. Data are shifted so that the ribosome P site corresponds to the first codon position (0–2 nt) on the plot (16 nt shift). The appearance of accumulating ribosomes (pausing) when Lys codons are positioned in the E, P, and A sites of the ribosome is indicated with a red arrow for the KKK motif. Replicate data sets are from a previous study (Guydosh and Green 2014). Yellow line indicates continuing pausing when the lysines move into the exit tunnel. Green underline indicates region of artifactual short read enrichment downstream from pause region in only one replicate.
In addition to these very immediate effects from lysine codons, we also saw evidence for weak stalling when the lysines were positioned deeper in the peptide exit tunnel (yellow line, Fig. 4), consistent with previous analyses that, while variable, show some pausing when polybasic peptides are positioned in the exit tunnel (Brandman et al. 2012; Weinberg et al. 2016).
Is translation of consecutive lysines or polybasic sequence sufficient to induce endonucleolytic cleavage?
We next asked whether the modest pauses induced by three consecutive lysines were sufficient to trigger upstream endonucleolytic cleavage of the mRNA. When we examined the short read data (darker traces, Fig. 4), we noticed an accumulation of density just “downstream” from the pause (black traces, green underline regions). This effect is likely due to an artifact (and not stalled ribosomes) because it was not observed in the replicate and it was also observed in DOM34 cells (Supplemental Fig. S2B), where we never observe substantial short-read accumulation in 3′UTRs. When we filtered our sequences for those including exclusively three AAA or AAG codons (Supplemental Fig. S2A), it became clear this effect is limited to AAA codons and could also be detected by mRNA-seq in this case (dark blue trace, green underline regions), arguing that the short read accumulation may not be related to translation but more likely stems from a sequencing/cloning artifact. Importantly, there was no short-read enrichment “upstream” of the polylysine sequence as we would have expected based on our SUP4-o analysis of authentic 3′ poly(A) translation (above). We conclude that short (3–4) runs of consecutive lysine residues do not trigger mRNA cleavage.
Since it appeared that modest stalling on short runs of lysine (or arginine) is insufficient to trigger upstream endonucleolytic cleavage and rescue by Dom34, we wondered whether longer runs of lysine are required. Given prior estimates of poly(A) tail length in yeast, it is conceivable that ribosomes would need to translate >10 lysine codons before reaching the end of the poly(A) tail (Subtelny et al. 2014). Interestingly, however, in the ribosome profiling data, we never observe footprints with more than ∼15 consecutive As on their 3′ ends suggesting that at most one lysine is typically incorporated after encountering poly(A) tail (Supplemental Fig. S3A); these observations are similar to those we made previously for primarily nontranslating ribosomes found in the 3′UTR of mRNAs in the dom34Δ strain (Guydosh and Green 2014). However, we cannot rule out the possibility that ribosomes do translate further into the poly(A) tail (and possibly reach the end of the transcript) because the absence of more A-rich reads could be explained by (i) the preferential dissociation of such sequences from ribosomes during sample preparation (KS Koutmou and R Green, unpubl.) and (ii) the poor ability of the Illumina machine to sequence runs of consecutive As.
We therefore tested whether a long run of nine AAA or AAG lysine codons would induce endonucleolytic cleavage when placed directly into the middle of an open reading frame. Since such long runs do not exist in the genome, we took advantage of a previously described reporter gene that included nine (either AAA or AAG) lysine codons inserted between thioredoxin-HA and mCherry protein ORFs (Koutmou et al. 2015). We placed this sequence under the control of a constitutive promoter and performed ribosome profiling (Supplemental Fig S3B). Long and short reads were collected together in a single gel slice and both were normalized against long read footprint density on the plasmid's URA3 marker.
The most striking observation in these data is that ribosomes appear to read through both iterated lysine motifs (9 AAA or AAG codons) since ribosome density before and after the insertion site is approximately equivalent. This observation rules out the possibility of any severe stalling or extensive ribosome rescue due to translation of the lysine tract. We also notice that while there are a modest number of reads in the insert region for the AAG construct, there are essentially no reads in this region for the AAA construct. We suspect that the aforementioned challenges of preparing libraries of ribosomes on poly(A) sequences continue to confound this analysis of pausing on polylysine/basic tracts.
Despite these limitations, we were able to ask whether endonucleolytic cleavage was triggered by translation of these sequences by looking at the density of short footprints in the regions just upstream of the poly-lysine insert. We did not observe strong enrichment of short (15–18) reads upstream of the poly-lysine tract as we observed when ribosomes read into the poly(A) tail. Given the lack of cleavage, it seems likely that either longer poly-lysine tracts or tracts positioned specifically at the end of an mRNA [and/or bound to poly(A) binding protein] are required to trigger a robust no-go decay effect.
Iterated nucleolytic cleavage is the major source of short footprints in the absence of the exosome
With this better understanding of the molecular signature of ribosome-mediated endonucleolytic cleavage, we next investigated the loci where short reads are found in the transcriptome in the absence of suppressor tRNA. Previously, we showed that some such reads are found at a known (rare) site of endonucleolytic cleavage (on HAC1, an endogenous target of Ire1). To ascertain the scope of short footprints across the transcriptome, we computed the ratio of short versus long reads that align to ORFs in the dom34Δ* relative to the WT* strain (Fig. 5A). We found the long and short read ratios covered a wide range of values, reflecting large variation in the number of ribosomes that require rescue by Dom34 on any given gene. Indeed, the positive correlation in both long and short reads in this analysis is consistent with the idea that ribosomes stall in poly(A) tracts (protecting long footprints) and induce endonucleolytic cleavage; the subsequent ribosomes then stall on the cleaved ends, generating the correlated number of 16-nt reads. The fact that we observe an enrichment in the long reads amid the normal background of translating ribosomes suggests that a substantial number of ribosomes accumulate on these particular transcripts at steady state when Dom34 is not present.
FIGURE 5.

Ribosome stalling and mRNA cleavage identifies premature polyadenylation sites genome-wide. (A) Enrichment ratio (dom34Δ:DOM34) of short (15–18 nt) reads is plotted as a function of the ratio of long (25–34 nt) reads in a ski2Δ background. Genes in the upper right quadrant correspond to those with relatively high levels of premature polyadenylation, and those in the lower left are those with relatively low levels of premature polyadenylation. Genes included in analysis had >20 rpkm in the short reads and >5 rpkm in the long reads in either strain. (B) Cumulative histograms of frame to which the 3′ ends of 15–18 reads map across individual genes (thresholded at 10 rpm). A baseline median value of 0.33 is obtained for mRNA-seq data, as anticipated. A great proportion of footprint reads in WT* strain maps to frame 0 (previously shown to be diagnostic for endonucleolytic activity upstream of ribosome stalls). In the absence of DOM34, even more ribosomes go without rescue, increasing the fraction of short footprint reads in frame 0 to ∼73% on average. This value is roughly equal to the ∼72% fraction found when 3′UTRs are translated in the presence of a suppressor tRNA and therefore suggests most short footprints in this strain result from NSD/NGD. (C) Examples of footprints mapped to genes (YAP1 and RNA14) with known premature polyadenylation sites (green data, from Pelechano et al. 2013). Data reveal enrichment in short and (to a lesser extent) long reads at and upstream of known poly(A) sites in the dom34Δ strain. To be included in analysis, a threshold of 10 rpkm was required in total for all three frames. (D) Ratio of short to long reads is plotted against two previously published ratios of ORF (premature) poly(A) reads to 3′UTR (at the end of the full-length transcript) poly(A) reads. Spearman correlation coefficients (R2) were computed for each comparison and argue that translation (and resultant endonucleolytic cleavage) of prematurely polyadenylated genes is widespread. Footprint data were thresholded to require a minimum of 5 rpkm for long reads or 20 rpkm for short reads.
To more accurately assess the fraction of short footprint ribosomes engaged in nonstop/no-go decay, we tallied the reading frame assignment for the 3′ ends of short footprints across all ORFs in a cumulative histogram for footprints from strains with and without DOM34 (and for mRNA-seq data as a control) (Fig. 5B). We reasoned that the number of reads mapping to frame 0, which we previously showed correlates with cleavage induced during no-go decay (Fig. 3B), can serve as a metric for the fraction of short reads originating from nonstop/no-go decay. As expected, the median mRNA-seq frame was ∼0.33 since all reads map equally to the three reading frames. Strikingly, around 54% of short reads mapped to frame 0 in the WT* strain, on average, and nearly 73% mapped there in the dom34Δ* strain, reflecting the preservation of stalled ribosomes on endo-cleaved mRNAs in the absence of Dom34. As we showed earlier (Fig. 3C) that ∼72% of the short footprints in the 3′UTR of the critical subset of genes (with a UAA stop codon and no further in frame stop codons) mapped to frame 0 when the suppressor tRNA induced translation of the poly(A) tail, our observation that 73% of short reads are in that same frame is consistent with the interpretation that the majority of short footprints in this strain are generated by ribosomes stalled at sites of endonucleolytic cleavages generated during nonstop/no-go decay in our dom34Δ* strain. We note that because the exosome was lacking in these strains (ski2Δ), we fail to observe the likely synergistic contributions of exonucleolytic decay that follow once the initial endonucleolytic cleavage has occurred. Nevertheless, since short footprints usually make up around 10% of total footprints in the dom34Δ strain, it is conceivable that ribosome rescue by Dom34 occurs with some regularity in the cell.
Premature polyadenylation is a major source of short footprints
Because premature polyadenylation events very clearly lead to endonucleolytic cleavage and the accumulation of short footprints, we compared our short read footprint data with existing data sets documenting premature polyadenylation. Two different studies using sophisticated sequencing approaches revealed that premature polyadenylation sites are surprisingly widespread in the yeast genome (Ozsolak et al. 2010; Pelechano et al. 2013). When we checked specific examples of genes with substantial premature polyadenylation in these earlier data sets, we found that the annotated sites largely matched the enrichment sites of short footprints in our dom34Δ* strain (Fig. 5C). And, as anticipated from our suppressor (SUP4-o) analysis, we also observed long read footprints enriched at these positions (sometimes containing a number of uncoded A residues).
To perform a more quantitative comparison of these data, we compared the number of premature poly(A) sites measured previously (Pelechano et al. 2013) [computed here as the ratio of ORF poly(A) reads to 3′UTR poly(A) reads] to the ratio of short:long reads in our dom34Δ* strain (Fig. 5D). We found significant correlation (Spearman R2 values of 0.25–0.3) between these different data sets and our own metric, implying that many of the short footprints are generated by the endonucleolytic cleavage triggered by translation of premature poly(A) tails. Though strong, the correlation was not perfect; we suggest that this may be because loss of the normal 3′UTR may result in reduction of the rate of initiation (Szostak and Gebauer 2013) and therefore reduces the number of ribosomes that respond to premature poly(A) to initiate NSD/NGD. Such transcripts would then undergo degradation by an alternative pathway.
DISCUSSION
In this study, we took advantage of our recently introduced short-read ribosome profiling methodology to find ribosomes stalled on mRNA 3′ ends and address the question of how translation of poly(A) sequences brings about degradation of the mRNA via the nonstop/no-go decay pathway. In a dom34Δski2Δ (dom34Δ*) background, we observed ribosomes trapped on endonucleolytically cleaved mRNAs hundreds of nucleotides upstream of a leading ribosome that translated into the poly(A) tail: We suggest this is the molecular signature of NSD/NGD that is exaggerated by the dom34Δ* background. These data are strongly consistent with the model (Fig. 6) where an initiating ribosome stalling event in the poly(A) tail recruits an endonuclease that cleaves the mRNA upstream. Normally, the exosome would likely play a major role in mRNA degradation subsequent to this initial endonucleolytic cleavage event, but in the absence of the exosome, repeated rounds of mRNA cleavage and ribosome stalling dominate, ultimately resulting in complete degradation of the mRNA. Dom34 plays a key role in rescuing the ribosomes stalled on the truncated ends in this scenario. In the presence of the exosome, the kinetics of ribosome-recruited endonucleolytic cleavage versus exosome recruitment and processivity determine the relative contributions of these pathways. Our observation of some short reads in the presence of SKI2 is a signature that the iterated cleavage pathway discernibly contributes to decay in the cell (Fig. 2A, heavy yellow trace).
FIGURE 6.
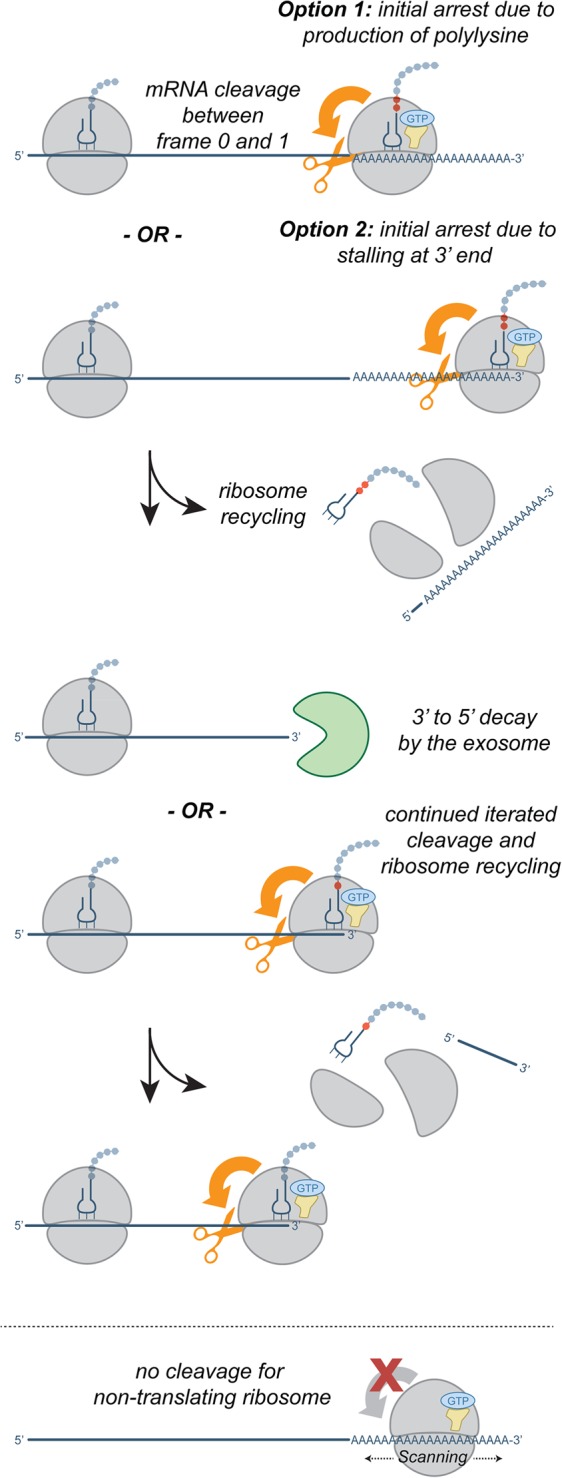
Model for translation-coupled mRNA cleavage during NSD/NGD. Ribosomes that translate the poly(A) tail and stall (top 2 rows) in the poly(A) tail (option 1) or at the end of the transcript (option 2). Stalled ribosomes recruit an endonuclease that cleaves the message upstream of the stalled ribosome (orange arrow). Upstream ribosomes in turn stall at these truncated ends, propagating the cleavage iteratively upstream. Once begun, this repetitive process may be interrupted by binding of the exosome to a truncated end. It can then carry out processive 3′–5′ decay until the transcript is degraded or the exosome collides with an upstream ribosome. Nontranslating ribosomes, such as those that fail to recycle properly at stop codons, do not bring about this iterative degradation pathway (bottom row).
Our data also constrain the model for how the ribosome is coupled to mRNA cleavage. To improve the resolution of the reading frame assignment of mapped reads, we introduced 3′ end assignment, a technique previously shown to be useful for improving reading frame detection in bacteria (Woolstenhulme et al. 2015). Similarly, in yeast, 3′ end assignment enhances the visualization of the reading frame for long (full-length) reads, and therefore promises to improve profiling resolution in the future. Interestingly, by using 3′ end assignment of short footprints, we found that most cleavage events occur in only one of the three reading frames of the coding sequence. This result is consistent with a model where the endonuclease that is recruited or activated by translation of the poly(A) tail is tightly associated with the stalled ribosome, likely on its upstream face (5′ end of the ribosome near the mRNA exit channel). We therefore predict that the mRNA is degraded into fragments of ∼16 nt, a length set by the distance between the A site and this upstream face of the 80S ribosome. These data rule out other models where the endonuclease is recruited separately to the mRNP or the poly(A) tail. Given this tight specificity, it seems unlikely that the downstream mRNA fragment is degraded through this iterated endonuclease pathway. We therefore assume that the 3′ mRNA cleavage product is degraded by another pathway. Importantly, we also found that ribosomes that are naturally present in the 3′UTR/poly(A) tail (i.e., in the absence of nonsense suppression) and are not thought to be translating (Guydosh and Green 2014) do not trigger upstream endonucleolytic cleavage. These observations indicate that some property of a translating ribosome is required to recruit the endonuclease.
In addition to an enrichment of 3′UTR short reads in the dom34Δ strain in the presence of a suppressor tRNA, we also found long reads were enriched in 3′UTRs as previously documented (Guydosh and Green 2014). There are at least two formal possibilities to explain the enrichment of ribosomes protecting long footprints. The ribosomes could translate to the end of the poly(A) tail and stall at the 3′ end of the message. Such ribosomes would protect a short footprint and the ribosomes observed to be stalled further upstream in the 3′UTR would result from the accumulation of ribosomes that follow. We were unable to provide direct support for this model because we detected little to no footprints (long or short) that are completely comprised of poly(A). The other possibility is that the translation of the repeated AAA codons into polylysine stalls the ribosome early within the poly(A) tract, leading to stacking of ribosomes protecting long footprints. Importantly, the observation that these reads are Dom34-dependent implies that Dom34 is functioning to rescue ribosomes positioned on an intact mRNA despite its known biochemical preference for truncated mRNAs (Pisareva et al. 2011; Shoemaker and Green 2011). Our previous work (Koutmou et al. 2015) suggests that poly(A) mRNA has unusual properties that may preclude efficient Lys-tRNALys binding, thus leaving the A site relatively unoccupied. Given the lack of significant ribosome accumulation (stalling) or upstream mRNA cleavage on translation of nine consecutive lysines in a synthetic ORF (Supplemental Fig. S3B), we conclude that either stalling becomes more severe with >9 lysines [as in a true poly(A) tail] or that stalling in the poly(A) tail is somehow different from stalling in an ORF-internal poly(A) sequence.
Additionally, the stalling we did observe on runs of consecutive lysine codons in the ORF provided an unexpected result. To our knowledge, analysis of ribosome profiling density on consecutive lysine codons has not been performed previously so it comes as a surprise that as few as three lysine codons positioned in the A, P, and E sites are sufficient to discernibly pause the ribosome. This finding argues that pausing begins before lysines are positioned deep in the exit tunnel, possibly due to unfavorable interactions already in the active site region. In addition, we looked for stalling due to lysines being positioned deeper in the exit tunnel and, consistent with the breadth of findings in the literature (Lu and Deutsch 2008; Brandman et al. 2012; Weinberg et al. 2016), found evidence for some weak stalling as the polybasic peptide passes through the tunnel. It would be of interest to know whether both of these types of stall recruit the RQC machinery and trigger tagging of the nascent chains with CAT tails (Shen et al. 2015; Yonashiro et al. 2016).
Finally, we addressed the question of how widespread no-go decay (i.e., decay initiated by an endonucleolytic cleavage) is in the cell. Prior studies have suggested the endonucleolytic cleavage plays only a small role in degradation of the bulk of most mRNAs (Harigaya and Parker 2012). While that may be true for most gene products, we provide key support for earlier findings that premature polyadenylation is widespread and affects most genes at some level (Fig. 5A). Furthermore, we found that these sites are a source of ribosome stalling and upstream mRNA cleavage, and therefore a major substrate for the nonstop/no-go decay pathway. The number of ribosomes engaged at these sites is not trivial (potentially ∼10% of the total cellular ribosomes under steady-state conditions in a dom34Δ background) and it is clear that these sites are the primary source of ribosomes stalled on truncated 3′ ends in the cell. When this fraction is combined with ribosomes on full-length footprints that are also rescued by Dom34 (such as those also on these premature poly(A) tails and those that accumulate in the canonical poly(A) tail after a period of nontranslation), the number becomes even more substantial. Indeed, even in yeast, dom34Δ cells exhibit slow growth under conditions of reduced ribosome availability (Bhattacharya et al. 2010) while in mammalian cells, loss of PELO (DOM34) is embryonic lethal (Adham et al. 2003). The widespread nature of these rescue events on poly(A) also implies that the RQC (or some other) pathway is burdened with the substantial critical task of processing large numbers of incomplete peptides for degradation.
We expect that the role of Dom34 becomes more critical under conditions where poly(A) translation is further induced. For example, neuronal activation and some cancers have been proposed to up-regulate premature polyadenylation events (Berg et al. 2012; Tian and Manley 2013). In addition, we recently found that ribosome recycling defects at stop codons lead to widespread 3′UTR translation (Young et al. 2015). 3′UTR translation is also known to increase under a variety of cellular stresses and has been hypothesized to be involved in promoting adaptation via C-terminal protein extensions (True and Lindquist 2000; Dunn et al. 2013). More broadly, the effects of nonstop/no-go decay are critical to understand for treatment of many diseases that result from mutation of a stop codon to a sense codon and in cases where drugs inducing low-level nonsense suppression are used in treatment of inheritable diseases caused by the introduction of premature termination codons in coding sequences (Klauer and van Hoof 2012). We anticipate ongoing work on the mechanism and consequences of poly(A) translation will be critical for future progress on therapies.
MATERIALS AND METHODS
Growth conditions
All cells were grown in YPD (BD Biosciences) media with the following exceptions: those expressing SUP4-o (CSM-His) and reporter strains (CSM-Ura). All media were sterile filtered and cultures were grown at 30°C. Cultures were usually harvested at an OD of ∼0.6 after approximately five doubling times.
Plasmids and strains
The expression vector for SUP4-o (pNG100) was created as previously described (Guydosh and Green 2014). The expression vectors for the Lys9 reporters were created by taking the Thrdx-HA-Lys9-mCherry bacterial expression construct (Koutmou et al. 2015) and cloning it into a p-ENTR/D-TOPO vector (Thermo-Fisher). This was then transferred (using an LR clonase reaction) into the pAG426GPD-ccdB destination vector (Alberti et al. 2007). This yielded a high-copy vector with the gene driven by a GPD constitutive promoter. This plasmid was called pNG103 for the AAA construct and pNG104 for the AAG construct. The sequence for the AAA construct is given in a separate file (Supplemental Material).
All yeast strains (Supplemental Table S1) were derived from the BY4741 or BY4742 background (derived from S288C). The dom34Δ* strains (yNG103a/b) were created previously (Guydosh and Green 2014) by mating dom34Δ yeast with ski2Δ yeast, sporulating the resultant diploid strain, and then dissecting tetrads. Strains yNG103a/b were obtained from the same tetrad. All new data sets included in this manuscript are from strain yNG103b. In Figure 4 and Supplemental Figure S2, previously published data are shown as replicates for short footprints from strain yNG103b or long footprints from yNG103a.
The strains expressing the Lys9 reporters were created by transforming the pNG103 and pNG104 plasmids into the dom34Δ* strain (yNG103b). The strains expressing SUP4-o were created by transforming them with the previously created centromeric pNG100 plasmid (Guydosh and Green 2014).
Deep-sequencing library preparation
Ribosome profiling libraries were prepared as previously described (Guydosh and Green 2014) by using a protocol very similar to that used by Ingolia et al. (2012). All RNA size separation gels (including those for mRNA-seq) were cut from 15–34 nt (or a smaller subset thereof). All samples were lysed and separated over sucrose gradients in the presence of 0.1 mg/mL CHX. Unlike samples in our previous study, new footprint samples here were subject to rRNA subtraction by using a yeast Ribo-Zero kit (Epicentre) prior to linker ligation. Samples for mRNA-seq were subject to subtraction after purification of total RNA. The 50°C incubation step for standard footprint preparation was skipped in the Ribo-Zero-modified protocol, as recommended by the manufacturer. One exception to this is that long footprints from the dom34Δ* SUP4-o and SUP4-o* strains were subtracted with custom-synthesized oligos after cDNA circularization. Previously published data that is analyzed here as replicate samples in Figure 4 and Supplemental Fig. S2 were also previously treated with custom complementary oligos for subtraction of rRNA as previously described (Guydosh and Green 2014). Sequencing was performed on an Illumina HiSeq2000 or HiSeq2500 at UC Riverside or the Johns Hopkins Institute of Genetic Medicine.
Deep-sequencing analysis
Analysis of footprints was essentially as previously described (Guydosh and Green 2014) with modifications as noted below. The R64-1-1 S288C reference genome assembly (SacCer3) was the reference genome used for analysis (Saccharomyces Genome Database Project). Demultiplexed sequences were processed to remove reads with any position with Phred score <20 or assigned N as a quality filter step. Following a search for the linker, contaminating ladder oligos were removed and alignment to the RNA gene database (http://downloads.yeastgenome.org/sequence/S288C_reference/rna/archive/rna_coding_R64-1-1_20110203.fasta.gz) was used to remove noncoding sequences. Following this step, a second round of subtraction for short, 15–18 nt, reads (or all reads for the 15–34 nt distribution in Fig. 1A) was performed by aligning to all the tRNA sequences from the noncoding RNA database that were extended with CCA on their 3′ ends. This enhanced the removal of cytoplasmic tRNA fragments. The remaining reads were mapped to the genome and those that failed to match were aligned to a database of splice junctions. Reads left over at this stage were then trimmed of consecutive 3′ As and realigned to the genome to obtain additional matches [proximal to poly(A) tails]. This 3′ A trimming step was not performed for 3′ read alignment. For alignment to the reporter sequences (Supplemental Fig. S3B), alignment to the full plasmid sequence was performed using bowtie. Read lengths were assessed with the FastQC software (Babraham Bioinformatics).
All reads that aligned to multiple coding sequences were discarded. Read occupancy was determined by giving one count per read (at the 5′ or 3′ end depending on the specific task) and in some cases shifted to align with various active sites in the ribosome (i.e., start of the A site) as described below. Read counts were then normalized by dividing by the total number of million mapped reads in a sample. Alignments were performed with Bowtie 0.12.7 or 1.01 (Langmead et al. 2009) and included the parameters: -y -a -m 1 -best -strata. All alignments to coding sequences allowed for no mismatches. All other analysis software was custom coded in the Python 2.7 programming language and Biopython. Plots were made with Igor Pro 6 (Wavemetrics).
ORFs marked dubious or those that overlapped with other transcripts were ignored in the analysis. Annotations for the length of 3′UTRs (Nagalakshmi et al. 2008) used coordinates from the R64-1-1 genome assembly and were downloaded from Saccharomyces Genome Database Project. 3′UTRs were generally extended 25 nt downstream from their annotated endpoints in order to ensure that all ribosomes that partially protected poly(A) sequences were accounted for (though not done for the size analysis in Fig. 1A). We define the start of the ribosome A site to be separated from the 5′ end of the footprint by 15 nt (short or long reads) and the 3′ end of the footprint by 12 nt (long reads). Read occupancy plots for example genes (Figs. 1B, 5C; Supplemental Fig. S1A) and position average plots (Fig. 4; Supplemental Fig. S2A–C) show reads shifted in this way. No shift was used for creation of average density plots (Fig. 2A,B). Gene quantitation (Figs. 3B,C, 5A,B,D) relied on a shift of 13 nt (center of P site) for 5′ assignment (all read lengths) and −2 for 3′ assignment (short reads) or −14 (long reads). However, the reading frame reported is not shifted.
Total gene ribosome occupancy was quantitated into density units of reads per kilobase per million mapped reads (rpkm) by taking reads mapping to an annotated sequence and dividing by the gene length in kilobases. For quantitation of ORFs, reads from the first and last five amino acids were not included to prevent known artifacts around start and stop codons from skewing results. In addition, to eliminate outliers due to difference in mating type between strains, we removed 12 mating-type related genes as well as DOM34 in the quantitation analyses in Figure 5A,D; Supplemental Figure S1B.
Gene-average plots (Figs. 2A,B, 3A) were constructed by taking the unweighted average of reads over the windows shown (note that the window for 3A is zoomed in to show detail from its original zone of 100 nt upstream and 300 nt downstream). Position-average plots were similarly created by averaging together (with equal weight) reads in a window for every occurrence of a particular motif in a gene (Fig. 4; Supplemental Fig. S2A–C).
Because we observed the appearance of seemingly random single-position read density spikes across coding and noncoding sequences in our short-read data sets for samples that had been treated with Ribo-Zero, we attribute this to contamination being introduced by the kit at these very short read lengths. In other data sets (not shown here), these contaminants disappear when Ribo-Zero subtraction is performed after linker ligation, consistent with the contaminants consisting of RNAs. To reduce the noise that this introduced for some analyses, we eliminated genes where only two positions accounted for a large proportion of the total read density. For average gene plots, a threshold of 0.5 was used, for quantitation and frame analysis, a threshold of 0.8 was used, except Figure 5A,D, where 0.3 was used.
DATA DEPOSITION
The NCBI GEO accession number for the sequencing data (debarcoded fastq files and wig files) reported in this paper is GSE85944.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Liana Lareau and Toshi Inada for helpful discussions, and Boris Zinshteyn for careful reading of the manuscript. This work was supported by the National Institutes of Health, National Institute of General Medical Sciences GM 059425 to R.G.; Howard Hughes Medical Institute for salary support to R.G.; and the National Institute of Diabetes and Digestive and Kidney Diseases Intramural Program for salary support to N.G.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.060418.116.
REFERENCES
- Adham IM, Sallam MA, Steding G, Korabiowska M, Brinck U, Hoyer-Fender S, Oh C, Engel W. 2003. Disruption of the Pelota gene causes early embryonic lethality and defects in cell cycle progression. Mol Cell Biol 23: 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Gitler AD, Lindquist S. 2007. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 24: 913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artieri CG, Fraser HB. 2014. Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation. Genome Res 24: 2011–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CA. 2010. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467: 470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, et al. 2012. U1 snRNP determines mRNA length and regulates isoform expression. Cell 150: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, McIntosh KB, Willis IM, Warner JR. 2010. Why Dom34 stimulates growth of cells with defects of 40S ribosomal subunit biosynthesis. Mol Cell Biol 30: 5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, et al. 2012. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151: 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Lim J, Ha M, Kim VN. 2014. TAIL-seq: genome-wide determination of poly(A) tail length and 3′ end modifications. Mol Cell 53: 1044–1052. [DOI] [PubMed] [Google Scholar]
- Charneski CA, Hurst LD. 2013. Positively charged residues are the major determinants of ribosomal velocity. PLOS Biol 11: e1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Muhlrad D, Hauryliuk V, Cheng Z, Lim MK, Shyp V, Parker R, Song H. 2010. Structure of the Dom34-Hbs1 complex and implications for no-go decay. Nat Struct Mol Biol 17: 1233–1240. [DOI] [PubMed] [Google Scholar]
- Chiabudini M, Conz C, Reckmann F, Rospert S. 2012. Ribosome-associated complex and Ssb are required for translational repression induced by polylysine segments within nascent chains. Mol Cell Biol 32: 4769–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R. 2006. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440: 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, Foo CK, Belletier NG, Gavis ER, Weissman JS. 2013. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife 2: e01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O'Donnell K, Guerrerio AL, Parker R, Dietz HC. 2002. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295: 2258–2261. [DOI] [PubMed] [Google Scholar]
- Guydosh NR, Green R. 2014. Dom34 rescues ribosomes in 3′ untranslated regions. Cell 156: 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbach F, Reichelt P, Rode M, Conti E. 2013. The yeast ski complex: crystal structure and RNA channeling to the exosome complex. Cell 154: 814–826. [DOI] [PubMed] [Google Scholar]
- Harigaya Y, Parker R. 2012. Global analysis of mRNA decay intermediates in Saccharomyces cerevisiae. Proc Natl Acad Sci 109: 11764–11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussmann JA, Patchett S, Johnson A, Sawyer S, Press WH. 2015. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLOS Genet 11: e1005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi K, Inada T. 2016. Ribosome-associated Asc1/RACK1 is required for endonucleolytic cleavage induced by stalled ribosome at the 3′ end of nonstop mRNA. Sci Rep 6: 28234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7: 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Song R, Regev A, Struhl K. 2015. Many lncRNAs, 5′UTRs, and pseudogenes are translated and some are likely to express functional proteins. eLife 4: e08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszkiewicz S, Hegde RS. 2016. Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol Cell 65: 743–750.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauer AA, van Hoof A. 2012. Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. Wiley Interdiscip Rev RNA 3: 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutmou KS, Schuller AP, Brunelle JL, Radhakrishnan A, Djuranovic S, Green R. 2015. Ribosomes slide on lysine-encoding homopolymeric A stretches. eLife 4: 405534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalinski E, Kogel A, Ebert J, Reichelt P, Stegmann E, Habermann B, Conti E. 2016. Structure of a cytoplasmic 11-subunit RNA exosome complex. Mol Cell 63: 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Akamatsu M, Dimitrova L, Ito T, Kato Y, Shirahige K, Inada T. 2010. Receptor for activated C kinase 1 stimulates nascent polypeptide-dependent translation arrest. EMBO Rep 11: 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzring DP, Wolf AS, Brule CE, Grayhack EJ. 2013. Translation of CGA codon repeats in yeast involves quality control components and ribosomal protein L1. RNA 19: 1208–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Deutsch C. 2008. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol 384: 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AN, Montealegre MC, Jimenez-Lopez C, Lorenz MC, van Hoof A. 2013. Alternative splicing and subfunctionalization generates functional diversity in fungal proteomes. PLOS Genet 9: e1003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaux S, Van Hoof A. 2006. Yeast transcripts cleaved by an internal ribozyme provide new insight into the role of the cap and poly(A) tail in translation and mRNA decay. RNA 12: 1323–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ. 2009. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136: 688–700. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM. 2010. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143: 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peach SE, York K, Hesselberth JR. 2015. Global analysis of RNA cleavage by 5′-hydroxyl RNA sequencing. Nucleic Acids Res 43: e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM. 2013. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497: 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisareva VP, Skabkin MA, Hellen CU, Pestova TV, Pisarev AV. 2011. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J 30: 1804–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requiao RD, de Souza HJ, Rossetto S, Domitrovic T, Palhano FL. 2016. Increased ribosome density associated to positively charged residues is evident in ribosome profiling experiments performed in the absence of translation inhibitors. RNA Biol 13: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabi R, Tuller T. 2015. A comparative genomics study on the effect of individual amino acids on ribosome stalling. BMC Genomics 16(Suppl 10): S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, von der Malsburg K, Hegde RS. 2013. Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol Cell 50: 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbik N, Chernova TA, Chernoff YO, Pestov DG. 2016. Distinct types of translation termination generate substrates for ribosome-associated quality control. Nucleic Acids Res 44: 6840–6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen PS, Park J, Qin Y, Li X, Parsawar K, Larson MH, Cox J, Cheng Y, Lambowitz AM, Weissman JS, et al. 2015. Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 347: 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Green R. 2011. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc Natl Acad Sci 108: E1392–E1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Eyler DE, Green R. 2010. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science 330: 369–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, Bartel DP. 2014. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 508: 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramoorthy E, Leonard M, Mak R, Liao J, Fulzele A, Bennett EJ. 2017. ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol Cell 65: 751–760.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak E, Gebauer F. 2013. Translational control by 3′-UTR-binding proteins. Brief Funct Genomics 12: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Manley JL. 2013. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci 38: 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- True HL, Lindquist SL. 2000. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407: 477–483. [DOI] [PubMed] [Google Scholar]
- Tsuboi T, Kuroha K, Kudo K, Makino S, Inoue E, Kashima I, Inada T. 2012. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol Cell 46: 518–529. [DOI] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. 2002. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295: 2262–2264. [DOI] [PubMed] [Google Scholar]
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, Bartel DP. 2016. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep 14: 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AS, Grayhack EJ. 2015. Asc1, homolog of human RACK1, prevents frameshifting in yeast by ribosomes stalled at CGA codon repeats. RNA 21: 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin SL, Walter P. 1988. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J 7: 3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolstenhulme CJ, Guydosh NR, Green R, Buskirk AR. 2015. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep 11: 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Tahara EB, Bengtson MH, Khokhrina M, Lorenz H, Chen KC, Kigoshi-Tansho Y, Savas JN, Yates JR, Kay SA, et al. 2016. The Rqc2/Tae2 subunit of the ribosome-associated quality control (RQC) complex marks ribosome-stalled nascent polypeptide chains for aggregation. eLife 5: e11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DJ, Guydosh NR, Zhang F, Hinnebusch AG, Green R. 2015. Rli1/ABCE1 recycles terminating ribosomes and controls translation reinitiation in 3′UTRs in vivo. Cell 162: 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.