

Abstract
Persistent organic pollutants (POPs) are lipophilic compounds that travel with lipids and accumulate mainly in adipose tissue. Recent human evidence links low-dose POPs to an increased risk of type 2 diabetes (T2D). Because humans are contaminated by POP mixtures and POPs possibly have nonmonotonic dose-response relations with T2D, critical methodological issues arise in evaluating human findings. This review summarizes epidemiological results on chlorinated POPs and T2D, and relevant experimental evidence. It also discusses how features of POPs can affect inferences in humans. The evidence as a whole suggests that, rather than a few individual POPs, background exposure to POP mixtures—including organochlorine pesticides and polychlorinated biphenyls—can increase T2D risk in humans. Inconsistent statistical significance for individual POPs may arise due to distributional differences in POP mixtures among populations. Differences in the observed shape of the dose-response curves among human studies may reflect an inverted U-shaped association secondary to mitochondrial dysfunction or endocrine disruption. Finally, we examine the relationship between POPs and obesity. There is evidence in animal studies that low-dose POP mixtures are obesogenic. However, relationships between POPs and obesity in humans have been inconsistent. Adipose tissue plays a dual role of promoting T2D and providing a relatively safe place to store POPs. Large prospective studies with serial measurements of a broad range of POPs, adiposity, and clinically relevant biomarkers are needed to disentangle the interrelationships among POPs, obesity, and the development of T2D. Also needed are laboratory experiments that more closely mimic real-world POP doses, mixtures, and exposure duration in humans.
-
I.
Introduction
-
II.
Human Evidence Linking POPs and T2D
-
A.
What are POPs?
-
B.
Earlier puzzling findings linking POPs and T2D
-
C.
Recent evidence linking POPs and T2D: cross-sectional and case-control studies
-
D.
Reverse causality due to disease progression bias
-
E.
Prospective evidence linking POPs and T2D
-
A.
-
III.
Experimental Evidence Linking POPs and T2D
-
IV.
POPs and Obesity
-
A.
POPs as obesogens
-
B.
Are obesogens unequivocally harmful?
-
C.
POPs and inflammation in adipose tissue
-
D.
POPs, gut microbiota, and obesity
-
E.
Can the obesity paradox be explained by POPs?
-
A.
-
V.
Methodological Issues Crucial for Human Studies
-
A.
Issue 1: inverted U-shaped associations
-
B.
Issue 2: mixtures
-
C.
Issue 3: how can we deal with lipid profiles?
-
A.
-
VI.
Possible Mechanisms
-
A.
Traditional endocrine disrupting-related mechanisms
-
B.
Mitochondrial dysfunction-related mechanisms
-
A.
-
VII.
Future Research Issues
-
A.
Human studies
-
B.
Experimental studies
-
A.
-
VIII.
Conclusion
I. Introduction
Over the past three decades, the number of people with type 2 diabetes (T2D) has more than doubled globally, making it one of the most important public health challenges to all nations (1). T2D and prediabetes are increasingly observed among children and adolescents (2). A common assumption is that lifestyle changes characterized by excess energy intake and a lack of exercise have led to the obesity epidemic and, in turn, to the diabetes epidemic. However, there is considerable evidence suggesting that individuals with similar degrees of obesity can have strikingly different risks of T2D (3). It is particularly noteworthy that whereas 80% of T2D patients are obese, approximately 75–80% of obese people never develop T2D (4). Insulin resistance, a prediabetic state, varies 6-fold among obese persons (5). Although causal relationships between genetic factors and T2D have been eagerly sought, the data from genome-wide association studies have shown that genetic variants might explain statistically only about 10% of the phenotypic variability (6). Thus, obesity itself is not a sufficient cause of T2D. Neither is genetics sufficient in the vast majority of cases.
Recently, evidence has linked environmental chemicals with obesity, insulin resistance, and T2D. In January 2011, the US National Toxicology Program and the National Institute of Environmental Health Sciences held a workshop that evaluated the science assessing exposure to certain chemicals with the development of these disorders (7). A main conclusion was that persistent organic pollutants (POPs) have generated particularly strong evidence as a risk factor for T2D in humans (8).
POPs have several unique features that distinguish them from other common chemicals. First, POPs include various lipophilic compounds that accumulate mainly in lipid-containing tissues like adipose tissue and move within the body bound to lipids. Metabolic disturbances of both adipose tissue and lipids are key to the pathophysiology of T2D (9). Therefore, an interesting question is whether the concurrent and continuous presence of POPs in these sites is harmless? Another interesting aspect is that the change of adipose tissue mass is one factor determining the pharmacodynamics of POPs in humans (10, 11).
The second feature is that POPs are always present as chemical mixtures due to mixing in the environment, food webs, and long-term retention in fat tissue. The same difficulties that pertain to evaluating any chemical mixture (12) are therefore relevant to the evaluation of POPs. There are positive correlations among concentrations of many POPs, although some pairs of POPs are weakly correlated (13). In studies performed in general populations with only background exposure to POPs, findings for a specific compound cannot be interpreted as due solely to that compound; rather, they likely reflect the properties of the POP mixture of which the compound is part. Therefore, focusing on individual POPs can be misleading. In this review, we often use the word “POPs” to denote a POP mixture. Also, organochlorine (OC) pesticides, polychlorinated biphenyls (PCBs), and dioxins are terms referring to chemical mixtures of each POP subclass.
A third key feature arises from the possibility of nonlinear and nonmonotonic dose-response relationships between POPs and T2D. The discussion on nonmonotonicity in POPs and T2D associations is a major focus of this paper. Traditional approaches to summarize results from epidemiological studies, including meta-analyses, typically pool single parameters, such as a linear slope or risk ratio between the highest and lowest categories of the exposure. However, these approaches are limited for describing nonlinear dose-response relations. As we will discuss in Section V, nonlinearities may lead to differing associations across the range of exposure, which in turn may lead to apparent inconsistencies across studies with different exposure ranges. Therefore, data analytic pooling may in many cases blur estimates of true associations.
Finally, there is no population group without any exposure to POPs; virtually everyone in modern society has some POP exposure, given contamination of the environment and food webs (14). Therefore, a major point in the evaluation of the health effects of POPs is how to form a reference group that is as close as possible to unexposed.
This review has two main purposes: first, to summarize the available epidemiological findings on POPs and T2D; and second, to discuss how features of POPs can affect inferences relevant for human health. The two aims are closely linked because the first purpose cannot be validly achieved without an appropriate understanding of the second. After a discussion of the epidemiological evidence, we will present experimental evidence from studies that used POP mixtures similar to the pattern of human internal POP contamination. Such studies provide insights into plausible mechanisms for the relationships between POPs and T2D.
Although a pathway starting with obesity, leading to insulin resistance, and ultimately to T2D is commonly regarded as the natural pathogenesis of T2D, we discuss issues related specifically to obesity after the section concerned with T2D. The current prevailing concept is that chemicals inducing obesity (obesogens) can also predispose individuals to T2D due to the role of obesity in the development of T2D. However, when assessing such relationships, it should be considered that adipose tissue mass may play a dual role in the biological effects of POPs, which will be discussed in Section IV.
II. Human Evidence Linking POPs and T2D
A. What are POPs?
POPs encompass a variety of lipophilic chemicals resistant to environmental degradation that bioaccumulate in food webs and living organisms (14). OC pesticides such as dichlorodiphenyltrichloroethane (DDT), lindane, chlordane, and hexachlorobenzene are typical examples of POPs. Other POPs are produced as industrial chemicals or byproducts, including PCBs, polychlorinated dibenzo-p-dioxins (PCDDs), polychlorinated dibenzofurans (PCDFs), and polybrominated diphenyl ethers. Characteristics of common POPs and their estimated half-lives are listed in Table 1.
Table 1.
List of Common Chlorinated POPs
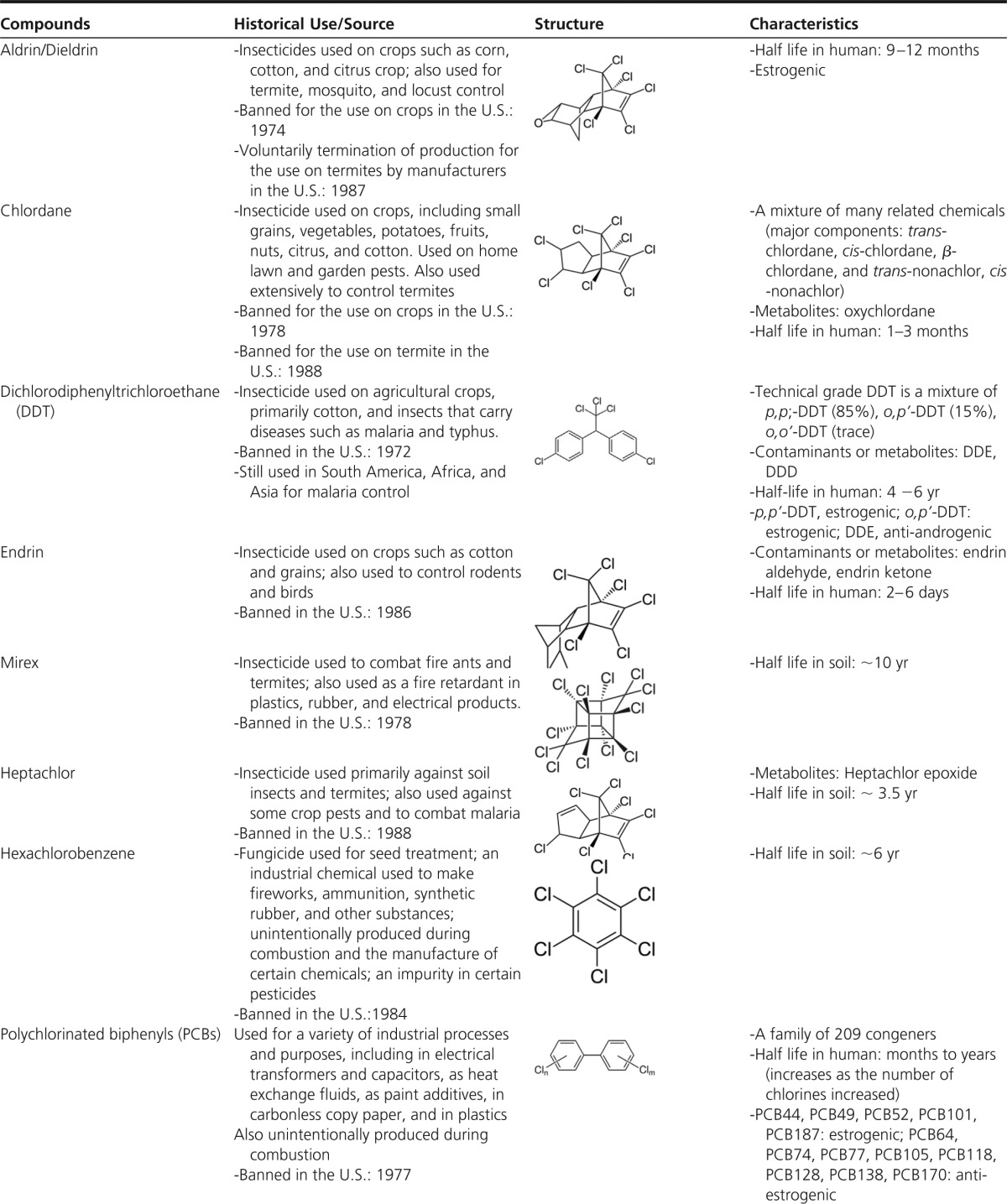
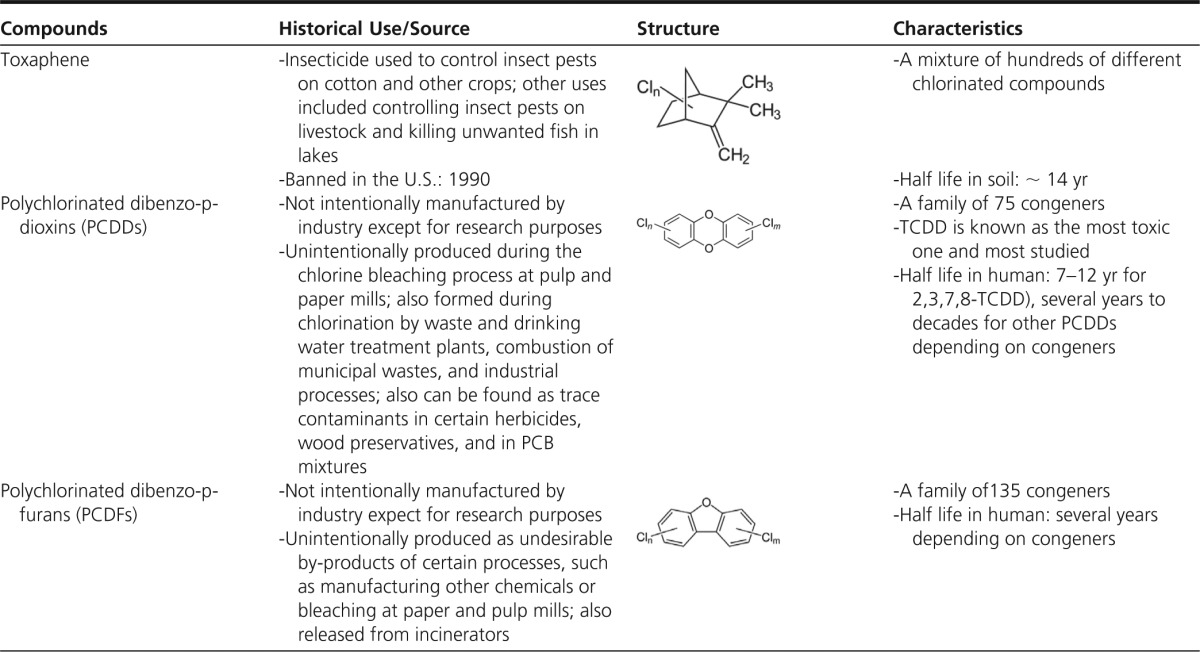
Abbreviation: DDD, dichlorodiphenyldichloroethane.
Source: All contents are extracted from the Agency for Toxic Substances and Disease Registry (http://www.atsdr.cdc.gov/).
Unless otherwise specified, information on half lives is primarily in humans. When there is no information on humans, half lives in soil are provided.
Among all POPs, those with chloride atoms have long been suspected to have a deleterious effect on humans and wild animals (14). They were thus banned several decades ago in most developed countries, and the emission of dioxins is strictly regulated in many countries (15). On the other hand, brominated POPs are still commonly used, and their exposure patterns are different from those of chlorinated POPs (16). In addition, because human evidence regarding brominated POPs is scarce compared to that for chlorinated POPs, we focus here on chlorinated POPs.
Despite regulation, the exposure to chlorinated POPs by the general population continues, mostly through consumption of fatty foods of animal origin (14). The resistance of POPs to chemical and metabolic degradation entails that they become more concentrated as they move up through food webs (17). Biomagnification can lead to concentrations in humans several orders of magnitude higher than in the general environment. In addition, POPs that accumulate in adipose tissue during life become a source of chronic internal exposure because they are continuously released from adipose tissue to the circulation and vital organs with lipid content (18, 19).
B. Earlier puzzling findings linking POPs and T2D
The first human evidence about the possible harmfulness of a chemical often comes to light after high-dose exposures in an occupational or accidental setting. Among various POPs, exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in workers or residents near accidental spills became the focus of researchers because TCDD was known to be highly toxic from traditional toxicological studies (20). Although direct comparison of results was difficult due to the diverse methodologies of studies, positive, inverse, and null associations were observed in the earliest human studies on TCDD and T2D (Table 2). There were also some puzzling findings, described below.
Table 2.
Summary of Epidemiological Findings on Associations Between TCDD and T2D in Workers in Occupational Settings or Residents Near Accidental Spills
| Settings | First Author, Year (Ref.) | Study Population | Assessment of T2D | Results |
|---|---|---|---|---|
| Occupational exposure to Agent Orange in Vietnam | Henriksen, 1997 (22)a | US Air Force veterans exposed to Agent Orange during aerial spraying of herbicides from fixed wing aircraft | Physician diagnosis | Positive association |
| Kim, 2003 (246) | Korean veterans exposed to Agent Orange | Self-report of physician diagnosis | Positive association | |
| Kang, 2006 (247) | US Army Chemical Corps veterans exposed to Agent Orange while handling herbicides around base camp and spraying from helicopters | Self-report of physician diagnosis | Positive association | |
| Occupational exposure in plants | Steenland, 1999 (248) | Workers at 12 US plants that produced TCDD-contaminated products | Mortality | No or inverse association |
| Calvert, 1999 (26) | Workers employed more than 15 y in the production of sodium trichlorophenol or one of its derivatives at 2 US plants | Fasting glucose or self-report of physician diagnosis | No or positive association | |
| Sweeney, 1997 (27) | ||||
| Vena, 1998 (249) | Workers in phenoxy herbicide and chlorophenol production plants in 12 countries | Mortality | No association | |
| Accidental exposure | Zober, 1994 (28) | Workers after autoclave accident during the production of trichlorophenol in Germany in 1953 | Hospital admissions | Inverse association |
| Bertazzi, 1998 (25) | Residents after accident of chemical plant in Seveso, Italy, in 1976 | Mortality | No association in high-exposure area. Positive association in women living in medium-exposure area |
The most extensive study was performed among US Air Force veterans of Operation Ranch Hand (Air Force Health Study), the unit responsible for aerial spraying of herbicides, including Agent Orange contaminated with TCDD, during the Vietnam War from 1961 to 1971 (21). Compared with other US Air Force veterans without exposure to Agent Orange, exposed veterans had a serum dioxin level approximately three times higher in serum collected in 1987; exposed veterans also had a 40% higher risk of fasting or 2-hour postprandial glucose abnormalities, as well as a 50% higher risk of T2D (22). Based on the combined evidence from reports about the Air Force Health Study, T2D was listed as a compensable disease for Vietnam veterans exposed to Agent Orange (23).
However, subsequent reports on the Air Force Health Study revealed puzzling results. The new analyses excluded veterans exposed to Agent Orange and included only the comparison group of veterans who never had contact with TCDD-contaminated herbicides in Vietnam (24). Their serum TCDD levels were within the range of background exposure typical of the US general population. Unexpectedly, dose-response relationships between TCDD and T2D tended to be clearer than in the earlier study, which compared veterans with Agent Orange exposure to the comparison group (22).
The chemical plant accident near the town of Seveso, Italy, in 1976 also provided an opportunity to examine the association between exposure to TCDD and T2D (25). Although the interpretation was limited because death due to T2D was used as the study outcome, one puzzling finding was that residents living in the medium-exposure area showed a higher risk of T2D mortality than those living in the high-exposure area (25). In addition, studies among workers with occupational exposures to TCDD in factories mainly showed no association with T2D relative to the respective general populations used as reference groups, although these workers had about 10 times higher serum levels of TCDD than Operation Ranch Hand veterans with exposure to Agent Orange (26–28).
These earlier findings may be interpreted as providing insufficient evidence to conclude that there was an association between TCDD exposure and T2D; traditional causal reasoning postulates higher risk of disease among higher exposure groups. Furthermore, data from the Air Force Health Study were recently reanalyzed, and the original conclusion of a positive association between Agent Orange contaminated with TCDD and T2D was questioned (29). This issue will be revisited in Section II.D.
C. Recent evidence linking POPs and T2D: cross-sectional and case-control studies
Despite some evidence linking TCDD with T2D from the Air Force Health Study, POPs like OC pesticides or PCBs without dioxin activity were not immediately considered possible risk factors for T2D in the general population. Perhaps the focus on TCDD to the exclusion of other POPs was partly the result of believing that the pathway for adverse health effects of TCDD, including diabetes, was known, namely binding to the aryl hydrocarbon receptor (AhR), an intracellular ligand-dependent transcriptional factor expressed in most tissues of mammals (30). AhR-binding affinity and toxic potency are highly correlated in different congeners of POPs with dioxin activity (31). Because OC pesticides and non-dioxin-like PCBs have no affinity to AhR (32), they were supposed to be less harmful than dioxins. In addition, because the association between TCDD and T2D was not consistent even among persons with high exposure, as discussed above, the traditional toxicological viewpoint implied that any relation of non-dioxin-like POPs (like OC pesticides and some common PCBs) with T2D would be smaller than that of TCDD, or even negligible.
More recently, cross-sectional and case-control epidemiological studies have reported strong associations of T2D with OC pesticides and PCBs (8). Cross-sectional studies evaluate the status of both exposure and disease at one specific time point (33); eg, serum concentrations of POPs and the presence of T2D are measured at the same time. Case-control studies compare past exposure to a suspected risk factor among persons with the disease of interest and among a control group of persons without the disease (33). Usually, information about suspected risk factors is collected to reflect the person's exposure or experience before disease development begins. In case-control studies of POPs and T2D, however, serum concentrations of POPs have often been measured after having selected for study the cases who had already been diagnosed with T2D; hence, the temporal relationship between such concentrations and the status of T2D has often been as difficult to determine as in cross-sectional studies. Given the long half-life of POPs, it is generally considered that serum concentrations of POPs are a good reflection of lifetime exposures, including those that occur long before blood was drawn for the study. However, a bias may arise when serum concentrations of POPs are measured after T2D onset because concentrations may then be altered by the disease and, hence, have no etiological significance (33), as we will discuss in Section II.D.
Most cross-sectional studies have been performed among the general population rather than among workers or veterans. Background exposure in the general population is characterized by being low dose and long term, mostly throughout the lifetime; and exposure is to various POP mixtures as opposed to high-dose exposures to one or several selected POPs in occupational or accidental exposure settings. Because the production and use of most OC pesticides and PCBs were banned several decades ago, their average absolute levels in recent epidemiological studies were low in most of the population compared to earlier years (34, 35).
As shown in Table 3, evidence on T2D, OC pesticides, and PCBs has been reported from the United States, Canada, Sweden, Finland, Spain, Belgium, Japan, Korea, and the Slovak Republic. Most cross-sectional studies have reported a positive association for at least one POP, although the details have differed, especially relative to which POPs showed statistical significance. One noteworthy exception is a lack of association in a cross-sectional study of Greenland Inuits; however, the participants had much higher serum concentrations of POPs than in other populations (36). This study will be discussed in Section V.A.2.
Table 3.
Summary of Findings of Cross-sectional and Case-Control Epidemiological Studies on Associations Between Chlorinated POPs and T2D in General Populations With Background Exposure
| Country | Author, Year (Ref.) | Study Participants | Assessment of T2D (No. of Cases) | Adjustment | Exposure -Measured Chemicals -Median Valuesa or Geometric Means | Resultsb |
|---|---|---|---|---|---|---|
| Cross-sectional studies | ||||||
| Belgium | Fierens, 2003 (252) | 257 men and women | Self-report of physician diagnosis (n = 10) | None | -17 PCDD/PCDF congeners -25.2 pg/g lipid TEQ-based summary measure -4 Dioxin-like PCB congeners (PCB77, PCB81, PCB126, PCB169) -7.2 pg/g lipid TEQ-based summary measure -12 Non-dioxin-like PCB congeners (PCB3, PCB8, PCB28, PCB52, PCB101, PCB118, PCB138, PCB153, PCB180, PCB194, PCB206, PCB209) -402 ng/g lipid for total of 12 PCBs |
Based on TEQ-based summary measure OR = 5.1 (higher 10th % vs lower 90th %) Based on TEQ-based summary measure OR = 13.3 (higher 10th % vs lower 90th %) Based on absolute concentration-based summary measure OR = 7.6 (higher 10th % vs lower 90th %) |
| Canada | Philibert, 2009 (253) | 101 First Nation community | Self-report of physician diagnosis (n = 25) | Age, sex, birthplace, smoking, total cholesterol, and triglycerides | -p,p′-DDE -3.1 ng/g -8 PCB congeners (PCB74, PCB99, PCB118, PCB138, PCB153, PCB170, PCB180, PCB187) -4.0 ng/g |
OR = 6.1 (>75th % vs ≤ 75th %) Based on absolute concentration-based summary measure OR = 4.9 (>75th % vs ≤ 75th %) |
| Finland | Airaksinen, 2011 (40) | 1988 men and women | Fasting glucose, 2-h glucose, or DM medication (n = 308) | Age, sex, waist circumference, and mean arterial pressure | -PCB153 | OR = 1.6 (highest vs lowest decile) |
| -310 ng/g lipid | ||||||
| -Oxychlordane | OR = 2.1 (highest vs lowest decile) | |||||
| -12 ng/g lipid | ||||||
| -Trans-nonachlor | OR = 2.2 (highest vs lowest decile) | |||||
| -32 ng/g lipid | ||||||
| -p,p′-DDE | OR = 1.8 (highest vs lowest decile) | |||||
| -610 ng/g lipid | ||||||
| Greenland | Jørgensen, 2008 (36) | 692 men and women | Fasting glucose, 2-h glucose, or DM medication (n = 71) | Age, sex, ethnicity, waist circumference, physical activity, alcohol consumption, smoking, education | -3 Dioxin-like PCB congeners (PCB105, PCB118, PCB156) -135 ng/g lipid for PCB118 -10 Non-dioxin-like PCB congeners (PCB28, PCB52, PCB99, PCB101, PCB128, PCB138, PCB153, PCB163, PCB170, PCB180) -808 ng/g lipid for PCB153 -OCPs (Aldrin, Mirex, hexachlorobenzene, β-hexachlorocyclohexane, α-chlordane, γ-chlordane, oxychlordane, trans-nonachlor, cis-nonachlor, DDT, p,p′-DDE) -1500 ng/g lipid for p,p′-DDE |
Based on rank-based summary measurec Statistically nonsignificant Based on rank-based summary measurec Statistically nonsignificant Based on rank-based summary measurec Statistically nonsignificant |
| Japan | Uemura, 2009 (254) | 1374 men and women | HbA1C or self-report of physician diagnosis (n = 65) | Age, sex, BMI, smoking, regional block, residential area, and survey year | -17 PCDDs and PCDF congeners -12.0 pg/g lipid TEQ-based summary measure -12 Dioxin-like PCBs (PCB77, PCB81, PCB105, PCB114, PCB118, PCB123, PCB126, PCB156, PCB157, PCB167, PCB169, PCB189) −7.6 pg/g lipid TEQ-based summary measure |
Based on TEQ-based summary measure OR = 2.2 (1st vs 3rd tertile) Based on TEQ-based summary measure OR = 3.1 (2nd vs 1st tertile) OR = 6.8 (3rd vs 1st tertile) |
| Japan | Tanaka, 2011 (255) | 117 men and women (participants of the Saku Control Obesity Program) | Fasting glucose, HbA1C, DM medication, or self-report of physician diagnosis (n = 32) | Age, sex, BMI, total cholesterol, and triglycerides | -PCB74 | Statistically nonsignificant |
| -0.029 ng/g | ||||||
| -PCB99 | Statistically nonsignificant | |||||
| -0.022 ng/g | ||||||
| -PCB118 | Statistically nonsignificant | |||||
| -0.054 ng/g | ||||||
| -PCB138 | Statistically nonsignificant | |||||
| -0.074 ng/g | ||||||
| -PCB146 | OR = 1.59 per 1 pg/g | |||||
| -0.022 ng/g | ||||||
| -PCB153 | Statistically nonsignificant | |||||
| -0.15 ng/g | ||||||
| -PCB156 | Statistically nonsignificant | |||||
| -0.016 ng/g | ||||||
| -PCB163/164 | OR = 0.77 per 1 pg/g | |||||
| -0.036 ng/g | ||||||
| -PCB170 | Statistically nonsignificant | |||||
| -0.020 ng/g | ||||||
| -PCB180 | OR = 1.09 per 1 pg/g | |||||
| -0.076 ng/g | ||||||
| -PCB182/187 | Statistically nonsignificant | |||||
| -0.039 ng/g | ||||||
| Slovak Republic | Ukropec, 2010 (256) | 2047 men and women | Fasting glucose, 2-h glucose, or DM medication (n = 296) | Age, sex, BMI | -15 PCB congeners (PCB28, PCB52, PCB101, PCB105, PCB115, PCB118, PCB123, PCB138 + 163, PCB153, PCB156 + 171, PCB157, PCB167, PCB170, PCB180, PCB189) -1123 ng/g lipid for total 15 PCBs - p,p′-DDE -1817 ng/g lipid - p,p′-DDT -49.5 ng/g lipid -Hexachlorobenzene -669 ng/g lipid -β-Hexachlorocyclohexane -46.5 ng/g lipid |
Based on absolute concentration-based summary measure OR = 1.77 (4th vs 1st quintile) OR = 1.86 (5th vs 1st quintile) OR = 1.85 (3rd vs 1st quintile) OR = 1.94 (5th vs 1st quintile) OR = 1.84 (3rd vs 1st quintile) OR = 2.51 (4th vs 1st quintile) OR = 2.49 (5th vs 1st quintile) Statistically nonsignificant Statistically nonsignificant |
| Spain | Gasull, 2012 (41) | 886 men and women | Fasting glucose, DM medication, or self-report of physician diagnosis (n = 143) | Age, sex, BMI, total cholesterol, and triglycerides | -7 PCB congeners (PCB28, PCB52, PCB101, PCB118, PCB138, PCB153, PCB180) -1.76 ng/g for total 7 congeners -Hexachlorobenzene -1.19 ng/g -β-Hexachlorocyclohexane -0.67 ng/g - p,p′-DDT -0.18 ng/g - p,p′-DDE -2.63 ng/g |
Based on rank-based summary measure OR = 2.4 (4th vs 1st quartile) OR = 2.8 (4th vs 1st quartile) Statistically nonsignificant Statistically nonsignificant Statistically nonsignificant |
| Sweden | Rylander, 2005 (257) | 380 fishermen and their wives | DM medication or self-report of physician diagnosis (n = 22) | Age, BMI (both were dropped from the final model) | -PCB153 -360 ng/g lipid for men -240 ng/g lipid for women - p,p′-DDE -570 ng/g lipid for men -590 ng/g lipid for women |
OR = 1.2 (per 100 ng/g lipid) for men Statistically nonsignificant for women Statistically nonsignificant for men OR = 1.05 (per 100 ng/g lipid) for women |
| Sweden | Rignell-Hydbom, 2007 (258) | 543 women | DM medication or self-report of physician diagnosis (n = 15) | Age, BMI (BMI was dropped from the final model) | -PCB153 -82 ng/g lipid - p,p′-DDE -140 ng/g lipid |
Statistically nonsignificant OR = 1.3 (per 100 ng/g lipid) |
| United States | Lee, 2006 (37) | 2016 men and women | Fasting glucose, nonfasting glucose, or self-report of physician diagnosis (n = 217) | Age, sex, race, poverty income ratio, BMI, and waist circumference | -PCB153 -48.5 ng/g lipid -Oxychlordane -20.3 ng/g lipid -Trans-nonachlor -28.7 ng/g lipid - p,p′-DDE -504.5 ng/g lipid -1,2,3,4,6,7,8-PCDD -49.3 pg/g lipid -1,2,3,4,6,7,8,9-PCDD -418.5 pg/g lipid |
OR = 2.5 (1st quartile vs nondetectable) OR = 4.3 (2nd quartile vs nondetectable) OR = 5.9 (3rd quartile vs nondetectable) OR = 5.9 (4th quartile to 90th % vs nondetectable) OR = 6.8 (≥90th % vs nondetectable) OR = 3.1 (3rd quartile vs nondetectable) OR = 4.5 (4th quartile to 90th % vs nondetectable) OR = 6.5 (≥90th % vs nondetectable) OR = 2.5 (2nd quartile vs nondetectable) OR = 4.9 (3rd quartile vs nondetectable) OR = 7.6 (4th quartile to 90th % vs nondetectable) OR = 11.8 (≥90th % vs nondetectable) OR = 2.3 (4th quartile to 90th % vs nondetectable) OR = 4.3 (≥90th % vs nondetectable) OR = 2.7 (≥90th % vs nondetectable) OR = 2.2 (2nd quartile vs nondetectable) OR = 2.7 (4th quartile to 90th % vs nondetectable) |
| United States | Everett, 2007 (259) | 1830 men and women | HbA1C or self-report of physician diagnosis (n not provided) | Age, sex, race, country of birth, education, poverty income ratio, BMI, waist circumference, physical activity | -1,2,3,6,7,8-PCDD -70.6 ng/g lipid -PCB126 -57.5 pg/g lipid - p,p′-DDT -23.7 ng/g lipid |
OR = 1.77 (2nd vs 1st tertile) OR = 1.67 (2nd vs 1st tertile) OR = 3.68 (3rd vs 1st tertile) OR = 2.69 (2nd vs 1st tertile) OR = 2.46 (3rd vs 1st tertile) |
| United States | Cox, 2007 (260) | 1303 Mexican American men and women | Self-report of physician diagnosis (n = 89) | Age, BMI, and alcohol consumption | -Hexachlorobenzene -1.34 ng/g -Trans-nonachlor -1.52 ng/g - p,p′-DDT -3.22 ng/g - p,p′-DDE -9.00 ng/g -β-Hexachlorocyclohexane -1.70 ng/g -Oxychlordane -1.22 ng/g -Dieldrin -1.50 ng/g |
Statistically nonsignificant OR = 2.9 (detectable vs nondectectable) OR = 1.9 (detectable vs nondectectable) OR = 2.4 (3rd vs 1st quartile) OR = 2.6 (4th vs 1st quartile) OR = 2.1 (detectable vs nondectectable) OR = 3.1 (detectable vs nondectectable) Statistically nonsignificant |
| United States | Cordu, 2007 (261) | 352 Native American men and women | Fasting glucose or DM medication (n = 71) | Age, sex, BMI, smoking | -101 PCB congeners -580 ng/g lipid for 101 totalPCBs -PCB153 -78.3 ng/g lipid -PCB74 -28.0 ng/g lipid -p,p′-DDE -350 ng/g lipid -Hexachlorobenzene -11.1 ng/g lipid -Mirex -12.4 ng/g lipid |
OR = 3.2 (3rd vs 1st tertile) OR = 2.4 (3rd vs 1st tertile) OR = 4.5 (3rd vs 1st tertile) OR = 6.2 (3rd vs 1st tertile) OR = 6.8 (3rd vs 1st tertile) Statistically nonsignificant |
| United States | Turyk, 2009 (262) | 503 men and women | HbA1C, DM medication, or self-report of physician diagnosis (n = 61) | Age, sex, BMI, total cholesterol, and triglycerides | -13 PCB congeners (PCB74, PCB99, PCB118, PCB146, PCB180, PCB194, PCB201, PCB206, PCB132/153/105, PCB138/163, PCB170/190, PCB182/187, PCB196/203) -2.17 ng/g for total 13 PCBs -2 Dioxin-like PCB congeners (PCB118, 67) -0.15 ng/g for total of 2 PCBs -p,p′-DDE -3.01 ng/g |
Statistically nonsignificant Based on absolute concentration-based summary measure OR = 2.1 (4th vs 1st tertile) OR = 3.6 (4th vs 1st tertile) |
| United States | Silverstone, 2012 (263) | 774 men and women (residents living in PCB- contaminated area) | Fasting glucose or self-report of physician diagnosis (n = 207) | Age, BMI, race/ethnicity, family history of DM, taking lipid-lowering medication, total cholesterol, and triglycerides | -35 PCB congeners (PCB28, 44, 49, 52, 66, 74, 87, 99, 101, 105, 110, 118, 128, 138 + 158, 146, 149, 151, 153, 156, 157, 167, 170, 172, 177, 178, 180, 183, 187, 189, 194, 195, 196 + 203, 199, 206, 209) -3.18 ng/g for total 35 |
Based on absolute concentration-based summary measure OR = 1.52 (per 1 SD increase) for women Statistically nonsignificant for men |
| Case-control study | ||||||
| Korea | Son, 2010 (264) | 40 cases and 40 controls | Fasting glucose or DM medication (n = 40) | Age, sex, BMI, alcohol consumption, smoking, total cholesterol, and triglycerides | - p,p′-DDE | OR = 26.6 (3rd vs 1st tertile) |
| -1.6 ng/g | ||||||
| -p,p′-DDD | OR = 10.8 (3rd vs 1st tertile) | |||||
| -0.027 ng/g | ||||||
| -p,p′-DDT | OR = 7.6 (3rd vs 1st tertile) | |||||
| -0.13 ng/g | ||||||
| -o,p′-DDT | Statistically nonsignificant | |||||
| -0.013 ng/g | ||||||
| -Oxychlordane | Statistically nonsignificant | |||||
| -0.047 ng/g | ||||||
| -Trans-nonachlor | Statistically nonsignificant | |||||
| -0.096 ng/g | ||||||
| -Heptachlor epoxide | Statistically nonsignificant | |||||
| -0.037 ng/g | ||||||
| -β-Hexachlorocyclohexane | Statistically nonsignificant | |||||
| -0.22 ng/g | ||||||
| -Hexachlorobenzene | OR = 28.4 (3rd vs 1st tertile) | |||||
| -0.10 ng/g | ||||||
| -Mirex | OR = 6.5 (3rd vs 1st tertile) | |||||
| -0.012 ng/g |
Abbreviations: DM, diabetes mellitus; HbA1C, glycosylated hemoglobin; OCP, OC pesticide; OR, odds ratio.
When only cutoff points by categories were provided in the original article, median values over all subjects were crudely estimated (the center value of 2nd tertile in tertile categories, the upper cutoff value of the 2nd quartile in quartile categories, the center value of 3rd quintile in quintile categories, etc). When median values of subjects with and without T2D were presented separately, values among subjects without T2D were cited.
Statistically significant ORs were presented; nonsignificant findings were not presented.
Summary measures were calculated by summing individual rank of individual POPs belonging to each category.
The most striking evidence among studies with a cross-sectional design suggesting that POPs are associated with T2D was found in the US general population using the 1999–2002 dataset from the US National Health and Examination Survey (NHANES) (37). NHANES is well designed to be nationally representative of the noninstitutionalized US civilian population. In this study, the six POPs (two PCDDs, one PCB, and three metabolites of OC pesticides) most commonly detected in the US general population were strongly positively associated with the prevalence of T2D after adjusting for known risk factors, including obesity. The adjusted odds ratios for trans-nonachlor, oxychlordane, p,p′-dichlorodiphenyldichloroethylene (DDE) (the main metabolite of DDT), and PCB153 comparing the highest to lowest exposure categories were 6.5, 11.8, 4.3, and 6.8, respectively. When participants were classified by a measure summarizing the combined exposure to the six POPs, the prevalence of T2D rose to 10 to 40 times higher as the summary measure levels increased compared with subjects classified in the lowest quartile of the summary measure. Such strength of association is unusual in epidemiological studies.
In this study (37), the magnitude of association was much stronger with trans-nonachlor, oxychlordane, p,p′-DDE, and PCB153, which do not have dioxin activity, than with the PCDDs, which do have dioxin-like activity. In fact, the POPs' toxic equivalency factors (TEFs), which measure the ability of a given dioxin-like contaminant to bind to the AhR, were not related to the strengths of the associations between POPs and T2D.
Another provocative finding was the interaction between POPs and obesity on the risk of T2D (37) (Figure 1A). The association between POPs and T2D was stronger among more obese persons. There was also a clear positive association between POPs and T2D among normal-weight persons (ie, with body mass index [BMI] < 25 kg/m2). However, obesity was not associated with T2D among persons in the lowest quartile of the POPs summary measure, and T2D itself was very rare among individuals who had a low POP summary score, even if their BMI was ≥30 kg/m2. These findings could mean that POPs that have accumulated in adipose tissue may play a more critical role in the pathogenesis of T2D than the adipose tissue itself (38, 39).
Figure 1.

Interaction between BMI and POPs estimating the prevalence of T2D. A, United States (37): The summary measure of six POPs was calculated by summing individual rank of six POPs (1,2,3,4,6,7,8-heptachlorodibenzo-p-dioxin, 1,2,3,4,6,7,8,9-octachlorodibenzo-p-dioxin, p,p′-DDE, oxychlordane, trans-nonachlor, and PCB153). The summary measure was classified into five quintiles from Q1 to Q5. Among persons with the lowest quintile (Q1) of the summary POPs, BMI was not associated with the risk of T2D, and T2D itself was very rare even among obese. In addition, the risk of diabetes increased with increasing concentrations of POPs even among lean persons. The highest risk was observed in persons with high POPs and high BMI. B, Finland (40): Because only results based on individual POP, not summary measures of POPs, were presented in the paper, we selected trans-nonachlor, which showed the strongest association with T2D for this figure. Serum concentrations of trans-nonachlor were divided into <0th, 10-<50th, 50-<90th, and ≥ 90th percentiles. The positive associations of T2D with POPs became stronger as BMI increased, similar to those in the United States. However, obesity was associated with T2D even among persons with the lowest levels of trans-nonachlor. C, Spain (41): The summary measure of four PCBs was calculated by summing individual rank of four PCBs (PCB118, PCB138, PCB153, and PCB180). The summary measure was classified into four quartiles from Q1 to Q4. All odds ratios were computed with Q1 and normal weight as the reference category, with models adjusted by age, sex, total cholesterol, and triglycerides. Results were similar with those from Finland.
Recent studies from Finland and Spain (40, 41) showed that the positive associations of T2D with POPs became stronger as BMI increased, in line with the findings from the NHANES dataset. Also, in each category of BMI the prevalence of T2D increased with increasing concentrations of POPs (Figure 1, B and C). However, obesity was associated with T2D among subjects with both high and low POP concentrations (40, 41). In Finland and Spain, serum concentrations of POPs—in particular, PCBs—were substantially higher than in the United States. When low and high POP groups were defined in each study, classifications were based on the relative distributions of POP concentrations within each population. In fact, groups in Finland and Spain with lower concentrations of POPs had higher concentrations than groups in the United States with the lowest levels. Therefore, the fact that the positive association between obesity and T2D was still observed even among low POPs groups in Finland and Spain may be attributable to a lack of subjects with very low concentrations of POPs similar to the low POP group in the United States. Alternatively, the complete lack of association between obesity and T2D among subjects with very low serum concentrations of POPs in the United States may be a chance finding that requires replication through studies examining a large number of people with relatively low serum concentrations of POPs across a wide range of obesity.
D. Reverse causality due to disease progression bias
Even when strong positive associations are observed in cross-sectional studies, attention should be paid to the possibility of “reverse causality,” which may be due to mechanisms like “disease progression bias” (33). There is a possibility that subclinical or overt progress of T2D might alter the metabolism of POPs, slowing their excretion from the body or increasing release of POPs from adipose tissue. Under this hypothesis, T2D would increase serum levels of POPs, rather than POPs increasing the risk of T2D (11).
The possibility of T2D slowing the excretion of POPs from the body was not supported by two studies that revealed no change in the rate of elimination of POPs from blood in relation to the presence or duration of T2D (42, 43). Nevertheless, given that patients with T2D are often obese, one mechanism that might lead to increased serum concentrations of POPs in T2D in cross-sectional studies is increased lipolysis, which releases POPs stored in adipose tissue (18).
This issue was addressed in a recent article that reanalyzed associations between TCDD and T2D from the Air Force Health Study among veterans who had multiple blood samples drawn over a 20-year period (29). The reanalysis suggested that increases of serum TCDD levels (compared to previous levels measured during the monitoring period) were slightly more common in veterans with diabetes (33%; 44 of 135) than in veterans without diabetes (26%; 95 of 405), thus identifying a possible reverse causal pathway. These authors also presented trends of increasing incidence of T2D according to deciles of serum concentrations of TCDD within Vietnam veterans exposed to Agent Orange; such reported incidences were similar to the trends over deciles in comparison veterans, despite large differences in TCDD concentrations between the two groups. The authors took these findings as evidence of reverse causality and further speculated that this finding applies to other persistent lipophilic chemicals such as OC pesticides or PCBs.
However, despite some evidence suggesting the existence of a reverse pathway (T2D causing elevated POPs), the magnitude of the reverse effect does not seem to be large, mainly because increases in serum TCDD levels during the monitoring period were common even among veterans without T2D. Furthermore, methodological issues like the inverted U-shaped association and/or the characteristics of POP mixtures need to be considered when interpreting findings of the Air Force Study, as will be done in Section V.
In general terms, the Air Force Health Study is classifiable as a prospective study. However, it is based on retrospective estimation of baseline levels of TCDD from about 1962 to 1972, derived from TCDD levels measured in 1987, and it evaluated associations with T2D diagnosed between the end of each veteran's Vietnam tour and 2002 (43). In some T2D patients, the TCDD measurement in 1987 could have been made after disease onset. Thus, similar to cross-sectional and case-control studies, in this study, it is difficult to evaluate temporality of presumed cause and effect. A full assessment of causality of POPs and T2D will require evidence from prospective epidemiological studies, as well as in vitro and in vivo experimental studies.
E. Prospective evidence linking POPs and T2D
Since publication of the cross-sectional associations of OC pesticides or PCBs with T2D, several prospective studies have been performed in general populations (42, 44–49). In prospective studies, researchers can be fairly certain that exposure precedes the development of diseases (33) because serum concentrations of POPs are measured in serum collected from individuals years before the development of T2D. In Table 4, we have summarized the main findings from six prospective studies performed in general populations with background exposures to POPs.
Table 4.
Prospective Studies Concerning Chlorinated POPs and the Risk of Incident T2D in General Populations With Background Exposure
| Country | First Author, Year (Ref.) | Study Participants: -No. of Subjects -Characteristics -Age at Baseline | Baseline Year | Follow-Up Period | Outcome: -Diagnosis -No. of Cases | Adjustment | Exposure -Measured Chemicals -Median Valuesa | Resultsb -Relative Risk (RR) (95% Confidence Interval) |
|---|---|---|---|---|---|---|---|---|
| Prospective cohort studies | ||||||||
| Sweden | Lee, 2011 (44) | -725 men and women -General population -70 y old |
2001–2004 | ∼5 y | -36 cases -Fasting glucose or DM medication |
Sex, BMI, smoking, alcohol consumption, physical activity, total cholesterol, and triglycerides | -PCB74, PCB99, PCB105, PCB118, PCB138, PCB153, PCB156, PCB157, PCB170, PCB180, PCB189, PCB194, PCB206, PCB209 -1.4 ng/g for PCB153 -p,p′-DDE, trans-nonachlor, hexachlorobenzene -2.0 ng/g for p,p′-DDE -Octachlorodibenzo-p -dioxin -0.0025 ng/g |
Based on rank-based summary measure of 14 PCBsc RR = 4.5 (0.9–23.5) (2nd vs 1st quintile) RR = 5.1 (1.0–26.0) (3rd vs 1st quintile) RR = 8.8 (1.8–42.7) (4th vs 1st quintile) RR = 7.5 (1.4–38.8) (5th vs 1st quintile) Based on rank-based summary measure of 3 OCPsc RR = 1.1 (0.3–4.5) (2nd vs 1st quintile) RR = 1.6 (0.4–5.8) (3rd vs 1st quintile) RR = 1.5 (0.4–5.8) (4th vs 1st quintile) RR = 3.4 (1.0–11.7) (5th vs 1st quintile) Statistically nonsignificant |
| United States | Vasiliu, 2006 (47) | -1384 men and women -Residents who consumed animal products that were exposed to PBBs -≥20 y old |
1976 | ∼25 y | -180 cases -Known DM |
Age, BMI, smoking, alcohol consumption | -Total PCBs -7 ng/g |
Only among women RR = 2.0 (1.1–3.8) (2nd vs 1st quartile) RR = 2.3 (1.2–4.3) (3rd vs 1st quartile) RR = 2.3 (1.3–4.3) (4th vs 1st quartile) |
| United States | Turyk, 2009 (42) | -471 men and women -Great Lakes Sport Fish Consumers -Mean age: ≈ 50 y old -3.2 ng/g |
1994–1995 | ∼11 y | -36 cases -2.4 ng/g -Self-report of physician diagnosis |
Age, sex, BMI | -Total PCBs -2.4 ng/g -PCB118 -0.15 ng/g -p,p′-DDE -3.2 ng/g |
Statistically nonsignificant Statistically nonsignificant RR = 5.5 (1.2–25.1) (2nd vs 1st tertile) RR = 7.1 (1.6–31.9) (3rd vs 1st tertile) |
| United States | Wu, 2013 (49) | -1095 women -Participants in two previous case-control studies (Breast Cancer Study and Non-Hodgkin Lymphoma Study) from the Nurses' Health Study -Mean age:: ≈ 59 y old |
1989–1990 | ∼19 y | -48 cases -Known DM |
Age, BMI, family history of DM, smoking, alcohol consumption, physical activity, case-control status | -PCB118, PCB138, PCB153, PCB180, total PCBs -104.5–106.1 ng/g lipid for PCB153 |
Statistically nonsignificant |
| -p,p′-DDE -773.6–989.6 ng/g lipid |
Statistically nonsignificant | |||||||
| -p,p′-DDT -53.1–43.7 ng/g lipid |
Statistically nonsignificant | |||||||
| -Hexachlorobenzene -29.8–37.0 ng/g lipid |
RR = 1.5 (0.6–3.9) (2nd vs 1st tertile) RR = 3.1 (1.3–7.7) (3rd vs 1st tertile) |
|||||||
| Nested case-control studies | ||||||||
| Sweden | Rignell-Hydbom, 2009 (46) | -372 cases and 371 controls (men and women) -General population -50–59 y old |
1995–2000 | ∼11 y | -372cases -Linkage with the Swedish inpatient and outpatient registers |
Age, calendar year, BMI (through matching) | -PCB153 -1.3 ng/g -p,p′-DDE -2.9 ng/g |
Statistically nonsignificant Only 39 cases diagnosed ≥ 7 y after baseline RR = 5.5 (1.2–25.0) (≥75th % vs <75th %) |
| United States | Lee, 2010 (45) | -90 cases and 90 control (men and women) -General population -18–30 y old |
1987–1988 | ∼18 y | -90 cases -Fasting glucose ≥126 mg/dL or known DM |
Age, sex, race, BMI, total cholesterol, and triglycerides |
-PCB74, PCB87, PCB99, PCB105, PCB118, PCB146, PCB153, PCB156, PCB157, PCB138–158, PCB167, PCB170, PCB178, PCB180, PCB183, PCB187, PCB194, PCB195, PCB199, PCB196–203, PCB206, PCB209 -0.4 ng/g for PCB153 -p,p′-DDE, p,p′-DDT, trans-nonachlor, oxychlordane, hexachlorobenzene, β-hexachlorocyclohexane, γ-hexachlorocyclohexane, mirex -3.3 ng/g for p,p′-DDE |
Based on rank-based summary measure of 22 PCBsc,d RR = 4.4 (1.3–14.8) (2nd vs 1st sextile) RR = 2.7 (0.8–8.8) (3rd vs 1st sextile) RR = 1.7 (0.5–5.7) (4th vs 1st sextile) RR = 1.2 (0.3–4.4) (5th vs 1st sextile) RR = 1.1 (0.3–4.6) (6th vs 1st sextile) Based on rank-based summary measure of 8 OCPsc,d Statistically nonsignificant Results on trans-nonachlor RR = 4.3 (1.5–12.6) (2nd vs 1st quartile) RR = 2.3 (0.7–7.4) (3rd vs 1st quartile) RR = 2.0 (0.6–6.9) (4th vs 1st quartile) |
Abbreviations: DM, diabetes mellitus; OCPs, OC pesticides; OR, odds ratio; PBB, polybrominated biphenyl.
When only cutoff points by categories were provided in the original article, median values over all subjects were crudely estimated (the center value of the 2nd tertile in tertile categories, the upper cutoff value of the 2nd quartile in quartile categories, the center value of 3rd quintile in quintile categories, etc). When median values of subjects with and without T2D were presented separately, values among subjects without T2D were cited. Median values of the Wu et al study (49) were shown as ranges because they used datasets from two case-control studies and presented concentrations of POPs respectively.
When there were statistically significant specific RRs, all RRs were presented with 95% confidence intervals.
Summary measures were calculated by summing individual rank of individual POPs belonging to each category.
Results not previously published. Findings were recalculated using the raw dataset because the original article reported results on summary measures over all POPs, not PCBs or OCPs separately. Summary measures were calculated by summing individual rank of individual POPs belonging to each category
Although all prospective studies concluded that some POPs or combinations of POPs predicted the future risk of T2D, no single POP significantly predicted T2D in all prospective studies. In the oldest cohort studies, just one or a few POPs were measured as a “surrogate marker” of all POPs (42, 46, 47), which is a significant limitation. By contrast, recent cohort studies have measured a much larger variety of POPs and used improved technology for such measurement (44, 45). Similar to cross-sectional or case-control studies, the interpretation of findings for individual POPs in human studies should also be cautious in prospective studies, given that humans are virtually always exposed to correlated POP mixtures. This issue will be discussed further in Section V.B.
Considering the characteristics of POPs as mixture, two prospective studies, Coronary Artery Risk Development in Young Adults (CARDIA) (45) and Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) (44), created a summary measure of POPs using the sum of ranks of various POPs rather than focusing on individual POPs. Results demonstrated that POP mixtures were associated with about three to five times higher T2D risk, depending on study subjects and exposure ranges. When the predictive power of POP mixture vs waist circumference was compared in the elderly, POPs were as strongly predictive of T2D as was waist circumference (Figure 2).
Figure 2.
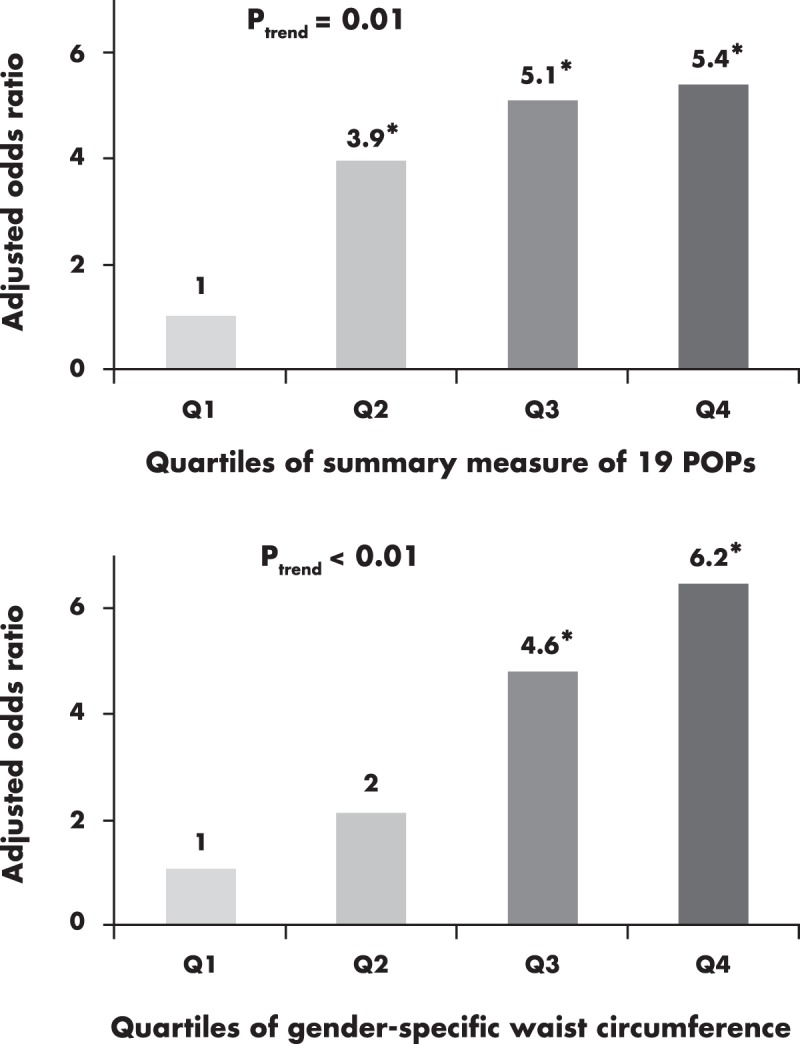
Comparison of strength of associations with T2D between a summary measure of 19 POPs and gender-specific waist circumference in the PIVUS (the Prospective Investigation of the Vasculature in Uppsala Seniors) cohort study (44). The summary measure was calculated by summing the individual rank of 19 POPs. Results were recalculated using the raw dataset because the original article (44) reported results on summary measures of subclasses of POPs (PCBs or OC pesticides), not all POPs. These findings use the methodology described in the original article (44) but have not been published before. Both POPs and gender-specific waist circumference were included in the same model and adjusted for gender, physical activity, cigarette smoking, alcohol consumption, total cholesterol, and triglyceride. Statistically significant odds ratios are indicated with asterisks.
In the CARDIA study, subjects were categorized by sextiles, not conventional quartiles, in an attempt to isolate subjects with very low concentrations of multiple POPs in the lowest sextile of the summary measure. When the quartile approach was used, there was no association between POP mixture and T2D (the adjusted odds ratios were 1.0, 1.4, 0.7, and 0.5). However, the association became significant and stronger with the sextile approach, with adjusted odds ratios of 1.0, 2.8, 3.7, 1.2, 1.3, and 1.0 (P value for quadratic term = .04) (Figure 3). We will also discuss this issue in Section V.B.
Figure 3.

Comparison of quartile vs sextile approaches to the associations between a summary measure of 31 POPs on T2D in the Coronary Artery Risk Development in Young Adults (CARDIA) cohort study (45). The summary measure was calculated by summing individual rank of 31 POPs. When the summary measure was classified into quartiles, there was no association between POPs and T2D. However, with classification into sextiles, low-dose POPs significantly predicted the future risk of T2D. The authors interpret this finding to have occurred because the sextile reference group was closer to a true hypothetical reference group without exposure to POPs than the quartile reference group. Results were adjusted for age, gender, race, BMI, total cholesterol, and triglyceride. Statistically significant odds ratios are indicated with an asterisk.
On the other hand, whereas the CARDIA study showed an inverted U-shaped association (45), the other prospective study, PIVUS (44), did not show a clear inverted U-shaped association (Figure 2). However, it did suggest nonlinearity; the risk of T2D was substantially increased with only a slight increase of concentrations of POPs within the lower dose range of POPs and only a slight increase in risk with increasing dose of POPs within higher doses of POPs.
One big difference between the CARDIA study and the PIVUS study was the age distribution of study subjects. At baseline, the PIVUS subjects were elderly adults aged 70 years, whereas the CARDIA subjects were young adults aged 18 to 30 years. Patients diagnosed with T2D at an advanced age have particular characteristics; they tend to be less obese and to have more β-cell dysfunction (50, 51). Thus, when the pathogenesis of T2D is classified into two stages, insulin resistance and insulin secretory defects, the relative importance of each stage might be different depending on the age at which T2D develops, which makes it difficult to produce exactly the same dose-response relations between these two prospective studies. In addition, physiological differences between the young and the elderly may play a mechanistic role, which will be discussed in Section V.A.
III. Experimental Evidence Linking POPs and T2D
In this section, we focus on experimental findings of subchronic/chronic exposure to low-dose POP mixtures that approximate current human exposure patterns. One recent in vivo and in vitro experimental study confirmed a causal relationship between POP mixtures and T2D- related metabolic dysfunction (52). In this experiment, scientists utilized POP-contaminated fish oil, not artificial mixtures of several POPs. Because POP-contaminated food is the main external exposure source of POPs for humans, this approach tends to mimic the current human exposure pattern in terms of mixture and doses of POPs. Adult rats were fed crude salmon oil containing environmental levels of POPs for 4 weeks. In this exposure period, the rats developed insulin resistance, visceral obesity, dyslipidemia, nonalcoholic fatty liver, and chronic low-grade inflammation, whereas animals fed salmon oil from the same source, but refined to achieve low POP concentrations, did not. Importantly, body burdens of POPs in these rats were similar to those observed in Northern Europeans at 40 to 50+ years of age (53), suggesting a strong relevance of these experimental findings to humans. Also, because this experiment was performed with adult rats, it suggests that chronic exposure to low-dose POP mixtures during adulthood can induce all of these T2D-related abnormalities.
These authors complemented their in vivo rat study with additional in vitro tests (52). They incubated differentiated 3T3-L1 adipocytes with artificially combined POP mixtures including OC pesticides, PCBs, PCDDs, and PCDFs within specified dose ranges. The authors observed reduced insulin action after treatment with both OC pesticides and PCBs, whereas PCDDs or PCDFs with dioxin activity had no effect on this endpoint. Interestingly, impaired insulin action was observed with 1 nm of mixed OC pesticides and PCBs; there were no clear dose-response relations on insulin action with 10 or 100 times higher concentrations, suggesting low-dose effects and nonmonotonic dose-response curves. The authors' approach is appropriate for the study of POP mixtures because each treatment consisted of several compounds belonging to specific subclasses of POPs. However, these treatments differed from the mixture in the POP-contaminated fish oil, which was a combination of all subclasses of POPs. Therefore, the relevance of findings from the in vitro aspect of this experiment to humans may be less than those of the in vivo animal experiments. Despite this limitation, this study provides an opportunity to evaluate what kinds of POPs might be more relevant to the development of T2D and what dose-response relationships might be expected.
Two further experimental studies were performed by the same researchers (54, 55). One study evaluated the effects of chronic consumption of farmed-salmon fillet in mice (54), whereas the other fed animals whale meat (55). Unlike their earlier experimental study that focused on crude fish oil contaminated with POPs, these studies evaluated effects of fish or mammal consumption as a whole food, considering both possible harmful effects of POPs contaminating food and possible beneficial effects of nutrients naturally contained in the food. Feeding adult mice the farmed-salmon fillet containing POP contaminants for 8 weeks caused visceral obesity and accelerated the development of insulin resistance and glucose intolerance (54). When POP levels in farmed-salmon fillet were reduced by decontamination, the experimental mice had a lower body burden of POPs, less visceral fat, and better insulin sensitivity and glucose tolerance (54). In contrast, surprisingly, feeding animals POP-contaminated whale meat improved insulin sensitivity and glucose tolerance and reduced body weight (55). Importantly, the extent of POP contamination of the whale meat was 10 to 15 times higher than that of the farmed-salmon fillet. Although the different nutrient profiles between salmon and whale meat could contribute to these contradictory results, the authors interpreted that low-dose POPs can be more harmful than high-dose POPs, similar to the expectation from epidemiological studies that show inverted U-shaped relationships.
IV. POPs and Obesity
A. POPs as obesogens
In 2002, Paula Baillie-Hamilton (56) proposed a hypothesis linking exposure to chemicals with obesity, and this premise is now gaining credence. Historically, the main purpose of measuring the weight of experimental animals was to gather basic information on their general health, and toxicologists were mainly concerned about weight loss as a sign of toxicity. When a wide range of chemical concentrations were tested in animals, weight gain effects were observed after treatment at low concentrations to a variety of chemicals including POPs. However, a significant amount of this evidence was ignored in early studies (56).
In 2006, weight gain effects of chemicals were reformulated under the “obesogen hypothesis,” a term coined by Bruce Blumberg (57). Obesogens can be functionally defined as chemicals that alter homeostatic metabolic setpoints, disrupt appetite controls, perturb lipid homeostasis to promote adipocyte hypertrophy, stimulate adipogenic pathways that enhance adipocyte hyperplasia, or otherwise alter adipocyte differentiation during development (57, 58). In particular, recent experimental studies have mainly focused on prenatal or early life exposure to chemicals during critical periods of cell and organ differentiation because these exposures can alter developmental programming of the endocrine controls of metabolism including the differentiation of adipocytes, resulting in obesity in the future (58). These proposed pathways include inappropriate modulation of nuclear receptor function; therefore, putative obesogens can be classified as endocrine-disrupting chemicals (EDCs).
Possible candidate obesogens encompass a wide range of compounds with different chemical and physical properties like organotins, organophosphates, bisphenol A, phthalates, and heavy metals (59). It is highly likely that there are many other chemicals in the environment that increase the risk of obesity but have yet to be recognized. Because a detailed analysis on various obesogens is outside the scope of this article, we will only focus on chlorinated POPs.
Earlier experimental studies on POPs mainly characterized a TCDD-related severe weight loss; one hallmark of TCDD systemic toxicity is a wasting syndrome (60). In vitro experiments have shown an inhibition of adipogenesis with the treatment of approximately 10 nm TCDD (61, 62). In fact, biological changes accompanying the wasting syndrome are diabetogenic phenotypes such as hyperlipidemia and decreased glucose uptake (63, 64). Because these treatment doses are close to overt toxicity levels, these findings are not relevant to the typical human population. However, the diabetogenic phenotype produced by inhibiting adipogenesis is important and will be discussed in Section IV.B.
On the other hand, a recent experimental study reported the importance of dose in inducing obesity (65). Similar to findings from other obesogens, treatment of cultured cells with low-dose PCB77 with dioxin activity (3.4 μm) or TCDD (0.1 nm, a concentration 100 times lower than the earlier experimental studies) induced adipogenesis, whereas higher doses of either compound inhibited it. In this experiment, the effects of PCB77 were abolished by an AhR antagonist; thus, the authors interpreted their findings as suggesting that low-dose dioxin-like chemicals can induce weight gain through AhR-mediated responses.
However, experimental studies on OC pesticides with no dioxin-like activities also showed effects on weight, indicating that there could be AhR-independent mechanisms of POPs. For example, a study of p,p′-DDT reported a concentration-dependent increased adipogenesis from 1 to 50 μm (66). However, endrin (a cyclidiene pesticide) caused a dose-dependent inhibition of adipocyte differentiation from 0.1 to 30 μm (67). Other cyclidiene pesticides like aldrin and dieldrin also showed adipogenesis-inhibiting effects (67). Neither p,p′-DDE nor oxychlordane (2 or 20 μm) had a significant effect on adipogenesis itself, but both promoted adipocyte fatty acid uptake in mature adipocytes, leading to the promotion of adipocyte hypertrophy (68).
Different effects on adipogenesis—depending on the kind and dose of compound studied—make it very difficult to assess the relevance of experimental findings on one specific POP to humans. Therefore, we focused on recent animal studies that have utilized POP mixtures with human relevant doses. These studies have revealed that chronic exposure to POP mixtures in rats or mice during adulthood led to increased visceral fat (52, 54). In another experiment using a zebrafish model, lifelong exposure to POP mixtures induced weight gain (69, 70). Therefore, whatever effects specific individual POPs may have shown in experimental settings, the net result of POP mixtures similar to current human exposure levels may be weight gain.
Despite the experimental evidence on POP mixtures from laboratory animals, human evidence on POPs is diverse (Table 5). When human studies are classified into early life exposure (including intrauterine) and adult exposures, results on the association between POPs and obesity are less consistent with early life exposure than with adult exposure, although direct comparisons among studies are difficult due to substantial methodological differences.
Table 5.
Associations Between Chlorinated POPs and Obesity in Human Studies
| Compound | Summary of Results |
||
|---|---|---|---|
| Positive Associations | Inverse Associations | Null Associations | |
| Prenatal or early exposure | |||
| PCBs | Hertz-Picciotto, 2005 (F) (265) | Blanck, 2002 (268) | Cupul-Uicab, 2010 (272) |
| Valvi, 2012 (F) (266) | Burns, 2012 (M) (269) | Gladen, 2000 (F) (273)a | |
| Verhulst, 2009 (267) | Jacobson, 1990 (270) | Gladen, 2000 (M) (273) | |
| Lamb, 2006 (F) (271) | Hertz-Picciotto, 2005 (M) (265) | ||
| Karmaus, 2009 (F) (274) | |||
| Lamb, 2006 (M) (271) | |||
| Mendez, 2011 (275) | |||
| Patandin, 1998 (276) | |||
| Valvi, 2012 (M) (266) | |||
| DDT | Gladen, 2000 (p,p′-DDE) (M) (273) Karmaus, 2009 (p,p′-DDE) (F) (274) Mendez, 2011 (275) Valvi, 2012 (p,p′-DDT) (M) (266)b Valvi, 2012 (p,p′-DDE) (F) (266)b Verhulst, 2009 (p,p′-DDE) (267)a |
Burns, 2012 (p,p′-DDE) (M) (269) | Cupul-Uicab, 2010 (p,p′-DDE) (M) (272) Cupul-Uicab, 2010 (272) Gladen, 2000 (p,p′-DDE) (F) (273) Gladen, 2004 (p,p′-DDE, p,p′-DDT) (M) (277) Jusko, 2006 (p,p′-DDE, p,p′-DDT, o,p′-DDT) (278) Valvi, 2012 (p,p′-DDE) (M) (266) Valvi, 2012 (p,p′-DDT) (W) (266) Warner, 2013 (p,p′-DDE, p,p′-DDT, o,p′-DDT) (279) |
| Hexachlorobenzene | Smink, 2008 (280) | Burns, 2012 (M) (269) | Cupul-Uicab, 2010 (272) |
| Mendez, 2011 (275) | |||
| Verhulst, 2009 (267) | |||
| β-Hexachlorocyclohexane | Burns, 2012 (M) (269) | Cupul-Uicab, 2010 (272) | |
| Mendez, 2011 (275) | |||
| Heptachlor epoxide | Cupul-Uicab, 2010 (272) | ||
| Chlordane | Cupul-Uicab, 2010 (oxychlordane, trans-nonachlor) (272) | ||
| Dieldrin | Cupul-Uicab, 2010 (272) | ||
| TEQ | Burns, 2012 (M) (269) | Patandin, 1998 (276) | |
| Verhulst, 2009 (267) | |||
| PCDDs/PCDFs | Burns, 2012 (M) (269) | ||
| Adult exposure, cross-sectional studies | |||
| PCBs | Glynn, 2003 (PCB105, PCB118) (281) Hovinga, 1993 (M) (282) Hue, 2007 (PCB138) (283 Lee, 2012 (PCB72, PCB99, PCB105, PCB118) (M) (78) Pelletier, 2002 (PCB28, PCB99, PCB118, PCB138, PCB153) (284) |
Bachelet, 2011 (285) De Roos, 2012 (PCB146, PCB153, PCB156, PCB170, PCB180, PCB183, PCB194, PCB196/203, PCB199, PCB206, PCB209) (286) Dirinck, 2011 (PCB153, PCB170, PCB180) (292) Glynn, 2003 (PCB156, PCB180) (281) Hue, 2007 (PCB153, PCB156, PCB170, PCB180, PCB187) (283) Ibarluzea, 2011 (PCB138, PCB153, PCB180) (287) Lee, 2006 (PCB153) (37) |
De Roos, 2012 (PCB74, PCB99, PCB105, PCB118, PCB138/158, PCB187) (286) Glynn, 2003 (PCB138, PCB153, PCB167) (281) Hovinga, 1993 (F) (282) Hue, 2007 (PCB99, PCB118) (283) Lee, 2012 (PCB74, PCB99, PCB105, PCB118) (F) (78) Lee, 2012 (PCB126, PCB138, PCB153, PCB156, PCB157) (M) (78) Pelletier, 2002 (PCB156, PCB170, PCB180, PCB187) (284) |
| Lee, 2012 (PCB126, PCB138, PCB153, PCB156, PCB157, PCB169, PCB170, PCB180, PCB189, PCB194, PCB206, PCB209) (F) (78) | |||
| Lee, 2012 (PCB169, PCB170, PCB180, PCB189, PCB194, PCB206, PCB209) (M) (78) | |||
| Sandanger, 2007 (PCB153) (288) | |||
| DDT | Bachelet, 2011 (p,p′-DDE) (285) Glynn, 2003 (p,p′-DDE) (281) Hovinga, 1993 (282) Hue, 2007 (p,p′-DDE) (283) Jakszyn, 2009 (p,p′-DDE) (289) Lee, 2006 (p,p′-DDE) (37) Lee, 2012 (p,p′-DDE) (M) (78) Lee, 2012 (p,p′-DDE) (F) (78) Pelletier, 2002 (p,p′-DDE) (284) Schildkraut, 1999 (p,p′-DDE) (290) |
Bradman, 2007 (p,p′-DDE, p,p′-DDT, o,p′-DDT) (291) De Roos, 2012 (p,p′-DDE) (286) Dirinck, 2011 (p,p′-DDE) (292) Ibarluzea, 2011 (p,p′-DDE) (287) Pelletier, 2002 (p,p′-DDT) (284) |
|
| Chlordane | Hue, 2007 (oxychlordane) (283) Lee, 2012 (trans-nonachlor) (M) (78) Pelletier, 2002 (oxychlordane, trans-nonachlor) (284) |
De Roos, 2012 (oxychlordane, trans-nonachlor) (286) Glynn, 2003 (281) Hue, 2007 (trans -nonachlor) (283) Lee, 2006 (oxychlordane, trans-nonachlor) (37) Lee, 2012 (trans-nonachlor) (F) (78) |
|
| Hexachlorobenzene | Glynn, 2003 (281) | Bradman, 2007 (291) | |
| Ibarluzea, 2011 (287) | Hue, 2007 (283) | ||
| Jakszyn, 2009 (289) | Lee, 2012 (F) (78) | ||
| Lee, 2012 (M) (78) | |||
| Pelletier, 2002 (284) | |||
| β-Hexachlorocyclohexane | Dirinck, 2011 (292) | Bradman, 2007 (291) | |
| Glynn, 2003 (281) | |||
| Hue, 2007 (283) | |||
| Ibarluzea, 2011 (287) | |||
| Jakszyn, 2009 (289) | |||
| Pelletier, 2002 (284) | |||
| Mirex | Pelletier, 2002 (284) | ||
| PCDDs/PCDFs | Lee, 2006 (HpCDD, OCDD) (37) | Lee, 2012 (OCDD) (F) (78) | |
| Lee, 2012 (OCDD) (M) (78) | |||
| Adult exposure, prospective studies | |||
| PCBs | Lee, 2011 (PCB153b,c, PCB156, PCB170b, PCB178b,c, PCB180b, PCB187b,c, PCB194, PCB195b,c, PCB199b,c, PCB196–203b, PCB206b, PCB209) (79) Lee, 2012 (PCB74b, PCB99b,c, PCB118b,c, PCB138b, PCB153b, PCB156) (F) (78) Lee, 2012 (PCB156c, PCB157b,c, PCB169c, PCB180, PCB189c, PCB209b) (M) (78) |
Lee, 2012 (PCB156, PCB157c, PCB169c, PCB170, PCB180, PCB189, PCB194, PCB206 PCB209b,c) (F) (78) Lee, 2012 (PCB180) (M) (78) |
Lee, 2011 (PCB74, PCB87, PCB99, PCB105, PCB118, PCB146, , PCB157, PCB138–158, PCB167, PCB183) (79) Lee, 2012 (PCB105, PCB126) (F) (78) Lee, 2012 (PCB74, PCB99, PCB105, PCB118, PCB126, PCB138, PCB153, PCB170, PCB194, PCB206) (M) (78) |
| DDT | Lee, 2011 (p,p′-DDEb, p,p′-DDT) (79) | Lee, 2012 (p,p′-DDE) (F) (78) | |
| Lee, 2012 (p,p′-DDEb) (M) (78) | |||
| Chlordane | Lee, 2011 (oxychlordane, trans-nonachlor) (79) | ||
| Lee, 2012 (trans -nonachlor) (F) (78) | |||
| Lee, 2012 (trans -nonachlor) (M) (78) | |||
| Hexachlorobenzene | Lee, 2011 (79) | ||
| Lee, 2012 (F) (78) | |||
| Lee, 2012 (M) (78) | |||
| β-Hexachlorocyclohexane | Lee, 2011 (79) | ||
| Mirex | Lee, 2011 (79) | ||
| PCDDs | Lee, 2012 (OCDD) (F) (78) | Lee, 2012 (OCDD) (M) (78) | |
Abbreviations: M, male; F, female; HpCDD, heptachlorodibenzo-p-dioxin; OCDD, octachlorodibenzo-p-dioxin. Data for summary of results are expressed as first author of study, year (specific compounds) (gender of subjects) (Ref.) When there were several compounds belonging to each POP, the kinds were listed. When male and female were separately reported, results by gender were presented using M or F. Otherwise, gender-specific results were not provided in original articles.
In addition to gender-specific analyses, these studies performed further stratified analyses on race or cigarette smoking and reported different results among subgroups.
Nonlinear dose-response relations.
POPs with marginal significances. They were considered only in prospective studies because a sample size or numbers of incident cases of these studies were small.
A recent review explored the relationships between EDCs and obesity in humans (71). Regarding POPs, it concluded that OC pesticides may play a role in the development of obesity, whereas PCBs showed variable results depending on dose, timing, and gender. This study did not clearly separate studies characterizing intrauterine exposures from those examining adult exposures, except in considering the former ones as prospective studies and the latter ones as cross-sectional studies. In fact, the conclusions of the review (71) may be more appropriate for cross-sectional findings focusing on adult exposure than for findings on intrauterine exposure. However, any cross-sectional association between POPs and obesity in humans cannot be interpreted as evidence for or against obesogenic effects of POPs for the reasons detailed below.
First, positive cross-sectional associations between POP body levels and obesity among adults can be observed even in the absence of any POP-related obesogen effects because obesity itself can lengthen the half-lives of POPs. Kinetic studies indicate that fat mass is among the major determinants of half-lives of POPs in humans (72). On the other hand, if lean and obese persons have equal total body burden of POPs, serum concentrations of POPs in obese persons are expected to be lower than in lean persons due to the dilution effects of adipose tissue mass, hence leading to the inverse cross-sectional associations (73).
Also, there are complicated dynamics between a change in adipose tissue volume and toxicokinetics of POPs. Weight loss is responsible for increasing serum concentrations of POPs due to the reduction in storage capacity in the adipose tissue compartment, which consequently leads to the release of POPs into blood (10, 74). In contrast, weight gain leads to decreasing serum concentrations of POPs through remobilization of POPs into adipose tissue (10, 74). Two persons who have the same weight at age 50 may have had substantially different histories of weight change, which may have affected differently their serum concentrations of POPs at this age.
However, as the interrelated pharmacodynamics between POPs and adipose tissue we mentioned above affect all lipophilic chemicals similarly, an interesting issue remains; cross-sectional associations tend to differ depending on the type of POP. OC pesticides tend to be positively associated or not associated with obesity; to date, no study has reported an inverse association. However, PCBs showed inverse associations in many studies (Table 5), particularly highly chlorinated PCB congeners, whereas less chlorinated PCBs were often positively associated with obesity.
Possible reasons for the contrasting associations with obesity between OC pesticides and PCBs have been discussed by other researchers, again from the viewpoint of pharmacodynamics (75). They suggest dilution effects of adipose tissue when there are active external sources of exposure to POPs, leading to inverse associations between serum concentrations of POPs and obesity. However, when there were fewer active exposure sources (eg, after banning of specific POPs), or the extent of external exposure was much lower than the amount already accumulated in the body, the direction of association between serum concentrations of POPs and obesity started to change from inverse to positive because of increased half-lives of POPs among obese persons. Different results between OC pesticides and PCBs may be due to the different balance between ongoing and past exposure levels. For example, average p,p′-DDE levels in 2000 were approximately one-tenth those in the 1970s, a much more precipitous decline than PCBs, whose concentrations are not declining in some populations (76, 77). Therefore, the dilution effect of increased adiposity may still be observed with PCBs.
Due to the fundamental limitations of cross-sectional studies, the possible obesogenic effects of POPs need to be evaluated in cohort studies. There were two recent prospective studies on POPs and obesity among adults (78, 79). In each study, approximately 20 POP compounds or metabolites were measured, with variable results. In particular, sample sizes or numbers of incident cases were small; thus, some POPs showed only marginal statistical significance although the patterns of associations were very similar to those of other POPs. Therefore, in these prospective studies it is appropriate to focus on the consistency rather than on statistical significance.
One prospective study among young adults observed that p,p′-DDE and highly chlorinated PCBs were associated with increased BMI 18 years later (79). Importantly, the dose-response curves between serum concentrations of these POPs and BMI were inverted and U-shaped; as serum concentrations of POPs at the baseline increased, BMI increased until a critical low dose; above this dose, BMI did not increase, and it even started to decrease as serum concentration of POPs increased. Except for p,p′-DDE, OC pesticides did not predict future BMI. Another prospective study of elderly adults reported various associations with obesity endpoints depending on the POP examined and gender. p,p′-DDE, dioxin, or less chlorinated PCBs predicted the future risk of abdominal obesity (78), with some of these POPs showing inverted U-shaped associations. However, highly chlorinated PCBs strongly inversely predicted abdominal obesity. Although the findings from these two prospective studies suggest possible obesogenic effects of some POPs, the shape of the associations and kinds of POPs were inconsistent.
Despite the importance of testing hypotheses on the relationships between POPs and obesity in humans, such tests are particularly difficult because of the major roles that both diet and physical activity play in causing obesity. Even when sophisticated statistical methods are applied, it is very difficult to completely eliminate the strong effects of diet and physical activity on obesity—eg, because of unavoidable measurement errors in estimating energy intake and physical activity in human beings and because information on diet and physical activity during follow-up is not commonly collected. An additional difficulty stems from the fact that humans are continuously exposed to POPs through fatty animal foods, an intake that is often also closely linked to obesity.
B. Are obesogens unequivocally harmful?
When POPs have obesogenic effects as discussed in the previous section, one may ask whether the development of insulin resistance and T2D after exposure to POPs is the result of the obesogenic effects of POPs or a direct effect of POPs. One interesting example is obesogens that act as PPARγ agonists (57, 80); PPARγ is a receptor that regulates glucose metabolism, lipid uptake, and fatty acid storage, and promotes adipogenesis. PPARγ-mediated proadipogenic effects of tributyltin and other EDCs have been proposed as the mechanistic basis for the environmental obesogen hypothesis (57). However, one group of PPARγ agonists, thiazolidinediones, are used to treat diabetes because PPARγ activation increases insulin sensitivity, even while these drugs promote fat accumulation (81). In this section, we will present evidence suggesting that the development of insulin resistance and T2D might not be directly related to the obesogenic effects of POPs.
As we stated in the introduction, obesity itself is not sufficient to cause insulin resistance and T2D. It is well-known that these metabolic abnormalities are accompanied by a state of chronic low-grade inflammation in adipose tissue (74). The presence of inflammation appears to be a main factor discriminating metabolically healthy obese and metabolically unhealthy lean persons (82). Which kinds of factors contribute to the inflammation of adipose tissue? Findings from experimental and human studies on adipose cell size provide a hint. In earlier studies with genetically obese mice or very obese persons, large adipose cell sizes were associated with the concentration of macrophages in the stromal vascular fraction of adipose tissue, which led to an inflammatory cascade (83, 84). Epidemiological studies also reported that enlarged mean adipocyte size is associated with increased risk of insulin resistance and T2D (85). However, there are recent reports that observed completely opposite associations; the increased proportion of small adipose cells, representing immature adipocytes with impaired ability to store triacylglycerol, was associated with inflammation in adipose tissue among moderately obese persons (86, 87). Also, impaired adipogenesis characterized with decreased expression of differentiation and/or fat storage genes was demonstrated in individuals with T2D or insulin resistance as compared with controls (88, 89).
Opposite results on adipose cell size can be at least partly explained by varying methodologies for measuring adipocyte size because earlier studies used a method that could not accurately separate small adipocytes (90). It is reported that the size distribution of adipocytes is bimodal, ie, cells primarily reside in either a large or a small cell fraction, with few in between (86). In this situation, the mean cell size estimated from total lipid extracted and total cell count is meaningless. Even after setting aside these methodological issues, the apparently contradictory results on adipocyte sizes may not be irreconcilable. An environment enriched with an excessive supply of food and lacking demand for physical activity requires so-called “adaptive responses” to buffer against metabolic imbalance. Normal adipose tissue functions adapt to this situation through adipocyte differentiation from precursor cells and maturation to triglyceride-loaded adipocytes of various sizes (91, 92). When adipose tissue expands in a healthy way, there is no insulin resistance. However, when there are inhibitors impairing adipocyte proliferation and/or differentiation, excess energy mainly leads to a serious enlargement of pre-existing adipocytes (93). In this situation, therefore, both an increased proportion of small immature adipocytes and an increased mean adipocyte size among mature adipocytes may coexist in adipose tissue.
Because there are limits to the size an adipocyte can achieve, if there is continuous excess energy intake, it will lead to consequent increase of fatty acid spillover into plasma and provide substrate availability for triglyceride synthesis in other tissues such as liver, skeletal muscle, myocardium, or even pancreas, increasing ectopic fat deposition, insulin resistance, and T2D (94). Therefore, certain factors that impair increased adipose tissue volume can be more problematic in inducing obesity-related metabolic dysfunction, under the current living conditions of excess energy intake (93). Supporting this idea, it is well-recognized that a lack of adipose tissue, the clinical state of lipodystrophy, is associated with insulin resistance and increased risk for development of T2D (95–98). The wasting syndrome and diabetogenic phenotype observed in TCDD-treated animals is an example of this type of T2D (60, 63, 64).
Under the situation of excess energy intake, hyperplasia and hypertrophy of adipocytes might be adaptive responses against metabolic imbalance, providing a means for the body to store extra energy. If this were true, chemical exposures that produce the same biological effects would not be harmful if they did not produce additional disturbances of other signaling pathways that are critical in the development of T2D. Given the same amount of excess energy intake, the presence of chemicals that inhibit adipogenesis—not obesogens that promote adipogenesis—could actually increase the risk of obesity-related metabolic dysfunction because they can exaggerate fat deposition in ectopic sites rather than within adipose fat pads. Although it may seem provocative, it may be time for a more nuanced approach; ie, it is perhaps too simplistic to suggest that the obesity-inducing effects of obesogens are themselves universally problematic. The worst-case scenario could be the combination of antiadipogenic chemicals and excess energy intake.
C. POPs and inflammation in adipose tissue
Regardless of obesogenic effects of POPs, POPs in adipose tissue can be involved in the inflammatory reaction in the adipose tissue, a critical condition for metabolically unhealthy adipose tissue. Direct proinflammatory effects of POPs have been observed with several individual POPs with AhR affinities like TCDD, PCB77, or PCB126 in both in vitro and in vivo experimental studies; note that doses in these studies were much higher than human exposure doses (61, 65, 99, 100). Recently, chronic low-grade inflammation of adipose tissue developed in mice after chronic consumption of farmed salmon containing mixed POPs at concentrations similar to human exposure doses (54). Furthermore, a more fundamental possibility can be proposed: the presence of poorly biodegradable chemicals like POPs in adipose tissue may contribute to the development of chronic inflammation in such tissue.
Adipose tissue participation in inflammation is attributed largely to the proinflammatory actions of bone marrow-derived white adipose tissue macrophages (74). According to animal and human studies, most macrophages tend to localize to dead adipocytes, where they fuse to form syncytia that sequester and scavenge the residual free adipocyte lipid droplet and ultimately form multinucleated giant cells (MGCs), a characteristic of local chronic inflammatory states (83, 84, 101). Adipocyte death increases in proportion to BMI or adipocyte hypertrophy, and such adipose tissue mainly exhibits ultrastructural features of necrosis (83, 84, 101). Unlike apoptosis, in which cell constituents are packaged into inflammation-suppressive apoptotic bodies, during the process of necrosis, cell contents are released into the extracellular space where they evoke an inflammatory response (102).
Macrophage activation consists of biochemical, morphological, and functional changes that result in the secretion of preformed and/or newly synthesized constituents, such as cytokines and chemokines, leading to the inflammatory response (103). Macrophage activation at sites of inflammation is typically transient, giving way to repair processes that reestablish local tissue function (103). However, at sites of resistant infectious agents (eg, tuberculosis) or poorly biodegradable tissue irritants (ie, foreign bodies), macrophages remain activated and fuse to form MGCs surrounding the unresolved site (104). In fact, MGCs are a regular feature of granulomatous inflammation and are found in various organs after exposure to some poorly soluble, persistent, and nondegradable foreign bodies (105). One typical example is MGCs in pulmonary diseases associated with inhalation of environmental or occupational particulates (106). Furthermore, MGCs are commonly found on the surfaces of implanted biomaterials (107). At these sites, MGCs actively phagocytose debris and can acutely produce inflammatory cytokines until the insult is either cleared by phagocytosis or encapsulated (108).
Researchers have proposed that MGCs in adipose tissue are present to scavenge the residual free lipid droplets released from the necrotic adipocyte (83, 84, 101). However, lipid is not the sole content of adipocytes; POPs are also released from necrotic adipocytes. A pure lipid droplet (absent any POPs or other xenobiotics) that is released from a necrotic adipocyte may be easily scavenged by a macrophage and is unlikely to lead to the formation of MGCs because the pure lipid droplet is a natural body product that can be catabolized and excreted. To the contrary, a lipid droplet contaminated with POPs may not be so easily cleared by a macrophage, because POPs are highly resistant to biodegradation (109). Thus, the release of biodegradation-resistant POPs from necrotic adipocytes could promote formation and maintenance of MGCs, similar to resistant infectious agents or poorly biodegradable tissue irritants, which finally lead to chronic inflammation status in adipose tissue. Besides adipose tissue, because POPs universally exist in other tissues with lipids, similar inflammatory reactions might present in these other tissues as well.
D. POPs, gut microbiota, and obesity
Growing evidence reveals the importance of gut microbiota in developing obesity and obesity-related metabolic dysfunctions through nutrition acquisition, energy harvest, and a myriad of host metabolic pathways (110). There has been a great effort to understand the structure and function of gut microbial communities with a hope to develop prebiotic and/or probiotic interventions that will enable manipulation of the gut microbiota (111).
Recently, a hypothesis paper was published exploring possible interactions between gut ecology and environmental chemicals (112), postulating that variations in gut microbiota are likely to affect toxicodynamics of chemicals. However, there has been little interest in the possibility that chemicals like POPs directly influence the composition of gut microbiota. Unlike other common chemicals that are mainly excreted in urine, the main excretion route of POPs is feces through bile acid and the colon mucosa (113). Therefore, although most POPs are stored in adipose tissue, human gut is continuously exposed to POPs.
In fact, some microorganisms are known to biodegrade a large number of organic chemical substances such as petroleum hydrocarbons, halogenated and nitroaromatic compounds, phthalate esters, solvents, and pesticides that pollute soil and aquatic environments (114). These microorganisms naturally replicate in places with chemical contamination as one way of autopurification and have also been artificially introduced to remove pollutants in environments (115). Because POPs are petroleum-based man-made chemicals, the continuous presence of these compounds in the human gut could alter the delicate balance of microorganisms present by increasing the number of microorganisms that survive via biodegradation of these chemicals. There is currently only one small-scale study that evaluated this possibility in humans (116); there were strong positive correlations between body burden of some POPs and methanogenic archaea levels in feces. Methanogenic archaea are a typical example of microorganisms that biodegrade PCBs or OC pesticides in the environment (117–119).
Methanogenic archaea may also be closely related to obesity. In mice, the presence of methanogenic archaea in the colon promotes obesity through improved efficiency of polysaccharide fermentation by the Bacteroidetes and the Firmicutes, the primary bacterial fermenters in the gut (120). The extent of methanogenic archaea colonization in the small bowel and the colon is predictive of the degree of weight gain in animal models, whereas total bacteria did not show this effect (121).
Despite clear experimental evidence from rodents, human studies on methanogenic archaea have reported inconsistent associations with obesity (116, 122–124). However, these discrepancies may be related to methodological issues because human studies measured methanogenic archaea in fecal samples. Because animal models revealed a higher abundance of methanogenic archaea in the small intestines, not the large intestines (121), methanogenic archaea measures in stool may not reflect their numbers in the remainder of the gut. In this sense, it is interesting that recent human studies measuring methane and hydrogen output via a breath test, a surrogate marker for colonization of methanogenic archaea, reported a consistent positive association with obesity (125, 126). Unlike methanogenic archaea in stools, methane and hydrogen on the breath test can better reflect the situation of the whole gut system, including both small and large intestines. The possible role of POPs affecting gut microbiota, which may itself be directly related to obesity, should be further studied.
E. Can the obesity paradox be explained by POPs?
Once POPs enter the bodies of humans or animals, they must be stored before excretion. Compared to other critical organs, adipose tissue, which is the natural lipid storage location, may be a relatively safe organ to accumulate POPs (19). Despite the side effects of increasing POP half-lives by storing them in adipose tissue and having a chronic internal exposure source of POPs, the accumulation of POPs in adipose tissue can reduce the acute burden of POPs on other critical organs. This idea is closely related to the concept that activation of the body's acute defense mechanisms can lead to long-term side effects (127), ie, there is a cost to “adaptation.”
It is well-known that weight loss intervention studies found clear improvements in many metabolic profiles of patients with insulin resistance and/or T2D (128, 129), although weight loss increases serum concentrations of POPs. Because obesity and POPs appear to interact in the risk of T2D, as was discussed in Section II.C, and any harmful effects due to POPs may require a long time to develop, a decrease in adipose tissue mass itself could lead to an improvement of the metabolic profiles among patients with T2D. However, long-term effects of weight loss may be different from short-term effects.
Although overweight and obesity are well-established independent risk factors for various chronic diseases, overweight and obese patients with various conditions (including T2D, cardiovascular diseases, cancer, and chronic kidney diseases) or obese elderly have a better prognosis than do patients with ideal body weight (130–133). This phenomenon is called the “obesity paradox.” Several mechanisms have been proposed for this phenomenon (130–133). Inaccurate assessment of body fatness by conventional BMI is one postulated mechanism. However, studies using directly measured body fat mass observed a similar obesity paradox (134). Other speculations include a survival effect, increased lean body mass, more aggressive treatment, or better nutritional reserve among obese patients or the elderly (130–133). However, these speculations lack supporting data.
POPs would help explain the obesity paradox if adipose tissue were shown to be a safe depot for POP storage relative to other critical organs. In this sense, the role of adipose tissue could well differ depending on POP levels. This hypothesis was recently tested in an elderly population showing the obesity paradox, using the NHANES 1999–2004 mortality follow-up database (135). Among subjects with relatively low serum concentrations of POPs, there was no obesity paradox: the risk of mortality among elderly with high fat mass was approximately two or three times higher than among those with low fat mass. On the other hand, the obesity paradox was strongly observed among elderly with high serum concentrations of POPs; among them, subjects with high fat mass showed a lower risk of mortality than those with low fat mass.
There is also experimental evidence suggesting possible protective effects of adipose tissue against POPs. For example, coplanar PCBs impaired glucose homeostasis in lean mice but not in obese mice; furthermore, obese mice developed impaired glucose homeostasis after weight loss (136). In rats, diet-induced obesity also significantly increased survival time after exposure to a lethal dose of TCDD (137).
To our knowledge, only one human study has evaluated the role of POPs in the obesity paradox (135); therefore, the findings on the obesity paradox need to be replicated in other cohort studies. Importantly, the different associations between obesity and mortality by POP levels may be observed or not, depending on the distribution of POPs in a specific population. For example, if most people in a population had high POP concentrations, there may be no observable difference in associations between obesity and mortality by POP levels. This factor needs to be considered in future cohort studies.
V. Methodological Issues Crucial for Human Studies
One dilemma in interpreting recent epidemiological findings on POPs is how the strengths of the associations of T2D with OC pesticides and PCBs observed in recent general population studies could be so large when the earlier studies on TCDD or PCBs with much higher doses revealed no association or only a modest increase in risk of T2D. Another apparently puzzling aspect is that the average body burden of many chlorinated POPs has declined worldwide over recent decades (138). By contrast, T2D has more recently been unfolding as a worldwide epidemic. If chlorinated POPs are really important in the development of T2D, how can these apparent mismatches be reconciled?
A. Issue 1: inverted U-shaped associations
1. How can inverted U-shaped associations affect human findings?
A central hypothesis and line of thinking to explain these seemingly contradictory processes is that a low-dose exposure might be more harmful than a high-dose exposure, producing an inverted U-shaped dose response. We will discuss two possible mechanisms that can explain the inverted U-shaped associations in Section VI. Here, we will show how epidemiological studies can be affected by inverted U-shaped associations.
Consistency of research findings is a key factor when making causal inferences between exposures and the risk of human diseases (139). In addition, a dose-response relationship is generally considered to be an important condition that supports a causal association (139). The traditional toxicological dogma, “the dose makes the poison,” has been actively debated in the field of toxicology and among environmental health scientists studying EDCs (140). However, a unique aspect of the inverted U-shaped association in human studies is that it can lead to varying association estimates among populations with different exposure distributions, even when the association is actually causal.
Unlike experimental studies, which can evaluate biological effects of a broad range of doses from zero to highly toxic levels, the exposure range in a specific human population is unique to that population and historical period; eg, it is influenced by the extent of industrialization, use of pesticides in agriculture, regulation of chemicals, and dietary patterns. The practical constraints inherent to studies in humans may directly influence epidemiological findings when an inverted U-shaped relationship truly exists.
Most importantly, human studies can observe different shapes of the dose-response relationship when there is an inverted U-shape over a wide dose range; inverted U-shaped, positive, inverse, and null associations are all possible depending on the exposure distribution of the population under study; this is illustrated and interpreted in Figure 4. In addition, although mean values of one specific POP may be generally similar between two populations, if the absolute concentrations in the reference category differ substantially among populations, the relative risk estimates will differ significantly as well.
Figure 4.

Hypothetical dose-response relationship between POPs and T2D. For simplicity, monotonically increasing doses are arbitrarily numbered starting from zero: 0–2 entails no significant response; doses 3–9 induce an increased risk of T2D; a decreased risk of T2D is observed from 10–20; toxicity (including strongly adverse effects) starts above dose 20. Let us assume that the dose range 0–20 represents a general population within environmental exposure to concentrations of chemicals within the presumed safety level, determined from the linear viewpoint of toxicology. The shape of the dose-response curve differs depending on the different distributions of POPs across populations. Population A shows an inverted U-shaped association (full range of doses 0–20); population B (doses 0–9) shows a strong positive association; population C (doses 5–15) shows a null association; and population D (doses 10–20) shows an inverse association. In addition, the shape of the dose-response curve can look different depending on the sensitivity of the physiological system. A young population is expected to show a sharper inverted U-shaped association (greater y-axis range), whereas an older population would show a blunted shape (less y-axis range). An additional feature made clear in Figure 4 is attenuation of risk estimation when there is a gradient of risk within the reference category. For example, in a population with exposure doses close to 0–12, relative risk for doses 8–12 (the “at risk” group) would have a much lower relative risk if the reference group were doses 0–7 than if doses 0–2 were taken as the reference group. In epidemiological studies, the most common shape of association we can observe may be the increase of risk within lower range of dose and flattening of the risk at higher doses.
The age distribution of each study may also influence the ability to unveil the whole spectrum of the dose-response relationship. Among the human studies on POPs and T2D, the only study that presented a clear inverted U-shaped association was CARDIA (45). One defining feature of this prospective study was that the participants had POPs measured as young adults. If POPs are involved in the development of T2D as EDCs or through mitochondrial dysfunction, this pattern would likely be clearer among young persons, who tend to have a more sensitive physiological system, such as the CARDIA subjects (young population in Figure 4). Considering that physiological system sensitivity decreases with aging (141), the inverted U-shaped dose-response curve can be blunted in elderly populations, as in the PIVUS subjects (44) (old population in Figure 4). Because most chronic diseases, including T2D, increase with age (142), in most epidemiological studies focusing on chronic diseases, incident cases are middle-aged or old. In such studies, therefore, inverted U-shaped associations may not be apparent.
2. Explanation for puzzling epidemiological findings on POPs
Inverted U-shaped associations can help to explain why the association from recent studies in general populations with low concentrations of POPs is more consistent and stronger than in earlier studies with occupational or accidental high exposure. Often, occupationally or accidentally highly exposed persons provide the first human evidence about possible harmfulness of a chemical. Such human studies compare the disease risk among persons who had a high exposure to a suspected chemical in an occupational or accidental setting with the risk of subjects “occupationally unexposed or accidentally unexposed” (the reference groups).
However, this study design is problematic when a low concentration of the chemical increases the risk and a higher concentration does not further increase the risk or even decreases the risk. In the presence of an inverted U-shaped relationship, this approach may miss an existing true association. On the other hand, epidemiological studies among general populations can discriminate much more finely among exposure levels and therefore identify gradients of risks according to POP levels.
In fact, the Air Force Health Study examining only comparison veterans with only background exposures found a clearer dose-response relationship between dioxin and T2D than did the original study, which compared veterans exposed to Agent Orange with those unexposed to it. Also, no association with T2D was seen among workers with high occupational exposures to TCDD or PCBs (26–28, 143, 144). All these earlier findings on high exposure populations are consistent with our hypothesis on the inverted U-shaped association between POPs and T2D.
A molecular epidemiological finding within the Air Force Health Study also supported the existence of an inverted U-shaped association, specifically between the GLUT4:NFkB ratio, which was selected as a biomarker for possible diabetogenic action of TCDD, and serum dioxin levels (145). In this study, inverse relationships were observed only among the comparison group without exposure to Agent Orange, particularly in those with genetic (family history of diabetes) and physiological (obesity) risk factors. Also, when both comparison and exposed veterans were examined, the shape of the dose-response curve between serum dioxin and GLUT4:NFkB ratio was consistent with an inverted U-shaped association, a result the authors called “somewhat surprising” (145).
The most recent cross-sectional studies of OC pesticides or PCBs with T2D in the general population showed positive associations, yet no such relationship was seen in an Inuit population highly exposed to POPs (exposures that are mostly due to a high intake of contaminated marine mammals), although this population has experienced a rapid increase in T2D prevalence over the last 30 years (36). In this study, the average body burden of POPs such as PCBs and chlordanes was about 10–12 times higher than in the current US general population, whereas DDE concentrations were twice as high (37). Therefore, it is plausible that the Inuit population does not include a true unexposed reference group (similar to population C in Figure 4); if almost all persons had high levels of POPs and the risk gradient were flat at higher exposure levels, it would be difficult to observe any positive association at the individual level, although the prevalence of T2D would be increased at the population level.
3. Inconsistent epidemiological findings on fish consumption
The inconsistency of the epidemiological evidence on effects of fish consumption in relation to T2D also illustrates several of the substantive and methodological issues that have been raised in this field of study. Some studies have reported beneficial effects of fish consumption on the risk of T2D (146–148), whereas other studies have reported that it increases such risk (149, 150). Interestingly, studies from Asian countries demonstrated beneficial effects, whereas those from North America or Europe showed detrimental effects on T2D (146). A recent review article on omega-3 fatty acid from seafood sources and T2D reported no harm or benefit on the risk of T2D, but omega-3 fatty acids from plant sources were possibly associated with a modestly lower risk (146, 151).
Fish, in particular oily species with omega-3 fatty acids, are a main dietary source of POPs in the general population (152). When two opposing forces, beneficial nutrients and harmful contaminants, simultaneously exist in the same food, inconsistent human data on the consumption of such food can be expected (153). In addition, if harmful effects of POPs on T2D truly occur at low doses, but beneficial effects of omega-3 fatty acids show linear dose-response relations, we can expect a nonlinear, or even nonmonotonic, dose-response relationship between fish consumption and T2D.
This hypothesis may explain different patterns of associations between studies from North America/Europe and Asia. Compared to persons living in North America or Europe, persons living in Asian countries tend to consume more fish. If the harmful effects due to POPs start to appear at low fish consumption and there is no further clear increase of risk with increasing doses of POPs, the harmful effects of low fish consumption may be difficult to observe among populations with high fish consumption like Asian people. In contrast, the possible beneficial effects due to omega-3 fatty acid could more prominently appear among a population with high fish consumption.
The widespread POP contamination of all animal fat, not just fish, should be considered when interpreting epidemiological findings concerning human consumption of fatty animal food. Moving forward, it will also be important to parse how much the animal fat itself, vs the POPs contained in this fat, contributes to chronic disease.
B. Issue 2: mixtures
A couple of recent animal studies have suggested that the low-dose POP mixture to which humans are currently exposed may be a more relevant research focus than studies examining single or even several specific POPs (52, 54). However, similar to traditional toxicological studies, most epidemiological studies were performed focusing on specific POPs, with interpretation of findings from the viewpoint of those POPs that emerge as statistically significant. However, one specific POP that is associated in a given study may partly or largely reflect the influence of other POPs rather than the role of that POP itself due to high correlations among serum concentrations of various POPs. In this sense, a recent meta-analysis on POPs and T2D, which focused on individual POPs, may be misleading (49).
Mixture issues become more complicated when combined with an inverted U-shaped association due to the common contamination of the reference group. Even Figure 4 presents an unrealistically simplified situation, using the example of just one chemical. In actuality, humans experience lifetime background exposure to several hundred POPs with various distributions. Given correlations among POPs and a multiplicity of POPs in background exposures, subjects who have low concentrations of a specific POP are likely to have higher concentrations of other POPs (13, 154). If these ranges of other POPs carry a high risk of T2D, the risk of T2D in the reference group for the specific POP would become high due to the contamination with other POPs. Consequently, relative risk for higher dose groups would be underestimated.
Unfortunately, an ideal reference group without any exposure to POPs does not exist in current human populations. Therefore, the isolation of a reference group that is as close as possible to having no exposure to POPs is critical. Summary POP measures using information on multiple POPs can be used to make a reference group that at least approximates such a true reference group that has low levels of as broad a set of POPs as possible.
In current human studies, there are several methods to form summary POP measures. In the field of toxicology, it has long been known that the AhR plays a major role in the onset of toxic effects of many POPs; this is the basis for the TEF concept that uses the relative potencies of individual congeners. TEFs multiplied by their concentrations are summed over all congeners in a mixture to achieve a sum-toxic potency, the toxic equivalent (TEQ) (31). Therefore, a TEF-based summary measure for POPs has been commonly used in many studies (31). Other summary measures have been formed according to underlying biological principles, such as PCB groupings by enzyme induction potential of cytochrome P450 subfamilies or estrogenic/neurotoxic, antiestrogenic, etc (155, 156). However, this kind of functional approach only makes sense when the association between POPs and T2D is mediated by the corresponding biological mechanism. Also, strong positive correlations among individual POPs can lead to positive correlations among different summary POP measures, leading to a similar interpretive problem as for individual POPs.
Given that we do not know the exact biological mechanisms between POPs and T2D, we have proposed two data-driven approaches for forming summary measures based on either absolute concentrations or individual POP rank. Absolute concentration-based summary measures have intuitive appeal, but there are large absolute concentration differences among POPs. Simple summation of these would be mainly influenced by the concentrations of a couple of POPs with the highest concentrations. For example, a concentration-based summary measure would be very similar to p,p′-DDE alone because this POP has concentrations that are several times higher than levels of other POPs. Alternatives exist to decrease the influence of specific POPs with high concentrations. One is to combine logarithms of absolute concentrations. Another is to combine standardized deviates (SD), in effect assuming that a common scale for POPs is the population SD of each. Scaling to the SD on the logarithmic scale is yet another option. Finally, rank-based summary measures lead to a common scale for all POPs, ignoring all aspects of absolute concentrations except their ordering. Rank-based and standardized deviate-based scalings generally lead to similar inferences about POPs and T2D.
To fulfill the purpose of isolating a reference group that is as close as possible to a true unexposed reference group, summary measures based on individual POP rank appear to perform well. At the very least, this procedure assures that reference subjects have relatively lower values of the multiple POPs in the summary measure. Even in this approach, there should be a further effort to isolate the best reference group, for example, by using small groups such as sextiles, rather than larger groups such as quartiles, as we presented in Figure 3.
C. Issue 3: how can we deal with lipid profiles?
In human studies on POPs, how lipid profiles are handled is an important methodological issue. Lipid-standardized concentrations (eg, the concentrations of serum POPs divided by total serum lipid content) have generally been reported in studies because POPs are predominantly carried in the lipid component of the blood and because they have been regarded as a measure that better reflects POP body burden than wet-weight concentrations (157). However, experimental studies have reported that low-dose POP mixtures significantly affected the expression of critical genes involved in lipid homeostasis (52) and ectopic fat accumulation in muscle and liver (52, 54). Epidemiological studies have also reported that exposures to POPs can disturb lipid metabolism, leading to dyslipidemia (79). Because dyslipidemia is also closely involved in the pathogenesis of T2D (158), in this situation, lipid concentrations are an intermediate factor within the causal chain linking POPs and T2D.
The problem of lipid standardization of lipophilic chemicals was considered in one simulation study (159), and wet-weight concentrations adjusting for triglycerides and total cholesterol as covariates in models were recommended as a less statistically biased approach than lipid-standardized concentrations. In fact, there is substantial variation in how serum lipid levels have been dealt with in epidemiological studies, as presented in Tables 3 and 4. Among the six prospective studies examining relationships between POPs and T2D we discussed above, three studies did not consider serum lipid levels in their analyses (42, 46, 47), one study used lipid-standardized concentrations (49), and two studies used wet concentrations adjusting for serum lipid levels (44, 45). However, any statistical adjustment for lipid levels would underestimate true associations between POPs and T2D if lipid levels are in the causal pathway between POPs and T2D. At the same time, failure to consider lipid levels in analyses would overestimate true associations. Therefore, we suspect that true risk estimates are intermediate between risk estimates based on wet concentrations and those adjusting for lipid profiles. Future epidemiological studies should primarily use wet concentrations of POPs in their analyses and present both results based on models with and without adjustment for lipid profiles.
VI. Possible Mechanisms
A. Traditional endocrine disrupting-related mechanisms
A central hypothesis and line of thinking to explain the findings in humans about POPs and T2D—which may be considered intuitively contradictory from a toxicological perspective because they disobey the expectation of a linear dose-response model—is that a low but persistent exposure might be more harmful than high-dose acute exposures if POPs are involved in the pathogenesis of T2D as EDCs (38). Knowledge is now sufficient to suggest that EDCs can have biological effects at low doses that are not predicted by their effects at higher doses, because these chemicals follow the same biological “rules” as natural hormones, which also often display nonmonotonic responses (160). It should be noted that EDCs target many endpoints, and thus the assumption that high doses are inherently “safe” is not true; although high doses may not be associated with a particular disease (ie, T2D), they do induce other forms of toxicity (161).
In biological systems, linear effects of hormones typically exist only up to a dose that occupies about 10% of receptors; at higher doses, occupancy rate does not linearly increase as the dose of the hormone increases (162). Furthermore, natural hormones display linear biological responses at much lower doses than those that produce linear responses in receptor occupancy. Thus, in the low-dose range, small increases in the concentration of hormones have large effects on the number of bound receptors, which also produces large biological effects; in contrast, in the high-dose range, the same increase in hormone concentration has small effects on the number of bound receptors, and further has very little effect on biological endpoints (160). Under high-dose exposure to hormones, there can be down-regulation of receptors as the dose further increases (163) as well as receptor desensitization (164–166). Thus, chemicals with endocrine-disrupting properties can produce nonmonotonic dose-response curves for certain biological endpoints.
Although POPs are well-known to be EDCs, it remains largely unknown what specific kinds of endocrine-disrupting properties of POPs may be involved in the pathogenesis of T2D. As a typical example of chemical mixtures, they have variable molecular and cellular targets and thus they cannot be considered to have a single mode of action. For example, DDT is known to have estrogen agonist activity (167), but its metabolite DDE has antiandrogenic properties (168). PCB products include mixtures of congeners with estrogenic and antiestrogenic effects (169, 170), and some PCBs alter thyroid hormone signaling (171). There are interactions between the AhR and estrogen receptor signaling pathways (172), suggesting that dioxins could have indirect effects on some estrogen-mediated endpoints as well.
A variety of hormones including estrogens, androgen, thyroid hormone, and glucocorticoids are involved in the homeostasis of glucose and lipid metabolism (173). Because humans are simultaneously exposed to various chemicals that have the capacity to function as antagonists or agonists to all these hormones (174), it is difficult to speculate about what kind of synergistic, additive, or antagonistic actions of EDC mixtures might occur. Importantly, biological effects of one EDC can differ widely depending on the presence of another EDC. For example, certain biological effects of tributyltin disappeared with a concurrent exposure to the DDT metabolite, DDE (175, 176). Other studies reveal that uterotrophic responses to relatively strong estrogens can be inhibited if weaker estrogens are coadministered, whereas other mixtures demonstrate intermediate uterotrophic responses to the effects of the individual components (177–179). The fact that hormone-related effects of EDCs are likely dependent on the endogenous hormonal milieu makes the issue even more complicated (180).
For many years, EDCs were commonly defined as chemicals that act as agonists or antagonists of nuclear receptors. However, this viewpoint is not sufficiently comprehensive; it is now accepted that EDCs can act via non-nuclear steroid membrane receptors as well as nonsteroid receptors (181). In addition, chemicals that can control the metabolism of endogenous hormones, including their breakdown, can also induce hormonal imbalances (182). Thus, any chemicals that are metabolized through the same metabolic pathways as endogenous hormones can act as EDCs, although these chemicals may not be direct agonists or antagonists of any receptor.
As a whole, experimental studies focusing on one EDC have demonstrated possible diabetogenic effects through traditional endocrine disruption-related mechanisms (183–186). However, the validity of extrapolating experimental findings in laboratory animals to humans is still largely unknown; although evolutionary theory predicts that chemicals will likely produce similar effects in various mammalian species, experimental studies involve carefully controlled exposures, typically to single compounds, whereas humans are exposed to a plethora of diverse mixtures with widely different endocrine-disrupting properties. There is also the possibility that the effects of POPs on T2D are not due to their endocrine-disrupting properties. A recent experimental study on POPs and insulin resistance suggested that xenosensor nuclear receptor-mediated mechanisms may be involved, not traditional hormonal receptor-mediated mechanisms (52); this is closely related to the second mechanism we discuss below.
B. Mitochondrial dysfunction-related mechanisms
Mitochondrial dysfunction has recently emerged as having a potentially pathogenic role in developing T2D (187, 188). Impaired mitochondrial oxidative activity may lead to ectopic fat accumulation in various organs including skeletal muscle, liver, and pancreas that can mediate insulin resistance and secretary dysfunction in pancreas β-cells (187, 188). At present, experimental studies have mainly focused on which molecular pathways explain the relationship between mitochondrial function and T2D; in contrast, few studies have examined the factors that can contribute to the development of mitochondrial dysfunction. Mutations in mitochondrial DNA, mutations in nuclear genes that code for a mitochondrial component, and aging are the most widely studied possible causes of mitochondrial dysfunction (189, 190). The traditional T2D risk factors (excess fat intake and/or physical inactivity) have also been reported to cause mitochondrial dysfunction (191, 192).
There is substantial evidence that a variety of environmental chemicals can damage mitochondria (193, 293). Many early toxicological studies demonstrated that DDT, DDT metabolites, PCBs, and TCDD all induced mitochondrial toxicity (194–196). One well-known and relevant example is the development of insulin-dependent diabetes after human intoxication with the rodenticide Vacor [3-(4-nitrophenyl)-1-(pyridin-3-ylmethyl)urea] which selectively destroys the insulin-producing β-cells in the pancreas (197). Vacor inhibits NADH:ubiquinone reductase activity of mitochondrial complex 1 in the pancreatic β-cells (198). It is therefore plausible that POPs could induce insulin resistance and T2D through mitochondrial toxicity. However, the doses of POPs in these early experimental studies were closer to the range of toxicity (194–196); thus, their findings might not be relevant to current POP exposure levels in humans. In addition, the inverted U-shaped association between POPs and T2D cannot be explained by this mechanism because there were clear monotonic dose-response relationships in the range of toxicity (194–196).
Therefore, a critical question is whether the current background levels of POPs can affect mitochondria. The recent animal study with subchronic exposure to a low-dose POP mixture similar to the background concentrations observed in many current human populations induced mitochondrial dysfunction (52). A comparison of gene expression profiles in the liver of rats fed the high-fat diet contaminated with POPs vs the high-fat diet without POPs demonstrated a significant reduction in the expression of several genes related to mitochondrial function such as PGC1α, citrate synthase, medium-chain acyl coenzyme A dehydrogenase, and succinate dehydrogenase. All of these findings suggest that chronic exposure to low-dose POP mixtures can gradually induce the impairment of mitochondrial function, finally leading to development of insulin resistance and T2D.
As we discussed in Section IV.C, chronic low-grade inflammation in adipose tissue is considered a key mechanism in the development of metabolic disorders like insulin resistance and T2D (199). In fact, mitochondrial dysfunction plays a fundamental role in chronic low-grade inflammation (200). In addition, recent studies have reported a diversity of adipose tissue macrophages depending on obesity status. Adipose tissue macrophages from obese animals with high-fat diets have a classically activated inflammatory phenotype, whereas adipose macrophages from healthy lean mice have alternatively activated macrophages that can protect adipocytes from inflammation (201). Mitochondrial dysfunction can inhibit the function of alternatively activated macrophages through the inhibition of IL-4 (202, 203). Therefore, if POPs can induce mitochondrial dysfunction, even without obesity, POP-induced phenotypic switch in adipose tissue macrophage polarization may be also possible, activating classic inflammatory responses.
The next critical question is how the inverted U-shaped association is possible if the mechanism linking POPs to T2D is mitochondrial dysfunction. Could the mitochondrial dysfunction caused by low-dose POPs be reversed by a somewhat increased dose of POPs? There is experimental evidence suggesting that, with increasing doses of chemicals, cells can increase their production of cytoprotective and restorative proteins including growth factors, phase II and antioxidant enzymes, and protein chaperones through the activation of enzymes such as kinases and deacetylases and transcription factors such as Nrf-2 and NF-κB (204). In particular, it is possible that an increasing dose of chemicals can improve mitochondrial function (205, 206). Furthermore, this mechanism can contribute to the protection against pancreatic β-cell death (207).
In one animal study, glutathione (GSH) content in various organs was measured in mice after treatment across a broad range of TCDD doses (0.15–150 ng/kg/d) (208). Increased GSH synthesis is one marker of cytoprotective and restorative responses (209, 210). Compared to control mice, very low-dose subchronic exposure to TCDD (0.15 ng/kg/d for 13 wk), which was estimated to be similar to current human background exposure, led to GSH depletion. However, increased doses of TCDD increased GSH levels. On the other hand, acute exposure did not show GSH depletion at low doses, but GSH levels increased with higher doses of TCDD. This finding suggests that only a chronic low-dose exposure to TCDD can lead to GSH depletion, but it can be reversed with increasing doses of TCDD. How then can intracellular GSH content be linked to mitochondrial dysfunction? Because mitochondria lack enzymes to synthesize GSH de novo, mitochondrial GSH originates from cytosolic GSH (211). Therefore, intracellular GSH depletion directly affects mitochondrial GSH content. Mitochondrial GSH is the main line of defense in the maintenance of the appropriate mitochondrial redox environment to avoid or repair oxidative modifications (212).
In the experimental study on TCDD (208), the authors did not further investigate the reason for GSH depletion with the very low-dose chronic exposure to TCDD. However, the depletion of GSH may not be specific to this POP. Chronic exposure to many chemicals can lead to intracellular GSH depletion because GSH is consumed through phase II conjugation of chemicals during the process of metabolism (213). In fact, the link between low-dose environmental POPs and T2D in the general population was first hypothesized from epidemiological findings based on serum γ-glutamyltransferase, which has been deemed a biomarker of cumulative exposure to various environmental pollutants that are conjugated by GSH and the status of chronic intracellular GSH depletion (213, 294).
Taken together, experimental studies suggest that chronic exposure to low-dose POP mixtures, similar to current human exposures, may favor the development of mitochondrial dysfunction through GSH depletion and, finally, insulin resistance and T2D. However, these effects might not obey linear dose-responses, perhaps because the activation of cytoprotective and restorative responses including the increase of GSH synthesis can reverse such adverse effects. As discussed in the context of endocrine-disrupting properties, although the increased dose of POPs may improve mitochondrial function, these high doses should not be considered “safe” or beneficial because of the likelihood of other harmful effects on other aspects of human physiology.
Another important point here is that increasing the POP concentration is not the only way to improve mitochondrial function. Mitochondria can be activated with well-accepted health promotion behaviors like exercise, consumption of phytochemicals, or calorie restriction (204, 214). Interestingly, all of these factors are traditionally known to be beneficial to prevent and/or control T2D (215–217). A recent in vitro and in vivo experimental study reported the protective effects of resveratrol, a plant polyphenol, against PCB-mediated impairment of glucose homeostasis in adipocytes through enhanced Nrf2 signaling (218), the same pathway in which certain levels of chemicals can improve mitochondrial function, as we discussed above (204).
VII. Future Research Issues
We have summarized the main issues that should be included in future research programs in Table 6. Details are explained below.
Table 6.
Suggestions for Main Items to Be Included in Research Agendas
| Human studies |
| More well-designed prospective studies on POPs and T2D |
| Focusing on the general population with low-dose exposure |
| Measurement of a broad range of POPs |
| Large sample size |
| Serial measurement of POPs, adiposity, and biomarkers |
| Disentangle the interrelations between obesity and POPs on T2D |
| Role of POPs on gut microbiota |
| Extended follow-up from early life (even in utero) exposure to adult exposure |
| Effects of gene-POP interactions on the risk of T2D |
| Known genetic factors of T2D |
| Xenobiotic-metabolizing genes |
| Comprehensive reevaluation of diet considering both nutrients and POPs |
| Role of POPs in the development of complications among patients with T2D |
| POPs and T1D |
| Develop methods to decrease body burden of POPs |
| Experimental studies |
| Use real-world scenarios in terms of dose, mixtures, duration of POPs |
| Consider using POP mixtures extracted from human adipose tissue |
| Continuous lifetime exposure starting from in utero exposure |
| Possible biological mechanisms underlying the inverted U-shaped associations |
| Effect of POP mixtures on pancreatic β-cells |
| Evaluation of mitochondrial dysfunction coupled with activation of cytoprotective and restorative responses |
| Evaluation of traditional endocrine-disrupting properties and related mechanisms |
| POPs and T1D |
A. Human studies
Although current human evidence linking POPs to T2D is substantial, more prospective studies are needed. Until now, most prospective studies were not specifically designed to test the hypothesis that POPs increase the risk of T2D. Because some studies used existing information on POPs that was collected for other reasons, a broad range of POPs was not considered. The common failure to measure the multiple POPs may lead to a serious underestimation of relative risks; this occurs despite the positive correlations among serum concentrations of some POPs, for reasons explained in Section V.B. Furthermore, POPs are likely to have additive and perhaps multiplicative interactions with other types of diabetogenic environmental pollutants. It is impressive that so little of this has been formally tested in humans and animals.
Future prospective studies should consider several methodological and scientific issues. First of all, studies should be performed in the general population with low concentrations of POPs because populations living in highly contaminated areas are not likely to have a valid reference group with close to null POP exposures. Second, a large sample size is recommended to ensure that the study includes a substantial number of subjects with very low levels of POPs who can act as an appropriate reference group. Third, although it is often assumed that one-time measurement of POPs appropriately reflects long-term exposure to POPs, serial measurements of POPs are recommended because serum concentrations of POPs are directly affected by weight changes, which are becoming more common in modern societies. Fourth, the interaction between POPs and obesity requires further investigation. Because there can be both pharmacodynamic and biological interactions between POPs and obesity, serial measurements of adiposity and clinically relevant endocrine biomarkers are also necessary to disentangle the complicated interrelationships between obesity and POPs. Related to this issue, future studies should assess the importance of POP total body burden. Such burden is essentially determined by POP exposure and body fat mass; hence, it may help explain the interactions between POPs and obesity that have been observed in epidemiological studies (37, 40, 41). Finally, the possible effects of POPs on gut microbiota and on inflammatory disorders ought to be incorporated as well in prospective studies.
It is also important to acknowledge what the available human studies indicate about critical periods of exposure to POPs. As shown in Section II, current human evidence suggests that chronic exposure to POP mixtures during adulthood may be sufficient to induce T2D. Although the individuals assessed in adulthood likely had early-life exposures to POPs, animal studies also support that early-life exposure to POPs might not be a necessary condition for T2D development (52, 54).
However, the hypothesis on the developmental origins of health and disease remains relevant (219), and research integrating POPs into that hypothesis would be scientifically important. Epidemiological studies starting from in utero exposure (or even prenatally in the subject's parents), continuing to early fetal, childhood, adolescence, and adult periods would be helpful to determine how important early-life exposure to POPs is to the development of metabolic dysfunction and how changes in exposure patterns during the lifetime affect metabolic function.
Although genome-wide association studies have identified a number of loci associated with T2D (220), the role of genetic factors in the relationships between POPs and T2D has not been investigated yet. Gene-environment interactions between known T2D genes and POPs on the development of T2D are plausible. In addition, genetic polymorphisms in xenobiotic-metabolizing enzymes need to be evaluated because they have influence on clearance and internal dose of POPs (221). Several small-scale studies have reported that a polymorphism of GSH S-transferase, one of the phase II xenobiotic-metabolizing enzymes, is associated with the risk of T2D (222–224). Therefore, the interaction between GSH S-transferase variants and POPs on the risk of T2D is another plausible hypothesis.
Contamination of foods from POPs also requires further study. A healthy diet plays a very important role in the prevention of the development of T2D and is also critical in the management of T2D. At present, dietary issues that have been considered include excess calorie intake, glycemic index, saturated fat intake, and others (225). Because POP-contaminated food is the main source of external exposure to POPs in the general population, studies focusing only on nutritional values of food deal with just one aspect of diet. It remains unknown to what extent animal food consumption might be partly detrimental due to the POPs these items contain. A more comprehensive approach considering both nutrients and pollutants like POPs is necessary to validly evaluate the effects of diet on T2D.
It is also necessary to evaluate the influence of POPs in the development of complications among patients with T2D. Hyperglycemia itself not only defines T2D but also is a cause of long-term diabetic complications (226). In one cross-sectional study, serum concentrations of POPs were associated with poorer glycemic control and the risk of peripheral neuropathy among patients with T2D (227). Also, epidemiological studies have reported that serum concentrations of POPs are associated with self-reported cardiovascular diseases and predicted the future risk of stroke in the general population (228, 229). Because all these diseases are common long-term complications of T2D, it is highly plausible that POPs affect the development of various such complications.
Another largely unexplored area is the possible association between POPs and type 1 diabetes (T1D) (230), an autoimmune disease destroying the pancreatic β-cells (231). Although some cross-sectional studies have suggested a possible positive association between POPs and T1D (232, 233), a prospective study from Sweden reported a decreased risk of T1D among children with high in utero exposure to POPs compared to those with low-dose exposures (234). If the link between chemicals and autoimmunity can also be explained by the endocrine-disrupting properties of these chemicals (235), low doses could be more harmful than high doses, producing an inverted U-shaped association. Because the Swedish study used serum collected in the 1970s, the distribution of POPs would be in the higher dose range of the inverted U-shaped association, as in population D in Figure 4. Therefore, future studies on this topic should also be performed among populations with low-dose exposure.
Finally, research and pilot projects are needed to identify and evaluate policies and interventions to combat the adverse effects of POPs. First of all, there are ways to decrease human exposures to POPs; whereas plans to implement the Stockholm Convention of POPs have been developed in some countries, others are not fulfilling their obligations as signatories of the Convention. Studies on the levels of POPs in the global environment show that emission sources of POPs have shifted from industrialized countries of the Northern Hemisphere to less developed countries in tropical and subtropical regions (236). These patterns may be due to late-implemented bans, the illegal use of restricted POPs, or the use of POPs in the control of malaria (237). In addition, many developing countries have large open dumping sites of municipal wastes with inappropriate control, which are a new source of POPs (238). Therefore, much more energetic public and private policies to combat the use and/or emission of POPs are needed.
However, in the past six decades, POPs have widely contaminated animal and human food webs across the planet. Under this situation, a plant-based diet is one option to prevent further exposure to POPs at the individual level because POP contamination is much lower in these diets compared to animal-based diets (239). There may also be ways to increase POP excretion from the body. Although POPs are excreted through feces, a substantial amount of POPs excreted through bile is reabsorbed through enterohepatic recirculation (184). Experimental animal studies suggest that dietary fiber may be efficient to absorb POPs in the gut and increase their excretion via feces (240, 241). Therefore, a plant-based diet with high dietary fiber may be helpful to decrease half-lives of POPs, hence decreasing body burden. These methods must be tested in well-designed large randomized clinical trials; if they were proven to be effective to prevent and/or manage T2D, they would hence provide further evidence on causal relations between POPs and T2D. It is interesting to note that a vegan diet was more effective than a conventional diet in the treatment of T2D, although this randomized, controlled clinical trial was performed without any consideration on POPs (242).
Importantly, under the assumption of an inverted U-shaped association, researchers should consider the possibility that some efforts to decrease POP body burden might lead to lower but more harmful concentrations for specific endpoints of concern. In this sense, the evaluation of benefits of decreasing body burden of POPs can be better tested in patients with T2D or prediabetic stages, focusing on improvement of glycemic control, because such patients can be regarded as already being in the range of harmful doses.
B. Experimental studies
Because most chlorinated POPs were banned several decades ago and evidence for a strong association between low-dose POP mixtures and T2D first emerged from human studies, laboratory research about chlorinated POPs does not seem to be as active as research on other environmental chemicals. Since human studies require large investments and long time commitments, more in vivo and in vitro experimental studies are needed. In particular, experimental studies with POP exposures that mimic real-world scenarios in terms of dose, mixtures, and duration are necessary to understand epidemiological findings observed in humans. The ultimate metabolic dysfunction phenotype (such as T2D) due to mixtures may differ considerably from studies of single chemicals in isolation. As a first step, researchers can evaluate whether similar or different biological effects are observed for specific POPs and how divergent the results are depending on the coexistence of other POPs with variable doses. If there are substantial differences in biological effects, laboratory researchers could consider using POP mixtures that are directly extracted from human adipose tissue in their experiments. Possible aging effects on the relations of POPs with the risk of T2D also require further experimental studies.
It is difficult to perform experimental studies of POP mixtures to explore underlying molecular mechanisms of the inverted U-shaped associations or the mixture effects that are observed in human studies. Therefore, experimental studies focusing on individual POPs over a broad range of doses and on mixtures of several POPs have clear merits. So far, many experimental studies on individual POPs have focused on POPs with dioxin activity, but more research on POPs without dioxin activity will also be helpful for understanding human findings on POPs.
In addition, in terms of exposure duration, animal studies only focusing on early-life exposures may not be relevant to humans because humans are continuously exposed to POPs before conception though death. It would be interesting to evaluate whether biological results of prenatal exposure to POPs without exposure after birth are different from those of prenatal exposure to POPs with continuous exposure after birth.
Additional studies are needed to examine whether chronic exposures to low-dose POPs affect pancreatic β-cells. When the pathogenesis of T2D is classified into two stages, insulin resistance and insulin secretory defects, the latter part is an important triggering factor for the progression of prediabetic states to full-blown T2D (243). A recent review article proposes possible mechanisms linking EDCs, including POPs, to the dysfunction of pancreatic β-cells (244); however, the exposure doses used in that study may not be relevant to current human exposure levels. More human relevant situation-based experimental studies are still needed.
VIII. Conclusion
The continuing worldwide increase in the occurrence of T2D is a great social, medical, and economic burden. Although obesity is the most well-known risk factor for T2D, a growing body of evidence links T2D to background exposure to environmental chemicals, in particular chlorinated POPs. As typical chemicals contaminating adipose tissues as well as all lipid compartments in biological systems, the available epidemiological and experimental evidence largely shows that background contamination from POP mixtures including OC pesticides and PCBs is strongly associated with the development of T2D. Inconsistencies concerning findings for specific individual POPs may arise due to different patterns of POP mixtures between and within populations despite high correlations among serum concentrations of many individual POPs. Differences in the shape of the dose-response curves among human studies may reflect an inverted U-shaped association secondary to mitochondrial dysfunction or their endocrine-disrupting properties. The bottom line is that low-dose effects of POPs appear to be quite real in humans. However, at present human evidence on POPs and obesity remains insufficient. Besides promoting T2D, adipose tissue might play dual roles by providing a relatively safe place to store POPs.
The overview and summary of our hypotheses on the relationships between POPs and T2D, with emphasis on possible mechanisms, are presented in Figure 5. At present, inflammation, ectopic fat accumulation, and mitochondrial dysfunction are considered key mechanisms that link obesity and T2D—without any consideration for POPs (94, 199, 245). However, these mechanisms can also be linked to POP exposure, and there is neither adipose tissue nor lipid uncontaminated with POPs in our world. Therefore, the scientific community should recognize POPs when exploring the pathophysiological roles of obesity in the development of insulin resistance and T2D. However, this hypothesis requires more evidence from experimental studies because it is possible that other mechanisms operate.
Figure 5.

Summary of postulated POPs and T2D relationship with possible mechanisms. Under the current paradigm (labeled “Traditional risk factors”), lifestyle changes characterized by excess energy intake and a lack of physical activity have led to the obesity and T2D epidemic. Inflammation in adipose tissue, lipotoxicity in liver, muscle, and pancreas, and mitochondrial dysfunction are considered to be primary mechanisms. However, adipose tissue also contains POPs because of the contaminated environment and food web. As depicted under an expanded paradigm (labeled “New risk factors”), POP mixtures consisting of several hundred compounds are stored in adipose tissue, are continuously released to circulation, and reach critical organs along with serum lipids. Human responses to POP mixtures may differ depending on levels of POPs. There can be low-dose POP mixtures that are not high enough to induce any chemical-specific responses. However, even at low doses POPs at least induce physiological responses like phase I, phase II, and phase III xenobiotic metabolism pathways aimed to increase the excretion of POPs. In particular, phase II glutathione conjugation can lead to chronic consumption and depletion of intracellular glutathione. Intracellular glutathione depletion can cause mitochondrial dysfunction that is closely related to inflammation and ectopic fat accumulation; all of these mechanisms are also known to play important roles in the pathogenesis of traditional obesity-related T2D. However, a somewhat increased dose of POP mixtures can activate increased synthesis of cytoprotective and restorative proteins, including glutathione synthesis. This activation may improve mitochondrial function, which in theory might decrease the risk of T2D. Therefore, we can expect the inverted U-shaped associations between POPs and T2D. Importantly, mitochondria function can be improved with other well-accepted health promotion behaviors like exercise, phytochemical consumption, or calorie restriction; they are traditionally known to be beneficial to prevent and/or control T2D. In the range of high-dose POPs, we can expect traditional toxicity responses. Many toxicological studies of POPs deal with this high range of POPs and focus on one specific POP. However, exposure patterns used in experimental settings are very different from human exposure patterns in terms of doses, mixtures, and exposure duration of POPs. Therefore, the relevance of experimental findings to humans is unclear. On the other hand, POPs can increase or decrease adipose hyperplasia and/or hypertrophy depending on dose or kinds of POPs. Various endocrine-disrupting mechanisms may be involved in these relationships. POPs can indirectly induce obesity through the influence on gut microbiota. However, in the risk of T2D, the worst-case scenario could be the combination of antiadipogenic chemicals and excess energy intake. Also, it is important to note that adipose tissue can play dual roles: promoting T2D, and providing a relatively safe place to store POPs.
The evidence about the potential influence of POPs in the pathogenesis of T2D can no longer be ignored. More prospective studies with serial measurement of a broad range of POPs and adiposity are needed to disentangle the interrelationships between obesity and POPs on the development of T2D. Also, we encourage laboratory researchers to design studies that resemble real-world scenarios as much as possible, in terms of dose, mixtures, and duration of POP exposure. However, experimental studies for the achievement of a better understanding of biological mechanisms behind the inverted U-shaped associations and mixture effects may require traditional approaches focusing on individual or sets of POPs. Experimental studies and studies in humans have long been known to complement each other. Researchers must continue to thoughtfully consider findings from other fields of study when they design research studies and interpret study findings.
Acknowledgments
This work was financially supported by Grant A111716 and Grant HI13C0715 of the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea and Grant BK21 PLUS KNU of the Biomedical Convergence Program for Creative Talent.
Disclosure Summary: The authors have nothing to declare.
Funding Statement
This work was financially supported by Grant A111716 and Grant HI13C0715 of the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea and Grant BK21 PLUS KNU of the Biomedical Convergence Program for Creative Talent.
Footnotes
- AhR
- aryl hydrocarbon receptor
- BMI
- body mass index
- CARDIA
- Coronary Artery Risk Development in Young Adults
- DDE
- dichlorodiphenyldichloroethylene
- DDT
- dichlorodiphenyltrichloroethane
- EDC
- endocrine-disrupting chemical
- GSH
- glutathione
- MGC
- multinucleated giant cells
- NHANES
- National Health and Nutrition Examination Survey
- OC
- organochlorine
- PCB
- polychlorinated biphenyl
- PCDD
- polychlorinated dibenzo-p-dioxins
- PCDF
- polychlorinated dibenzofurans
- PIVUS
- Prospective Investigation of the Vasculature in Uppsala Seniors
- POP
- persistent organic pollutant
- SD
- standardized deviates
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- T1D
- type 1 diabetes
- T2D
- type 2 diabetes
- TEF
- toxic equivalency factor
- TEQ
- toxic equivalent.
References
- 1. Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat Rev Endocrinol. 2012;8(4):228–236. [DOI] [PubMed] [Google Scholar]
- 2. Weigensberg MJ, Goran MI. Type 2 diabetes in children and adolescents. Lancet. 2009;373(9677):1743–1744. [DOI] [PubMed] [Google Scholar]
- 3. Sims EA. Are there persons who are obese, but metabolically healthy? Metabolism. 2001;50(12):1499–1504. [DOI] [PubMed] [Google Scholar]
- 4. Gregg EW, Cheng YJ, Narayan KM, Thompson TJ, Williamson DF. The relative contributions of different levels of overweight and obesity to the increased prevalence of diabetes in the United States: 1976–2004. Prev Med. 2007;45(5):348–352. [DOI] [PubMed] [Google Scholar]
- 5. McLaughlin T, Abbasi F, Lamendola C, Reaven G. Heterogeneity in the prevalence of risk factors for cardiovascular disease and type 2 diabetes mellitus in obese individuals: effect of differences in insulin sensitivity. Arch Intern Med. 2007;167(7):642–648. [DOI] [PubMed] [Google Scholar]
- 6. Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS? Ann NY Acad Sci. 2010;1212:59–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thayer KA, Heindel JJ, Bucher JR, Gallo MA. Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120(6):779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor KW, Novak RF, Anderson HA, et al. . Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: a national toxicology program workshop review. Environ Health Perspect. 2013;121(7):774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002;23(2):201–229. [DOI] [PubMed] [Google Scholar]
- 10. Lim JS, Son HK, Park SK, Jacobs DR Jr, Lee DH. Inverse associations between long-term weight change and serum concentrations of persistent organic pollutants. Int J Obes (Lond). 2011;35(5):744–747. [DOI] [PubMed] [Google Scholar]
- 11. Longnecker MP. Pharmacokinetic variability and the miracle of modern analytical chemistry. Epidemiology. 2006;17(4):350–351. [DOI] [PubMed] [Google Scholar]
- 12. Kortenkamp A. Low dose mixture effects of endocrine disrupters: implications for risk assessment and epidemiology. Int J Androl. 2008;31(2):233–240. [DOI] [PubMed] [Google Scholar]
- 13. Porta M, Pumarega J, Gasull M. Number of persistent organic pollutants detected at high concentrations in a general population. Environ Int. 2012;44:106–111. [DOI] [PubMed] [Google Scholar]
- 14. Fisher BE. Most unwanted. Environ Health Perspect. 1999;107(1):A18–A23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karlaganis G, Marioni R, Sieber I, Weber A. The elaboration of the ‘Stockholm convention’ on persistent organic pollutants (POPs): a negotiation process fraught with obstacles and opportunities. Environ Sci Pollut Res Int. 2001;8(3):216–221. [DOI] [PubMed] [Google Scholar]
- 16. Frederiksen M, Vorkamp K, Thomsen M, Knudsen LE. Human internal and external exposure to PBDEs–a review of levels and sources. Int J Hyg Environ Health. 2009;212(2):109–134. [DOI] [PubMed] [Google Scholar]
- 17. Suedel BC, Boraczek JA, Peddicord RK, Clifford PA, Dillon TM. Trophic transfer and biomagnification potential of contaminants in aquatic ecosystems. Rev Environ Contam Toxicol. 1994;136:21–89. [DOI] [PubMed] [Google Scholar]
- 18. Irigaray P, Newby JA, Lacomme S, Belpomme D. Overweight/obesity and cancer genesis: more than a biological link. Biomed Pharmacother. 2007;61(10):665–678. [DOI] [PubMed] [Google Scholar]
- 19. La Merrill M, Emond C, Kim MJ, et al. . Toxicological function of adipose tissue: focus on persistent organic pollutants. Environ Health Perspect. 2013;121(2):162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schecter A, Birnbaum L, Ryan JJ, Constable JD. Dioxins: an overview. Environ Res. 2006;101(3):419–428. [DOI] [PubMed] [Google Scholar]
- 21. Wolfe WH, Michalek JE, Miner JC, et al. . Health status of Air Force veterans occupationally exposed to herbicides in Vietnam. I. Physical health. JAMA. 1990;264(14):1824–1831. [PubMed] [Google Scholar]
- 22. Henriksen GL, Ketchum NS, Michalek JE, Swaby JA. Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand. Epidemiology. 1997;8(3):252–258. [DOI] [PubMed] [Google Scholar]
- 23. Institute of Medicine. Veterans and Agent Orange: herbicide/dioxin exposure and type 2 diabetes. Washington, DC: The National Academy Press; 2000. [Google Scholar]
- 24. Longnecker MP, Michalek JE. Serum dioxin level in relation to diabetes mellitus among Air Force veterans with background levels of exposure. Epidemiology. 2000;11(1):44–48. [DOI] [PubMed] [Google Scholar]
- 25. Bertazzi PA, Bernucci I, Brambilla G, Consonni D, Pesatori AC. The Seveso studies on early and long-term effects of dioxin exposure: a review. Environ Health Perspect. 1998;106(suppl 2):625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calvert GM, Sweeney MH, Deddens J, Wall DK. Evaluation of diabetes mellitus, serum glucose, and thyroid function among United States workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Occup Environ Med. 1999;56(4):270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sweeney MH, Calvert GM, Egeland GA, Fingerhut MA, Halperin WE, Piacitelli LA. Review and update of the results of the NIOSH medical study of workers exposed to chemicals contaminated with 2,3,7,8-tetrachlorodibenzodioxin. Teratog Carcinog Mutagen. 1997–1998;17(4–5):241–247. [PubMed] [Google Scholar]
- 28. Zober A, Ott MG, Messerer P. Morbidity follow up study of BASF employees exposed to 2,3,7, 8-tetrachlorodibenzo-p-dioxin (TCDD) after a 1953 chemical reactor incident. Occup Environ Med. 1994;51(7):479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kerger BD, Scott PK, Pavuk M, Gough M, Paustenbach DJ. Re-analysis of Ranch Hand study supports reverse causation hypothesis between dioxin and diabetes. Crit Rev Toxicol. 2012;42(8):669–687. [DOI] [PubMed] [Google Scholar]
- 30. Remillard RB, Bunce NJ. Linking dioxins to diabetes: epidemiology and biologic plausibility. Environ Health Perspect. 2002;110(9):853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van den Berg M, Birnbaum LS, Denison M, et al. . The 2005 World Health Organization reevaluation of human and mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol Sci. 2006;93(2):223–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van den Berg M, Peterson RE, Schrenk D. Human risk assessment and TEFs. Food Addit Contam. 2000;17(4):347–358. [DOI] [PubMed] [Google Scholar]
- 33. Porta M. A dictionary of epidemiology. 5th ed New York: Oxford University Press; 2008. [Google Scholar]
- 34. Norén K, Meironyté D. Certain organochlorine and organobromine contaminants in Swedish human milk in perspective of past 20–30 years. Chemosphere. 2000;40(9–11):1111–1123. [DOI] [PubMed] [Google Scholar]
- 35. Porta M, Puigdomènech E, Ballester F, et al. . Monitoring concentrations of persistent organic pollutants in the general population: the international experience. Environ Int. 2008;34(4):546–561. [DOI] [PubMed] [Google Scholar]
- 36. Jørgensen ME, Borch-Johnsen K, Bjerregaard P. A cross-sectional study of the association between persistent organic pollutants and glucose intolerance among Greenland Inuit. Diabetologia. 2008;51(8):1416–1422. [DOI] [PubMed] [Google Scholar]
- 37. Lee DH, Lee IK, Song K, et al. . A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999–2002. Diabetes Care. 2006;29(7):1638–1644. [DOI] [PubMed] [Google Scholar]
- 38. Lee DH, Jacobs DR Jr, Porta M. Could low-level background exposure to persistent organic pollutants contribute to the social burden of type 2 diabetes? J Epidemiol Community Health. 2006;60(12):1006–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Porta M. Persistent organic pollutants and the burden of diabetes. Lancet. 2006;368(9535):558–559. [DOI] [PubMed] [Google Scholar]
- 40. Airaksinen R, Rantakokko P, Eriksson JG, Blomstedt P, Kajantie E, Kiviranta H. Association between type 2 diabetes and exposure to persistent organic pollutants. Diabetes Care. 2011;34(9):1972–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gasull M, Pumarega J, Téllez-Plaza M, et al. . Blood concentrations of persistent organic pollutants and prediabetes and diabetes in the general population of Catalonia. Environ Sci Technol. 2012;46(14):7799–7810. [DOI] [PubMed] [Google Scholar]
- 42. Turyk M, Anderson H, Knobeloch L, Imm P, Persky V. Organochlorine exposure and incidence of diabetes in a cohort of Great Lakes sport fish consumers. Environ Health Perspect. 2009;117(7):1076–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Michalek JE, Ketchum NS, Tripathi RC. Diabetes mellitus and 2,3,7,8-tetrachlorodibenzo-p-dioxin elimination in veterans of Operation Ranch Hand. J Toxicol Environ Health A. 2003;66(3):211–221. [DOI] [PubMed] [Google Scholar]
- 44. Lee DH, Lind PM, Jacobs DR Jr, Salihovic S, van Bavel B, Lind L. Polychlorinated biphenyls and organochlorine pesticides in plasma predict development of type 2 diabetes in the elderly: the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) study. Diabetes Care. 2011;34(8):1778–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee DH, Steffes MW, Sjödin A, Jones RS, Needham LL, Jacobs DR Jr. Low dose of some persistent organic pollutants predicts type 2 diabetes: a nested case-control study. Environ Health Perspect. 2010;118(9):1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rignell-Hydbom A, Lidfeldt J, Kiviranta H, et al. . Exposure to p,p'-DDE: a risk factor for type 2 diabetes. PLoS One. 2009;4(10):e7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vasiliu O, Cameron L, Gardiner J, Deguire P, Karmaus W. Polybrominated biphenyls, polychlorinated biphenyls, body weight, and incidence of adult-onset diabetes mellitus. Epidemiology. 2006;17(4):352–359. [DOI] [PubMed] [Google Scholar]
- 48. Wang SL, Tsai PC, Yang CY, Guo YL. Increased risk of diabetes and polychlorinated biphenyls and dioxins: a 24-year follow-up study of the Yucheng cohort. Diabetes Care. 2008;31(8):1574–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu H, Bertrand KA, Choi AL, et al. . Persistent organic pollutants and type 2 diabetes: a prospective analysis in the Nurses' Health Study and Meta-analysis. Environ Health Perspect. 2013;121(2):153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Biggs ML, Mukamal KJ, Luchsinger JA, et al. . Association between adiposity in midlife and older age and risk of diabetes in older adults. JAMA. 2010;303(24):2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab. 2003;284(1):E7–E12. [DOI] [PubMed] [Google Scholar]
- 52. Ruzzin J, Petersen R, Meugnier E, et al. . Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ Health Perspect. 2010;118(4):465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kiviranta H, Tuomisto JT, Tuomisto J, Tukiainen E, Vartiainen T. Polychlorinated dibenzo-p-dioxins, dibenzofurans, and biphenyls in the general population in Finland. Chemosphere. 2005;60(7):854–869. [DOI] [PubMed] [Google Scholar]
- 54. Ibrahim MM, Fjære E, Lock EJ, et al. . Chronic consumption of farmed salmon containing persistent organic pollutants causes insulin resistance and obesity in mice. PLoS One. 2011;6(9):e25170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ibrahim MM, Fjære E, Lock EJ, et al. . Metabolic impacts of high dietary exposure to persistent organic pollutants in mice. Toxicol Lett. 2012;215(1):8–15. [DOI] [PubMed] [Google Scholar]
- 56. Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8(2):185–192. [DOI] [PubMed] [Google Scholar]
- 57. Grün F, Watanabe H, Zamanian Z, et al. . Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20(9):2141–2155. [DOI] [PubMed] [Google Scholar]
- 58. Richter CA, Birnbaum LS, Farabollini F, et al. . In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol. 2007;24(2):199–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Newbold RR, Padilla-Banks E, Jefferson WN, Heindel JJ. Effects of endocrine disruptors on obesity. Int J Androl. 2008;31(2):201–208. [DOI] [PubMed] [Google Scholar]
- 60. Seefeld MD, Corbett SW, Keesey RE, Peterson RE. Characterization of the wasting syndrome in rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol. 1984;73(2):311–322. [DOI] [PubMed] [Google Scholar]
- 61. Alexander DL, Ganem LG, Fernandez-Salguero P, Gonzalez F, Jefcoate CR. Aryl-hydrocarbon receptor is an inhibitory regulator of lipid synthesis and of commitment to adipogenesis. J Cell Sci. 1998;111:3311–3322. [DOI] [PubMed] [Google Scholar]
- 62. Phillips M, Enan E, Liu PC, Matsumura F. Inhibition of 3T3–L1 adipose differentiation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Cell Sci. 1995;108:395–402. [DOI] [PubMed] [Google Scholar]
- 63. Brewster DW, Matsumura F. TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) reduces lipoprotein lipase activity in the adipose tissue of the guinea pig. Biochem Biophys Res Commun. 1984;122(2):810–817. [DOI] [PubMed] [Google Scholar]
- 64. Enan E, Liu PC, Matsumura F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes reduction of glucose transporting activities in the plasma membranes of adipose tissue and pancreas from the guinea pig. J Biol Chem. 1992;267(28):19785–19791. [PubMed] [Google Scholar]
- 65. Arsenescu V, Arsenescu RI, King V, Swanson H, Cassis LA. Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect. 2008;116(6):761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Moreno-Aliaga MJ, Matsumura F. Effects of 1,1,1-trichloro-2,2-bis(p-chlorophenyl)-ethane (p,p'-DDT) on 3T3–L1 and 3T3–F442A adipocyte differentiation. Biochem Pharmacol. 2002;63(5):997–1007. [DOI] [PubMed] [Google Scholar]
- 67. Moreno-Aliaga MJ, Matsumura F. Endrin inhibits adipocyte differentiation by selectively altering expression pattern of CCAAT/enhancer binding protein-α in 3T3–L1 cells. Mol Pharmacol. 1999;56(1):91–101. [DOI] [PubMed] [Google Scholar]
- 68. Howell G 3rd, Mangum L. Exposure to bioaccumulative organochlorine compounds alters adipogenesis, fatty acid uptake, and adipokine production in NIH3T3–L1 cells. Toxicol In Vitro. 2011;25(1):394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lyche JL, Nourizadeh-Lillabadi R, Almaas C, et al. . Natural mixtures of persistent organic pollutants (POP) increase weight gain, advance puberty, and induce changes in gene expression associated with steroid hormones and obesity in female zebrafish. J Toxicol Environ Health A. 2010;73(15):1032–1057. [DOI] [PubMed] [Google Scholar]
- 70. Lyche JL, Nourizadeh-Lillabadi R, Karlsson C, et al. . Natural mixtures of POPs affected body weight gain and induced transcription of genes involved in weight regulation and insulin signaling. Aquat Toxicol. 2011;102:197–204. [DOI] [PubMed] [Google Scholar]
- 71. Tang-Péronard JL, Andersen HR, Jensen TK, Heitmann BL. Endocrine-disrupting chemicals and obesity development in humans: a review. Obes Rev. 2011;12(8):622–636. [DOI] [PubMed] [Google Scholar]
- 72. Milbrath MO, Wenger Y, Chang CW, et al. . Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environ Health Perspect. 2009;117(3):417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wolff MS, Anderson HA, Britton JA, Rothman N. Pharmacokinetic variability and modern epidemiology–the example of dichlorodiphenyltrichloroethane, body mass index, and birth cohort. Cancer Epidemiol Biomarkers Prev. 2007;16(10):1925–1930. [DOI] [PubMed] [Google Scholar]
- 74. Dirtu AC, Dirinck E, Malarvannan G, et al. . Dynamics of organohalogenated contaminants in human serum from obese individuals during one year of weight loss treatment. Environ Sci Technol. 2013;47(21):12441–12449. [DOI] [PubMed] [Google Scholar]
- 75. Wolff MS, Britton JA, Teitelbaum SL, et al. . Improving organochlorine biomarker models for cancer research. Cancer Epidemiol Biomarkers Prev. 2005;14(9):2224–2236. [DOI] [PubMed] [Google Scholar]
- 76. Addison RF, Stobo WT. Trends in organochlorine residue concentrations and burdens in grey seals (Halichoerus grypus) from Sable Is., NS, Canada, between 1974 and 1994. Environ Pollut. 2001;112(3):505–513. [DOI] [PubMed] [Google Scholar]
- 77. Maes J, Belpaire C, Goemans G. Spatial variations and temporal trends between 1994 and 2005 in polychlorinated biphenyls, organochlorine pesticides and heavy metals in European eel (Anguilla anguilla L.) in Flanders, Belgium. Environ Pollut. 2008;153(1):223–237. [DOI] [PubMed] [Google Scholar]
- 78. Lee DH, Lind L, Jacobs DR Jr, Salihovic S, van Bavel B, Lind PM. Associations of persistent organic pollutants with abdominal obesity in the elderly: The Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) study. Environ Int. 2012;40:170–178. [DOI] [PubMed] [Google Scholar]
- 79. Lee DH, Steffes MW, Sjödin A, Jones RS, Needham LL, Jacobs DR Jr. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS One. 2011;6(1):e15977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kirchner S, Kieu T, Chow C, Casey S, Blumberg B. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol Endocrinol. 2010;24(3):526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Olefsky JM. Treatment of insulin resistance with peroxisome proliferator-activated receptor γ agonists. J Clin Invest. 2000;106(4):467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Blüher M. The distinction of metabolically ‘healthy’ from ‘unhealthy’ obese individuals. Curr Opin Lipidol. 2010;21(1):38–43. [DOI] [PubMed] [Google Scholar]
- 83. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cinti S, Mitchell G, Barbatelli G, et al. . Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46(11):2347–2355. [DOI] [PubMed] [Google Scholar]
- 85. Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia. 2000;43(12):1498–1506. [DOI] [PubMed] [Google Scholar]
- 86. McLaughlin T, Deng A, Yee G, et al. . Inflammation in subcutaneous adipose tissue: relationship to adipose cell size. Diabetologia. 2010;53(2):369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. McLaughlin T, Sherman A, Tsao P, et al. . Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia. 2007;50(8):1707–1715. [DOI] [PubMed] [Google Scholar]
- 88. Dubois SG, Heilbronn LK, Smith SR, Albu JB, Kelley DE, Ravussin E. Decreased expression of adipogenic genes in obese subjects with type 2 diabetes. Obesity (Silver Spring). 2006;14(9):1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yang X, Jansson PA, Nagaev I, et al. . Evidence of impaired adipogenesis in insulin resistance. Biochem Biophys Res Commun. 2004;317(4):1045–1051. [DOI] [PubMed] [Google Scholar]
- 90. Hauner H. Adipose tissue inflammation: are small or large fat cells to blame? Diabetologia. 2010;53(2):223–225. [DOI] [PubMed] [Google Scholar]
- 91. Halvorsen YD, Bond A, Sen A, et al. . Thiazolidinediones and glucocorticoids synergistically induce differentiation of human adipose tissue stromal cells: biochemical, cellular, and molecular analysis. Metabolism. 2001;50(4):407–413. [DOI] [PubMed] [Google Scholar]
- 92. Shepherd PR, Gnudi L, Tozzo E, Yang H, Leach F, Kahn BB. Adipose cell hyperplasia and enhanced glucose disposal in transgenic mice overexpressing GLUT4 selectively in adipose tissue. J Biol Chem. 1993;268(30):22243–22246. [PubMed] [Google Scholar]
- 93. Danforth E., Jr Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26(1):13. [DOI] [PubMed] [Google Scholar]
- 94. Abate N. Adipocyte maturation arrest: a determinant of systemic insulin resistance to glucose disposal. J Clin Endocrinol Metab. 2012;97(3):760–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275(12):8456–8460. [DOI] [PubMed] [Google Scholar]
- 96. Reitman ML, Mason MM, Moitra J, et al. . Transgenic mice lacking white fat: models for understanding human lipoatrophic diabetes. Ann NY Acad Sci. 1999;892:289–296. [DOI] [PubMed] [Google Scholar]
- 97. Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401(6748):73–76. [DOI] [PubMed] [Google Scholar]
- 98. Garg A. Lipodystrophies. Am J Med. 2000;108(2):143–152. [DOI] [PubMed] [Google Scholar]
- 99. Kim MJ, Pelloux V, Guyot E, et al. . Inflammatory pathway genes belong to major targets of persistent organic pollutants in adipose cells. Environ Health Perspect. 2012;120(4):508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li W, Vogel CF, Matsumura F. Studies on the cell treatment conditions to elicit lipolytic responses from 3T3–L1 adipocytes to TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Cell Biochem. 2007;102(2):389–402. [DOI] [PubMed] [Google Scholar]
- 101. Xu H, Barnes GT, Yang Q, et al. . Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Leist M, Jäättelä M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2(8):589–598. [DOI] [PubMed] [Google Scholar]
- 103. Duffield JS. The inflammatory macrophage: a story of Jekyll and Hyde. Clin Sci (Lond). 2003;104(1):27–38. [DOI] [PubMed] [Google Scholar]
- 104. Anderson JM. Multinucleated giant cells. Curr Opin Hematol. 2000;7(1):40–47. [DOI] [PubMed] [Google Scholar]
- 105. Williams GT, Williams WJ. Granulomatous inflammation–a review. J Clin Pathol. 1983;36(7):723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Agustí C, Xaubet A, Arriols R, Marín A, Montserrat JM, Agustí-Vidal A. Multinuclear giant cells in bronchoalveolar lavage in interstitial lung diseases. Respiration. 1987;51(4):307–311. [DOI] [PubMed] [Google Scholar]
- 107. Smetana K Jr, Slavík J, Vancová E, et al. . Fusion of macrophages on an implant surface is associated with down-regulated expression of ligands for galectin-1 and -3 in the rat. Biomaterials. 1998;19(19):1799–1805. [DOI] [PubMed] [Google Scholar]
- 108. Hernandez-Pando R, Bornstein QL, Aguilar Leon D, Orozco EH, Madrigal VK, Martinez Cordero E. Inflammatory cytokine production by immunological and foreign body multinucleated giant cells. Immunology. 2000;100(3):352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fries GF. Transport of organic environmental contaminants to animal products. Rev Environ Contam Toxicol. 1995;141:71–109. [DOI] [PubMed] [Google Scholar]
- 110. Musso G, Gambino R, Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med. 2011;62:361–380. [DOI] [PubMed] [Google Scholar]
- 111. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Snedeker SM, Hay AG. Do interactions between gut ecology and environmental chemicals contribute to obesity and diabetes? Environ Health Perspect. 2012;120(3):332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rozman K. Intestinal excretion of toxic substances. Arch Toxicol Suppl. 1985;8:87–93. [DOI] [PubMed] [Google Scholar]
- 114. Megharaj M, Ramakrishnan B, Venkateswarlu K, Sethunathan N, Naidu R. Bioremediation approaches for organic pollutants: a critical perspective. Environ Int. 2011;37(8):1362–1375. [DOI] [PubMed] [Google Scholar]
- 115. Atlas RM. Biodegradation of hydrocarbons in the environment. Basic Life Sci. 1988;45:211–222. [DOI] [PubMed] [Google Scholar]
- 116. Lee HS, Lee JC, Lee IK, et al. . Associations among organochlorine pesticides, Methanobacteriales, and obesity in Korean women. PLoS One. 2011;6(11):e27773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Baczynski TP, Grotenhuis T, Knipscheer P. The dechlorination of cyclodiene pesticides by methanogenic granular sludge. Chemosphere. 2004;55(5):653–659. [DOI] [PubMed] [Google Scholar]
- 118. Natarajan MR, Nye J, Wu WM, Wang H, Jain MK. Reductive dechlorination of PCB-contaminated raisin river sediments by anaerobic microbial granules. Biotechnol Bioeng. 1997;55(1):182–190. [DOI] [PubMed] [Google Scholar]
- 119. Quintero JC, Moreira MT, Feijoo G, Lema JM. Anaerobic degradation of hexachlorocyclohexane isomers in liquid and soil slurry systems. Chemosphere. 2005;61(4):528–536. [DOI] [PubMed] [Google Scholar]
- 120. Samuel BS, Gordon JI. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci USA. 2006;103(26):10011–10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mathur R, Kim G, Morales W, et al. . Intestinal Methanobrevibacter smithii but not total bacteria is related to diet-induced weight gain in rats. Obesity (Silver Spring). 2013;21(4):748–754. [DOI] [PubMed] [Google Scholar]
- 122. Million M, Maraninchi M, Henry M, et al. . Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int J Obes (Lond). 2012;36(6):817–825. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 123. Schwiertz A, Taras D, Schäfer K, et al. . Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring). 2010;18(1):190–195. [DOI] [PubMed] [Google Scholar]
- 124. Zhang H, DiBaise JK, Zuccolo A, et al. . Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA. 2009;106(7):2365–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Basseri RJ, Basseri B, Pimentel M, et al. . Intestinal methane production in obese individuals is associated with a higher body mass index. Gastroenterol Hepatol (NY). 2012;8(1):22–28. [PMC free article] [PubMed] [Google Scholar]
- 126. Mathur R, Amichai M, Chua KS, Mirocha J, Barlow GM, Pimentel M. Methane and hydrogen positivity on breath test is associated with greater body mass index and body fat. J Clin Endocrinol Metab. 2013;98(4):E698–E702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Barouki R. Linking long-term toxicity of xeno-chemicals with short-term biological adaptation. Biochimie. 2010;92(9):1222–1226. [DOI] [PubMed] [Google Scholar]
- 128. Norris SL, Zhang X, Avenell A, et al. . Long-term effectiveness of weight-loss interventions in adults with pre-diabetes: a review. Am J Prev Med. 2005;28(1):126–139. [DOI] [PubMed] [Google Scholar]
- 129. Norris SL, Zhang X, Avenell A, et al. . Long-term non-pharmacologic weight loss interventions for adults with type 2 diabetes. Cochrane Database Syst Rev. 2005;2:CD004095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Janssen I, Mark AE. Elevated body mass index and mortality risk in the elderly. Obes Rev. 2007;8(1):41–59. [DOI] [PubMed] [Google Scholar]
- 131. Oreopoulos A, Kalantar-Zadeh K, Sharma AM, Fonarow GC. The obesity paradox in the elderly: potential mechanisms and clinical implications. Clin Geriatr Med. 2009;25(4):643–659, viii. [DOI] [PubMed] [Google Scholar]
- 132. Romero-Corral A, Montori VM, Somers VK, et al. . Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: a systematic review of cohort studies. Lancet. 2006;368(9536):666–678. [DOI] [PubMed] [Google Scholar]
- 133. Zamboni M, Mazzali G, Zoico E, et al. . Health consequences of obesity in the elderly: a review of four unresolved questions. Int J Obes (Lond). 2005;29(9):1011–1029. [DOI] [PubMed] [Google Scholar]
- 134. Lavie CJ, Milani RV, Artham SM, Patel DA, Ventura HO. The obesity paradox, weight loss, and coronary disease. Am J Med. 2009;122(12):1106–1114. [DOI] [PubMed] [Google Scholar]
- 135. Hong NS, Kim KS, Lee IK, et al. . The association between obesity and mortality in the elderly differs by serum concentrations of persistent organic pollutants: a possible explanation for the obesity paradox. Int J Obes (Lond). 2012;36(9):1170–1175. [DOI] [PubMed] [Google Scholar]
- 136. Baker NA, Karounos M, English V, et al. . Coplanar polychlorinated biphenyls impair glucose homeostasis in lean C57BL/6 mice and mitigate beneficial effects of weight loss on glucose homeostasis in obese mice. Environ Health Perspect. 2013;121(1):105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tuomisto JT, Pohjanvirta R, Unkila M, Tuomisto J. TCDD-induced anorexia and wasting syndrome in rats: effects of diet-induced obesity and nutrition. Pharmacol Biochem Behav. 1999;62(4):735–742. [DOI] [PubMed] [Google Scholar]
- 138. Solomon GM, Weiss PM. Chemical contaminants in breast milk: time trends and regional variability. Environ Health Perspect. 2002;110(6):A339–A347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Weed DL. Interpreting epidemiological evidence: how meta-analysis and causal inference methods are related. Int J Epidemiol. 2000;29(3):387–390. [PubMed] [Google Scholar]
- 140. Melnick R, Lucier G, Wolfe M, et al. . Summary of the National Toxicology Program's report of the endocrine disruptors low-dose peer review. Environ Health Perspect. 2002;110(4):427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Noth RH, Mazzaferri EL. Age and the endocrine system. Clin Geriatr Med. 1985;1(1):223–250. [PubMed] [Google Scholar]
- 142. Eyre H, Kahn R, Robertson RM, et al. . Preventing cancer, cardiovascular disease, and diabetes: a common agenda for the American Cancer Society, the American Diabetes Association, and the American Heart Association. Circulation. 2004;109(25):3244–3255. [DOI] [PubMed] [Google Scholar]
- 143. Prince MM, Hein MJ, Ruder AM, Waters MA, Laber PA, Whelan EA. Update: cohort mortality study of workers highly exposed to polychlorinated biphenyls (PCBs) during the manufacture of electrical capacitors, 1940–1998. Environ Health. 2006;5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ruder AM, Hein MJ, Nilsen N, et al. . Mortality among workers exposed to polychlorinated biphenyls (PCBs) in an electrical capacitor manufacturing plant in Indiana: an update. Environ Health Perspect. 2006;114(1):18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Fujiyoshi PT, Michalek JE, Matsumura F. Molecular epidemiologic evidence for diabetogenic effects of dioxin exposure in U.S. Air force veterans of the Vietnam war. Environ Health Perspect. 2006;114(11):1677–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wu JH, Micha R, Imamura F, et al. . Omega-3 fatty acids and incident type 2 diabetes: a systematic review and meta-analysis. Br J Nutr. 2012;107(suppl 2):S214–S227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kaushik M, Mozaffarian D, Spiegelman D, Manson JE, Willett WC, Hu FB. Long-chain omega-3 fatty acids, fish intake, and the risk of type 2 diabetes mellitus. Am J Clin Nutr. 2009;90(3):613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. van Woudenbergh GJ, van Ballegooijen AJ, Kuijsten A, et al. . Eating fish and risk of type 2 diabetes: a population-based, prospective follow-up study. Diabetes Care. 2009;32(11):2021–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Nanri A, Mizoue T, Noda M, et al. . Fish intake and type 2 diabetes in Japanese men and women: the Japan Public Health Center-based Prospective Study. Am J Clin Nutr. 2011;94(3):884–891. [DOI] [PubMed] [Google Scholar]
- 150. Villegas R, Xiang YB, Elasy T, et al. . Fish, shellfish, and long-chain n-3 fatty acid consumption and risk of incident type 2 diabetes in middle-aged Chinese men and women. Am J Clin Nutr. 2011;94(2):543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ruzzin J, Jacobs DR. The secret story of fish: decreasing nutritional value due to pollution? Br J Nutr. 2012;108(3):397–399. [DOI] [PubMed] [Google Scholar]
- 152. Schafer KS, Kegley SE. Persistent toxic chemicals in the US food supply. J Epidemiol Community Health. 2002;56(11):813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Lee DH, Jacobs DR Jr. Inconsistent epidemiological findings on fish consumption may be indirect evidence of harmful contaminants in fish. J Epidemiol Community Health. 2010;64(3):190–192. [DOI] [PubMed] [Google Scholar]
- 154. Porta M. Human contamination by environmental chemical pollutants: can we assess it more properly? Prev Med. 2012;55(6):560–562. [DOI] [PubMed] [Google Scholar]
- 155. McFarland VA, Clarke JU. Environmental occurrence, abundance, and potential toxicity of polychlorinated biphenyl congeners: considerations for a congener-specific analysis. Environ Health Perspect. 1989;81:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Wolff MS, Camann D, Gammon M, Stellman SD. Proposed PCB congener groupings for epidemiological studies. Environ Health Perspect. 1997;105(1):13–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. López-Carrillo L, Torres-Sánchez L, López-Cervantes M, Blair A, Cebrián ME, Uribe M. The adipose tissue to serum dichlorodiphenyldichloroethane (DDE) ratio: some methodological considerations. Environ Res. 1999;81(2):142–145. [DOI] [PubMed] [Google Scholar]
- 158. Howard BV. Insulin resistance and lipid metabolism. Am J Cardiol. 1999;84(1A):28J–32J. [DOI] [PubMed] [Google Scholar]
- 159. Schisterman EF, Whitcomb BW, Louis GM, Louis TA. Lipid adjustment in the analysis of environmental contaminants and human health risks. Environ Health Perspect. 2005;113(7):853–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, et al. . Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev. 2012;33(3):378–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Vandenberg LN, Colborn T, Hayes TB, et al. . Regulatory decisions on endocrine disrupting chemicals should be based on the principles of endocrinology. Reprod Toxicol. 2013;38:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Welshons WV, Thayer KA, Judy BM, Taylor JA, Curran EM, vom Saal FS. Large effects from small exposures. I. Mechanisms for endocrine-disrupting chemicals with estrogenic activity. Environ Health Perspect. 2003;111(8):994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Medlock KL, Lyttle CR, Kelepouris N, Newman ED, Sheehan DM. Estradiol down-regulation of the rat uterine estrogen receptor. Proc Soc Exp Biol Med. 1991;196(3):293–300. [DOI] [PubMed] [Google Scholar]
- 164. Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Prog Horm Res. 1996;51:319–351; discussion 352–313. [PubMed] [Google Scholar]
- 165. Lohse MJ. Molecular mechanisms of membrane receptor desensitization. Biochim Biophys Acta. 1993;1179(2):171–188. [DOI] [PubMed] [Google Scholar]
- 166. Shankaran H, Wiley HS, Resat H. Receptor downregulation and desensitization enhance the information processing ability of signalling receptors. BMC Syst Biol. 2007;1:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Robison AK, Stancel GM. The estrogenic activity of DDT: correlation of estrogenic effect with nuclear level of estrogen receptor. Life Sci. 1982;31(22):2479–2484. [DOI] [PubMed] [Google Scholar]
- 168. Mills LJ, Gutjahr-Gobell RE, Haebler RA, et al. . Effects of estrogenic (o,p'-DDT; octylphenol) and anti-androgenic (p,p'-DDE) chemicals on indicators of endocrine status in juvenile male summer flounder (Paralichthys dentatus). Aquat Toxicol. 2001;52(2):157–176. [DOI] [PubMed] [Google Scholar]
- 169. Jansen HT, Cooke PS, Porcelli J, Liu TC, Hansen LG. Estrogenic and antiestrogenic actions of PCBs in the female rat: in vitro and in vivo studies. Reprod Toxicol. 1993;7(3):237–248. [DOI] [PubMed] [Google Scholar]
- 170. Ma R, Sassoon DA. PCBs exert an estrogenic effect through repression of the Wnt7a signaling pathway in the female reproductive tract. Environ Health Perspect. 2006;114(6):898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Giera S, Bansal R, Ortiz-Toro TM, Taub DG, Zoeller RT. Individual polychlorinated biphenyl (PCB) congeners produce tissue- and gene-specific effects on thyroid hormone signaling during development. Endocrinology. 2011;152(7):2909–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Swedenborg E, Pongratz I. AhR and ARNT modulate ER signaling. Toxicology. 2010;268(3):132–138. [DOI] [PubMed] [Google Scholar]
- 173. Neel BA, Sargis RM. The paradox of progress: environmental disruption of metabolism and the diabetes epidemic. Diabetes. 2011;60(7):1838–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Woodruff TJ, Zota AR, Schwartz JM. Environmental chemicals in pregnant women in the United States: NHANES 2003–2004. Environ Health Perspect. 2011;119(6):878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Makita Y, Omura M, Ogata R. Effects of perinatal simultaneous exposure to tributyltin (TBT) and p,p'-DDE [1,1-dichloro-2,2-bis(p-chlorophenyl) ethylene) on male offspring of Wistar rats. J Toxicol Environ Health A. 2004;67(5):385–395. [DOI] [PubMed] [Google Scholar]
- 176. Makita Y, Omura M, Tanaka A, Kiyohara C. Effects of concurrent exposure to tributyltin and 1,1-dichloro-2,2 bis (p-chlorophenyl) ethylene (p,p'-DDE) on immature male Wistar rats. Basic Clin Pharmacol Toxicol. 2005;97(6):364–368. [DOI] [PubMed] [Google Scholar]
- 177. Charles GD, Gennings C, Tornesi B, et al. . Analysis of the interaction of phytoestrogens and synthetic chemicals: an in vitro/in vivo comparison. Toxicol Appl Pharmacol. 2007;218(3):280–288. [DOI] [PubMed] [Google Scholar]
- 178. Charles GD, Gennings C, Zacharewski TR, Gollapudi BB, Carney EW. An approach for assessing estrogen receptor-mediated interactions in mixtures of three chemicals: a pilot study. Toxicol Sci. 2002;68(2):349–360. [DOI] [PubMed] [Google Scholar]
- 179. Tinwell H, Ashby J. Sensitivity of the immature rat uterotrophic assay to mixtures of estrogens. Environ Health Perspect. 2004;112(5):575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Sheehan DM, Willingham E, Gaylor D, Bergeron JM, Crews D. No threshold dose for estradiol-induced sex reversal of turtle embryos: how little is too much? Environ Health Perspect. 1999;107(2):155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, et al. . Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30(4):293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kretschmer XC, Baldwin WS. CAR and PXR: xenosensors of endocrine disrupters? Chem Biol Interact. 2005;155(3):111–128. [DOI] [PubMed] [Google Scholar]
- 183. Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. The estrogenic effect of bisphenol A disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ Health Perspect. 2006;114(1):106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Alonso-Magdalena P, Ropero AB, Carrera MP, et al. . Pancreatic insulin content regulation by the estrogen receptor ER α. PLoS One. 2008;3(4):e2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Alonso-Magdalena P, Vieira E, Soriano S, et al. . Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect. 2010;118(9):1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Newbold RR, Padilla-Banks E, Jefferson WN. Environmental estrogens and obesity. Mol Cell Endocrinol. 2009;304(1–2):84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev. 2010;31(3):364–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(2):92–103. [DOI] [PubMed] [Google Scholar]
- 189. Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91(23):10771–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13(12):878–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Anderson EJ, Lustig ME, Boyle KE, et al. . Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119(3):573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Menshikova EV, Ritov VB, Toledo FG, Ferrell RE, Goodpaster BH, Kelley DE. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am J Physiol Endocrinol Metab. 2005;288(4):E818–E825. [DOI] [PubMed] [Google Scholar]
- 193. Meyer JN, Leung MC, Rooney JP, et al. . Mitochondria as a target of environmental toxicants. Toxicol Sci. 2013;134(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Bagchi M, Stohs SJ. In vitro induction of reactive oxygen species by 2,3,7,8-tetrachlorodibenzo-p-dioxin, endrin, and lindane in rat peritoneal macrophages, and hepatic mitochondria and microsomes. Free Radic Biol Med. 1993;14(1):11–18. [DOI] [PubMed] [Google Scholar]
- 195. Pardini RS. Polychlorinated biphenyls (PCB): effect on mitochondrial enzyme systems. Bull Environ Contam Toxicol. 1971;6(6):539–545. [DOI] [PubMed] [Google Scholar]
- 196. Pardini RS, Heidker JC, Baker TA, Payne B. Toxicology of various pesticides and their decomposition products on mitochondrial electron transport. Arch Environ Contam Toxicol. 1980;9(1):87–97. [DOI] [PubMed] [Google Scholar]
- 197. Karam JH, Lewitt PA, Young CW, et al. . Insulinopenic diabetes after rodenticide (Vacor) ingestion: a unique model of acquired diabetes in man. Diabetes. 1980;29(12):971–978. [DOI] [PubMed] [Google Scholar]
- 198. Esposti MD, Ngo A, Myers MA. Inhibition of mitochondrial complex I may account for IDDM induced by intoxication with the rodenticide Vacor. Diabetes. 1996;45(11):1531–1534. [DOI] [PubMed] [Google Scholar]
- 199. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. [DOI] [PubMed] [Google Scholar]
- 200. López-Armada MJ, Riveiro-Naveira RR, Vaamonde-García C, Valcárcel-Ares MN. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13(2):106–118. [DOI] [PubMed] [Google Scholar]
- 201. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Ferger AI, Campanelli L, Reimer V, et al. . Effects of mitochondrial dysfunction on the immunological properties of microglia. J Neuroinflammation. 2010;7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Vats D, Mukundan L, Odegaard JI, et al. . Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Mattson MP. Dietary factors, hormesis and health. Ageing Res Rev. 2008;7(1):43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol. 2010;45(6):410–418. [DOI] [PubMed] [Google Scholar]
- 206. Sano M, Fukuda K. Activation of mitochondrial biogenesis by hormesis. Circ Res. 2008;103(11):1191–1193. [DOI] [PubMed] [Google Scholar]
- 207. Li N, Stojanovski S, Maechler P. Mitochondrial hormesis in pancreatic β cells: does uncoupling protein 2 play a role? Oxid Med Cell Longev. 2012;2012:740849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Slezak BP, Hatch GE, DeVito MJ, et al. . Oxidative stress in female B6C3F1 mice following acute and subchronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol Sci. 2000;54(2):390–398. [DOI] [PubMed] [Google Scholar]
- 209. Goto S, Naito H, Kaneko T, Chung HY, Radák Z. Hormetic effects of regular exercise in aging: correlation with oxidative stress. Appl Physiol Nutr Metab. 2007;32(5):948–953. [DOI] [PubMed] [Google Scholar]
- 210. Utley WS, Mehendale HM. Evidence for stimulated glutathione synthesis by phenobarbital pretreatment during an oxidative challenge in isolated hepatocytes. J Biochem Toxicol. 1991;6(2):101–113. [DOI] [PubMed] [Google Scholar]
- 211. Mårtensson J, Lai JC, Meister A. High-affinity transport of glutathione is part of a multicomponent system essential for mitochondrial function. Proc Natl Acad Sci USA. 1990;87(18):7185–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Marí M, Morales A, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa JC. Mitochondrial glutathione: features, regulation and role in disease. Biochim Biophys Acta. 2013;1830(5):3317–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Lee DH, Jacobs DR Jr. Is serum γ-glutamyltransferase a marker of exposure to various environmental pollutants? Free Radic Res. 2009;43(6):533–537. [DOI] [PubMed] [Google Scholar]
- 214. Radak Z, Chung HY, Goto S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic Biol Med. 2008;44(2):153–159. [DOI] [PubMed] [Google Scholar]
- 215. Hansen BC. Dietary considerations for obese diabetic subjects. Diabetes Care. 1988;11(2):183–188. [DOI] [PubMed] [Google Scholar]
- 216. Horton ES. Role and management of exercise in diabetes mellitus. Diabetes Care. 1988;11(2):201–211. [DOI] [PubMed] [Google Scholar]
- 217. Leiherer A, Mündlein A, Drexel H. Phytochemicals and their impact on adipose tissue inflammation and diabetes. Vascul Pharmacol. 2013;58(1–2):3–20. [DOI] [PubMed] [Google Scholar]
- 218. Baker NA, English V, Sunkara M, Morris AJ, Pearson KJ, Cassis LA. Resveratrol protects against polychlorinated biphenyl-mediated impairment of glucose homeostasis in adipocytes. J Nutr Biochem. 2013;24(12):2168–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Boekelheide K, Blumberg B, Chapin RE, et al. . Predicting later-life outcomes of early-life exposures. Environ Health Perspect. 2012;120(10):1353–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;363(24):2339–2350. [DOI] [PubMed] [Google Scholar]
- 221. Wormhoudt LW, Commandeur JN, Vermeulen NP. Genetic polymorphisms of human N-acetyltransferase, cytochrome P450, glutathione-S-transferase, and epoxide hydrolase enzymes: relevance to xenobiotic metabolism and toxicity. Crit Rev Toxicol. 1999;29(1):59–124. [DOI] [PubMed] [Google Scholar]
- 222. Bid HK, Konwar R, Saxena M, Chaudhari P, Agrawal CG, Banerjee M. Association of glutathione S-transferase (GSTM1, T1 and P1) gene polymorphisms with type 2 diabetes mellitus in north Indian population. J Postgrad Med. 2010;56(3):176–181. [DOI] [PubMed] [Google Scholar]
- 223. Hori M, Oniki K, Ueda K, et al. . Combined glutathione S-transferase T1 and M1 positive genotypes afford protection against type 2 diabetes in Japanese. Pharmacogenomics. 2007;8(10):1307–1314. [DOI] [PubMed] [Google Scholar]
- 224. Yalin S, Hatungil R, Tamer L, et al. . Glutathione S-transferase gene polymorphisms in Turkish patients with diabetes mellitus. Cell Biochem Funct. 2007;25(5):509–513. [DOI] [PubMed] [Google Scholar]
- 225. Ajala O, English P, Pinkney J. Systematic review and meta-analysis of different dietary approaches to the management of type 2 diabetes. Am J Clin Nutr. 2013;97(3):505–516. [DOI] [PubMed] [Google Scholar]
- 226. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):837–853. [PubMed] [Google Scholar]
- 227. Lee DH, Jacobs DR Jr, Steffes M. Association of organochlorine pesticides with peripheral neuropathy in patients with diabetes or impaired fasting glucose. Diabetes. 2008;57(11):3108–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Ha MH, Lee DH, Jacobs DR. Association between serum concentrations of persistent organic pollutants and self-reported cardiovascular disease prevalence: results from the National Health and Nutrition Examination Survey, 1999–2002. Environ Health Perspect. 2007;115(8):1204–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Lee DH, Lind PM, Jacobs DR Jr, Salihovic S, van Bavel B, Lind L. Background exposure to persistent organic pollutants predicts stroke in the elderly. Environ Int. 2012;47:115–120. [DOI] [PubMed] [Google Scholar]
- 230. Howard SG, Lee DH. What is the role of human contamination by environmental chemicals in the development of type 1 diabetes? J Epidemiol Community Health. 2012;66(6):479–481. [DOI] [PubMed] [Google Scholar]
- 231. Narendran P, Estella E, Fourlanos S. Immunology of type 1 diabetes. QJM. 2005;98(8):547–556. [DOI] [PubMed] [Google Scholar]
- 232. Langer P, Tajtáková M, Guretzki HJ, et al. . High prevalence of anti-glutamic acid decarboxylase (anti-GAD) antibodies in employees at a polychlorinated biphenyl production factory. Arch Environ Health. 2002;57(5):412–415. [DOI] [PubMed] [Google Scholar]
- 233. Longnecker MP, Klebanoff MA, Brock JW, Zhou H. Polychlorinated biphenyl serum levels in pregnant subjects with diabetes. Diabetes Care. 2001;24(6):1099–1101. [DOI] [PubMed] [Google Scholar]
- 234. Rignell-Hydbom A, Elfving M, Ivarsson SA, et al. . A nested case-control study of intrauterine exposure to persistent organochlorine pollutants in relation to risk of type 1 diabetes. PLoS One. 2010;5(6):e11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Chalubinski M, Kowalski ML. Endocrine disrupters–potential modulators of the immune system and allergic response. Allergy. 2006;61(11):1326–1335. [DOI] [PubMed] [Google Scholar]
- 236. Tanabe S, Minh TB. Dioxins and organohalogen contaminants in the Asia-Pacific region. Ecotoxicology. 2010;19(3):463–478. [DOI] [PubMed] [Google Scholar]
- 237. Wong MH, Leung AO, Chan JK, Choi MP. A review on the usage of POP pesticides in China, with emphasis on DDT loadings in human milk. Chemosphere. 2005;60(6):740–752. [DOI] [PubMed] [Google Scholar]
- 238. Someya M, Ohtake M, Kunisue T, et al. . Persistent organic pollutants in breast milk of mothers residing around an open dumping site in Kolkata, India: specific dioxin-like PCB levels and fish as a potential source. Environ Int. 2010;36(1):27–35. [DOI] [PubMed] [Google Scholar]
- 239. Schecter A, Cramer P, Boggess K, et al. . Intake of dioxins and related compounds from food in the U.S. population. J Toxicol Environ Health A. 2001;63(1):1–18. [DOI] [PubMed] [Google Scholar]
- 240. Aozasa O, Ohta S, Nakao T, Miyata H, Nomura T. Enhancement in fecal excretion of dioxin isomer in mice by several dietary fibers. Chemosphere. 2001;45(2):195–200. [DOI] [PubMed] [Google Scholar]
- 241. Sera N, Morita K, Nagasoe M, Tokieda H, Kitaura T, Tokiwa H. Binding effect of polychlorinated compounds and environmental carcinogens on rice bran fiber. J Nutr Biochem. 2005;16(1):50–58. [DOI] [PubMed] [Google Scholar]
- 242. Barnard ND, Cohen J, Jenkins DJ, et al. . A low-fat vegan diet and a conventional diabetes diet in the treatment of type 2 diabetes: a randomized, controlled, 74-wk clinical trial. Am J Clin Nutr. 2009;89(5):1588S–1596S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(3):193–205. [DOI] [PubMed] [Google Scholar]
- 244. Hectors TL, Vanparys C, van der Ven K, et al. . Environmental pollutants and type 2 diabetes: a review of mechanisms that can disrupt β cell function. Diabetologia. 2011;54(6):1273–1290. [DOI] [PubMed] [Google Scholar]
- 245. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–387. [DOI] [PubMed] [Google Scholar]
- 246. Kim JS, Lim HS, Cho SI, Cheong HK, Lim MK. Impact of Agent Orange exposure among Korean Vietnam veterans. Ind Health. 2003;41(3):149–157. [DOI] [PubMed] [Google Scholar]
- 247. Kang HK, Dalager NA, Needham LL, et al. . Health status of Army Chemical Corps Vietnam veterans who sprayed defoliant in Vietnam. Am J Ind Med. 2006;49(11):875–884. [DOI] [PubMed] [Google Scholar]
- 248. Steenland K, Piacitelli L, Deddens J, Fingerhut M, Chang LI. Cancer, heart disease, and diabetes in workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Natl Cancer Inst. 1999;91(9):779–786. [DOI] [PubMed] [Google Scholar]
- 249. Vena J, Boffetta P, Becher H, et al. . Exposure to dioxin and nonneoplastic mortality in the expanded IARC international cohort study of phenoxy herbicide and chlorophenol production workers and sprayers. Environ Health Perspect. 1998;106(suppl 2):645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Michalek JE, Pavuk M. Diabetes and cancer in veterans of Operation Ranch Hand after adjustment for calendar period, days of spraying, and time spent in Southeast Asia. J Occup Environ Med. 2008;50(3):330–340. [DOI] [PubMed] [Google Scholar]
- 251. Steenland K, Calvert G, Ketchum N, Michalek J. Dioxin and diabetes mellitus: an analysis of the combined NIOSH and Ranch Hand data. Occup Environ Med. 2001;58(10):641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Fierens S, Mairesse H, Heilier JF, et al. . Dioxin/polychlorinated biphenyl body burden, diabetes and endometriosis: findings in a population-based study in Belgium. Biomarkers. 2003;8(6):529–534. [DOI] [PubMed] [Google Scholar]
- 253. Philibert A, Schwartz H, Mergler D. An exploratory study of diabetes in a First Nation community with respect to serum concentrations of p,p'-DDE and PCBs and fish consumption. Int J Environ Res Public Health. 2009;6(12):3179–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Uemura H, Arisawa K, Hiyoshi M, et al. . Associations of environmental exposure to dioxins with prevalent diabetes among general inhabitants in Japan. Environ Res. 2008;108(1):63–68. [DOI] [PubMed] [Google Scholar]
- 255. Tanaka T, Morita A, Kato M, et al. . Congener-specific polychlorinated biphenyls and the prevalence of diabetes in the Saku Control Obesity Program (SCOP). Endocr J. 2011;58(7):589–596. [DOI] [PubMed] [Google Scholar]
- 256. Ukropec J, Radikova Z, Huckova M, et al. . High prevalence of prediabetes and diabetes in a population exposed to high levels of an organochlorine cocktail. Diabetologia. 2010;53(5):899–906. [DOI] [PubMed] [Google Scholar]
- 257. Rylander L, Rignell-Hydbom A, Hagmar L. A cross-sectional study of the association between persistent organochlorine pollutants and diabetes. Environ Health. 2005;4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Rignell-Hydbom A, Rylander L, Hagmar L. Exposure to persistent organochlorine pollutants and type 2 diabetes mellitus. Hum Exp Toxicol. 2007;26(5):447–452. [DOI] [PubMed] [Google Scholar]
- 259. Everett CJ, Frithsen IL, Diaz VA, Koopman RJ, Simpson WM Jr, Mainous AG 3rd. Association of a polychlorinated dibenzo-p-dioxin, a polychlorinated biphenyl, and DDT with diabetes in the 1999–2002 National Health and Nutrition Examination Survey. Environ Res. 2007;103(3):413–418. [DOI] [PubMed] [Google Scholar]
- 260. Cox S, Niskar AS, Narayan KM, Marcus M. Prevalence of self-reported diabetes and exposure to organochlorine pesticides among Mexican Americans: Hispanic health and nutrition examination survey, 1982–1984. Environ Health Perspect. 2007;115(12):1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Codru N, Schymura MJ, Negoita S, Akwesasne Task Force on Environment, Rej R, Carpenter DO. Diabetes in relation to serum levels of polychlorinated biphenyls and chlorinated pesticides in adult Native Americans. Environ Health Perspect. 2007;115(10):1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Turyk M, Anderson HA, Knobeloch L, Imm P, Persky VW. Prevalence of diabetes and body burdens of polychlorinated biphenyls, polybrominated diphenyl ethers, and p,p'-diphenyldichloroethene in Great Lakes sport fish consumers. Chemosphere. 2009;75(5):674–679. [DOI] [PubMed] [Google Scholar]
- 263. Silverstone AE, Rosenbaum PF, Weinstock RS, et al. . Polychlorinated biphenyl (PCB) exposure and diabetes: results from the Anniston Community Health Survey. Environ Health Perspect. 2012;120(5):727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Son HK, Kim SA, Kang JH, et al. . Strong associations between low-dose organochlorine pesticides and type 2 diabetes in Korea. Environ Int. 2010;36(5):410–414. [DOI] [PubMed] [Google Scholar]
- 265. Hertz-Picciotto I, Charles MJ, James RA, Keller JA, Willman E, Teplin S. In utero polychlorinated biphenyl exposures in relation to fetal and early childhood growth. Epidemiology. 2005;16(5):648–656. [DOI] [PubMed] [Google Scholar]
- 266. Valvi D, Mendez MA, Martinez D, et al. . Prenatal concentrations of polychlorinated biphenyls, DDE, and DDT and overweight in children: a prospective birth cohort study. Environ Health Perspect. 2012;120(3):451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Verhulst SL, Nelen V, Hond ED, et al. . Intrauterine exposure to environmental pollutants and body mass index during the first 3 years of life. Environ Health Perspect. 2009;117(1):122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Blanck HM, Marcus M, Rubin C, et al. . Growth in girls exposed in utero and postnatally to polybrominated biphenyls and polychlorinated biphenyls. Epidemiology. 2002;13(2):205–210. [DOI] [PubMed] [Google Scholar]
- 269. Burns JS, Williams PL, Sergeyev O, et al. . Serum concentrations of organochlorine pesticides and growth among Russian boys. Environ Health Perspect. 2012;120(2):303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Jacobson JL, Jacobson SW, Humphrey HE. Effects of exposure to PCBs and related compounds on growth and activity in children. Neurotoxicol Teratol. 1990;12(4):319–326. [DOI] [PubMed] [Google Scholar]
- 271. Lamb MR, Taylor S, Liu X, et al. . Prenatal exposure to polychlorinated biphenyls and postnatal growth: a structural analysis. Environ Health Perspect. 2006;114(5):779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Cupul-Uicab LA, Hernández-Avila M, Terrazas-Medina EA, Pennell ML, Longnecker MP. Prenatal exposure to the major DDT metabolite 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene (DDE) and growth in boys from Mexico. Environ Res. 2010;110(6):595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Gladen BC, Ragan NB, Rogan WJ. Pubertal growth and development and prenatal and lactational exposure to polychlorinated biphenyls and dichlorodiphenyl dichloroethene. J Pediatr. 2000;136(4):490–496. [DOI] [PubMed] [Google Scholar]
- 274. Karmaus W, Osuch JR, Eneli I, et al. . Maternal levels of dichlorodiphenyl-dichloroethylene (DDE) may increase weight and body mass index in adult female offspring. Occup Environ Med. 2009;66(3):143–149. [DOI] [PubMed] [Google Scholar]
- 275. Mendez MA, Garcia-Esteban R, Guxens M, et al. . Prenatal organochlorine compound exposure, rapid weight gain, and overweight in infancy. Environ Health Perspect. 2011;119(2):272–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Patandin S, Koopman-Esseboom C, de Ridder MA, Weisglas-Kuperus N, Sauer PJ. Effects of environmental exposure to polychlorinated biphenyls and dioxins on birth size and growth in Dutch children. Pediatr Res. 1998;44(4):538–545. [DOI] [PubMed] [Google Scholar]
- 277. Gladen BC, Klebanoff MA, Hediger ML, et al. . Prenatal DDT exposure in relation to anthropometric and pubertal measures in adolescent males. Environ Health Perspect. 2004;112(17):1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Jusko TA, Koepsell TD, Baker RJ, et al. . Maternal DDT exposures in relation to fetal and 5-year growth. Epidemiology. 2006;17(6):692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Warner M, AguilarSchall R, Harley KG, Bradman A, Barr D, Eskenazi B. In utero DDT and DDE exposure and obesity status of 7-year-old Mexican-American children in the CHAMACOS cohort. Environ Health Perspect. 2013;121(5):631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Smink A, Ribas-Fito N, Garcia R, et al. . Exposure to hexachlorobenzene during pregnancy increases the risk of overweight in children aged 6 years. Acta Paediatr. 2008;97(10):1465–1469. [DOI] [PubMed] [Google Scholar]
- 281. Glynn AW, Granath F, Aune M, et al. . Organochlorines in Swedish women: determinants of serum concentrations. Environ Health Perspect. 2003;111(3):349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Hovinga ME, Sowers M, Humphrey HE. Environmental exposure and lifestyle predictors of lead, cadmium, PCB, and DDT levels in Great Lakes fish eaters. Arch Environ Health. 1993;48(2):98–104. [DOI] [PubMed] [Google Scholar]
- 283. Hue O, Marcotte J, Berrigan F, et al. . Plasma concentration of organochlorine compounds is associated with age and not obesity. Chemosphere. 2007;67(7):1463–1467. [DOI] [PubMed] [Google Scholar]
- 284. Pelletier C, Després JP, Tremblay A. Plasma organochlorine concentrations in endurance athletes and obese individuals. Med Sci Sports Exerc. 2002;34(12):1971–1975. [DOI] [PubMed] [Google Scholar]
- 285. Bachelet D, Truong T, Verner MA, et al. . Determinants of serum concentrations of 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene and polychlorinated biphenyls among French women in the CECILE study. Environ Res. 2011;111(6):861–870. [DOI] [PubMed] [Google Scholar]
- 286. De Roos AJ, Ulrich CM, Sjodin A, McTiernan A. Adiposity, body composition, and weight change in relation to organochlorine pollutant plasma concentrations. J Expo Sci Environ Epidemiol. 2012;22(6):617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Ibarluzea J, Alvarez-Pedrerol M, Guxens M, et al. . Sociodemographic, reproductive and dietary predictors of organochlorine compounds levels in pregnant women in Spain. Chemosphere. 2011;82(1):114–120. [DOI] [PubMed] [Google Scholar]
- 288. Sandanger TM, Sinotte M, Dumas P, et al. . Plasma concentrations of selected organobromine compounds and polychlorinated biphenyls in postmenopausal women of Québec, Canada. Environ Health Perspect. 2007;115(10):1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Jakszyn P, Goñi F, Etxeandia A, et al. . Serum levels of organochlorine pesticides in healthy adults from five regions of Spain. Chemosphere. 2009;76(11):1518–1524. [DOI] [PubMed] [Google Scholar]
- 290. Schildkraut JM, Demark-Wahnefried W, DeVoto E, Hughes C, Laseter JL, Newman B. Environmental contaminants and body fat distribution. Cancer Epidemiol Biomarkers Prev. 1999;8(2):179–183. [PubMed] [Google Scholar]
- 291. Bradman AS, Schwartz JM, Fenster L, Barr DB, Holland NT, Eskenazi B. Factors predicting organochlorine pesticide levels in pregnant Latina women living in a United States agricultural area. J Expo Sci Environ Epidemiol. 2007;17(4):388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Dirinck E, Jorens PG, Covaci A, et al. . Obesity and persistent organic pollutants: possible obesogenic effect of organochlorine pesticides and polychlorinated biphenyls. Obesity. 2011;19(4):709–714. [DOI] [PubMed] [Google Scholar]
- 293. Lim S, Cho YM, Park KS, Lee HK. Persistent organic pollutants, mitochondrial dysfunction, and metabolic syndrome. Ann NY Acad Sci. 2010;1201:166–176. [DOI] [PubMed] [Google Scholar]
- 294. Lee DH, Steffes MW, Jacobs DR Jr. Can persistent organic pollutants explain the association between serum gamma-glutamyltransferase and type 2 diabetes? Diabetologia. 2008;51(3):402–407. [DOI] [PubMed] [Google Scholar]