

Abstract
Prostate cancer (PC) is the second leading cause of cancer related deaths in US men. Androgen deprivation therapy (ADT) improves clinical outcome, but tumors often recur and progress to androgen independent prostate cancer (AIPC) which no longer responds to ADT. The progression to AIPC is due to genetic alterations that allow PC cancer cells to grow in the absence of androgen. Here we performed an insertional mutagenesis screen using a replication-incompetent lentiviral vector (LV) to identify the genes that promote AIPC in an orthotopic mouse model. Androgen sensitive PC cells, LNCaP, were mutagenized with LV and injected into the prostate of male mice. After tumor development, mice were castrated to select for cells that proliferate in the absence of androgen. Proviral integration sites and nearby dysregulated genes were identified in tumors developed in an androgen deficient environment. Using publically available datasets, the expression of these candidate androgen independence genes in human PC tissues were analyzed. A total of 11 promising candidate AIPC genes were identified: GLYATL1, FLNA, OBSCN, STRA13, WHSC1, ARFGAP3, KDM2A, FAM83H, CLDN7, CNOT6 and B3GNT9. Seven out the 11 candidate genes; GLYATL1, OBSCN, STRA13, KDM2A, FAM83H, CNOT6 and B3GNT6, have not been previously implicated in PC. An in vitro clonogenic assay showed that knockdown of KDM2A, FAM83H and GLYATL1 genes significantly inhibited the colony forming ability of LNCaP cells. Additionally, we showed that a combination of four genes, OBSCN, FAM83H, CLDN7 and ARFGAP3 could significantly predicted the recurrence risk in PC patients after prostatectomy (P=5.3×10-5).
Keywords: Retrovirus, Genome integration, Gene dysregulation, Biomarker, Prostate cancer
INTRODUCTION
Prostate cancer (PC) is the most commonly diagnosed cancer in men and the second leading cause of cancer related deaths in United States. The five year survival rate of PC is almost 100% if diagnosed early. However the rate drops to ~30% if diagnosed late or once it has metastasized to distant organs [1]. Localized primary PC is treated by prostatectomy or radiotherapy, but often patients develop metastatic disease. PC progression is dependent on androgen levels, therefore halting the androgen supply to the cancerous cells using anti-androgen drugs (androgen deprivation therapy, ADT) has been the frontline option for treatment of metastatic PC [2-4]. Though patients initially respond positively to ADT, in almost all patients the tumor recurs. The recurrent tumor, clinically referred as androgen-independent prostate cancer (AIPC), is aggressive and no longer responds to ADT. AIPC is usually lethal, with an average life expectancy of less than 5 years [5, 6]. The molecular basis for this progression from androgen sensitive to AIPC after treatment is one of the most extensively studied areas in the field of PC. Several genes have been linked to the progression of PC into AIPC, but the molecular mechanisms responsible for AIPC progression are still poorly defined. High-throughput techniques have been used to discover genes that are altered/mutated and differentially expressed between androgen sensitive and androgen insensitive PC. However, differentiating gene mutations that actually drive AIPC from bystander mutations is a major challenge [7, 8]. Identifying the driver genetic mutations responsible for AIPC is critical for improving our ability to predict recurrence and for developing new therapeutics to increase the life expectancy of PC patients [9].
Over the past few decades insertional mutagenesis screens have been used to discover cancer related genes [10]. The majority of the previous insertional mutagenesis screens have used either replicating retroviruses or transposons to induce mutation and cause cancer. These mutagenic elements also function as molecular tags to identify the common insertions sites (CISs) and genes nearby integration sites whose dysregulation might trigger cancer initiation and progression. A major drawback of using replicating retroviruses or transposons is that these mutagenic elements can replicate after integration resulting in multiple integrations after the cells have progressed to a cancerous state [11]. Multiple late integrations make it difficult to differentiate integrations actually involved in causing cancer (driver gene mutation) from integrations that occur after cancer progression (passenger gene mutations). By contrast replication-incompetent retroviruses do not replicate after integrating into the genome and therefore do not introduce additional insertions. This reduces passenger insertions. Recent studies have used replication incompetent retroviral vectors as mutagens to identify driver genes involved in the initiation and progression of leukemia, liver, breast, pancreatic and PC [7, 8, 12].
Here we report for the first time an insertional mutagenesis screen in an orthotopic xenograft mouse model to identify genes involved in AIPC. In this approach, mutagenized-LNCaP cells were directly injected into the prostate of male immunodeficient mice. After tumors formed, mice were castrated to select for androgen independent tumors in vivo, modeling what occurs in PC patients. This human xenograft PC orthotopic model has the advantage that genes promoting AIPC in the prostate microenvironment can potentially be identified [13, 14]. We identified several interesting genes using this screen and established their ability to predict recurrence after ADT.
MATERIALS AND METHODS
Cell line Culture, Vector Production, and Transduction
The androgen-dependent human prostate carcinoma cell line LNCaP-FGC (ATCC CRL-1740) was transduced with LV vector LV-SFFVEGFP as previously described [7]. LV-SFFVEGFP, contains self-inactivating long terminal repeats (LTRs) and a spleen focus forming virus (SFFV) promoter driving enhanced green fluorescent protein (EGFP), and WPRE (woodchuck hepatitis virus posttranscriptional regulatory element) (Figure 1A). It also contains an R6Kγ origin of replication and kanamycin resistance gene.
Figure 1.
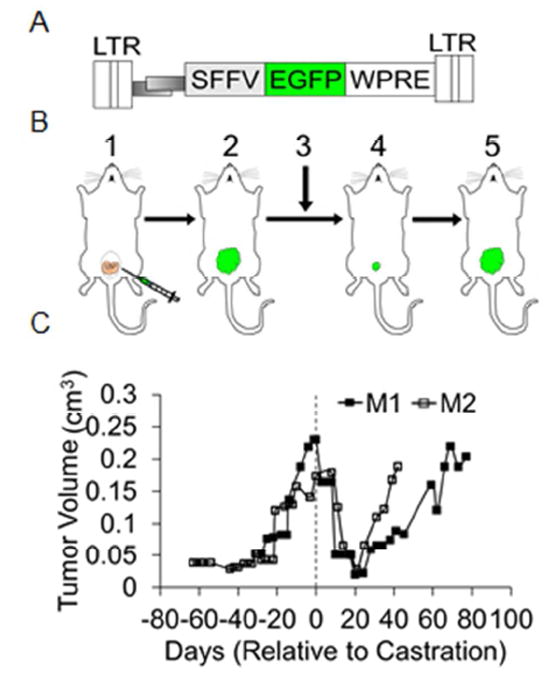
LV-insertional mutagenesis screen to identify genes involved in AIPC using a orthotopic mouse model. A) Schematic representation of the lentiviral vector (LV) used to mutagenize LNCaP cells. SFFV: spleen focus forming virus promoter, EGFP: enhanced green fluorescent protein, WPRE: woodchuck hepatitis virus posttranscriptional regulatory element. LTR: long terminal repeats. B) Orthotopic model showing 1) injection of LV-mutagenized LNCaP cells into the prostate gland of a male NSG mouse, 2) tumor development, 3) surgical castration, 4) regression of tumor and 5) re-growth of androgen-independent tumor. C) In vitro tumor growth: Tumor volumes at various time points before and after castration in mice injected with LV-mutagenized LNCaP cells were plotted. Dotted line indicates the castration time.
Orthotopic PC Mouse Model
All animal procedures were reviewed and approved by the Washington State University Institutional Animal Care and Use Committee and institutional guidelines for the humane use of animals in research were followed. Male 4-8 week old NSG mice were obtained from the Jackson Laboratory (Bar Harbor, Maine). For orthotopic injection, mice were anesthetized via isoflurane gas. A small incision of approximately 1 cm was made in the lower abdomen through the skin and peritoneum to expose the prostate gland. 1×106 LV-mutagenized LNCaP cells in 20 μL RPMI 1640 plus 5% FBS plus 100 μL Matrigel (BD Biosciences, Bedford, MA) were inoculated via orthotopic injection into the dorsal or ventral prostate using a 27 gauge needle. Wound clips were placed over the incision to seal the wound for recovery. Mice were initially monitored daily for 3 days following surgery, and then every 3 days over the course of the experiment. Tumor growth was monitored and measured by vernier calipers every 3 days. To provide an androgen-deficient environment for the selection and growth of androgen-independent tumors, mice were castrated via the scrotal approach. Following castration, tumor growth was monitored. Once primary tumors reached sizes larger than their size prior to castration, tumor tissue was harvested. Genomic DNA was obtained from tumor tissue using the Puregene Cell and Tissue Kit (Qiagen Inc., Valencia, CA).
MGS-PCR Sequencing and Identification of Integration Sites
Integration sites were identified using the deep sequencing technique known as modified genomic sequencing (MGS)-PCR as previously described [15] except that a hydroshear device (Digilab, Marlborough, MA) was used for DNA shearing. Primary tumors from two castrated mice were collected and processed for MGS-PCR sequencing. Approximately one to four million sequence reads were obtained per tumor. Forward and reverse sequence reads were paired to extend sequence read lengths using PEAR sequencing pairing software [16]. VISA, vector integration site analysis server [17] (https://visa.pharmacy.wsu.edu/bioinformatics/) was used to identify vector-chromosome junctions and determine integration site locations within the human genome (hg38) as well as identify nearby genes and promoters. Custom PERL programming was used to further identify the closest genes with transcription start sites within 50 kbp of insertions. Only alignments that had a LTR-chromosome junction and met additional criteria as previously described [18] were considered as provirus integration sites.
Candidate Gene Identification and Analysis
cDNA microarray datasets available in the Oncomine database (http://www.oncomine.com) [19] were used to systematically assess the differential gene expression in normal versus PC patients. We performed a meta-analysis of 16 datasets [20-35] to determine the expression pattern of the genes identified in our screen. cBioportal of cancer genomics was used to examine the genetic alteration of the candidate genes in PC patient samples [36, 37].To investigate the clinical outcome and prognostic relevance of candidate genes we used the online biomarker validation tool Survexpress (http://bioinformatica.mty.itesm.mx:8080/Biomatec/SurvivaX.jsp) [38]. Seven PC datasets are available in SurvExpress and for our analysis we only used the two datasets [20, 39] that contained all of our candidate gene records and had data for recurrence after prostatectomy.
Doxycycline-induced shRNA lentiviral vector
LV plasmid expressing doxycycline-inducible shRNA sequence, pTRIPZ vectors, were purchased from Dharamcon (Pittsburgh PA). pTRIPZ shRNA (pTsh)-LV vectors were produced as described previously [7]. LNCaP cells were transduced with pTsh-LV vectors at an MOI of 2.5 and were puromcyin (2 μg/ml) selected for 1-2 weeks. For shRNA induction, the pTsh-LV vector transduced cells were cultured in media containing doxycycline (500 ng/ml). While the pTsh-LV vector transduced LNCaP cells cultured in absence of doxycycline was used control. Knockdown efficiency was confirmed by RT-PCR and western blot analysis. Total RNA was isolated from pTsh-LV vector transduced cells cultured in presence or absence of doxycycline using TRIzol reagent (Invitrogen, Carsbad, CA). cDNA synthesis and RT-PCR analysis was performed as described previously [7]. Sequences of the primers used in the current study are: GLYATL1: Forward primer (For) 5’- GGCCTCAAAAGCAGGAGATG-3’, reverse primers (Rev) 5’-AGCCACTCTTATCCCCTCACC-3’; FAM83H: For 5’-GCTACAGCTTCATGTGGT CCT-3’, Rev 5’- CATACGGAGCCAGGGCATAG-3’; STRA13: For 5’-GGTCTTCGTTGTG GAAGCAGC-3’, Rev 5’-ATCAGAGGCCGCTGGAAACA-3’; KDM2A: For 5’-GAAAGGTC TTCTGGCTCATCC-3’, Rev 5’-TGAATCCAGCCTGAGGGAATG-3’, and GAPDH: For 5’- GATTTGGTCGTATTGGGCGC-3’, rev 5’-AAATGAGCCCCAGCCTTCTC-3’. For western blot analysis, total cell lysate was isolated from pTsh-LV vector transduced LNCaP cells cultured in media with or without doxycycline for at least 72 hr. Thirty μg of total protein was resolved on SDS-PAGE, transferred onto PVDF membrane and probed with specific antibodies at a dilution of 1:1000. Blots were washed and incubated in species specific secondary IgG conjugated to HRP, developed using ECL western blot substrate (Thermo Scientific, Rockford, IL) and imaged using ChemiDoc+ XRS (Bio-Rad, Hercules CA). Antibodies used in the present study were: FAM83H (Novus Biologicals, Littleton, CO), KDM2A, GLYATL1, β-actin (Santa Cruz Biotechnology, Santa Cruz CA). All the HRP conjugated secondary antibodies were purchased from Santa Cruz Biotechnology.
Clonogenic assay
To determine the effect of the candidate gene knockdown on proliferation rate of LNCaP cells an in vitro clonogenic assay was performed [40]. Briefly, 2000 pTsh-LV vector transduced LNCaP cells were plated in 6-well plate and cultured for 3 weeks in media with or without doxycycline. Cells were fixed with methanol, stained with 0.1% crystal violet solution, rinsed with water, air dried and number of colonies were counted.
Cell proliferation recovery assay
To investigate the effect of the candidate gene knockdown on proliferation of LNCaP cells in androgen depleted media, we used proliferation recovery assay that was previously used to demonstrate the association of genes with AIPC [41]. Briefly, we pre-cultured 2 × 105 pTsh-LV vector transduced LNCaP cells in charcoal treated serum for eight days that induces a proliferation arrest. After eight days, cells were counted and 105 cells were re-cultured in complete media supplemented with or without doxycycline for another seven days allowing the cells to recover from the proliferative arrest. Number of cells at the end point were enumerated to determine the effect of candidate gene knockdown on the proliferation recovery rate.
Statistical analysis
To confirm the effects of doxycycline-induced shRNA knockdown effect on the clonogenic assay and proliferation recovery assay a student’s t-test was used. P-values <0.05 were considered to be significant.
RESULTS
LV Mutagenized LNCaP Cells Induce Androgen Independent Orthotopic Tumors in Mice
We previously showed that we could identify novel AIPC genes in vitro and in subcutaneous tumors using a replication-incompetent LV [7]. In this study we used LV to induce mutations in LNCaP cells, an androgen dependent PC cell line, and investigate the genes associated with androgen independent growth. LNCaP cells transform into androgen independent cells (LNCaP-AI), potentially by acquiring genetic alterations (activation of oncogenes and suppression of tumor suppressor genes) [42, 43]. Here we performed a screen to identify AIPC genes using orthotopic PC tumors. LV-mutagenized LNCaP cells were injected into NSG mice, forming tumors approximately 20 days post injection (Figure 1). Once the tumor volumes reached 0.2 cm3, mice were surgically castrated. Post-castration the tumors regressed, as measured by the tumor volumes (Figure 1B and 1C). As expected, after 3-4 weeks of initial regression the tumors re-grew in the castrated mice, modeling what occurs in human PC patients treated with ADT. AIPC tumors that regrew in castrated mice were collected for analysis of LV-vector integration sites and nearby candidate genes.
LV Proviral Integration Site Analysis and Identification of Nearby Genes
Genomic DNA isolated from primary tumor samples were analyzed for provirus integration sites using MGS-PCR [15]. Sequence reads were mapped to the human genome (hg38) using the VISA bioinformatics server [17]. From over 4,657,000 total sequence reads, only 459,979 sequence reads met our selection criteria [17, 18] and were considered as provirus integration sites. A total of 394 unique proviral integration sites were recovered from the two tumors. The unique integration sites were captured at varying frequencies ranging from one to 78,139 times. To prioritize the integration sites, the top 20% of captured integration sites from each tumor, comprising a total of 79 unique integration sites, were selected for additional analysis. Genes near the provirus integrations were determined by two criteria: 1) genes containing an integration and 2) the three closest genes with transcription start sites within 50 kb of a provirus integration site.
Meta-analysis of Genes Near Vector Proviruses to Identify Candidate PC Genes
To identify which genes near vector proviruses are candidate AIPC genes, we explored their expression in PC patient tumors using publically available microarray data (Oncomine) [19]. Oncomine comprises a collection of more than 16 gene-expression datasets related to PC alone. To reduce the biases of an individual study/dataset, a meta-analysis of 16 different datasets was performed to determine the differential expression pattern of the genes in normal human prostate tissues and human PC tissues [20-35]. Only genes with p-values <0.01 and gene rank less than 2000 were considered as candidate genes. Eight promising candidate genes that were differentially expressed in PC patients were identified by meta-analysis: GLYATL1, FLNA, STRA13, WHSC1, FAM38H, CLDN7, CNOT6 and B3GNT6 (Table 1 and supplementary figure 1). Genes GLYATL1, STRA13, WHSC1, FAM83H, CLDN7 and CNOT6 were significantly overexpressed in PC tissues compared to normal tissue, whereas FLNA and B3GNT6 were under expressed in PC tissue (Table 1). Supplementary figure 2 shows the eight proviral integration sites mapped on the human genome using the University of California, Santa Cruz (UCSC) genome browser [44].
Table 1.
Meta-analysis of the candidate gene across 16 different datasets. Column 1 and 2 indicates chromosome and the candidate gene in/near integration. Column 3 indicates whether the LV provirus integrated within gene or has a gene TSS within the distance indicated. Column 4 and 5 indicates the expression of the candidate gene in PC patients and the corresponding p-value from the meta-analysis. Column 6 indicates whether the gene was previously associated with PC or other cancers.
| Chromosome | Candidate Gene | Integration in/near gene | Expression pattern | p-value | Previously implicated with prostate cancer (other cancers*) |
|---|---|---|---|---|---|
| Chr11 | GLYATL1 | In | Over | 0.0000178 | No* |
| ChrX | FLNA | 2.1 kb | Under | 0.001 | Yes |
| Chr17 | STRA13 | 22.7 kb | Over | 0.001 | No* |
| Chr8 | FAM83H | 10.8 kb | Over | 0.005 | No* |
| Chr4 | WHSC1 | In | Over | 0.005 | Yes |
| Chr17 | CLDN7 | 5.1 kb | Over | 0.006 | Yes |
| Chr5 | CNOT6 | In | Over | 0.008 | No |
| Chr16 | B3GNT9 | 9.4 kb | Under | 0.009 | No |
Gene was reported to be implicated with other cancer
Identification of CISs and Nearby Genes
In past insertional mutagenesis studies, identification of CISs has been the most common method to identify loci or genes dysregulated by provirus integrations [10]. In order to identify CISs, we analyzed the top 30% of captured unique integrations sites and looked for integration sites that existed within a 100 kb window of one another. We identified three CIS within chromosome 1, 11 and 22. In chromosome 1 two integration sites were observed in the gene OBSCN, which are separated by ~30.3 kb (Table 2, Supplementary figure 3A). In chromosome 11 proviruses integrated at three distinct sites within a window of 78.6 kb in the gene KDM2A (Table 2, Supplementary figure 3B). In chromosome 22 proviruses were observed in the ARFGAP3 gene twice within a window of 5.9 kb (Table 2, Supplementary figure 3C). These CISs (within OBSCN, KDM2A and ARFGAP3 genes) had no other gene transcription start sites within 50 kb of the CISs (Table 2). These three genes were included in our list of candidate AIPC genes based on the presence of a CIS. Oncomine analysis showed that KDM2A and OBSCN genes were under expressed, while ARFGAP3 gene was overexpressed in PC patients samples compared to normal prostate tissues samples (Supplementary figure 1).
Table 2.
List of CISs identified by insertional site analysis. Columns 1 and 2 are the chromosome number and number of integrations within a range of 100 kb. Column 3 and 4 are integration site positions in the chromosome and distance between the integration sites. Column 5 lists the genes in/near the CISs. Column 6 shown the expression of candidate genes in PC sample based on meta-analysis of datasets in Oncomine. Column 7 indicates whether the gene was previously reported to be associated with PC or other cancers.
| Chr | No. of Integrations | Position of the integration (bp) | Maxiumium distance between integration sites (bp) | Candidate genes | Expression in PC | Previously implicated with prostate cancer (other cancers*) |
|---|---|---|---|---|---|---|
| 1 | 2 | 228264130 | 30313 | OBSCN | Under | No* |
| 228233817 | ||||||
|
| ||||||
| 11 | 3 | 67123540 | 78662 | KDM2A | Under | No* |
| 67132312 | ||||||
| 67202202 | ||||||
|
| ||||||
| 22 | 2 | 42817724 | 5923 | ARFGAP3 | Over | Yes |
| 42823647 | ||||||
Gene was reported to be implicated with other cancer
Candidate Genes are Recurrently Altered in PC Patients
To investigate whether the candidate genes we identified are genetically altered in PC patients, we examined distinct genetic alterations including mutations, copy number variations and mRNA expression levels in PC patients in the TCGA dataset (http://cancergenome.nih.gov/) using tools available in cBioPortal of Cancer Genomics. We chose the TCGA dataset because it has all three of these data types, and it has a large number of patient tumors. This analysis showed that all 11 candidate genes had genetic alterations in PC patients. Among the candidate genes, we observed that FAM83H gene was most frequently altered. 39 out of 257 (15%) of PC patients showed genetic alteration in FAM83H, out of which 36 tumors had either copy number gain (amplification) or mRNA upregulation, while the remaining three tumors each had a copy number loss, missense mutation or truncated mutation in FAM83H gene (Figure 2). For comparison, TCGA data showed 21% and 31% of tumors have alterations in TP53 and PTEN, respectively. Analysis of the TCGA dataset showed that 11-4% of prostate tumor samples had genetic alterations in CLDN7, KDM2A, ARFGAP3, OBSCN, CNOT6, WHSC1, B3GNT9, FLNA, STRA13 and GLYATL1, respectively (Figure 2). We also queried the occurrence of genetic alteration in PC patients in five other datasets [20, 21, 45-47] available in cBioPortal. Alteration frequencies of the candidate genes in six datasets is shown in supplementary figure 4. We observed that FAM83H was recurrently altered in PC patients across all the six datasets available in cBioPortal (Supplementary figure 4). OBSCN, CLDN7 and STRA13 were frequently altered among PC patients in at least 4 datasets (Supplementary figure 4). In summary we demonstrated that all 11 genes identified in our screen were recurrently altered in PC patients, implicating these genes in PC progression.
Figure 2.
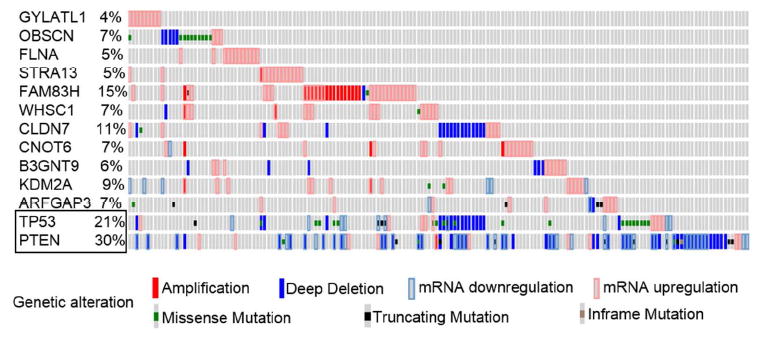
cBioportal data showing distinct genetic alteration in candidate genes in 257 PC patients using data from TCGA dataset. Genetic alteration of TP53 and PTEN (boxed), were included as the most frequently altered genes in PC patients. Each patient sample is represented by a bar and each color indicates specific genetic alteration, as shown below the figure. Only patients with alterations were shown (from 257 samples). The frequency of gene alteration is represented in percentage.
Validation of the Candidate Genes
To confirm the implication of the novel candidate genes in PC progression, we selected four top candidate genes (GLYATL1, FAM83H, STRA13 and KDM2A) identified in our insertional mutagenesis screen and determined the effect of gene knockdown on clonogenic ability and proliferation rate in androgen depleted conditions. RT-PCR analysis confirmed efficient knockdown of KDM2A, FAM83H and GLYATL1 mRNA levels in the doxycycline induced pTsh-LV vector transduced LNCaP cells compared to respective control cells (Supplementary Figure 5). Whereas, STRA13 pTsh-LV vectors tested in the present study had no effect on STRA13 mRNA levels. Further, western blot analysis confirmed that doxycycline induction reduced KDM2A, FAM83H and GLYATL1 protein levels in pTsh-LV vector transduced LNCaP cells compared to respective control cells (Figure 3 A-C).
Figure 3. Validation of selected candidate genes using clonogenic assay and proliferation assay.
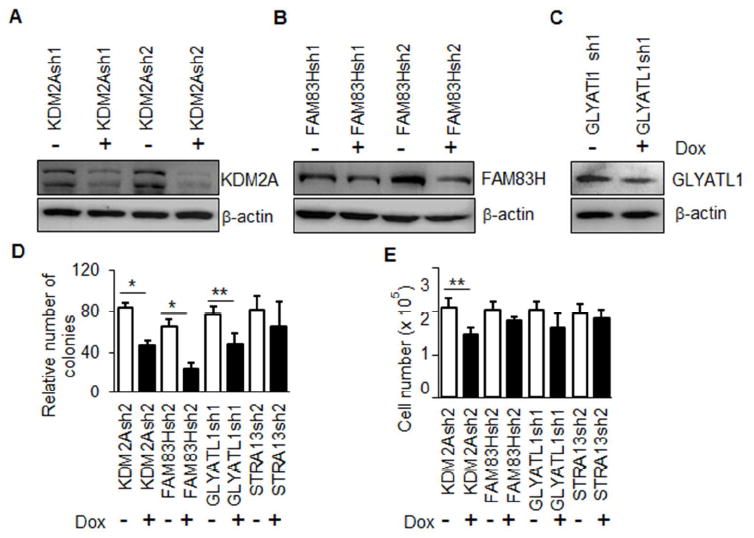
LNCaP cells were transduced with pTRIPZ LV vector expressing inducible shRNA sequences (pTsh-LV vector) targeting the candidate gene. Gene knockdown was confirmed by western blot analysis of the total cell lysate isolated from transduced LNCaP cells cultured with and without doxycycline for at least 72 hr and immune-probed specifically for A) KDM2A, B) FAM83H and C) GLYATL1. All the blots were stripped and re-probed for β-actin as an internal protein control. D) In vitro clonogenic assay showing the effect of knocking down KDM2A, FAM83H and GLYATL1 on LNCaP cells. Number of colonies in doxycycline treated and untreated LNCaP cells were counted and represented graphically. Open (white) and closed (black) columns represent pTsh-LV vector transduced cells cultured in absence and presence of doxycycline, respectively. Error bars in each column indicates the SD from triplicates, * p value is <0.01 and ** p value is less than <0.05. E) Proliferation recovery rate of pTsh-LV vector transduced was determined to associate the candidate gene with AIPC. LNCaP cells were pre-cultured in charcoal treated serum for 8 days and counted and re-seeded 105 cells in complete media. After 7 days the cells were counted to determine the proliferative ability of cells. Open (white) and closed (black) columns represent cells cultured in absence and presence of doxycycline, respectively. Error bars in each column indicates the SD from triplicates, * p value is <0.05.
In vitro clonogenic assay is a widely used technique to determine the survival and proliferative ability of the cells, including cancer and cancer-stem cells [48]. Indefinite proliferative ability of the single cells to grow into a colony is a characteristic biological property of cancer cells including cancer-stem cells. Fedr et al, [48] demonstrated the ability of the clonogenic assay in characterizing different phenotype and biological properties of stem cells and cancer stem cells. Our clonogenic assay indicated that suppression of KDM2A, FAM83H and GLYATL1 significantly inhibited the colony forming ability of the LNCaP cells (Figure 3D). We observed that knockdown of KDM2A, FAM83H and GLYATL1 showed reduced number of colonies by 53%, 62% and 37%, respectively when compared to the respective control cells. As expected, STRA13 pTsh-LV vector transduced LNCaP cells cultured in presence or absence of doxycycline showed no effect on the colony numbers (Figure 3D). This result confirm that the candidate gene identified in our insertional mutagenesis screen could significantly effect proliferation and survival of LNCaP cells.
One of the key step involved in AIPC progression is the recovery of the androgen sensitive cells from the adverse effects induced by androgen depletion, including proliferative arrest [49]. In order to investigate the association of the candidate genes with AIPC progression, we used proliferation recovery assay described by Barakat et al, [41]. We observed that KDM2A knockdown has significantly reduced the ability of LNCaP cells to recover from proliferation arrest induced by pre-culturing the cells in androgen depletion conditions. While FAM83H and GLYATL1 knockdown had no effect on the recovery rate of the LNCaP cells. Collectively these results indicate that KDM2A, FAM83H and GLYATL1 could promote proliferation of LNCaP cells, but KDM2A might have an impact on progression of androgen dependent PC to AIPC.
Candidate Genes Predicted the Recurrence Risk of PC patients After Treatment
Prostate serum antigen (PSA) is the most widely used biomarker for screening, diagnosis and prognostication of PC. However, lack of sensitivity and specificity of PSA results in over-diagnosis and overtreatment in a large number of PC patients [50]. Given the heterogeneous nature of PC, a single biomarker cannot provide accurate diagnosis and prediction information among diverse PC patients. Therefore much research now is focused on identify new biomarkers that could discriminate various forms of disease, indolent from aggressive, and set the clinical parameter for treatment. With “-omics” technologies several novel clinical markers for PC diagnosis and prognosis have been discovered [50]. Thus we investigated the prognostic value of the candidate genes in predicting clinical outcome of PC patients using SurvExpress [38]. Based on the differential expression of gene(s) in PC patients, the tool stratifies the patient samples into low and high risk groups and derives Kaplan-Meier curves defining the specified risk of the patients. Out of 7 published PC datasets, 2 contained records of all 11 candidate genes and had expression data from recurrent tumors following radical prostatectomy [20, 39]. The dataset generated by Taylor et al. [20] had the largest number of patient tumor samples (140) and was thus used to determine the prognostic value of the candidate genes in predicting recurrence risk in cancer patients after prostatectomy. To identify gene sets that would efficiently predict clinical outcome we analyzed prognostic values for each gene independently as well as 2-gene, 3-gene and 4-gene combinations. The combination of OBSCN, FAM83H, CLDN7 and ARFGAP3 was the most promising combination that predicted the recurrence risk of PC patient after treatment with statistically significant values: p-value=5.3 × 10-5, concordance index=74.37 and risk hazard ratio=6.12 (Figure 4 and Table 3). Several other gene combination, especially the ones with CLDN7 and ARFGAP3 genes, significantly predicted the clinical outcome of PC patients (Supplementary table 1). Moreover, the best 4-gene combination (OBSCN, FAM83H, CLDN7 and ARFGAP3) was able to stratify the cohorts available in the second database [39] and significantly predicted the clinical outcome of PC patients, p=0.006281 (Supplementary figure 5). Overall we were able to demonstrate that the candidate genes identified in our screen are of prognostic value that can predict the recurrence risk of PC patients.
Figure 4.
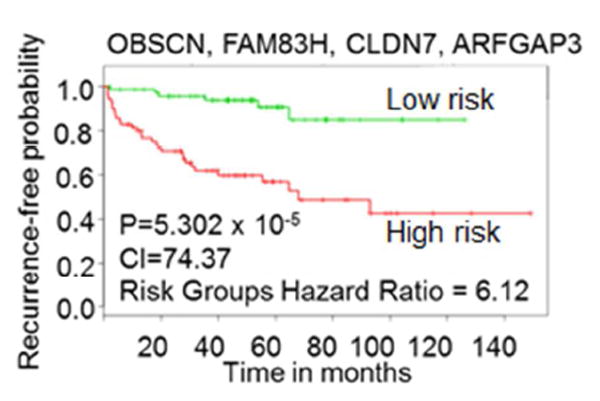
Kaplan-Meier curves showing the ability of the candidate genes to predict the recurrence risk in PC patients. The combination of OBSCN, FAM83H, CLDN7 and ARFGAP3 genes stratified the cohorts into high risk (red) and low risk (green) groups based on the expression levels of candidate genes. The combination of four genes showed a high ability to predict the recurrence risk in the PC cancer patients treated by prostatectomy (p-value=5.3×10-5).
Table 3.
Relative ability of the candidate genes to predict the recurrence risk in PC patients. All 11 candidate genes and combinations of at least four-candidate genes were analyzed for their prognostic significance in predicting clinical outcome in PC patients. Only top 10 combinations were represented in this table.
| Gene combinations | p-Value | Concordance Index | Risk groups hazard ratio |
|---|---|---|---|
| OBSCN + FAM83H + CLDN7 + ARFGAP3 | 0.00005302 | 74.37 | 6.12 |
| OBSCN + CLDN7 + B3GNT9 + ARFGAP3 | 0.00007988 | 75.14 | 5.3 |
| WHSC1 + CLDN7 + KDM2A + ARFGAP3 | 0.00008705 | 74.51 | 5.24 |
| OBSCN + CLDN7 + ARFGAP3 | 0.000101 | 74.78 | 5.16 |
| OBSCN + CLDN7 + CNOT6 + ARFGAP3 | 0.000101 | 74.81 | 5.16 |
| CLDN7 + ARFGAP3 | 0.0001031 | 74.05 | 5.16 |
| FLNA + FAM83H + CLDN7 + CNOT6 | 0.0001047 | 74.92 | 4.57 |
| CLDN7 + CNOT6 + ARFGAP3 | 0.0001238 | 74.56 | 5.06 |
| FAM83H + CLDN7 + CNOT6 + ARFGAP3 | 0.0001238 | 74.43 | 5.06 |
| FAM83H + CLDN7 | 0.00013 | 71.05 | 4.63 |
Concordance index: Indicator whether subjects with higher risk prediction will experience the event after subjects of lower risk.
Risk groups hazard ratio: Used to interpret the chance of an event occurring in the higher-risk population divided by the chance of an event occurring in the lower-risk population.
DISCUSSION
Insertional mutagenesis screens are valuable tools for discovering novel genes associated with cancer [51]. Here for the first time, we have performed a LV-mediated insertional mutagenesis screen in an orthotopic mouse model to discover genes associated with PC. Injection of LV-mutagenized LNCaP cells into the mouse prostate efficiently developed tumors in mouse prostates approximately 3 weeks after injections and these tumors regressed after castration. Similar to PC progression post ADT in cancer patients [2, 4, 52] we observed that tumors reemerged in the castrated mice. Re-emergence of AIPC from androgen responsive PC has been implicated due to various mechanism, including acquiring genetic alteration that support progression of AIPC [5]. We reasoned that tumors that develop in castrated mice have a selective advantage to proliferate in an androgen deficient environment. We hypothesize that these tumors gain this advantage due to provirus integrations dysregulating nearby genes, thereby triggering AIPC. Analyzing the proviral integration sites, we identified 11 promising candidate genes. Three of the candidate genes were identified in/near CISs and another eight candidate genes were identified by performing meta-analysis of all genes in/near the 79 tumor unique integrations sites from androgen independent primary tumor samples. Meta-analysis enabled us to prioritize the candidate genes that are differentially expressed in human PC samples. In addition to differentially expression, cancer-associated genes are mostly known to be genetically altered in cancer patients. It has been shown that PC progression is most often the result of chromosomal rearrangements and gene mutations [53]. The loss/mutations of TP53 and PTEN are common genetic alterations found in AIPC [54, 55]. In addition to PTEN and TP53, several other genetic alterations were reported to be important for AIPC emergence [53]. Our analysis demonstrated that at least four of our candidate genes (FAM83H, OBSCN, CLDN7 and STRA13) were recurrently altered in PC patients (Supplementary figure 2), suggesting that dysregulation of these candidate genes might have triggered AIPC in our screen.
The candidate genes identified in our screen had various functions: cytoskeleton organization (FLNA, CLD7, FAM83H and OBSCN), epigenetic regulation (WSHC1, KDM2A), protein glycosylation (B3GNT9, GLYATL1), DNA damage repair (STRA13), protein transport (ARFGAP3) and RNA regulation (CNOT6) (Supplementary table 2). Of the 11 candidate genes, only ARFGAP3, CLDN7, FLNA and WHSC1 were previously shown to be involved with PC progression. Whereas, OBSCN, KDM2A, FAM83H, GLYATL1 and STRA13 genes were associated with other cancer types. B3GNT9 and CNOT6 genes are novel candidate genes which to our knowledge have not been previously linked to any cancer. B3GNT9 gene encodes a protein with galactosyltransferase activity. CNOT6 encodes the catalytic component of the CCR4-NOT core transcriptional regulation complex, which has a 3’-5’ RNase activity and play a role in miRNA-mediated repression, mRNA degradation, and transcriptional regulation.
Of the genes which were previously implicated with PC, ARFGAP3 was reported as a novel androgen-regulated gene that promotes PC cell proliferation and migration [56]. Using a reporter gene assay, the authors showed that ARFGAP3 enhanced androgen receptor-mediated transactivation activity of PSA in LNCaP cells [56]. FLNA, actin-binding protein that links actin with membrane glycoproteins, was previously reported to correlate with proliferation and invasive properties of several human cancers including melanoma [57], renal [58], breast [59], lung [60], leukemia [61], gastric [62] and PC [63]. Castoria et al. (2011) and Giovannelli et al. (2014) demonstrated that FLNA/androgen receptor complex activates signaling associated with cell migration and motility in PC [64, 65]. Moreover, nuclear localization of FLNA was reported to enhance androgen responsiveness of PC [66]. WHSC1, a chromatin binding protein with histone-lysine N-methyltransferase activity, was reported to epigenetically regulate the expression of TWIST and other metastatic-related genes to promote progression of prostate [67] and lung cancers [68]. Recently, WHSC1 was reported to promote squamous cell carcinoma of the head and neck via regulating NIMA-related kinase-7 activity through H3K36me2 mediated regulation [69]. CLDN7, a membrane protein and component of tight junctions, was reported to be differentially expressed in several cancers [70]. CLDN7 was shown to regulate prostate specific antigen (PSA) expression in LNCaP cells [71, 72]. Identifying these four genes (ARFGAP3, CLDN7, FLNA and WHSC1) which were previously implicated with PC establishes the ability of our insertional mutagenesis approach to discover PC-associated genes.
FAM83H, the candidate gene most recurrently altered in PC, was reported to associate with casein kinase1α to regulate keratin cytoskeleton rearrangement and contribute to progression of colorectal cancer [73]. GLYATL1 encodes glycine N-acyltransferase like1 protein and is reported to play a role in carcinogenesis in liver cancer [74]. STRA13 is a double-strand DNA binding protein that interacts with Fanconi anemia (FA) nuclear core complex which regulates DNA damage response and repair and genome maintenance [29, 75]. OBSCN is highly mutated in various cancers including breast [76] and colorectal cancers [77]. Loss of OBSCN was reported to disrupt cell-cell contact and enhance mesenchymal transitions in breast carcinoma [76]. KDM2A binds to CpG islands and demethylates histone residues in H3K36 protein, highlighting its role in heterochromatin modulations and gene regulation by epigenetic modifications. KDM2A expression was reported to promote cell growth and migration in gastric cancer [78] and activate ERK1/2 signaling promoting lung tumorigenesis and metastasis [79]. Frescas et al. (2008) reported low levels of KDM2A in PC tissues using Oncomine. Low levels of KDM2A were suggested to contribute in centromeric rearrangements and mitotic aberration that play a crucial role in PC progression [80]. The above described function of KDM2A combined with our observations showing the enrichment of clones with CISs targeting KDM2A gene, suggests that dysregulation of KDM2A might have played a crucial role in progression of PC in an androgen deficient environment.
We wanted to determine the direct implications of the candidate genes on AI growth. For this, four genes, which include three top candidate genes (GLYATL1, STRA13, FAM83H) and a gene that had CISs (KDM2A) were selected for validation. An additional criteria was that these genes were not been previously implicated with PC progression. First an in vitro clonogenic assay was performed to determine the effect on proliferation of LNCaP and next investigated the implications gene knockdown on AIPC progression. Significant reduction in the clonogenic ability of the LNCaP cells by knocking down KDM2A, FAM83H and GLYATL1 suggest that these three genes might be associated in promoting proliferation of LNCaP cells. We were the first to show that KDM2A, FAM83H and GLYATL1 knockdown reduces proliferation rate of PC cells. Further, in the proliferation recovery assay it was KDM2A gene knockdown that had a significant effect on recovery, indicating that KDM2A might play a role in recovering of PC cells from the proliferative arrest induced by castration. Interestingly, our proviral integration site analysis revealed CISs in the KDM2A gene and each of these integration site was recovered at a high frequency (See Table 2). These results suggest that the clones with a dysregulation of KDM2A gene by the proviral integration might had a growth advantage and were enriched in the castrated mice. Previously KDM2A was reported to be upregulated in gastric and lung cancers, and the knockdown of KDM2A was shown to suppress tumorigenicity and invasion of gastric cancer [78] and non-small cell lung cancer cells [79]. Though FAM83H and GLYATL1 had not shown any effect in the proliferative recovery assay, implication of these gene in AIPC progression needs further investigations. Migration and invasion studies could shed some more light regarding the association of the candidate genes in AIPC progression.
In addition to identifying genes related to PC progression, our study identified that these candidate genes were prognostic for recurrence risk in PC patients after treatment. The dataset we used for our prognostication studies [20] measured recurrence risk based on PSA levels. Two of our candidate genes, ARFGAP3 and CLDN7, were previously shown to regulate PSA in PC cells [71, 72]. This could be the reason why the prognostic value of combinations comprising these two genes showed a highly predictive ability to predict the clinical outcome of PC patients (see table 3; 8 out of the top 10 combinations uses ARFGAP3 and CLDN7). As expected using multiple biomarkers improved the predictive accuracy of PC over the use a single biomarker.
Taken together our studies identified novel genes which are differentially expressed and recurrently altered in PC samples. However these genes need further study to confirm their involved in AIPC. Importantly, the candidate genes identified in our screen predicted the recurrence risk in PC patients after prostatectomy, suggesting genetic testing of these candidate genes might predict recurrence risk in the patients. Moreover, our validation demonstrated that suppression of KDM2A could potential block the emergence of AIPC cells following ADT, which might be used as a potential therapeutic target in AIPC. Further, future studies on these genes might be explored to determine if they can be used to predict the therapeutic options to have the best clinical outcome.
Supplementary Material
Acknowledgments
We would like to thank Ellyn Schinke for technical assistance and Jonah Hocum for creating scripts used to find candidate genes. This study was supported by NIH NCI Grant CA173598.
Abbreviations
- PC
prostate cancer
- AIPC
androgen independent prostate cancer
- LV
lentiviral vector
- ADT
androgen deprivation therapy
- SFFV
spleen focus forming virus
- EGFP
enhanced green fluorescent protein
- MGS-PCR
modified genome sequencing-polymerase chain reaction
- VISA
vector integration site analysis
- CISs
common integration sites
- PSA
prostate serum antigen
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Eisenberger MA, Blumenstein BA, Crawford ED, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–1042. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 3.Bennett NC, Gardiner RA, Hooper JD, et al. Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol. 2010;42:813–827. doi: 10.1016/j.biocel.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 4.Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. Jama. 2005;294:238–244. doi: 10.1001/jama.294.2.238. [DOI] [PubMed] [Google Scholar]
- 5.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 6.Caffo O. The treatment of metastatic castration-resistant prostate cancer. Recenti Prog Med. 2015;106:35–39. doi: 10.1701/1740.18958. [DOI] [PubMed] [Google Scholar]
- 7.Schinke EN, Bii V, Nalla A, et al. A novel approach to identify driver genes involved in androgen- independent prostate cancer. Molecular cancer. 2014;13:120. doi: 10.1186/1476-4598-13-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranzani M, Cesana D, Bartholomae CC, et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat Methods. 2013;10:155–161. doi: 10.1038/nmeth.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altieri DC, Languino LR, Lian JB, et al. Prostate cancer regulatory networks. Journal of cellular biochemistry. 2009;107:845–852. doi: 10.1002/jcb.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uren AG, Kool J, Berns A, et al. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24:7656–7672. doi: 10.1038/sj.onc.1209043. [DOI] [PubMed] [Google Scholar]
- 11.Powers JM, Trobridge GD. Identification of Hematopoietic Stem Cell Engraftment Genes in Gene Therapy Studies. J Stem Cell Res Ther. 2013;2013 doi: 10.4172/2157-7633.S3-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ranzani M, Annunziato S, Calabria A, et al. Mol Ther. United States: 2014. Lentiviral vector-based insertional mutagenesis identifies genes involved in the resistance to targeted anticancer therapies; pp. 2056–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, An Z, Geller J, et al. High-malignancy orthotopic nude mouse model of human prostate cancer LNCaP. The Prostate. 1999;39:182–186. doi: 10.1002/(sici)1097-0045(19990515)39:3<182::aid-pros6>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 14.Stephenson RA, Dinney CP, Gohji K, et al. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84:951–957. doi: 10.1093/jnci/84.12.951. [DOI] [PubMed] [Google Scholar]
- 15.Beard BC, Adair JE, Trobridge GD, et al. Methods in molecular biology. Vol. 1185. Clifton, NJ: 2014. High-throughput genomic mapping of vector integration sites in gene therapy studies; pp. 321–344. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Kobert K, Flouri T, et al. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hocum JD, Battrell LR, Maynard R, et al. MOLECULAR THERAPY. Vol. 22. NATURE PUBLISHING GROUP; 75 VARICK ST, 9TH FLR, NEW YORK, NY 10013-1917 USA: 2014. VISA-Vector Integration Site Analysis Server: A Web- Based Server To Rapidly Identify Retroviral Integration Sites From Next-Generation Sequencing; pp. S84–S84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trobridge GD, Miller DG, Jacobs MA, et al. Foamy virus vector integration sites in normal human cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1498–1503. doi: 10.1073/pnas.0510046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhodes DR, Yu J, Shanker K, et al. Neoplasia. Vol. 6. New York, NY: 2004. ONCOMINE: a cancer microarray database and integrated data- mining platform; pp. 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arredouani MS, Lu B, Bhasin M, et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin Cancer Res. 2009;15:5794–5802. doi: 10.1158/1078-0432.CCR-09-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holzbeierlein J, Lal P, LaTulippe E, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lapointe J, Li C, Higgins JP, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaTulippe E, Satagopan J, Smith A, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 26.Luo JH, Yu YP, Cieply K, et al. Gene expression analysis of prostate cancers. Mol Carcinog. 2002;33:25–35. doi: 10.1002/mc.10018. [DOI] [PubMed] [Google Scholar]
- 27.Liu P, Ramachandran S, Ali Seyed M, et al. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 2006;66:4011–4019. doi: 10.1158/0008-5472.CAN-05-3055. [DOI] [PubMed] [Google Scholar]
- 28.Magee JA, Araki T, Patil S, et al. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 2001;61:5692–5696. [PubMed] [Google Scholar]
- 29.Singh TR, Saro D, Ali AM, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell. 2010;37:879–886. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomlins SA, Mehra R, Rhodes DR, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 31.Vanaja DK, Cheville JC, Iturria SJ, et al. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–3882. [PubMed] [Google Scholar]
- 32.Varambally S, Yu J, Laxman B, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Wallace TA, Prueitt RL, Yi M, et al. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68:927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 34.Welsh JB, Sapinoso LM, Su AI, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- 35.Yu YP, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 36.Gao J, Aksoy BA, Dogrusoz U, et al. Sci Signal. United States: 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal; p. pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, et al. PLoS One. United States: 2013. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis; p. e74250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gulzar ZG, McKenney JK, Brooks JD. Increased expression of NuSAP in recurrent prostate cancer is mediated by E2F1. Oncogene. 2013;32:70–77. doi: 10.1038/onc.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci. 2014;10:627–642. doi: 10.7150/ijbs.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barakat DJ, Zhang J, Barberi T, et al. CCAAT/Enhancer binding protein beta controls androgen- deprivation-induced senescence in prostate cancer cells. Oncogene. 2015 doi: 10.1038/onc.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu G, Wu J, Zhou L, et al. Characterization of the small RNA transcriptomes of androgen dependent and independent prostate cancer cell line by deep sequencing. PLoS One. 2010;5:e15519. doi: 10.1371/journal.pone.0015519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Liu Q, Xu G, et al. Comparative RNA-seq analysis reveals potential mechanisms mediating the conversion to androgen independence in an LNCaP progression cell model. Cancer Lett. 2014;342:130–138. doi: 10.1016/j.canlet.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 44.Kent WJ. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baca SC, Prandi D, Lawrence MS, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hieronymus H, Schultz N, Gopalan A, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A. 2014;111:11139–11144. doi: 10.1073/pnas.1411446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbieri CE, Baca SC, Lawrence MS, et al. Nat Genet. United States: 2012. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer; pp. 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fedr R, Pernicova Z, Slabakova E, et al. Automatic cell cloning assay for determining the clonogenic capacity of cancer and cancer stem-like cells. Cytometry A. 2013;83:472–482. doi: 10.1002/cyto.a.22273. [DOI] [PubMed] [Google Scholar]
- 49.Burton DG, Giribaldi MG, Munoz A, et al. Androgen deprivation-induced senescence promotes outgrowth of androgen-refractory prostate cancer cells. PLoS One. 2013;8:e68003. doi: 10.1371/journal.pone.0068003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Velonas VM, Woo HH, dos Remedios CG, et al. Current Status of Biomarkers for Prostate Cancer. Int J Mol Sci. 2013;14:11034–11060. doi: 10.3390/ijms140611034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ranzani M, Annunziato S, Adams DJ, et al. Cancer gene discovery: exploiting insertional mutagenesis. Mol Cancer Res. 2013;11:1141–1158. doi: 10.1158/1541-7786.MCR-13-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Amm J, Aragon-Ching JB. The changing landscape in the treatment of metastatic castration- resistant prostate cancer. Ther Adv Med Oncol. 2013:25–40. doi: 10.1177/1758834012458137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho H, Herzka T, Stahlhut C, et al. Rapid in vivo validation of candidate drivers derived from the PTEN-mutant prostate metastasis genome. Methods. 2015 doi: 10.1016/j.ymeth.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin P, Liu YN, Pierce R, et al. Am J Pathol. United States: Published by Elsevier Inc; 2011. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition; pp. 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schlomm T, Iwers L, Kirstein P, et al. Mod Pathol. United States: 2008. Clinical significance of p53 alterations in surgically treated prostate cancers; pp. 1371–1378. [DOI] [PubMed] [Google Scholar]
- 56.Obinata D, Takayama K, Urano T, et al. ARFGAP3, an androgen target gene, promotes prostate cancer cell proliferation and migration. Int J Cancer. 2012;130:2240–2248. doi: 10.1002/ijc.26224. [DOI] [PubMed] [Google Scholar]
- 57.Zhang K, Zhu T, Gao D, et al. Filamin A expression correlates with proliferation and invasive properties of human metastatic melanoma tumors: implications for survival in patients. J Cancer Res Clin Oncol. 2014;140:1913–1926. doi: 10.1007/s00432-014-1722-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun GG, Wei CD, Jing SW, et al. Interactions between filamin A and MMP-9 regulate proliferation and invasion in renal cell carcinoma. Asian Pac J Cancer Prev. 2014;15:3789–3795. doi: 10.7314/apjcp.2014.15.8.3789. [DOI] [PubMed] [Google Scholar]
- 59.Tian HM, Liu XH, Han W, et al. Differential expression of filamin A and its clinical significance in breast cancer. Oncol Lett. 2013;6:681–686. doi: 10.3892/ol.2013.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uramoto H, Akyurek LM, Hanagiri T. A positive relationship between filamin and VEGF in patients with lung cancer. Anticancer Res. 2010;30:3939–3944. [PubMed] [Google Scholar]
- 61.Nguyen le XT, Chan SM, Ngo TD, et al. Interaction of TIF-90 and filamin A in the regulation of rRNA synthesis in leukemic cells. Blood. 2014;124:579–589. doi: 10.1182/blood-2013-12-544726. [DOI] [PubMed] [Google Scholar]
- 62.Sun GG, Sheng SH, Jing SW, et al. An antiproliferative gene FLNA regulates migration and invasion of gastric carcinoma cell in vitro and its clinical significance. Tumour Biol. 2014;35:2641–2648. doi: 10.1007/s13277-013-1347-1. [DOI] [PubMed] [Google Scholar]
- 63.Sun GG, Lu YF, Zhang J, et al. Filamin A regulates MMP-9 expression and suppresses prostate cancer cell migration and invasion. Tumour Biol. 2014;35:3819–3826. doi: 10.1007/s13277-013-1504-6. [DOI] [PubMed] [Google Scholar]
- 64.Castoria G, D’Amato L, Ciociola A, et al. Androgen-induced cell migration: role of androgen receptor/filamin A association. PLoS One. 2011;6:e17218. doi: 10.1371/journal.pone.0017218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giovannelli P, Di Donato M, Auricchio F, et al. Analysis of the androgen receptor/filamin a complex in stromal cells. Methods Mol Biol. 2014;1204:109–121. doi: 10.1007/978-1-4939-1346-6_10. [DOI] [PubMed] [Google Scholar]
- 66.Mooso BA, Vinall RL, Tepper CG, et al. Enhancing the effectiveness of androgen deprivation in prostate cancer by inducing Filamin A nuclear localization. Endocr Relat Cancer. 2012;19:759–777. doi: 10.1530/ERC-12-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ezponda T, Popovic R, Shah MY, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene. 2013;32:2882–2890. doi: 10.1038/onc.2012.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuo CH, Chen KF, Chou SH, et al. Lung tumor-associated dendritic cell-derived resistin promoted cancer progression by increasing Wolf-Hirschhorn syndrome candidate 1/Twist pathway. Carcinogenesis. 2013;34:2600–2609. doi: 10.1093/carcin/bgt281. [DOI] [PubMed] [Google Scholar]
- 69.Saloura V, Cho HS, Kiyotani K, et al. WHSC1 Promotes Oncogenesis through Regulation of NIMA- Related Kinase-7 in Squamous Cell Carcinoma of the Head and Neck. Mol Cancer Res. 2015;13:293–304. doi: 10.1158/1541-7786.MCR-14-0292-T. [DOI] [PubMed] [Google Scholar]
- 70.Hewitt KJ, Agarwal R, Morin PJ. The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer. 2006;6:186. doi: 10.1186/1471-2407-6-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng JY, Yu D, Foroohar M, et al. Regulation of the expression of the prostate-specific antigen by claudin-7. J Membr Biol. 2003;194:187–197. doi: 10.1007/s00232-003-2038-4. [DOI] [PubMed] [Google Scholar]
- 72.Wang Q, Zheng JY, Kreth J, et al. Urology. United States: 2009. Regulation of prostate-specific antigen expression by the junctional adhesion molecule A; pp. 1119–1125. [DOI] [PubMed] [Google Scholar]
- 73.Kuga T, Kume H, Kawasaki N, et al. J Cell Sci. England: 2013. A novel mechanism of keratin cytoskeleton organization through casein kinase Ialpha and FAM83H in colorectal cancer; pp. 4721–4731. [DOI] [PubMed] [Google Scholar]
- 74.Matsuo M, Terai K, Kameda N, et al. Biochem Biophys Res Commun. United States: Elsevier Inc; 2012. Designation of enzyme activity of glycine-N-acyltransferase family genes and depression of glycine-N-acyltransferase in human hepatocellular carcinoma; pp. 901–906. [DOI] [PubMed] [Google Scholar]
- 75.Yan Z, Delannoy M, Ling C, et al. Mol Cell. United States: Elsevier Inc; 2010. A histone-fold complex and FANCM form a conserved DNA- remodeling complex to maintain genome stability; pp. 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shriver M, Stroka KM, Vitolo MI, et al. Loss of giant obscurins from breast epithelium promotes epithelial-to-mesenchymal transition, tumorigenicity and metastasis. Oncogene. 2014 doi: 10.1038/onc.2014.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huhn S, Bevier M, Pardini B, et al. Colorectal cancer risk and patients’ survival: influence of polymorphisms in genes somatically mutated in colorectal tumors. Cancer Causes Control. 2014;25:759–769. doi: 10.1007/s10552-014-0379-1. [DOI] [PubMed] [Google Scholar]
- 78.Huang Y, Liu Y, Yu L, et al. Histone demethylase KDM2A promotes tumor cell growth and migration in gastric cancer. Tumour Biol. 2015;36:271–278. doi: 10.1007/s13277-014-2630-5. [DOI] [PubMed] [Google Scholar]
- 79.Wagner KW, Alam H, Dhar SS, et al. KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J Clin Invest. 2013;123:5231–5246. doi: 10.1172/JCI68642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frescas D, Guardavaccaro D, Kuchay SM, et al. Cell Cycle. United States: 2008. KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state; pp. 3539–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.