

Abstract
The mitochondrial permeability transition pore was originally described in the 1970’s as a Ca2+ activated pore and has since been attributed to the pathogenesis of many diseases. Here we evaluate how each of the current models of the pore complex fit to what is known about how Ca2+ regulates the pore, and any insight that provides into the molecular identity of the pore complex. We also discuss the central role of Ca2+ in modulating the pore’s open probability by directly regulating processes, such as ATP/ADP balance through the tricarboxylic acid cycle, electron transport chain, and mitochondrial membrane potential. We review how Ca2+ influences second messengers such as reactive oxygen/nitrogen species production and polyphosphate formation. We discuss the evidence for how Ca2+ regulates post-translational modification of cyclophilin D including phosphorylation by glycogen synthase kinase 3 beta, deacetylation by sirtuins, and oxidation/nitrosylation of key residues. Lastly we introduce a novel view into how Ca2+ activated proteolysis through calpains in the mitochondria may be a driver of sustained pore opening during pathologies such as ischemia reperfusion injury.
Keywords: Mitochondrial Permeability Transition Pore, Calcium, Reactive Oxygen Species, Glycogen Synthase Kinase 3 Beta, Cyclophilin D, Calpain
1. Introduction
1.1 The discovery of the mitochondrial mega-channel and its characteristics
Mitochondria not only provide the majority of ATP for cells, they are also the central nexus for executing cell signaling for survival or death by Ca2+-induced swelling and uncoupling. The swelling and uncoupling of isolated mitochondria were first observed in the 1950’s and 1960’s using electron microscopy, and light scattering; the term permeability transition was coined in the 1970’s upon the observations that matrix Ca2+ induced an increase in the permeability of the inner mitochondria membrane of bovine hearts (Lehninger 1959; Crofts and Chappell 1965; Hackenbrock and Caplan 1969; Hunter et al. 1976). This permeability was shown to be non-specific because both neutral sucrose and charged salts could pass through with a maximum capacity of 1.5 kD (Hunter et al. 1976).
When the mitochondrial permeability transition pore (mPTP) opens, its large conductance can result in rapid swelling from the osmotic pressure of matrix solutes, rupture of the outer mitochondria membrane (OMM), collapse of the mitochondria membrane potential (ΔΨm), and depletion of cellular ATP, ultimately leading to necrotic cell death (Halestrap 2010; Kinnally et al. 2011). Interestingly, in addition to the large conductance, the mPTP has also been found to open transiently (flickering) while in sub-conductance states (Kinnally et al. 1989; Petronilli et al. 1999). After researchers discovered that cyclosporine A (CsA) is an effective inhibitor for pore opening, they learned by studies in intact cells that this transient opening of mPTP has several important physiological functions. In particular, it serves as a rapid mitochondrial efflux mechanism for excess reactive oxygen species (ROS) and Ca2+ tied to the activities of the electron transport chain (ETC) (Wang et al. 2008; Kwong and Molkentin 2015).
1.2 Ca2+ as the master regulator of mPTP opening
Several experimental findings substantiated the central role of Ca2+ in triggering the opening of mPTP by (Hunter et al. 1976): 1) preventing ionic inducers of swelling, such as phosphate, arsenate, and oleic acid, with Ca2+ chelation or blocking of Ca2+ entry into the mitochondrial matrix using ruthenium red or lanthanum, and 2) altering the order of addition of Ca2+ and inhibitors of swelling, such as phosphate carrier inhibitor MalNEt, Sr2+, and Mg2+. It was determined that these inhibitors exert their action by either preventing Ca2+ accumulation into the mitochondria (MalNEt & Sr2+) or displacing Ca2+ from binding to the mPTP complex (Mg2+).
Mechanistically, the existence of Ca2+ binding sites within the mPTP complex indicated that Ca2+ was a master regulator of mPTP opening. Careful studies by (Haworth and Hunter 1979).} first indicated that the rate of Ca2+ induction of mPTP opening had a Hill coefficient of 1.85, and the Km for Ca2+ and the Vmax of mPTP opening was independently modulated by Mg2+, ADP and NADH.
The goal of this review is to provide an overview of current models of the structure and molecular components of the mPTP and its regulators and of how these account for the role of Ca2+ as the master regulator. We will discuss in depth how Ca2+ directly regulates the pore as well as how Ca2+ helps integrate downstream processes that can affect mPTP opening such as mitochondrial energetics, ROS production, protein-mediated signaling, and, the proteolysis of key matrix pore regulatory proteins, which is, to our knowledge, a novel perspective.
2 Regulation of mitochondrial Ca2+
2.1 Mitochondrial Ca2+ influx mechanisms
In the resting state, the ΔΨm is the driving force for mitochondrial Ca2+ influx, which in turn is regulated by matrix Ca2+. As such, these two components are intrinsically linked (see section 6.1). The OMM was initially thought to be freely permeable to Ca2+ through the voltage dependent anion channel (VDAC). VDAC reconstituted in a lipid bilayer favors Ca2+ permeability in the closed state of VDAC; with gating independent of the presence of Ca2+ suggesting that the gating state of VDAC is relevant to Ca2+ conductance (Tan and Colombini 2007). Additionally, VDAC overexpression in HeLa cells enhanced matrix Ca2+ uptake following agonist induced endoplasmic reticulum Ca2+ release by allowing a more rapid transfer of Ca2+ from the cytosol to the inner mitochondrial membrane (IMM) channels (Rapizzi et al. 2002).
Ca2+ conductance across the IMM was originally thought to be a single mechanism highly sensitive to ruthenium red and lanthanides (Gunter and Pfeiffer 1990). Only recently has this ruthenium and lanthanide sensitive protein been identified the gene CCDC109A, or mitochondrial calcium uniporter (MCU) (Baughman et al. 2011; De Stefani et al. 2012). The MCU protein is regulated by both post-translational modification and its association with auxiliary subunits essential MCU regulator (EMRE), mitochondrial calcium uptake (MICU)1–3, and mitochondrial calcium uniporter regulator 1 (MCUR1) (Joiner et al. 2012; Mallilankaraman et al. 2012; Csordás et al. 2013; Sancak et al. 2013; O-Uchi et al. 2014a). Interestingly knockout of the MCU core protein does not alter basal ΔΨm, mitochondrial matrix Ca2+ content, mitochondrial morphology, or yield any apparent phenotype under resting conditions (Baughman et al. 2011; De Stefani et al. 2012; Luongo et al. 2015). Indeed, in a cardiac specific knockout of MCU cells were normal but protected from ischemia reperfusion (I/R) injury through decreased mPTP opening; knockout cells were also unable to upregulate energy supply with increased demand during β-adrenergic stimulation through the Ca2+ enhanced activity of the tricarboxylic acid (TCA) cycle (Luongo et al. 2015). In addition to MCU, there are other mitochondrial Ca2+ influx mechanisms, including the mitochondrial ryanodine receptor Type 1 (mRYR1), rapid mode of uptake (RaM), and Leucine Zipper-EF-Hand Containing Transmembrane Protein (Letm1) mCa1&2; all have unique biophysical properties other than MCU (Sparagna et al. 1995; Beutner 2001; Michels et al. 2009; Jiang et al. 2009).
2.2 Mitochondrial Ca2+ efflux mechanisms
Given that ΔΨm drives Ca2+ influx, one would expect mitochondria to eventually reach a steady state with ΔΨm; however this would result in a [Ca2+] of 10−1M that is incompatible with mitochondrial function (De Stefani et al. 2013). Experimental evidence for Ca2+ efflux was provided when isolated mitochondria were incubated with Ca2+ until a steady state was reached, ruthenium red was added, and nearly all Ca2+ was released into the extra-mitochondrial environment. Since this occurred independent of ΔΨm, two observations were made shortly thereafter: 1) the existence of a 2H+/Ca2+ antiporter, thought to be electrically neutral; and 2) the existence of a Ca2+ efflux pathway that was strongly stimulated by Na+ or Li+ in isolated heart mitochondria (Crompton et al. 1977; Pozzan et al. 1977). The molecular identity of the 2H+/Ca2+ exchanger has not yet been established, but the Na+/Ca2+ exchanger was identified as the Na+/Ca2+ Li+-permeable exchanger (NCLX) that operates at a stoichiometry of 3 or 4 Na+/1 Ca2+ resulting in an import of 1–2 net positive charges into the matrix and favored by ΔΨm (Palty et al. 2010). The evidence that the mPTP itself acts as a Ca2+ efflux mechanism is discussed in section 3.4.
2.3 Mitochondrial matrix Ca2+ buffering
Since [Ca2+]mt largely exists as a steady state balance between the influx and efflux mechanisms, as described above, an anion buffers a majority of excess Ca2+ when it enters the mitochondria in order to maintain ΔΨm, pH, and [Ca2+]mt. Inorganic phosphate (Pi) and its polymerized form polyphosphate (PolyP) largely mediate this Ca2+ buffering system. The buffering system of mitochondria depends on the mode of Ca2+ entry; a small acute entry of Ca2+ below a mitochondrial “set point” will rapidly increase [Ca2+]mt to act as a second messenger for targets such as the TCA cycle. However, a large but sustained entry of Ca2+ above the “set point” allows Ca2+ to interact with the phosphate buffering system (Wei et al. 2012). It has become apparent that large amounts of Ca2+ (50–500 nmols/mg) can be buffered without significantly changing [Ca2+]mt (Chalmers and Nicholls 2003). This buffering largely depends on the matrix pH; as Pi enters the mitochondria as H2PO4− it must lose 2 H+ ions to the alkaline environment to become PO43− where it can then interact with Ca2+ to become Ca3(PO4)2 (Nicholls and Chalmers 2004). Support for this this buffering hypothesis includes evidence that a rapid dissociation occurs upon addition of a protonophore/acidification and that Ca2+ and Pi can exit through their respective carriers or the mPTP. It appears that PolyP is also able to interact with and disrupt the irreversible precipitation of this complex, increasing the [Ca2+]mt. Indeed, in cells depleted of matrix PolyP, the detectable [Ca2+] is lower and resistant to mPTP opening (Solesio et al. 2016a). However, these measurements rely on total fluorescence of calcium probes in a population of cells or mitochondria. It is worth noting that the relevant [Ca2+]mt-mediated signaling my be constrained to microdomains within the mitochondria matrix, and this matrix Ca2+ cannot be effectively measured using these techniques. So while the total pool of measured [Ca2+]mt remains in balance, the [Ca2+]mt restricted to the immediate vicinity of the mPTP complex is physiologically relevant.
3. Characteristics of the mPTP
3.1 Conductance properties of the mPTP
The mPTP is a non-selective pore that can pass both ionic and nonionic substrates (Hunter et al. 1976). Studies using different sizes of polyethylene glycol identified that solutes up to 1500 Daltons are able to pass through the pore, which closely matched the mathematically modeled pore size of 1.4 nm (Massari and Azzone 1972; Haworth and Hunter 1979). The mPTP displays multiple subconductance states from ~200–700 pS with the majority occurring in the 500–700 pS range (Zorov et al. 1992). Its maximum conductance state is ~1.0–1.3 nS (Kinnally et al. 1989; Petronilli et al. 1989). The multi-conductance nature of the mPTP suggests that the molecular nature of the pore likely is a multi-subunit complex that can oligomerize to varying degrees, resulting in the observed ranges of conductance, with the majority occurring in the partially oligomerized state. Aside from Ca2+ as an activator, a low ΔΨm can also increase open probability, beginning at ~−40mV (Zorov et al. 1992). An important property of the mPTP is that its opening is reversible upon addition of ADP and/or restoration of the Mg2+/Ca2+ratio (Haworth and Hunter 1979), which leads to re-establishing ΔΨm (Petronilli et al. 1999). This reversibility is important physiologically because it allows the mPTP to have both sustained and transient opening, which can initiate a necrotic signaling pathway or help maintain normal cellular function, respectively. Interestingly, these transient openings operate at a subconductance state and appear to allow solutes no larger than 300–600 Daltons to pass. The different modes of opening could provide a means for selective signaling and indicate that transient openings could be a result from lower oligomerization configurations of the pore (Ichas et al. 1997; Lu et al. 2015).
3.2 Inhibitors of the mPTP
Without a definitively identified molecular structure to date, direct pharmacological blockers of the pore component of the mPTP are not known. The most well-known of the mPTP inhibitors is the small immunosuppressive compound CsA, which blocks cyclophilin D (CypD) from binding to the pore complex (Fournier et al. 1987; Crompton et al. 1988). Another known class of chemical inhibitors of the mPTP are benzodiazepenes (Kinnally et al. 1993). Interestingly, studies showed that the peripheral benzodiazepine receptor on the mitochondria was not required for pore opening or benzodiazepine sensitivity of the mPTP, suggesting a second site of action (Šileikytė et al. 2014), possibly the Oligomycin sensitivity conferring protein (OSCP) of the FoF1 ATP Synthase (Giorgio et al. 2013).
The competition by other divalent cations, such as Sr2+, Mn2+, and Mg2+, act in two ways: 1) they compete with or inhibit Ca2+ entrance into the mitochondria (Vainio et al. 1970), and 2) they compete with Ca2+ for a shared site on the mPTP in the matrix (Km ~10−5M) regardless of total ion concentration (Szabo et al. 1992; Bernardi et al. 1993). Interestingly, when divalent ions, including Ca2+, are added after matrix Ca2+ loading and subsequent uptake inhibition they play an inhibitory role (IC50 ~0.2mM) independent of internal matrix Ca2+ load. This suggests that there is an external (inter mitochondria membrane space (IMS) or OMM) binding site, and the sub-mitochondrial localization of Ca2+ can have profound effects on mPTP open probability (Bernardi et al. 1993).
The direct effect of adenine nucleotides on the mPTP has long been observed as a powerful inhibitor of mPTP opening (Hunter and Haworth 1979). The ADP/ATP ratio is important because it can affect the conformation of the adenine nucleotide translocator (ANT), a proposed IMM pore component (Haworth and Hunter 2000). Indeed, when ANT is inhibited with bongkrekic acid, which locks the ANT in the “m” (matrix-facing) conformation, it opposes mPTP opening (Hunter and Haworth 1979; Halestrap et al. 1997). Lastly, a low affinity ADP binding site independent of ANT in the matrix was identified (Novgorodov et al. 1994). Indeed, when coupled with Mg2+, ADP binds the FoF1 ATP Synthase and prevents its hydrolytic activity helping to maintain ΔΨm (Feniouk and Yoshida 2008).
In de-energized mitochondria, a high level of NADH protects Ca2+-induced mPTP in a manner independent of the ETC that could not be augmented with external NADH, suggesting a matrix site of action (Hunter and Haworth 1979). In a condition where NADH was fully oxidized to NAD+, Ca2+-induced mPTP opening was still inhibited as long as ΔΨm was maintained (Hunter and Haworth 1979). However, collapse of ΔΨm with FCCP is not sufficient to induce pore opening because it can be opposed by reduction of NAD+ to NADH with β-hydroxybutyrate (Hunter and Haworth 1979). These results demonstrate that both NADH and hyperpolarization of ΔΨm can independently inhibit Ca2+-induced mPTP opening.
Acidic matrix pH has long been known to strongly regulate mPTP opening by displacing Ca2+ from the trigger site of the mPTP, with the optimum pH for opening residing at 7.4 (Haworth and Hunter 1979; Halestrap 1991; Bernardi et al. 1992; Szabo et al. 1992). The inhibition of the mPTP by low pH is likely due to protonation of histidine residues on the mPTP complex treatment of energized or uncoupled mitochondria with diethyl pyrocarbonate or potassium thiocyanate respectively can restore pore opening even at an acidic matrix pH 6.5 (Nicolli et al. 1993). Notably, the histidine residues do not appear to be on CypD because CsA treatment was still able to prevent pore opening (Bernardi et al. 1993).
In summary, these older studies suggest that mPTP inhibition acts primarily through interference with Ca2+. The divalent competitive ions, MalNEt, and ANT conformation prevent Ca2+ entry to the matrix or modulate direct Ca2+ binding to the pore.
3.3 Activators of the mPTP
The primary mechanism of mPTP opening is Ca2+ overload in the mitochondrial matrix. The mechanism requires as little as 100–200 nmols of Ca2+/mg of bovine heart mitochondria (Hackenbrock and Caplan 1969; Hunter et al. 1976). One of the longest known agents to increase pore open probability is atractyloside, which acts by locking the ANT in the “c” conformation, apparently creating a net positive charge on the matrix facing side of the IMM that, while reducing Ca2+ influx, can also reduce the rate of Ca2+ dissociation from the matrix faceing IMM and create a higher membrane surface microdomain of Ca2+ to interact with the mPTP pore trigger site (Hunter and Haworth 1979; Rottenberg and Marbach 1990).
mPTP opening is also influenced by mitochondrial morphology. Mitochondrial fission, regulated by dynamin-like protein1 (DLP1) can increase the pore open probability of the mPTP. In a model of ischemia reperfusion (I/R) injury overexpression of dominant negative DLP1, mitochondrial fusion protein mitofusin2 (MFN2), or treatment with mdivi (a pharmacological inhibitor of DLP1), were all able to induce mitochondrial elongation, reduced infarct size, and delayed onset of mPTP opening(Ong et al. 2010). In a model of hyperglycemia mitochondrial fragmentation was found to be required for excessive ROS production, mPTP opening, and cell death(Yu et al. 2008). Indeed, it has been shown that increased cytosolic Ca2+ can induce translocation of DLP1 to the mitochondria (Cribbs and Strack 2007; Hom et al. 2010). However, matrix Ca2+ has been shown to also be required for fission to proceed after DLP1 transolcation has occurred, suggesting Ca2+ is a major regulator for mitochondrial morphology, ROS Production, and mPTP activity (Breckenridge et al. 2003; Hom et al. 2007).
Second messengers such as ROS and reactive nitrogen species (RNS) can also modulate mPTP activity (Halestrap et al. 1997). Pharmacological agents that increase pore activity by increasing the level of ROS and RNS inside the cell include tert-butylhydroperxide (TBH), phenylarsine oxide (PhA sO), diamide, and peroxynitrite (ONOO−) hydrogen peroxide (H2O2) and superoxide (O2•−) (Hunter and Haworth 1979; Halestrap et al. 1997(Scarlett et al. 1996). ROS and RNS can potentiate mPTP opening indirectly by modulating the electron transport chain or by directly modifying the pore (see section 6.2). In intact cells, ROS can further potentiate mPTP opening by modulating intracellular Ca2+ levels available to be taken up by the mitochondria by activating plasma membrane influx and SR/ER IP3 and ryanodine receptors, and by inhibiting sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps (Hool and Corry 2007; Song et al. 2010; Santos et al. 2011; Song et al. 2011; O-Uchi et al. 2014b)
Pi is capable of sensitizing mPTP opening, through a mechanism that is not yet entirely known (Crompton and Costi 1988); (Gunter and Pfeiffer 1990). Its sensitizing role is likely due to the formation of polyP, a mPTP inducer and disruptor of the Ca2+ buffering system (Pavlov et al. 2005; Abramov et al. 2007)(see section 2.3).
Table I summarizes the known characteristics of the mPTP.
Table I.
Established Characteristics of the mPTP
| Characteristic | Value/Status | Reference | Comment |
|---|---|---|---|
| Size | 1.4 nm | (Massari and Azzone 1972) | |
| Permeability | ≤ 1500 Daltons | (Haworth and Hunter 1979) | |
| Subconductance states | < 500 pS | (Kinnally et al. 1989; Szabo et al. 1992) | |
| Maximum Conductance | 1.0–1.3 nS | (Kinnally et al. 1989); (Petronilli et al. 1989) | |
| Specificity | Nonspecific-ionic and non-ionic solutes | (Hunter et al. 1976) | |
| Reversibility | Yes | (Hunter et al. 1976) | Transient openings can be protective |
| Inhibitors: | Known Value | Reference | Comment |
| Cyclosporine A | 1 μM | (Fournier et al. 1987; Crompton et al. 1988) | Disrupts CypD binding to the mPTP |
| Benzodiazepines | 10 nM Ro5-4864 | (Kinnally et al. 1993) | |
| Mg2+, | 2 mM (0.8–1.5 mM is normal matrix [Mg2+]) | (Hunter et al. 1976; Bernardi et al. 1992) | Mg2+ competes for matrix binding site with Ca2+ |
| Sr2+, Mn2+ | 2mM | (Hunter et al. 1976; Bernardi et al. 1992) | Competes with Ca2+ for entry through the MCU |
| ADP | 6 μM | (Hunter and Haworth 1979) | |
| Bongkrekic Acid | 20 μM/0.5 mg/1 mL mitochondrial protein | (Hunter and Haworth 1979; Rottenberg and Marbach 1990) | ANT “m” conformation/negative IMM matrix surface change |
| NADH>NAD+ | (Hunter and Haworth 1979) | ||
| Energized State (ΔΨm) | (Hunter and Haworth 1979) | ||
| pH | < 7.0 | (Haworth and Hunter 1979; Halestrap 1991) | Optimum opening pH is 7.2–7.4 |
| Activators: | Value | Reference | Comment |
| Matrix Ca2+ | 100–200 nmols/1mg/mL mitochondria. | (Hackenbrock and Caplan 1969; Hunter et al. 1976; Chalmers and Nicholls 2003) | Paradoxically mPTP is independent of measured total [Ca2+]m |
| Atractyloside | 25 μM/0.5 mg/1 mL mitochondria protein | (Hunter and Haworth 1979; Rottenberg and Marbach 1990) | ANT “c”-conformation/positive IMM matrix surface change |
| CypD | Bound to mPTP | (Connern and Halestrap 1994; Tanveer et al. 1996) | Blocked by CsA |
| Mitochondrial Morphology | Fragmented | (Ong et al. 2010) | |
| ROS/RNS | 1 mM TBH, 100 μM diamide, 20 μM PheArs, 250μM ONOO− | (Hunter and Haworth 1979; Halestrap et al. 1997) (Scarlett et al. 1996) | |
| Pi | 10 mM extramitochondrial, | (Crompton and Costi 1988) | Acts as inhibitor in CypD KO mouse |
3.4 Role of mPTP in cellular function and disease
As mentioned in Section 1.1, the mPTP has two modes of opening: transient and sustained. The evidence for the transient opening of mPTP as a physiological efflux pathway began when Altschuld et al demonstrated that treating cardiomyocytes with the mPTP inhibitor CsA in the presence of 45Ca2+ total cellular Ca2+ increased within 15 minutes without inducing hypercontraction, an effect that could be prevented with the MCU blocker ruthenium red. It was determined the increased mitochondrial Ca2+ was due to a loss of efflux through the mPTP, as cardiomyocytes preloaded with 45Ca2+ retained the 45Ca2+ when treated with CsA (Altschuld et al. 1992). This idea was strengthened when neurons from control and CypD knockout (ppif−/−) mice were treated with either ATP or KCl to induce Ca2+ influx. In both control and ppif−/− neurons, those treated with ATP or KCl alone showed a similar increase in [Ca2+]mt. However, when treated with both reagents simultaneously, the ppif−/− mitochondria had significantly higher levels of Ca2+, suggesting that the mPTP as an efflux mechanism is only important when the matrix Ca2+ load exceeds the capacity of the NCLX (Barsukova et al. 2011).
The physiological role of the mPTP pore as a Ca2+ efflux mechanism, however, is still under debate. Using HeLa cells treated with a NCLX blocker prior to mitochondrial Ca2+ uptake of histamine released Ca2+, overexpression or a 70% siRNA knockdown of the c subunit of the FoF1 ATP Synthase, a hypothesized pore component, resulted in no change in the rate of mitochondrial Ca2+ efflux (De Marchi et al. 2014). Furthermore, no difference in Ca2+ efflux was observed during Ca2+ overload using a mitochondria calcium uniporter (MCU) overexpression model followed by mPTP inhibition using CsA or by mPTP sensitization using H2O2 (De Marchi et al. 2014). These experiments both may suffer from too many uncontrolled variables, 70% knockdown of the pore may still leave a sufficient amount for pore formation. Additionally, other studies suggest the MCU expression level does not regulate basal [Ca2+]m (Elrod et al. 2010).
The transient pore opening has been shown to be associated with a transient depolarization of ΔΨm (Petronilli et al. 1999; Jou 2011). Using a mitochondria-targeted circularly permuted yellow fluorescence protein (cpYFP), transient openings, of the mPTP (so-called mitoflashes) were associated with a sudden burst of O2•− generation that is linked to the activity of the electron transport chain and initiation of local ROS signaling (Wang et al. 2008). While there is still ongoing debate about how sensitive cpYFP is to ROS vs pH, numerous recent reports have shown stochastic oscillations of mitochondrial O2•− and pH in a wide spectrum of cells (Quatresous et al. 2012; Schwarzländer et al. 2014). While the exact mechanism responsible for these transient openings of the mPTP and the generation of mitochondrial O2•− and pH flashes is still unclear, their physiological and pathophysiological roles are beginning to be elucidated. Indeed, the frequency of these flashes has been associated with metabolism, ageing, diabetes, wound healing, as well as with playing a possible role in cell differentiation and stemness (Hom et al. 2011; Shen et al. 2014; Vega-Naredo et al. 2014; Ding et al. 2015).
The mPTP has been implicated in a large number of diseases across multiple organ systems. Most notably the mPTP is the major regulator of the extent of I/R injury when a vessel’s blood flow is occluded and subsequently restored. Indeed, inhibiting the mPTP in the heart can prevent up to 50% of the final infarct size, (Yellon and Hausenloy 2007). The mPTP’s role in I/R is also evident in the brain, liver, intestine, and kidney (Shiga et al. 1992; Konukoglu et al. 1998; Soda et al. 1999; Mizuta et al. 1999; Saxton et al. 2002; Feldkamp et al. 2009; Park et al. 2011; Gill et al. 2012; Cho et al. 2013). An abundance of evidence indicates that the mPTP plays a critical role in I/R injury, which can be prevented using pharmacological means, or by pre- and post-conditioning (periods of brief ischemia that prevents Ca2+ overload through transient mPTP opening and reperfusion that desensitizes the mPTP of the ischemic tissue (Javadov et al. 2003; Hausenloy et al. 2004; Feng et al. 2005; Clarke et al. 2008; Gomez et al. 2008; Inserte et al. 2009). In clinical trials, treatment with CsA at reperfusion reduced levels of the biomarkers troponin and mitochondrial creatine kinase (mtCK) and reduced total infarct size (Piot et al. 2008). In many neurological disorders dysregulated Ca2+ and ROS levels appear to have some component of injury mediated by increased mPTP opening, including excitotoxicity (Pivovarova and Andrews 2010), epilepsy (Jung et al. 2012), Huntington’s disease (Quintanilla et al. 2013), Alzheimer’s disease (Du et al. 2011), Parkinson’s disease (Martin et al. 2014b), Amyotrophic Lateral Sclerosis (Martin et al. 2014a), and Multiple Sclerosis (Savino et al. 2013). Conversely, it appears that susceptibility to mPTP opening is reduced in cancer, leading to cell resilience. In rats exposed to a carcinogenic substance (e.g., 2-acetylaminofluorene), desensitization of the mPTP in hepatocytes is one of the first steps observed prior to transformation (Klöhn et al. 2003). While an exact mechanism is unknown, and different cancers evolve separate strategies to survive, it appears multiple cancer types reduce mPTP sensitivity by modulating their metabolism, Ca2+ handling, ROS levels, and expression of key pore modulators; see (Bonora and Pinton 2014) for an in depth review.
4. Models of the mPTP
While the pathological and physiological role of the mPTP has been well studied, the molecular identity of the pore forming unit and its essential regulators remains controversial. Initially, the mPTP was thought to reside at contact sites (points at which the OMM and IMM meet) formed by the VDAC in the OMM and the ANT residing in the IMM, respectively, and with CypD residing in the matrix. A series of genetic knockout studies ruled out VDAC, ANT, and CypD as essential pore forming units. Subsequently the phosphate carrier (PiC), FoF1 ATP Synthase, and, most recently, a metalloproteinase, Spastic Paraplegia 7 (SPG7), have been proposed as the core pore-forming component.
Despite the controversy surrounding what makes up the pore itself, a few proteins have been identified as key regulators of the pore. Most important among these is CypD, which was identified as a key pore regulator through its CsA sensitive interaction with the pore complex. Both glycogen synthase kinase 3 beta (GSK-3β) and sirtuin 3 (SIRT3) have been shown to be important for mPTP opening in the context of I/R injury by modifying CypD post-transationally, leading to increased sensitivity to Ca2+ overload (see sections 5.2 &5.3).
Since this review is focused on the molecular basis for the Ca2+ sensitivity of the mPTP, this review will focus on the proposed models of the mPTP, through the lens of how well they fit the known characteristics of Ca2+-mediated regulation. It will also focus on what they can tell us about how Ca2+ regulates the mPTP both directly through its components and indirectly through regulating known modulators of the mPTP.
4.1 Classical model of the pore
In the early 1990’s, researchers observed that several ligands of the mitochondrial benzodiazepine receptor (TSPO) were able to modulate mPTP opening (Kinnally et al. 1993), and TSPO was also shown to co-purify with both VDAC and ANT (Halestrap and Davidson 1990) (McEnery et al. 1992). This evidence led to the idea that the mPTP could be located at contact sites between the OMM and IMM. Subsequently, evidence that hexokinase (HK) II, a primarily cytosolic protein, and mtCK in the IMS co-purified with ANT and VDAC. This complex had conductance patterns similar to the mPTP with ADP and N-methyl-4Val-Cyclosprorine sensitivity suggested a multi-protein complex assembled to form the mPTP (Beutner et al. 1996). Later, researchers found that this HKII-co-purified complex also include Bcl-2 family proteins by using 2D PAGE electrophoresis and Western blotting of reconstituted liposomes containing a functional mPTP complex (Marzo et al. 1998). Unfortunately, it was difficult to identify the key pore forming subunits from those that merely served regulatory roles due to an inability to selectively reconstitute each protein. Nevertheless, these studies suggested that Bcl-2 associated X protein (Bax), HKII, VDAC, mtCK, TSPO, ANT, and CypD formed the mPTP complex at membrane contact sites where VDAC and ANT formed the core pore in the OMM and IMM, respectively (Figure 1A) (reviewed in (Zamzami and Kroemer 2001)).
Fig. 1.
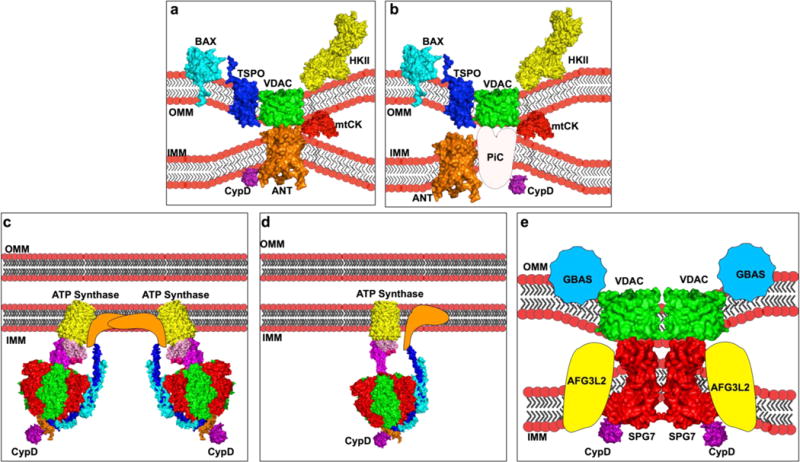
Models of the mPTP. A representative diagram of the proposed models of the mPTP through time. Where possible, known 3-dimensional (3D) structures obtained from the Protein Data Bank (http://www.rcsb.org/pdb) are shown. a. Classical model composed of BCL-2-associated-X-protein (BAX), Voltage Dependent Anion Channel (VDAC), the Perepheral Benzodiazepiene receptor (TSPO), Hexokinase II (HKII) on the Outer Mitochondrial Membrane (OMM), Mitochondrial Creatine Kinase (mtCK) in the intermembrane space, Adenine Nucleotide Transporter (ANT) in the inner mitochondrial membrane (IMM), and mitochondrial cyclophilin D (CypD) bound to ANT in the matrix. b. Phosphate Carrier model composed of BAX, VDAC, TSPO, HKII, mtCK, ANT, and CypD, bound to the phosphate carrier (PiC) c. Dimers of the FoF1 ATP Synthase. d. Uncoupling of the FoF1 ATP Synthase, and e. Spastic Paraplegia 7 composed of VDAC, and Glioblastoma Amplified Sequence (GBAS) on the OMM, Spastic Paraplegia 7 (SPG7) on the IMM, and matrix CypD bound to (SPG7)
However, subsequent mouse genetic knockout studies have called this model into question. Isolated mitochondria from hepatocytes with genetic ablation of ANT1&2, still exhibited mPTP opening, albeit with increased resistance to Ca2+ overload, and loss of sensitivity to ANT-dependent mPTP modulators, ADP, bongkrekic acid, and atractyloside (Kokoszka et al. 2004). Knockout of VDAC provided no protection in isolated mitochondria to Ca2+ induced mPTP opening and even exacerbated ionomycin-induced mPTP opening (Baines et al. 2007). Knockout of TSPO also showed no resistance to Ca2+-induced mPTP opening while maintaining benzodiazepine sensitivity suggesting that the TSPO ligands used previously must have an additional site(s) of action (Šileikytė et al. 2014). Finally, similar to treatment with CsA, genetic ablation of CypD showed a large resistance to Ca2+-induced mPTP opening, but the mPTP was still able to open and maintain oxidative stress-induced sensitization to pore opening, suggesting that CypD is a critical regulator, but not a core pore component of the mPTP (Baines et al. 2005; Nakagawa et al. 2005; Basso et al. 2005; Schinzel et al. 2005). While initially this model appeared to account for diverse array of molecules known to regulate the mPTP (See table I), the above-mentioned genetic knockouts studies have suggested that other proteins must be able to form the pore of mPTP complex without these constituents.
4.2 Phosphate carrier
Interest increased in the possible role of the phosphate carrier protein PiC as an essential component of the mPTP complex when evidence emerged of CypD binding to the PiC in a CsA sensitive manner. Sensitivity of the PiC to sulfhydryl reagents, such as N-ethylmaleimide (NEM) and diamide, also made it a promising candidate as a core pore forming mPTP constituent (Leung et al. 2008) (Figure 1B). Further increasing interest, Leung et al. also demonstrated through mass spectroscopy that the ANT antibody they had used previously also recognized the PiC, possibly explaining the failure of ANT knockout to block mPTP opening (Leung et al. 2008). The PiC was also an attractive candidate due to the ability of Pi to activate mPTP opening (Crompton and Costi 1988). However, subsequent patch clamping of the PiC displayed currents of only 20–30 pS, too low a conductance for it to be considered a core pore-forming constituent of the mPTP. Additionally, genetic overexpression or knockdown did not affect mPTP pore function, as measured by Ca2+ retention capacity (CRC) (Varanyuwatana and Halestrap 2012; Gutiérrez-Aguilar et al. 2014). Genetic deletion of PiC in the heart resulted in a more resistant CRC phenotype, but the findings still suggest that it is not the obligate pore-forming component of the mPTP (Kwong et al. 2014). It should be noted that the cardiolipin present in the IMM may play a key role for both PiC and ANT to function as regulators of the mPTP (Kadenbach et al. 1982; Hoffmann et al. 1994; Pestana et al. 2009).
4.3 FoF1 ATP Synthase
Evidence has been mounting that the FoF1 ATP Synthase may be a critical component of the pore-forming unit of the mPTP (Figure 1C&D). Initially selected as a candidate by searching for CypD binding partners, Giorgio et al. had shown that CypD interacts with the FoF1 ATP Synthase in a CsA (decreased binding) and Pi (increased binding) dependent manner (Giorgio et al. 2009). They later demonstrated that the direct binding partner of CypD with the FoF1 ATP Synthase was OSCP and that this interaction could also be blocked by a benzodiazepine receptor agonist Bz-423 (Giorgio et al. 2013). Resuspending these gel-purified ATP-Synthase complexes into lipid bilayers, they demonstrated that only the dimers have a Ca2+, Bz-423, and PhAsO activated channel conductance of approximately 500 pS, similar to the half-conductance state of the mPTP (Giorgio et al. 2013). The dimer’s open channel probability was significantly reduced in the presence of ADP as well as Mg2+. The model suggests that CypD binding to OSCP induces a conformational change in the lateral stalk of the FoF1 ATP Synthase. This increases the affinity of the divalent binding site normally occupied by Mg2+ to favor binding to Ca2+ (Bernardi et al. 2015). It remains to be determined if or how subsequent binding of Ca2+ regulates dimerization or permeability of the IMM.
Removal of the dimerization interface subunits e (TIM11) and g (ATP20) in yeast enhanced the CRC required for mPTP opening (Carraro et al. 2014). While the pore still opened in dimerless mutants, these findings also demonstrated that transient dimers can still form, as evidenced by cysteine crosslinking with CuCl2-induced dimer formation observed by blue native PAGE.
Independently, Alavian et al. also identified the FoF1 ATP Synthase as the mPTP pore forming unit; however, the proposed mechanism of action between the two models is quite different. They resuspended purified c-subunit oligomers and observed a voltage-sensitive current of about 100 pS reaching peak conductance of 1.5–2 nS similar to the mPTP (Alavian et al. 2014). They were able to block this current using an antibody targeted to the c-subunit. Unlike in the studies of Bernardi et al., purified c-subunit monomers showed infrequent channel conductance that was greatly enhanced when recombinant CypD or Ca2+ was added (Alavian et al. 2014). They hypothesize that the uncoupling of the F1 from the Fo complex of the FoF1 ATP Synthase is critical for pore formation, as addition of the β-subunit blocked the conductance of the purified c-subunit pore. Similar to Pinton’s group, they demonstrated that knockdown of the c-subunit enhanced resistance to mPTP opening, whereas mPTP opening was sensitized in cells overexpressing a glycine to valine mutant of the c-subunit, which causes the Fo ring to become larger (Bonora et al. 2013). By inducing mPTP opening in isolated mitochondria with Ca2+ and using immunocapture of the FoF1 ATP Synthase, Alavian et al. identified the presence of the c-subunit in the supernatant which could be reduced by pretreatment with CsA or ADP, suggesting that the F1 and Fo subunits were disassociated during mPTP opening. However, this may just be a result of mitochondrial fragments that were too small to be pelleted by ultracentrifugation in the percoil gradient.
While these two models are at odds in terms of the mechanism of pore formation, they both offer strong evidence that the FoF1 ATP Synthase plays a critical role in terms of pore opening by introducing a model that allows for a matrix-side Mg2+ competitive Ca2+ site, ADP/ATP sensitivity, benzodiazepine target and similar conductance to those reported for the mPTP. These models both merit further study to elucidate a unified mechanism.
4.4 Spastic paraplegia 7
Most recently, Shanmughapriya et al. identified SPG7 as a candidate mPTP component using an unbiased shRNA silencing approach (Shanmughapriya et al. 2015) (Figure 1E). Screening for 128 mitochondrial proteins to identify loss of function in the CRC of permeabilized HEK 293T cells, SPG7 was one of 14 candidate genes. Knockdown of the FoF1 ATP Synthase c-subunit or PiC conferred no resistance to mPTP opening in this study. The researchers subsequently treated these 14 knockdown cell lines with H2O2, followed by CRC assessments. The results indicated that only SPG7, VDAC, and CypD were indifferent to H2O2 sensitization (reduced CRC). Using both co-immunoprecipitation and a yeast two-hybrid assay with various mutants of SPG7, the Madesh group determined that the c-terminal region of SPG7 was essential for CsA sensitive CypD binding. However, the authors failed to show the CRC properties of this CypD non-interacting Δct-SPG7. Instead they mutated the CsA- binding domain of CypD and demonstrated that it was unable to bind SPG7 and conferred resistance to mPTP opening. However, this CsA-insensitive mutant also contained the mutation R97G, which renders it an isomerase-deficient mutant and confounds their results (Baines et al. 2005). CRISPR/Cas9-mediated knockout and reintroduction of a non-functional SPG7 mutant demonstrated that the enzymatic activity of SPG7 was not required to elicit protection against mPTP opening.
Several major questions remain to be addressed concerning this study. Similarly to ppif−/− null cells, the SPG7 knockout cells still maintain a Ca2+-sensitive transition, suggesting that SPG7 functions as a pore regulatory protein not as the core pore-forming complex. It is possible that SPG7 plays a structural or scaffolding role by bringing key components together, but unlikely that SPG7 itself forms the 1.4 nm IMM channel that solutes can pass through. The authors have yet to show any conductance states of a reconstituted SPG7-containing pore complex in a lipid bilayer. However the authors provide strong genetic and functional evidence that SPG7 plays an important role in mPTP formation. How Ca2+, Pi, ADP/ATP, or any of the other known modulators affect this protein is yet to be determined.
Due to the contentious nature and conflicting evidence of the molecular components of the mPTP, it is possible that the pore complex may be an amalgamation of all these components together as part of the ATP synthasome, as recently reviewed by (Halestrap and Richardson 2015; Morciano et al. 2015). The mPTP has also been proposed to arise from PolyP interaction and association with the proteinaceous components whereby the PolyP provides Ca2+ and voltage sensitivity and selectivity of the pore as reviewed by (Solesio et al. 2016b). According to available evidence, the mPTP complex must account for the known biophysical and structural properties and, at a minimum, contain a matrix side divalent cation binding site that is inhibitory when bound to Mg2+ and activating when bound to Ca2+(where the site favors Ca2+ binding through CypD isomerase activity on the pore). The mPTP must also account for an external (IMS/OMM) binding site that is inhibitory when occupied by any divalent metal cation and contain histidine residues that are protonated at pH <6.8 resulting in pore inhibition.
5. Major Protein Regulators of the mPTP
While there are many models of the mPTP complex itself, there is a consensus regarding the key regulators of pore activity. Here, we will focus on the role of the outer membrane proteins as well as CypD, and we will focus on key modulators of pore activity that work together to modulate the mPTP.
5.1 Outer mitochondrial membrane proteins
Since mPTP activity can be observed in mitoplasts (resealed IMM with no OMM), we can assume that OMM proteins play a primarily regulatory role in mPTP opening. Initially, the OMM was implicated with the possibility of VDAC being a core component and ligands for TSPO sensitizing mPTP opening, but this was shown to be not a direct action on TSPO itself and genetic knockout of VDAC proved ineffective in preventing pore opening (Szabo and Zoratti 1993; Johnson et al. 2005; Sundberg et al. 2009; Giorgio et al. 2013). However VDAC can regulate the rate of Ca2+ entry into to the IMS (see section 2.1). While not pore components themselves, the Bcl2 family of proteins are capable of regulating the mPTP sensitivity (Marzo et al. 1998). A double knockout of Bax and Bad conferred resistance to mPTP induced cell death, independent of their ability to oligomerize. However, the IMM mPTP machinery was still functional and the protection was attributed to preventing OMM rupture and release of IMS components (Whelan et al. 2012; Karch et al. 2013).
Another OMM inhibitor of mPTP opening is HKII. The hydrophobic N-terminus of hexokinase I & II inserts into the OMM and tethers it to the outer surface of the mitochondria (Xie and Wilson 1988; Gelb et al. 1992; Chiara et al. 2008). The binding of HKII to the OMM is regulated in part by phosphorylation of HKII at Thr 463 by AKT and by GSK-3β mediated phosphorylation on Thr 51 of its binding partner VDAC1 (Pastorino and Hoek 2003; Pastorino 2005; Miyamoto et al. 2008; Roberts et al. 2013). HKII binding to the mitochondria largely protects against mPTP opening by decreasing OMM permeability and thus preventing release of IMS components (Pasdois et al. 2013). The presence of Glucose-6-phosphate, coupled to the acidic environment during ischemia induces HKII to dissociate from the OMM (Pasdois et al. 2013). This increases the availability of VDAC to bind to and stabilize recruitment of Bax and Bak proteins to the OMM (Shimizu et al. 1999). Additionally, loss of cytochrome c from the IMS will increase ROS by increasing electron backup at Complex I and III, promoting O2•− formation (Pasdois et al. 2011; Markevich and Hoek 2015). HKII may also mediate OMM permeability to cytochrome c by modulating the morphology of the mitochondria cristae by stabilizing the contact sites (Pasdois et al. 2013).
5.2 Post-translational modification of CypD
Mitochondrial CypD is a peptidyl-prolyl cis-trans isomerase encoded by the PPIF gene. Its binding sensitizes the mPTP to opening by causing a conformational change in the PTP complex, increasing the affinity of Ca2+ over other divalent metals in the matrix Ca2+ binding site (Baines et al. 2005; Bernardi et al. 2015). Here we will discuss factors that regulate CypD’s activity and binding to the mPTP.
Pi was shown to enhance the sensitivity to mPTP opening with increased Pi leading to increased sensitivity in mammalian mitochondria (Out et al. 1971; Hunter et al. 1976; Gunter and Pfeiffer 1990). Recently, however, Pi was shown to inhibit mPTP opening in ppif−/− mouse liver mitochondria, suggesting that its activating role may occur through effects on CypD (Basso et al. 2008). In agreement with this hypothesis, Pi increased co-immunoprecipitation between OSCP and CypD suggesting a role for Pi in modulating CypD binding affinity and supporting the model of the FoF1 ATP Synthase as the mPTP (Giorgio et al. 2009; Giorgio et al. 2013).
CypD binding activity is also modulcated by a second mechanism: S-nitrosylation (SNO) modifies a reactive thiol function on CypD by NO•. Global SNO was shown to be increased during ischemic pre-conditioning and treatment with S-nitrosoglutathione (GSNO), a NO donor, was shown to be protective during I/R injury (Sun et al. 2007; Kohr et al. 2011b; Kohr et al. 2011a). After researchers treated mouse hearts with GSNO and used a resin to enrich and capture proteins with S-nitrosylated cysteine residues subsequently analyzed with LC-Tandem mass spectroscopy, they identified CypD to be modified by SNO at cysteines 103, 156, and 202 (Kohr et al. 2011a). Subsequent mutation of the analogous site in human CypD at C203 to serine conferred protection similar to ppif knockout (Nguyen et al. 2011). Since C203S mimics CypD ablation and not reintroduction of wild-type CypD without addition of GSNO, the findings suggests that it is not the SNO modification itself that causes protection, but rather that modification by SNO is mutually exclusive with other forms of cysteine modification, such as oxidation or formation of disulfide bridges (Sun et al. 2006; Sun and Murphy 2010). Indeed it was suggested that C203 is a redox-sensitive residue and both ischemic and temperature preconditioning of hearts showed reduced carbonylation of total protein, a general marker for oxidative stress (Khaliulin et al. 2004; Khaliulin et al. 2007; Linard et al. 2009). Finally, it should be noted that C203 is not conserved across residues and is absent in bovine CypD where the mPTP was first functionally characterized and then subsequently shown to have a CsA sensitive component; these results suggest other factors at play regulate CypD binding to the mPTP (Hunter et al. 1976; Inoue et al. 1993; Giorgio et al. 2009).
CypD has also been identified as a substrate for kinases. Rasola et al. showed that cancer cell lines resistant to mPTP opening displayed constitutive ERK activation, reducing phosphorylation of CypD mediated by a small pool of GSK-3β in the matrix (Rasola et al. 2010). Similarly, Traba et al. demonstrated that a reduction of GSK-3β activity and corresponding reduction of associated phosphorylation of overexpressed FLAG-CypD correlated with increased CRC (Traba et al. 2012). The evidence in cancer cells likely also applies to normal tissue, as supported by the finding that NSAID-induced kidney injury involving mPTP opening is accompanied by a GSK-3β mediated phosphorylation of CypD (Bao et al. 2012). The exact phosphorylation site on CypD has not been identified yet, but in silico analysis of the GSK-3β phosphorylation motif suggests there are several Ser/Thr candidate sites on CypD (Bernardi et al. 2015). It has also not yet been identified whether this phosphorylation regulates binding of CypD to the mPTP or its isomerase activity.
Lastly, CypD has been identified as a target of acetylation. A study of ageing-related activation of the mPTP showed that sirt3−/− mice display increased susceptibility to mPTP opening with age. This was correlated with an increased acetylation of lysine 166 on CypD (Hafner et al. 2010). Independently, Shulga et al. demonstrated that exposure to ethanol sensitize the mPTP, possibly through reduced SIRT3 activity, and this sensitization was accompanied by a corresponding increase in acetylated CypD, which increased its binding to ANT1 (Shulga and Pastorino 2010). Shulga et al. also demonstrated that CypD acetylation at Lysine 145 is responsible for its association with ANT1, which can also be regulated by SIRT3 (Shulga et al. 2010). Bochatan et al., in a model of ischemic post-conditioning demonstrated that SIRT3 is essential for protection by increasing the activity of SIRT3 and deacetylation of K166 on CypD. In contrast, reduction of SIRT3 activity appears to maintain HKII binding to the OMM in a CypD-dependent manner that may reduce mPTP sensitivity (Verma et al. 2013). It is assumed that SIRT3 is activated during postconditioning as a result of the changes in mitochondrial NAD+/NADH ratio, as NAD is greatly reduced after hypoxia (Bochaton et al. 2015).
In summary, it needs to be determined what post translational modifications are important for regulating CypD function and whether they modulate binding to the pore and/or isomerase activity. Acetylation and nitrosylation appear to be responsible for a protective localization/reduced isomerase activity, whereas phosphorylation or oxidation may be responsible for binding to the pore itself and inducing conformational pore changes that increase affinity for Ca2+ binding and subsequent pore opening.
5.3 CypD Interacting Proteins
Cells treated with H2O2 signaled a fraction of the transcription factor p53 to translocate into the mitochondria matrix where it interacted with CypD and induced mPTP opening (Vaseva et al. 2012). Several groups subsequently demonstrated a CypD-p53 signaling axis involved in a necrotic cell death pathway (Zhao et al. 2013; Chen et al. 2013; Zhen et al. 2014). However, important questions remain as to how important this signaling pathway is in mPTP regulation. Isolated mitochondria display Ca2+-induced mPTP opening, whereas the proposed p53-dependent signaling axis is independent of Ca2+ (Karch and Molkentin 2012). Bergeaud et al. demonstrated that there is a basal level of p53 in the mitochondrial matrix that interacts with OSCP to stabilize the FoF1 ATP Synthase complex and reduce ROS production (Bergeaud et al. 2013). It would be expected that the reduced ROS production should desensitize mPTP opening since the p-53 signaling was Ca2+ independent.
There is strong evidence that GSK-3β plays a major role in regulating mPTP opening through both direct and indirect mechanisms. Inhibition of GSK-3β serves as an important integrator for many protective signals of mPTP opening, such as during ischemic preconditioning and post-conditioning (Tong et al. 2002; Juhaszova et al. 2004; Gomez et al. 2008; Rasola et al. 2010; Miura and Tanno 2010; Zhai et al. 2011; Tanno et al. 2014). The role of GSK-3β in mPTP regulation was first identified using GSK-3β inhibitors LiCl and SB216763 (Tong et al. 2002). However, pre-treatment with insulin, which inhibits GSK-3β, did not afford protection from ischemia-reperfusion injury, suggesting other signaling pathways may be involved (Clarke et al. 2008). It later became apparent that a subset of GSK-3β was present in the mitochondria, as was demonstrated by trypsin digestion of isolated mitochondria and co-immunoprecipitation with mitochondrial matrix components (Xi et al. 2009; Rasola et al. 2010). It was later shown that the N-terminus of GSK-3β has a weak mitochondrial matrix targeting sequence that is partially dependent on GSK-3β kinase activity and binding to VDAC2 (Tanno et al. 2014). This brings into question if the effect of GSK-3β on the mPTP using transgenic models to alter GSK-3β activity is due to an alteration in mitochondrial localization and not the enzymatic activity itself of GSK-3β on proposed cytosolic targets. Indeed ischemic preconditioning did not increase translocation of GSK-3β suggesting its protective effect must be partially due to a subset of the enzyme already present in the mitochondria if the effect mediated by phosphorylation of CypD (Clarke et al. 2008). Fascinatingly, the activity of GSK-3β appears to play an opposing role during differing stages of ischemia reperfusion injury; GSK-3β activity is beneficial during the ischemic phase by promoting autophagy during nutrient starvation, however it is detrimental during the reperfusion phase through activation of the mPTP (Zhai et al. 2011). It should also be noted that GSK-3β in its basal state is an active kinase and, as such, CypD phosphorylation must be balanced by mitochondrial phosphatases. It suggests that a modification of GSK-3β increases its activity past its basal state during the reperfusion phase that tips the balance to phosphorylated CypD during reperfusion. What may regulate this switch in GSK-3β behavior during reperfusion is further discussed in section 6.4.
6 Ca2+ as key to regulation of the mPTP
6.1 Ca2+ and modulation of the ATP/ADP balance
Ca2+ can modulate the ATP/ADP balance primarily through two mechanisms. First, Ca2+ can regulate the activity of the Krebs cycle by enhancing activity of pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase (McCormack and Denton 1989). The enhanced activity of these enzymes ultimately increases mitochondrial NADH as substrate for complex I of the electron transport chain. The increased substrate availability increases proton motive force and ATP production through complex V (McCormack et al. 1990). Second, Ca2+ can regulate complex V directly to increase ATP production, although the exact mechanism is unclear (Das and Harris 1990). It was shown in rat liver mitochondria that the β subunit has a low sensitivity to Ca2+, but that may not be physiologically relevant given the low [Ca2+] maintained by the extensive buffering system discussed in section 2.3 (Hubbard and McHugh 1996). Additionally, based on sequence homology to Troponin T, the Fo subunit e, which lies between the Fo ring and F1 rotor, has a hypothesized IMS-facing Ca2+ binding motif from amino acids 34–65; treatment with an antibody raised against that peptide to block Ca2+ binding increased ATPase activity in mitoplasts but not in submitochondrial vesicles (IMM vesicles with the matrix side facing out) (Arakaki et al. 2001). Ca2+ can bind to the IMS side of the Fo c-subunit as determined by radio-labeled 45Ca2+ and SDS PAGE; binding of Ca2+ blocked proton translocation in bacteria (Van Walraven et al. 2002). If the FoF1 ATP Synthase is indeed the mPTP, then either of these sites accessible to the IMS could be the external inhibitory divalent metal binding site that was discussed in section 3.2.
6.2 Ca2+ and ROS/RNS production
Ca2+ can also modulate both ROS and RNS since ROS generation is tightly coupled to ETC activity (Sohal and Allen 1985; Perez-Campo et al. 1998). In the mitochondria, ROS generation primarily occurs as O2•− production during the Q cycle from the ubisemiquinone intermediate (QH•) at complex III (St-Pierre et al. 2002; Muller et al. 2003; Turrens 2003; Markevich and Hoek 2015). Additional sites of O2•−− production are electron slippage entering complex II and production at complex I (Turrens 1997; Trachootham et al. 2008). Once produced, O2•− is rapidly converted to H2O2 by spontaneous dismutation or catalyzed by superoxide dismutase 2 (SOD2) (Brookes 2004a). The H2O2 can further be converted to OH• if Fe2+ is available. O2•− will also interact non-enzymatically with NO• to produce ONOO− (Valko et al. 2007; Trachootham et al. 2008). NO• production comes from a variety of nitric oxide synthase (NOS) complexes including endothelial NOS (NOS3), neuronal NOS (NOS1), inducible NOS (NOS2), and possibly a mitochondrial NOS (mtNOS) whose existence is still debated (Ghafourifar and Cadenas 2005; Lacza et al. 2006; Oess et al. 2006; Moody and Calvert 2011). Since NO• is largely membrane permeable, its production at any cellular location can be assumed to have free access to the mitochondrial milieu (O-Uchi et al. 2014b). Ca2+ is required to activate NOS3 and NOS1, which can then further increase mitochondrial NOS and ROS production (Alderton et al. 2001). Indeed, NO• can inhibit Complex IV which leads to an increase in ROS production (Brookes and Darley-Usmar 2002; Brookes 2004b; Zaobornyj and Ghafourifar 2012). Complex I inhibition can also be achieved when NO• and high levels of matrix Ca2+ are present to stimulate O2•− production and formation of ONOO− which in turn causes SNO of Complex I (Jekabsone et al. 2003). ONOO− production at the mitochondria can also inhibit Complex III and V, which as discussed previously can lead to respiratory block and additional ROS production (Radi et al. 1994). Interestingly ONOO− treatment of isolated liver mitochondria loaded with Ca2+ resulted in rapid permeability transition that could be blocked by CsA (Packer et al. 1997). Lastly, Ca2+ can compete with cytochrome c for cardiolipin binding sites on the IMM which can cause a respiratory block at complex III leading to ROS production (Grijalba et al. 1999; Ott et al. 2002). Overall it appears that Ca2+ is central to regulating RNS and ROS production by the ETC. Under conditions in which the scavenging systems become overwhelmed, the ROS and RNS will feedback to block the ETC complexes and encourage additional ROS/RNS production, collapse of ΔΨm, or encourage pore activity by modifying mitochondrial proteins such as CypD or the pore itself.
6.3 Ca2+ and activation of proteolytic enzymes in the mitochondria
More recently, proteolysis of mitochondrial proteins is being recognized as an important mechanism to modulate mitochondrial energetics and trigger mitochondrial dysfunction. Calpains are a large family Ca2+-dependent cysteine proteases (for review, see (Sorimachi and Ono 2012)). Typically, calpains require tens of μM (Calpain I) to 1 mM (Calpain II) of [Ca2+] to be activated in vitro. This suggests other mechanisms must be in play in situ such as proximity to membranes and localization to microdomains of high [Ca2+] (Shao et al. 2006; Sorimachi and Ono 2012). So far, calpains I, II, IV, and X have been identified to be partially localized in the mitochondria (Arrington et al. 2006; Chen et al. 2011; Shintani-Ishida and Yoshida 2015). Calpain activation occurs during reperfusion due to the Ca2+ overload and a restoration of pH (Hernando et al. 2010).
The exact submitochondrial localization of the classical calpains I and II are still in debate. Calpain I has been identified in the IMS where it will cleave AIF and NCLX, thereby increasing apoptosis and matrix Ca2+ levels (Kar et al. 2010). More recently, Chen et al. separated mitochondrial compartments using digitonin, combined with ultra-centrifugation identified calpain I to be in the matrix along with the small subunit calpain IV, and the endogenous calpain inhibitor calpastatin (Chen and Lesnefsky 2015). It was also identified that upon reperfusion the mitochondrial ND6 subunit of complex I is digested in a calpain-dependent manner (Arrington et al. 2006; Chen and Lesnefsky 2015; Shintani-Ishida and Yoshida 2015). However, the specific residues, and site of action of calpain-mediated cleavage is yet to be identified leaving the possibility of an IMS site of action. Calpain II was identified in the IMS of liver mitochondria, and in the matrix of mouse heart mitochondria using a crude fractionation protocol (Ozaki et al. 2009; Shintani-Ishida and Yoshida 2015). The specificity of each antibody to recognize calpain I vs II is also an issue (Ozaki et al. 2007). A more detailed subfractionation or digestion method using CRISPR/Cas9 knockout lines of each gene could shed light on to the true localization of calpain II, which does not have a strong mitochondrial target signal as compared to calpain I (Claros and Vincens 1996). Interestingly chemical inhibition of calpain has been identified as an effective strategy in blunting I/R injury (Chen et al. 2002; Maekawa et al. 2003; Khalil et al. 2005; Inserte et al. 2006). Unfortunately chemical inhibitors do not allow us to differentiate between isoforms or localization of the effect, causing some confusion as to which isoform is digesting what target, and how protection is afforded. Recently Ni et al. demonstrated overexpression of matrix targeted calpain I or calpastatin could dramatically increase/decrease ROS production and transient mPTP opening, respectively, indicating modulation of mPTP open probability (Ni et al. 2015). These authors also demonstrated that calpain I could cleave the α- subunit of the FoF1 ATP Synthase. This may be the reason why Alavian et al. identified a dissociation between the F1 and Fo subunits when mitochondria underwent permeability transition via Ca2+ overload. However, no lower bands were observed when probed for the α subunit, but only a functional complex V was subjected to SDS page after immunocapture.
Calpain I has also been shown to be able to truncate GSK-3β during excitotoxicity of neurons or oxidative stress of H9c2 myoblasts (Goñi-Oliver et al. 2007; Ma et al. 2012; Feng et al. 2013; Jin et al. 2015). While the reports differ on the exact site(s) of truncation it appears they all hyperactivate GSK-3β by removing one or both inhibitory phosphorylation site(s) at Ser9 and/or Thr390. These experiments were all done in whole cell lysates. However, in our hands truncation of GSK-3β can be observed primarily in the mitochondrial matrix (Hurst et al. 2015). Since GSK-3β inhibition is only beneficial during reperfusion and calpain I is activated during reperfusion, it is tantalizing to hypothesize that truncation of GSK-3β in the mitochondria may cause its hyperactivation and promote phosphorylation of CypD and its binding to the mPTP pore to induce sustained pore opening (Hurst et al. 2015).
Mounting evidence suggests that Ca2+ is central to regulation of the mPTP and its modulators through direct effects on the pore itself and through regulating the ADP/ATP balance, mitochondrial ΔΨm, ROS/RNS levels, and proteolytic events. This is summarized in Figure 2.
Fig.2.
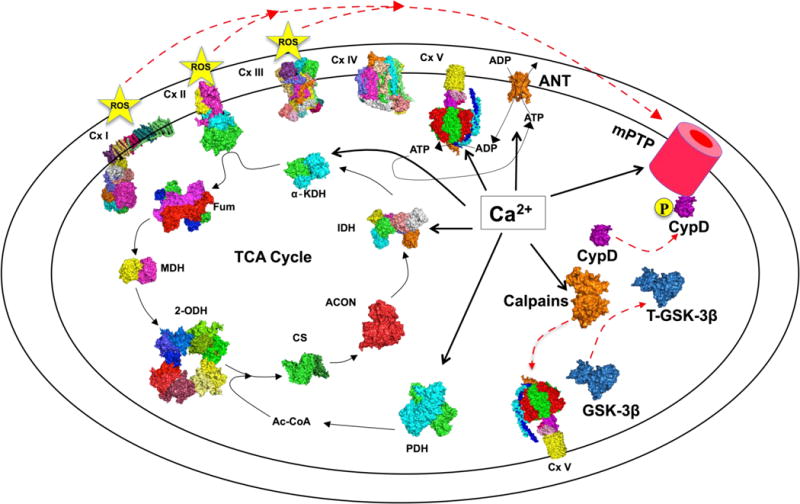
Ca2+ as the central regulator of the mPTP. A representative diagram of the regulatory role of Ca2+ in the mitochondria. Where possible, known 3-dimensional (3D) structures obtained from the Protein Data Bank (http://www.rcsb.org/pdb) are shown. Thick solid lines denote proteins on which calcium exerts a direct effect. Thin lines are metabolic processes regulated by Ca2+, and dashed lines are processes that promote mPTP opening that are regulated by calcium. GSK-3β,Glycogen Synthase 3 Beta; CypD, mitochondrial Cyclophilin D; Cx I –V, Complex I –V; PDH, Pyruvate Dehydrogenase; CS Citrate Synthase; ACON, Aconitase; IDH, Isocitrate Dehydrogenase; α-KDH alpha Ketoglutarate Dehydrogenase; Fum, Fumarase; MDH, Malate Dehydrogenase; 2-ODH, 2-Oxoglutarate Dehydrogenase
7. Summary and concluding remarks
The past 60 years has led to many remarkable discoveries on the nature and regulation of the mPTP. Overall, Ca2+ remains the central regulator, ever since its discovery as the original stimulus to induce pore opening and the demonstration of direct binding to the mPTP. Ca2+ has many indirect roles regulating the mPTP; by coupling energy demand and supply, mitochondrial morphology, ROS production, and kinase activity, which can all converge on modulation of the mPTP. Ca2+ at physiological levels will stimulate transient opening of the pore, but during Ca2+ overload the balance tips from physiology to pathology, which leads to the sustained mPTP opening that is at the crux of many diseases and pathologies. The definitive identification of the molecular identity of the pore-forming component can lead to the development of specific inhibitors and treatment for many ailments in which Ca2+ handling, mitochondrial dysfunction, and sustained mPTP opening are central etiologies.
Acknowledgments
We would like to acknowledge Jennifer Wilson, for her constructive comments as well as our funding from the National Instute of Health : 2R01HL093671, 1R01HL122124, & 1RO1114760 to S-S. Sheu; T32AA007463 to J. Hoek & S. Hurst, and R01AA018873 to J.Hoek.
Abbreviations
- 2-ODH
2-Oxoglutarate dehydrogenase
- ACON
Aconitase
- ANT
Adenine nucleotide translocator
- α-KDH
Alpha ketoglutarate dehydrogenase
- CRC
Calcium retention capacity
- cpYFP
Circularly permuted yellow fluorescence protein
- CS
Citrate synthase
- CxI-V
Complex I-V
- CypD
Cyclophilin D
- CsA
Cyclosporine A
- DLP1
Dynamin-like protein 1
- ETC
Electron transport chain
- EMRE
Essential MCU regulator
- Fum
Fumarase
- GBAS
Glioblastoma Amplified Sequence
- GSK-3β
Glycogen synthase kinase 3 beta
- HK
Hexokinase
- IMM
Inner mitochondrial membrane
- Pi
Inorganic phosphate
- I/R
Ischemia reperfusion
- IDH
Isocitrate dehydrogenase
- Letm1
Leucine Zipper-EF-Hand Containing Transmembrane Protein
- NCLX
Na+/Ca2+ Li+-permeable exchanger
- MDH
Malate dehydrogenase
- MFN2
Mitofusin 2
- TSPO
Mitochondrial benzodiazepine receptor
- MCU
Mitochondrial calcium uniporter
- MCUR 1
Mitochondrial calcium uniporter regulator 1
- MICU
Mitochondrial calcium uptake
- mtCK
Mitochondrial creatine kinase
- mPTP
Mitochondrial permeability transition pore
- ΔΨm
Mitochondria membrane potential
- mRYR1
Mitochondrial ryanodine receptor Type 1
- NEM
N-ethylmaleimide
- NOS
Nitric oxide synthase
- OSCP
Oligomycin sensitivity conferring protein
- OMM
Outer mitochondria membrane
- PhA sO
Phenylarsine oxide
- PiC
Phosphate carrier
- PolyP
Polyphosphate
- PDH
Pyruvate Dehydrogenase
- RaM
Rapid mode of uptake
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SNO
S-nitrosylation
- GSNO
S-nitrosoglutathione
- SERCA
Sarco/endoplasmic reticulum Ca2+-ATPase
- SIRT3
Sirtuin 3
- SPG7
Spastic Paraplegia 7
- SOD2
Superoxide dismutase 2
- TCA
Tricarboxylic acid
- VDAC
Voltage dependent anion channel
References
- Abramov AY, Fraley C, Diao CT, et al. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proceedings of the National Academy of Sciences. 2007;104:18091–18096. doi: 10.1073/pnas.0708959104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, Beutner G, Lazrove E, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proceedings of the National Academy of Sciences. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuld RA, Hohl CM, Castillo LC, et al. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am J Physiol. 1992;262:H1699–704. doi: 10.1152/ajpheart.1992.262.6.H1699. [DOI] [PubMed] [Google Scholar]
- Arakaki N, Ueyama Y, Hirose M, et al. Stoichiometry of subunit e in rat liver mitochondrial H(+)-ATP synthase and membrane topology of its putative Ca(2+)-dependent regulatory region. Biochim Biophys Acta. 2001;1504:220–228. doi: 10.1016/s0005-2728(00)00248-6. [DOI] [PubMed] [Google Scholar]
- Arrington DD, Van Vleet TR, Schnellmann RG. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am J Physiol, Cell Physiol. 2006;291:C1159–71. doi: 10.1152/ajpcell.00207.2006. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, et al. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H, Ge Y, Zhuang S, et al. Inhibition of glycogen synthase kinase-3β prevents NSAID-induced acute kidney injury. Kidney Int. 2012;81:662–673. doi: 10.1038/ki.2011.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsukova A, Komarov A, Hajnóczky G, et al. Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur J Neurosci. 2011;33:831–842. doi: 10.1111/j.1460-9568.2010.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, et al. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Basso E, Petronilli V, Forte MA, Bernardi P. Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J Biol Chem. 2008;283:26307–26311. doi: 10.1074/jbc.C800132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeaud M, Mathieu L, Guillaume A, et al. Mitochondrial p53 mediates a transcription-independent regulation of cell respiration and interacts with the mitochondrial F1 F0-ATP synthase. Cell Cycle. 2013;12:2781–2793. doi: 10.4161/cc.25870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Rasola A, Forte M, Lippe G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol Rev. 2015;95:1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Vassanelli S, Veronese P, et al. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem. 1992;267:2934–2939. [PubMed] [Google Scholar]
- Bernardi P, Veronese P, Petronilli V. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. I. Evidence for two separate Me2+ binding sites with opposing effects on the pore open probability. J Biol Chem. 1993;268:1005–1010. [PubMed] [Google Scholar]
- Beutner G. Identification of a Ryanodine Receptor in Rat Heart Mitochondria. Journal of Biological Chemistry. 2001;276:21482–21488. doi: 10.1074/jbc.M101486200. [DOI] [PubMed] [Google Scholar]
- Beutner G, Ruck A, Riede B, et al. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Letters. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7. [DOI] [PubMed] [Google Scholar]
- Bochaton T, Crola-Da-Silva C, Pillot B, et al. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. Journal of Molecular and Cellular Cardiology. 2015;84:61–69. doi: 10.1016/j.yjmcc.2015.03.017. [DOI] [PubMed] [Google Scholar]
- Bonora M, Bononi A, De Marchi E, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Pinton P. The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front Oncol. 2014;4:302. doi: 10.3389/fonc.2014.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. The Journal of Cell Biology. 2003;160:1115–1127. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes P, Darley-Usmar VM. Hypothesis: the mitochondrial NO(*) signaling pathway, and the transduction of nitrosative to oxidative cell signals: an alternative function for cytochrome C oxidase. Free Radic Biol Med. 2002;32:370–374. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- Brookes PS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. AJP: Cell Physiology. 2004a;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Brookes PS. Mitochondrial nitric oxide synthase. MITOCH. 2004b;3:187–204. doi: 10.1016/j.mito.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Carraro M, Giorgio V, Šileikyte J, et al. Channel Formation by Yeast F-ATP Synthase and the Role of Dimerization in the Mitochondrial Permeability Transition. Journal of Biological Chemistry. 2014;289 doi: 10.1074/jbc.C114.559633. jbc.C114.559633-15985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers S, Nicholls DG. The Relationship between Free and Total Calcium Concentrations in the Matrix of Liver and Brain Mitochondria. J Biol Chem. 2003;278:19062–19070. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- Chen B, Xu M, Zhang H, et al. Cisplatin-induced non-apoptotic death of pancreatic cancer cells requires mitochondrial cyclophilin-D-p53 signaling. Biochemical and Biophysical Research Communications. 2013;437:526–531. doi: 10.1016/j.bbrc.2013.06.103. [DOI] [PubMed] [Google Scholar]
- Chen M, Won D-J, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277:29181–29186. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- Chen Q, Lesnefsky EJ. Heart mitochondria and calpain 1: Location, Function, and Targets. BBA - Molecular Basis of Disease. 2015:1–34. doi: 10.1016/j.bbadis.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Chen Q, Paillard M, Gomez L, et al. Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia–reperfusion. Biochemical and Biophysical Research Communications. 2011;415:533–538. doi: 10.1016/j.bbrc.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara F, Castellaro D, Marin O, et al. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho T-H, Aguettaz P, Campuzano O, et al. Pre- and post-treatment with cyclosporine A in a rat model of transient focal cerebral ischaemia with multimodal MRI screening. Int J Stroke. 2013;8:669–674. doi: 10.1111/j.1747-4949.2012.00849.x. [DOI] [PubMed] [Google Scholar]
- Clarke SJ, Khaliulin I, Das M, et al. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circulation Research. 2008;102:1082–1090. doi: 10.1161/CIRCRESAHA.107.167072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J. 1994;302(Pt 2):321–324. doi: 10.1042/bj3020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofts AR, Chappell JB. Calcium Ion Accumulation and volume changes of isolated liver mitochondria. Reversal of calcium ion-induced swelling. Biochem J. 1965;95:387–392. doi: 10.1042/bj0950387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur J Biochem. 1988;178:489–501. doi: 10.1111/j.1432-1033.1988.tb14475.x. [DOI] [PubMed] [Google Scholar]
- Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Künzi M, Carafoli E. The calcium-induced and sodium-induced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. Eur J Biochem. 1977;79:549–558. doi: 10.1111/j.1432-1033.1977.tb11839.x. [DOI] [PubMed] [Google Scholar]
- Csordás G, Golenár T, Seifert EL, et al. MICU1 Controls Both the Threshold and Cooperative Activationof the Mitochondrial Ca. Cell Metabolism. 2013;17:976–987. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AM, Harris DA. Control of mitochondrial ATP synthase in heart cells: inactive to active transitions caused by beating or positive inotropic agents. Cardiovascular Research. 1990;24:411–417. doi: 10.1093/cvr/24.5.411. [DOI] [PubMed] [Google Scholar]
- De Marchi E, Bonora M, Giorgi C, Pinton P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium. 2014;56:1–13. doi: 10.1016/j.ceca.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, et al. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2012;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Rizzuto R, Pozzan T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu Rev Biochem. 2013;85 doi: 10.1146/annurev-biochem-060614-034216. annurev–biochem–060614–034216–32. [DOI] [PubMed] [Google Scholar]
- Ding Y, Fang H, Shang W, et al. Mitoflash altered by metabolic stress in insulin-resistant skeletal muscle. J Mol Med. 2015;93:1119–1130. doi: 10.1007/s00109-015-1278-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Zhang W, et al. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2011;32:398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod JW, Wong R, Mishra S, et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–3687. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldkamp T, Park JS, Pasupulati R, et al. Regulation of the mitochondrial permeability transition in kidney proximal tubules and its alteration during hypoxia-reoxygenation. Am J Physiol Renal Physiol. 2009;297:F1632–46. doi: 10.1152/ajprenal.00422.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Lucchinetti E, Ahuja P, et al. Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3beta. Anesthesiology. 2005;103:987–995. doi: 10.1097/00000542-200511000-00013. [DOI] [PubMed] [Google Scholar]
- Feng Y, Xia Y, Yu G, et al. Cleavage of GSK-3β by calpain counteracts the inhibitory effect of Ser9 phosphorylation on GSK-3β activity induced by H 2O 2. J Neurochem. 2013;126:234–242. doi: 10.1111/jnc.12285. [DOI] [PubMed] [Google Scholar]
- Feniouk BA, Yoshida M. Regulatory mechanisms of proton-translocating F(O)F (1)-ATP synthase. Results Probl Cell Differ. 2008;45:279–308. doi: 10.1007/400_2007_043. [DOI] [PubMed] [Google Scholar]
- Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. 1987;19:297–303. doi: 10.1007/BF00762419. [DOI] [PubMed] [Google Scholar]
- Gelb BD, Adams V, Jones SN, et al. Targeting of hexokinase 1 to liver and hepatoma mitochondria. Proc Natl Acad Sci USA. 1992;89:202–206. doi: 10.1073/pnas.89.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26:190–195. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Gill RS, Manouchehri N, Lee T-F, et al. Cyclosporine treatment improves mesenteric perfusion and attenuates necrotizing enterocolitis (NEC)-like intestinal injury in asphyxiated newborn piglets during reoxygenation. Intensive Care Med. 2012;38:482–490. doi: 10.1007/s00134-011-2436-5. [DOI] [PubMed] [Google Scholar]
- Giorgio V, Bisetto E, Soriano ME, et al. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. Journal of Biological Chemistry. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V, Stockum von S, Antoniel M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proceedings of the National Academy of Sciences. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez L, Paillard M, Thibault H, et al. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–2768. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- Goñi-Oliver P, Lucas JJ, Avila J, Hernandez F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem. 2007;282:22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- Grijalba MT, Vercesi AE, Schreier S. Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry. 1999;38:13279–13287. doi: 10.1021/bi9828674. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–86. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-Aguilar M, Douglas DL, Gibson AK, et al. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. Journal of Molecular and Cellular Cardiology. 2014;72:316–325. doi: 10.1016/j.yjmcc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock CR, Caplan AI. Ion-induced ultrastructural transformations in isolated mitochondria. The energized uptake of calcium. The Journal of Cell Biology. 1969;42:221–234. doi: 10.1083/jcb.42.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner AV, Dai J, Gomes AP, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. Calcium-dependent opening of a non-specific pore in the mitochondrial inner membrane is inhibited at pH values below 7. Implications for the protective effect of low pH against chemical and hypoxic cell damage. Biochem J. 1991;278(Pt 3):715–719. doi: 10.1042/bj2780715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38:841–860. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Richardson AP. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. Journal of Molecular and Cellular Cardiology. 2015;78:129–141. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–1717. doi: 10.1161/01.CIR.0000126294.81407.7D. [DOI] [PubMed] [Google Scholar]
- Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Archives of Biochemistry and Biophysics. 1979;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- Haworth RA, Hunter DR. Control of the mitochondrial permeability transition pore by high-affinity ADP binding at the ADP/ATP translocase in permeabilized mitochondria. J Bioenerg Biomembr. 2000;32:91–96. doi: 10.1023/a:1005568630151. [DOI] [PubMed] [Google Scholar]
- Hernando V, Inserte J, Sartório CL, et al. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. Journal of Molecular and Cellular Cardiology. 2010;49:271–279. doi: 10.1016/j.yjmcc.2010.02.024. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, Stöckl A, Schlame M, et al. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J Biol Chem. 1994;269:1940–1944. [PubMed] [Google Scholar]
- Hom J, Yu T, Yoon Y, et al. Regulation of mitochondrial fission by intracellular Ca2+ in rat ventricular myocytes. Biochim Biophys Acta. 2010;1797:913–921. doi: 10.1016/j.bbabio.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom JR, Gewandter JS, Michael L, et al. Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J Cell Physiol. 2007;212:498–508. doi: 10.1002/jcp.21051. [DOI] [PubMed] [Google Scholar]
- Hom JR, Quintanilla RA, Hoffman DL, et al. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Developmental Cell. 2011;21:469–478. doi: 10.1016/j.devcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool LC, Corry B. Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2007;9:409–435. doi: 10.1089/ars.2006.1446. [DOI] [PubMed] [Google Scholar]
- Hubbard MJ, McHugh NJ. Mitochondrial ATP synthase F1-beta-subunit is a calcium-binding protein. FEBS Letters. 1996;391:323–329. doi: 10.1016/0014-5793(96)00767-3. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Archives of Biochemistry and Biophysics. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- Hurst S, Gomez L, Jhun B, et al. Truncation of GSK-3β in Cardiac Mitochondria is the Master Switch of the mPTP 2015 [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Inoue T, Yoshida Y, Isaka Y, Tagawa K. Isolation of mitochondrial cyclophilin from bovine heart. Biochemical and Biophysical Research Communications. 1993;190:857–863. doi: 10.1006/bbrc.1993.1127. [DOI] [PubMed] [Google Scholar]
- Inserte J, Barba I, Hernando V, Garcia-Dorado D. Delayed recovery of intracellular acidosis during reperfusion prevents calpain activation and determines protection in postconditioned myocardium. Cardiovascular Research. 2009;81:116–122. doi: 10.1093/cvr/cvn260. [DOI] [PubMed] [Google Scholar]
- Inserte J, Garcia-Dorado D, Hernando V, et al. Ischemic preconditioning prevents calpain-mediated impairment of Na+/K+-ATPase activity during early reperfusion. Cardiovascular Research. 2006;70:364–373. doi: 10.1016/j.cardiores.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Javadov SA, Clarke S, Das M, et al. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. The Journal of Physiology. 2003;549:513–524. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekabsone A, Ivanoviene L, Brown GC, Borutaite V. Nitric oxide and calcium together inactivate mitochondrial complex I and induce cytochrome c release. Journal of Molecular and Cellular Cardiology. 2003;35:803–809. doi: 10.1016/s0022-2828(03)00137-8. [DOI] [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, Yin X, Yu D, et al. Truncation and activation of GSK-3β by calpain I: a molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci Rep. 2015;5:8187. doi: 10.1038/srep08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KM, Chen X, Boitano A, et al. Identification and validation of the mitochondrial F1F0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem Biol. 2005;12:485–496. doi: 10.1016/j.chembiol.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Joiner M-LA, Koval OM, Li J, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou M-J. Melatonin preserves the transient mitochondrial permeability transition for protection during mitochondrial Ca2+ stress in astrocyte. Journal of Pineal Research. 2011;50:427–435. doi: 10.1111/j.1600-079X.2011.00861.x. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim S-H, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Yang H, Kim BS, et al. The immunosuppressant cyclosporin A inhibits recurrent seizures in an experimental model of temporal lobe epilepsy. Neurosci Lett. 2012;529:133–138. doi: 10.1016/j.neulet.2012.08.087. [DOI] [PubMed] [Google Scholar]
- Kadenbach B, Mende P, Kolbe HV, et al. The mitochondrial phosphate carrier has an essential requirement for cardiolipin. FEBS Letters. 1982;139:109–112. doi: 10.1016/0014-5793(82)80498-5. [DOI] [PubMed] [Google Scholar]
- Kar P, Samanta K, Shaikh S, et al. Archives of Biochemistry and Biophysics. Archives of Biochemistry and Biophysics. 2010;495:1–7. doi: 10.1016/j.abb.2009.12.020. [DOI] [PubMed] [Google Scholar]
- Karch J, Kwong JQ, Burr AR, et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife. 2013;2:e00772. doi: 10.7554/eLife.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch J, Molkentin JD. Is p53 the long-sought molecular trigger for cyclophilin D-regulated mitochondrial permeability transition pore formation and necrosis? Circulation Research. 2012;111:1258–1260. doi: 10.1161/CIRCRESAHA.112.280990. [DOI] [PubMed] [Google Scholar]
- Khalil PN, Neuhof C, Huss R, et al. Calpain inhibition reduces infarct size and improves global hemodynamics and left ventricular contractility in a porcine myocardial ischemia/reperfusion model. Eur J Pharmacol. 2005;528:124–131. doi: 10.1016/j.ejphar.2005.10.032. [DOI] [PubMed] [Google Scholar]
- Khaliulin I, Clarke SJ, Lin H, et al. Temperature preconditioning of isolated rat hearts–a potent cardioprotective mechanism involving a reduction in oxidative stress and inhibition of the mitochondrial permeability transition pore. The Journal of Physiology. 2007;581:1147–1161. doi: 10.1113/jphysiol.2007.130369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliulin I, Schwalb H, Wang P, et al. Preconditioning improves postischemic mitochondrial function and diminishes oxidation of mitochondrial proteins. Free Radic Biol Med. 2004;37:1–9. doi: 10.1016/j.freeradbiomed.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Kinnally KW, Campo ML, Tedeschi H. Mitochondrial channel activity studied by patch-clamping mitoplasts. J Bioenerg Biomembr. 1989;21:497–506. doi: 10.1007/BF00762521. [DOI] [PubMed] [Google Scholar]
- Kinnally KW, Peixoto PM, Ryu S-Y, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta. 2011;1813:616–622. doi: 10.1016/j.bbamcr.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnally KW, Zorov DB, Antonenko YN, et al. Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc Natl Acad Sci USA. 1993;90:1374–1378. doi: 10.1073/pnas.90.4.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöhn P-C, Soriano ME, Irwin W, et al. Early resistance to cell death and to onset of the mitochondrial permeability transition during hepatocarcinogenesis with 2-acetylaminofluorene. Proc Natl Acad Sci USA. 2003;100:10014–10019. doi: 10.1073/pnas.1633614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr MJ, Aponte AM, Sun J, et al. Characterization of potential S-nitrosylation sites in the myocardium. AJP: Heart and Circulatory Physiology. 2011a;300:H1327–35. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr MJ, Sun J, Aponte A, et al. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circulation Research. 2011b;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konukoglu D, Taşci I, Cetinkale O. Effects of cyclosporin A and ibuprofen on liver ischemia-reperfusion injury in the rat. Clin Chim Acta. 1998;275:1–8. doi: 10.1016/s0009-8981(97)00089-2. [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Davis J, Baines CP, et al. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014;21:1209–1217. doi: 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metabolism. 2015;21:206–214. doi: 10.1016/j.cmet.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacza Z, Pankotai E, Csordás A, et al. Mitochondrial NO and reactive nitrogen species production: does mtNOS exist? Nitric Oxide. 2006;14:162–168. doi: 10.1016/j.niox.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Lehninger AL. Reversal of various types of mitochondrial swelling by adenosine triphosphate. J Biol Chem. 1959;234:2465–2471. [PubMed] [Google Scholar]
- Leung AWC, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linard D, Kandlbinder A, Degand H, et al. Redox characterization of human cyclophilin D: identification of a new mammalian mitochondrial redox sensor? Archives of Biochemistry and Biophysics. 2009;491:39–45. doi: 10.1016/j.abb.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Lu X, Kwong J, Molkentin JD, Bers DM. Individual Cardiac Mitochondria Undergo Rare Transient Permeability Transition Pore Openings. Circulation Research CIRCRESAHA. 2015;115:308093–16. doi: 10.1161/CIRCRESAHA.115.308093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. CellReports. 2015;12:23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Liu S, Huang Q, et al. Site-specific Phosphorylation Protects Glycogen Synthase Kinase-3 from Calpain-mediated Truncation of Its N and C Termini. Journal of Biological Chemistry. 2012;287:22521–22532. doi: 10.1074/jbc.M111.321349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa A, Lee J-K, Nagaya T, et al. Overexpression of calpastatin by gene transfer prevents troponin I degradation and ameliorates contractile dysfunction in rat hearts subjected to ischemia/reperfusion. Journal of Molecular and Cellular Cardiology. 2003;35:1277–1284. doi: 10.1016/s0022-2828(03)00238-4. [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Cárdenas C, Doonan PJ, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14:1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markevich NI, Hoek JB. Computational modeling analysis of mitochondrial superoxide production under varying substrate conditions and upon inhibition of different segments of the electron transport chain. Biochim Biophys Acta. 2015;1847:656–679. doi: 10.1016/j.bbabio.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Fancelli D, Wong M, et al. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front Cell Neurosci. 2014a;8:433. doi: 10.3389/fncel.2014.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Semenkow S, Hanaford A, Wong M. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant α-synuclein transgenic mice. Neurobiol Aging. 2014b;35:1132–1152. doi: 10.1016/j.neurobiolaging.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, et al. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J Exp Med. 1998;187:1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari S, Azzone GF. The equivalent pore radius of intact and damaged mitochondria and the mechanism of active shrinkage. Biochim Biophys Acta. 1972;283:23–29. doi: 10.1016/0005-2728(72)90094-1. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Denton RM. The role of Ca2+ ions in the regulation of intramitochondrial metabolism and energy production in rat heart. Mol Cell Biochem. 1989;89:121–125. [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels G, Khan IF, Endres-Becker J, et al. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation. 2009;119:2435–2443. doi: 10.1161/CIRCULATIONAHA.108.835389. [DOI] [PubMed] [Google Scholar]
- Miura T, Tanno M. Mitochondria and GSK-3β in Cardioprotection Against Ischemia/Reperfusion Injury. Cardiovasc Drugs Ther. 2010;24:255–263. doi: 10.1007/s10557-010-6234-z. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- Mizuta K, Ohmori M, Miyashita F, et al. Effect of pretreatment with FTY720 and cyclosporin on ischaemia-reperfusion injury of the liver in rats. J Pharm Pharmacol. 1999;51:1423–1428. doi: 10.1211/0022357991777065. [DOI] [PubMed] [Google Scholar]
- Moody BF, Calvert JW. Emergent role of gasotransmitters in ischemia-reperfusion injury. Med Gas Res. 2011;1:3. doi: 10.1186/2045-9912-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morciano G, Giorgi C, Bonora M, et al. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. Journal of Molecular and Cellular Cardiology. 2015;78:142–153. doi: 10.1016/j.yjmcc.2014.08.015. [DOI] [PubMed] [Google Scholar]
- Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry. 2003;42:6493–6499. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Stevens MV, Kohr M, et al. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. Journal of Biological Chemistry. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni R, Zheng D, Xiong S, et al. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type-1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy. Diabetes db150963. 2015 doi: 10.2337/db15-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Chalmers S. The integration of mitochondrial calcium transport and storage. J Bioenerg Biomembr. 2004;36:277–281. doi: 10.1023/B:JOBB.0000041753.52832.f3. [DOI] [PubMed] [Google Scholar]
- Nicolli A, Petronilli V, Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation. Biochemistry. 1993;32:4461–4465. doi: 10.1021/bi00067a039. [DOI] [PubMed] [Google Scholar]
- Novgorodov SA, Gudz TI, Brierley GP, Pfeiffer DR. Magnesium ion modulates the sensitivity of the mitochondrial permeability transition pore to cyclosporin A and ADP. Archives of Biochemistry and Biophysics. 1994;311:219–228. doi: 10.1006/abbi.1994.1230. [DOI] [PubMed] [Google Scholar]
- O-Uchi J, Jhun BS, Xu S, et al. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid Redox Signal. 2014a;21:863–879. doi: 10.1089/ars.2013.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O-Uchi J, Ryu S-Y, Jhun BS, et al. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid Redox Signal. 2014b;21:987–1006. doi: 10.1089/ars.2013.5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oess S, Icking A, Fulton D, et al. Subcellular targeting and trafficking of nitric oxide synthases. Biochem J. 2006;396:401–409. doi: 10.1042/BJ20060321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S-B, Subrayan S, Lim SY, et al. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- Ott M, Robertson JD, Gogvadze V, et al. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Out TA, Kemp A, Souverijn JH. The effect of bongkrekic acid on the Ca 2+ -stimulated oxidation in rat-liver mitochondria and its relation to the efflux of intramitochondrial adenine nucleotides. Biochim Biophys Acta. 1971;245:299–304. doi: 10.1016/0005-2728(71)90148-4. [DOI] [PubMed] [Google Scholar]
- Ozaki T, Tomita H, Tamai M, Ishiguro S-I. Characteristics of mitochondrial calpains. J Biochem. 2007;142:365–376. doi: 10.1093/jb/mvm143. [DOI] [PubMed] [Google Scholar]
- Ozaki T, Yamashita T, Ishiguro S-I. Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochim Biophys Acta. 2009;1793:1848–1859. doi: 10.1016/j.bbamcr.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Packer MA, Scarlett JL, Martin SW, Murphy MP. Induction of the mitochondrial permeability transition by peroxynitrite. Biochem Soc Trans. 1997;25:909–914. doi: 10.1042/bst0250909. [DOI] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proceedings of the National Academy of Sciences. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Pasupulati R, Feldkamp T, et al. Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am J Physiol Renal Physiol. 2011;301:F134–50. doi: 10.1152/ajprenal.00033.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdois P, Parker JE, Griffiths EJ, Halestrap AP. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem J. 2011;436:493–505. doi: 10.1042/BJ20101957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdois P, Parker JE, Halestrap AP. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J Am Heart Assoc. 2013;2:e005645–e005645. doi: 10.1161/JAHA.112.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG. Activation of Glycogen Synthase Kinase 3 Disrupts the Binding of Hexokinase II to Mitochondria by Phosphorylating Voltage-Dependent Anion Channel and Potentiates Chemotherapy-Induced Cytotoxicity. Cancer Research. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem. 2003;10:1535–1551. doi: 10.2174/0929867033457269. [DOI] [PubMed] [Google Scholar]
- Pavlov E, Zakharian E, Bladen C, et al. A large, voltage-dependent channel, isolated from mitochondria by water-free chloroform extraction. Biophys J. 2005;88:2614–2625. doi: 10.1529/biophysj.104.057281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Campo R, López-Torres M, Cadenas S, et al. The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. J Comp Physiol B, Biochem Syst Environ Physiol. 1998;168:149–158. doi: 10.1007/s003600050131. [DOI] [PubMed] [Google Scholar]
- Pestana CR, Silva CHTP, Pardo-Andreu GL, et al. Ca(2+) binding to c-state of adenine nucleotide translocase (ANT)-surrounding cardiolipins enhances (ANT)-Cys(56) relative mobility: a computational-based mitochondrial permeability transition study. Biochim Biophys Acta. 2009;1787:176–182. doi: 10.1016/j.bbabio.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Petronilli V, Miotto G, Canton M, et al. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V, Szabo I, Zoratti M. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Letters. 1989;259:137–143. doi: 10.1016/0014-5793(89)81513-3. [DOI] [PubMed] [Google Scholar]
- Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Andrews SB. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010;277:3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzan T, Bragadin M, Azzone GF. Disequilibrium between steady-state Ca2+ accumulation ratio and membrane potential in mitochondria. Pathway and role of Ca2+ efflux. Biochemistry. 1977;16:5618–5625. doi: 10.1021/bi00644a036. [DOI] [PubMed] [Google Scholar]
- Quatresous E, Legrand C, Pouvreau S. Mitochondria-targeted cpYFP: pH or superoxide sensor? The Journal of General Physiology. 2012;140:567–570. doi: 10.1085/jgp.201210863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Jin YN, Bernhardi von R, Johnson GVW. Mitochondrial permeability transition pore induces mitochondria injury in Huntington disease. Mol Neurodegener. 2013;8:45. doi: 10.1186/1750-1326-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Archives of Biochemistry and Biophysics. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- Rapizzi E, Pinton P, Szabadkai G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. The Journal of Cell Biology. 2002;159:613–624. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Sciacovelli M, Chiara F, et al. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proceedings of the National Academy of Sciences. 2010;107:726–731. doi: 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. Journal of Biological Chemistry. 2013;288:23798–23806. doi: 10.1074/jbc.M113.482026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg H, Marbach M. Regulation of Ca 2+ transport in brain mitochondria. II. The mechanism of the adenine nucleotides enhancement of Ca 2+ uptake and retention. Biochimica et Biophysica Acta (BBA)- …. 1990;1016:87–98. doi: 10.1016/0005-2728(90)90010-2. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science. 2013;342:1379–1382. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos CXC, Anilkumar N, Zhang M, et al. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savino C, Pelicci P, Giorgio M. The P66Shc/mitochondrial permeability transition pore pathway determines neurodegeneration. Oxid Med Cell Longev. 2013;2013:719407–7. doi: 10.1155/2013/719407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton NE, Barclay JL, Clouston AD, Fawcett J. Cyclosporin A pretreatment in a rat model of warm ischaemia/reperfusion injury. J Hepatol. 2002;36:241–247. doi: 10.1016/s0168-8278(01)00248-3. [DOI] [PubMed] [Google Scholar]
- Scarlett JL, Packer MA, Porteous CM, Murphy MP. Alterations to glutathione and nicotinamide nucleotides during the mitochondrial permeability transition induced by peroxynitrite. Biochemical Pharmacology. 1996;52:1047–1055. doi: 10.1016/0006-2952(96)99426-5. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzländer M, Wagner S, Ermakova YG, et al. The “mitoflash” probe cpYFP does not respond to superoxide. Nature. 2014;514:E12–4. doi: 10.1038/nature13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmughapriya S, Rajan S, Hoffman NE, et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol Cell. 2015:1–41. doi: 10.1016/j.molcel.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Chou J, Baty CJ, et al. Spatial localization of m-calpain to the plasma membrane by phosphoinositide biphosphate binding during epidermal growth factor receptor-mediated activation. Molecular and Cellular Biology. 2006;26:5481–5496. doi: 10.1128/MCB.02243-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen E-Z, Song C-Q, Lin Y, et al. Mitoflash frequency in early adulthood predicts lifespan in Caenorhabditis elegans. Nature. 2014;508:128–132. doi: 10.1038/nature13012. [DOI] [PubMed] [Google Scholar]
- Shiga Y, Onodera H, Matsuo Y, Kogure K. Cyclosporin A protects against ischemia-reperfusion injury in the brain. Brain Res. 1992;595:145–148. doi: 10.1016/0006-8993(92)91465-q. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Shintani-Ishida K, Yoshida K-I. Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion. Int J Cardiol. 2015;197:26–32. doi: 10.1016/j.ijcard.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Shulga N, Pastorino JG. Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. Journal of Cell Science. 2010;123:4117–4127. doi: 10.1242/jcs.073502. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. Journal of Cell Science. 2010;123:894–902. doi: 10.1242/jcs.061846. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Soda Y, el-Assal ON, Yu L, Nagasue N. Suppressed endothelin-1 production by FK506 and cyclosporin A in ischemia/reperfusion of rat small intestine. Surgery. 1999;125:23–32. [PubMed] [Google Scholar]
- Sohal RS, Allen RG. Relationship between metabolic rate, free radicals, differentiation and aging: a unified theory. Basic Life Sci. 1985;35:75–104. doi: 10.1007/978-1-4899-2218-2_4. [DOI] [PubMed] [Google Scholar]
- Solesio ME, Demirkhanyan L, Zakharian E, Pavlov EV. Contribution of inorganic polyphosphate towards regulation of mitochondrial free calcium. BBA - General Subjects. 2016a;1860:1317–1325. doi: 10.1016/j.bbagen.2016.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solesio ME, Elustondo PA, Zakharian E, Pavlov EV. Inorganic polyphosphate (polyP) as an activator and structural component of the mitochondrial permeability transition pore. Biochem Soc Trans. 2016b;44:7–12. doi: 10.1042/BST20150206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y-H, Cho H, Ryu S-Y, et al. L-type Ca(2+) channel facilitation mediated by H(2)O(2)-induced activation of CaMKII in rat ventricular myocytes. Journal of Molecular and Cellular Cardiology. 2010;48:773–780. doi: 10.1016/j.yjmcc.2009.10.020. [DOI] [PubMed] [Google Scholar]
- Song Y-H, Choi E, Park S-H, et al. Sustained CaMKII activity mediates transient oxidative stress-induced long-term facilitation of L-type Ca(2+) current in cardiomyocytes. Free Radic Biol Med. 2011;51:1708–1716. doi: 10.1016/j.freeradbiomed.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Sorimachi H, Ono Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovascular Research. 2012;96:11–22. doi: 10.1093/cvr/cvs157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu SS, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J Biol Chem. 1995;270:27510–27515. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen R-F, et al. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circulation Research. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circulation Research. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Steenbergen C, Murphy E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid Redox Signal. 2006;8:1693–1705. doi: 10.1089/ars.2006.8.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg TB, Swenson L, Wahl DR, et al. Apoptotic signaling activated by modulation of the F0F1-ATPase: implications for selective killing of autoimmune lymphocytes. J Pharmacol Exp Ther. 2009;331:437–444. doi: 10.1124/jpet.109.156422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Bernardi P, Zoratti M. Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem. 1992;267:2940–2946. [PubMed] [Google Scholar]
- Szabo I, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Letters. 1993;330:201–205. [Google Scholar]
- Šileikytė J, Blachly-Dyson E, Sewell R, et al. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)) Journal of Biological Chemistry. 2014;289:13769–13781. doi: 10.1074/jbc.M114.549634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2007;1768:2510–2515. doi: 10.1016/j.bbamem.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno M, Kuno A, Ishikawa S, et al. Translocation of GSK-3β, a Trigger of Permeability Transition, Is Kinase Activity-dependent and Mediated by Interaction with VDAC2. Journal of Biological Chemistry. 2014;289 doi: 10.1074/jbc.M114.563924. jbc.M114.563924–29296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanveer A, Virji S, Andreeva L, et al. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur J Biochem. 1996;238:166–172. doi: 10.1111/j.1432-1033.1996.0166q.x. [DOI] [PubMed] [Google Scholar]
- Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase–dependent pathway is cardioprotective. Circulation Research. 2002;90:377–379. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- Traba J, Del Arco A, Duchen MR, et al. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca(2+) buffering. Cell Death Differ. 2012;19:650–660. doi: 10.1038/cdd.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Lu W, Ogasawara MA, et al. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. The Journal of Physiology. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainio H, Mela L, Chance B. Energy dependent bivalent cation translocation in rat liver mitochondria. Eur J Biochem. 1970;12:387–391. doi: 10.1111/j.1432-1033.1970.tb00863.x. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Van Walraven HS, Scholts MJC, Zakharov SD, et al. pH-dependent Ca2+ binding to the F0 c-subunit affects proton translocation of the ATP synthase from Synechocystis 6803. J Bioenerg Biomembr. 2002;34:455–464. doi: 10.1023/a:1022518109371. [DOI] [PubMed] [Google Scholar]
- Varanyuwatana P, Halestrap AP. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. MITOCH. 2012;12:120–125. doi: 10.1016/j.mito.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Marchenko ND, Ji K, et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Naredo I, Loureiro R, Mesquita KA, et al. Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells. Cell Death Differ. 2014;21:1560–1574. doi: 10.1038/cdd.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma M, Shulga N, Pastorino JG. Sirtuin-3 modulates Bak- and Bax-dependent apoptosis. Journal of Cell Science. 2013;126:274–288. doi: 10.1242/jcs.115188. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang W, Fang H, Groom L, et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei A-C, Liu T, Winslow RL, O’Rourke B. Dynamics of matrix-free Ca2+ in cardiac mitochondria: two components of Ca2+ uptake and role of phosphate buffering. The Journal of General Physiology. 2012;139:465–478. doi: 10.1085/jgp.201210784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan RS, Konstantinidis K, Wei A-C, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proceedings of the National Academy of Sciences. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi J, Wang H, Mueller RA, et al. Mechanism for resveratrol-induced cardioprotection against reperfusion injury involves glycogen synthase kinase 3beta and mitochondrial permeability transition pore. Eur J Pharmacol. 2009;604:111–116. doi: 10.1016/j.ejphar.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie GC, Wilson JE. Rat brain hexokinase: the hydrophobic N-terminus of the mitochondrially bound enzyme is inserted in the lipid bilayer. Archives of Biochemistry and Biophysics. 1988;267:803–810. doi: 10.1016/0003-9861(88)90090-2. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- Yu T, Sheu S-S, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovascular Research. 2008;79:341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- Zaobornyj T, Ghafourifar P. Strategic localization of heart mitochondrial NOS: a review of the evidence. AJP: Heart and Circulatory Physiology. 2012;303:H1283–93. doi: 10.1152/ajpheart.00674.2011. [DOI] [PubMed] [Google Scholar]
- Zhai P, Sciarretta S, Galeotti J, et al. Differential Roles of GSK-3 During Myocardial Ischemia and Ischemia/Reperfusion. Circulation Research. 2011;109:502–511. doi: 10.1161/CIRCRESAHA.111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L-P, Ji C, Lu P-H, et al. Oxygen glucose deprivation (OGD)/re-oxygenation-induced in vitro neuronal cell death involves mitochondrial cyclophilin-D/P53 signaling axis. Neurochem Res. 2013;38:705–713. doi: 10.1007/s11064-013-0968-5. [DOI] [PubMed] [Google Scholar]
- Zhen Y-F, Wang G-D, Zhu L-Q, et al. P53 dependent mitochondrial permeability transition pore opening is required for dexamethasone-induced death of osteoblasts. J Cell Physiol. 2014;229:1475–1483. doi: 10.1002/jcp.24589. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Kinnally KW, Perini S, Tedeschi H. Multiple conductance levels in rat heart inner mitochondrial membranes studied by patch clamping. Biochim Biophys Acta. 1992;1105:263–270. doi: 10.1016/0005-2736(92)90203-x. [DOI] [PubMed] [Google Scholar]