

Abstract
A tissue-selective estrogen complex (TSEC), combining a selective estrogen receptor modulator, bazedoxifene (BZA), with conjugated equine estrogen (CEE), represents a novel strategy of menopausal hormone therapy without involving a progestin. We hypothesized that the antiestrogenic properties of BZA can also block the estrogenic effects of CEE on breast tissue and thereby prevent breast cancer in women. To test our hypothesis, the effects of estradiol (E2), CEE, and BZA on mammary gland and breast cancer xenografts were assessed in mouse models. In immature castrate mice, BZA completely blocked CEE- or E2-stimulated ductal and terminal end bud growth of mammary gland as well as estrogen-responsive gene expression. As a positive control, E2 stimulated tumor growth in nude mice bearing MCF-7 xenografts. This effect was completely blocked by BZA as were E2-stimulated expression of PR, pS2 (trefoil factor 1), cMyc, and AREG; the enhancement of Ki67 and proliferating cell nuclear antigen (PCNA); and the antiapoptotic effect. CEE was much less potent than E2 in stimulating Ki67, reducing apoptosis, and stimulating gene expression, but all effects were blocked by BZA. Unexpectedly, CEE alone, even at high doses, did not stimulate tumor growth. As confirmation of its absorption and deconjugation, CEE caused a 6-fold increase in uterine weight and stimulation of gene expression. These data support our hypothesis that the net effect of the CEE/BZA TSEC is to block estrogen action in benign and malignant breast tissue. These findings provide a rationale for a clinical study to determine whether this TSEC prevents breast cancer in women.
The Endocrine Society's Scientific Statement on menopausal hormone therapy (MHT) concluded that an estrogen plus a progestogen increases the risk of breast cancer, whereas estrogens alone do not (1–4). These data suggest that progestogens are critical elements necessary for the adverse effects of MHT on breast cancer. Accordingly, major efforts are underway to develop MHT regimens that do not include a progestogen (5). The rationale for use of a progestogen in women with an intact uterus is to oppose the estrogenic effect on uterine stimulation and to prevent uterine cancer (6, 7). Selective estrogen receptor modulators (SERMs) have been developed which antagonize the effects of estrogen on the uterus (6, 8). Based on these findings, the concept arose that the combination of a SERM with an estrogen might abrogate uterine stimulation while relieving other menopausal symptoms. The complexes containing a SERM and an estrogen have been termed tissue-selective estrogen complexes (TSECs) (5). One such TSEC, containing bazedoxifene (BZA) and conjugated equine estrogens (CEEs), has undergone extensive clinical testing (6, 7, 9–15). This TSEC retards bone loss, enhances vaginal maturation, and reduces hot flashes while not causing an increase in cardiovascular events or endometrial cancer.
An important, unresolved issue regarding the TSEC containing BZA/CEE is its specific effects on breast tissue. If the blocking effects of BZA on breast parallel those on the uterus (6), this TSEC might potentially reduce the risk of breast cancer in a manner analogous to the effects of tamoxifen and raloxifene (16). No systematic studies have previously examined the effects of BZA in combination with CEE on human breast tumor xenografts in mice. Our companion study provides data regarding the actions of this TSEC on MCF-7 breast cancer cells in vitro (17). The present study examines actions on benign breast development in immature mice and on the growth of MCF-7 xenografts in nude mice (18).
This study tested the hypothesis that BZA will antagonize the effect of estradiol (E2) and CEE on breast development in ovariectomized mice and on estrogen-stimulated breast xenografts. The study design used quantitative morphometry to assess the individual and combined effects of E2, CEE, and BZA on ductal and terminal end bud development (18) and on proliferation, apoptosis, and gene expression in mice. Experiments on human tissue examined the effects of these compounds on wild-type MCF-7 and LTED-MCF-7 xenografts. The data demonstrate that BZA effectively abrogates the estrogenic effects of CEE and E2 on normal and malignant breast tissue. A surprising finding was that CEE did not stimulate xenograft growth, whereas it exerted estrogenic effects on uterus, benign breast tissue, gene expression, and tumor histology. These data, taken together, provide evidence that BZA can block the effects of E2 and CEE on benign mammary tissue as well as xenografts and provides a rationale for a clinical trial to determine whether the CEE/BZA TSEC prevents breast cancer.
Materials and Methods
Animal experiments
United States Department of Agriculture and Association for the Assessment and Accreditation of Laboratory Animal Care guidelines were followed, and the studies were approved by the Institutional Animal Care and Use Committee of University of Virginia. Four-week-old, C57B/L6 female mice, obtained from Charles River Laboratories (Germantown, MD), were ovariectomized and administered E2, CEE, or BZA alone and in combination. They were killed 4 wk later to assess breast development and gene expression. For breast cancer xenograft studies, 5,000,000 wild-type, human MCF-7 or “hypersensitive” LTED-MCF-7 cells mixed in 0.1 ml of Matrigel (19) were injected into mammary nipples of oophorectomized nude mice of the same age. Tumor volumes were measured using calipers once a week starting on the 7th d of cell inoculation. In C57B/L6 mice, E2 was given by daily injection (sc) at the dose of 5 μg/kg in dimethylsulfoxide/PBS (1:1) solution. In nude mice, SILASTIC (Dow Corning Corp., Midland, MI) implants were used to “clamp” plasma E2 levels at 80 pg/ml as previously described (20). SILASTIC implants were exchanged with new ones after 8 wk. CEE and BZA were administered orally (by gavage) in a vehicle of 2% Tween 80 and 0.5% methylcellulose. PremarinR at a dose 3 mg/kg and BZA at 2 mg/kg were administered according to the protocol of Peano et al. (21). In their studies, 3 mg/kg CEE elicited an increase in uterine wet weight and mammary gland stimulation, and 2 mg/kg BZA was the minimum fully effective antagonist dose. CEE and BZA were administered for a total of 12 wk in the current studies.
Parameters
Body weights were assessed weekly for 12 wk, after which the mice were euthanized by an overdose of ketamine/xylazine. No differences in body weight were observed among treatment groups (data not shown). The uteri were excised, trimmed of adherent fat, and weighed after expressing any luminal fluid. Representative single inguinal mammary glands were excised, fixed in Carnoy's solution, and stained for morphological analysis. Tumors were removed and weighed with an equal number fixed in formalin for histological analysis and the other quick frozen in liquid nitrogen and stored at −80 C for subsequent RNA isolation.
Whole-mount analysis
Our previously published methods were used to prepare whole mounts of mammary glands, and BIO-QUANT computer software was employed to analyze them (18). For histologic analysis, tumors were excised and fixed in formalin for hematoxylin and eosin (H&E), terminal deoxynucleotidyl transferase 2′-deoxyuridine, 5′-triphosphate nick end labeling (TUNEL), Ki67, and proliferating cell nuclear antigen (PCNA) staining. For detection of apoptotic cells by TUNEL, we used ApopTag Peroxidase In Situ Apoptosis Detection kit (Millipore, Temecula, CA), which labels apoptotic cells using terminal deoxynucleotidyl transferase for specific staining. Ki67 was revealed by immunocytochemistry using rabbit anti-Ki67 antibody from Novus Biologicals (Littleton, CO) and for PCNA, a monoclonal antibody from Cell Signaling Technology (Danvers, MA).
PCR analysis
Total RNA was extracted and purified using QIAGEN RNeasy Mini kit (QIAGEN, Valencia, CA). cDNA was synthesized using Bio-Rad cDNA Synthesis kit (Bio-Rad, Hercules, CA). Transcription of estrogen-regulated genes was determined by quantitative real time PCR (qPCR) using SYBR Green method. Glyceraldehyde-3-phosphate dehydrogenase was used as a housekeeping gene for quantification. Relative mRNA copies were compared with vehicle control using the comparative cycle threshold (ΔΔCt) method. The primer pairs for qPCR of mouse mammary gland cDNA and tumor cDNA are listed in Supplemental Material, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org.
Statistics
For analysis of tumor growth, the log slope method of Heitjan et al. (22) was used as described. Specifically, each tumor volume is converted to its log derivative, and a slope of the log of volumes over the period of observation is computed and used to perform Student's t test on the slopes. As discussed by Heitjan et al. (22), this method enhances the power of analysis by combining the information from each single volume into an integrated slope. Student's t test was used for other comparisons.
Results
Bioassay of estrogen effects
Because CEE represents a mixture of various estrogenic and potentially antiestrogenic substances, we considered it important to determine its in vivo estrogenic potency in comparison with E2, using a standard uterine weight bioassay (20, 23, 24). CEE stimulated uterine weight to approximately 80% of the weight induced by 5 μg/kg E2. The effects of both CEE and E2 were significantly blocked with BZA but not to the levels in vehicle-treated animals (Fig. 1).
Fig. 1.

Uterine wet weight of C57B/L6 mice. The animals were treated for 4 wk with sc 5 μg/kg E2 alone and in combination with 2 mg/kg BZA; 3 mg/kg CEE alone and in combination with 2 mg/kg BZA. Columns are average uterine wet weight in milligrams ± sem; a, P < 0.001 compared with vehicle control (V); b and c, P < 0.001 compared with E2 and CEE treatment, respectively.
Estrogenic effects on benign mammary glands
Whole-mount evaluation
We initially examined the effects of E2, CEE, and BZA, alone and in combination, on normal mammary tissue as a means to assess breast development in ovariectomized animals (18). Representative whole-mount photographs illustrating mammary gland morphology are shown in Fig. 2. Both CEE and E2 stimulated ductal length and terminal end bud formation. BIO-QUANT computer assessment demonstrated that CEE and E2 stimulated these parameters to the same extent and BZA blocked the effects of both estrogens (Fig. 3, A and B). With H&E staining, the same changes were visually apparent but were not quantitated (see Supplemental Fig. 1).
Fig. 2.

Whole mounts of mammary glands of ovariectomized (OVX) C57BL/6 mice treated with E2, CEE, and BZA. Magnification, ×7. Distance between ruler hatch marks, 1 mm. Inset magnification, ×20.
Fig. 3.

A, Total duct length in the mammary gland whole mounts after 4 wk of treatment with sc E2 at 5 μg/kg alone and in combination with 2 mg/kg BZA; 3 mg/kg CEE alone and in combination with BZA 2 mg/kg. Columns are average total duct length in millimeters ± sem; a, P < 0.01 compared with vehicle control (V); b and c, P < 0.01 compared with E2 and CEE group, respectively. B, Terminal end bud (TEB) count in the mammary gland whole mounts. Columns are average TEB count ± sem; a, P < 0.01 compared with vehicle control (V); b, P < 0.05 compared with E2 treatment; c, P < 0.01 compared with CEE treatment. C, Quantification of Ki67 staining of mammary glands. Columns represent average percentage of Ki67 positive cells ± sem (nine mice per group, three slides per mouse). *, P < 0.05; **, P < 0.01 compared with vehicle control (V); a, P < 0.01 compared with E2 group; b, P < 0.05 compared with CEE group. D, Quantification of PCNA staining of mammary glands. Columns are average percentage of positive cells ± sem (nine mice per group, three slides per mouse). *, P < 0.05; **, P < 0.01 compared with vehicle control (V); a, P < 0.01 compared with E2 group; b, P < 0.05 compared with CEE group.
Cell proliferation
Quantification of Ki67 staining provided a means to assess the degree of cell proliferation. Both E2 and CEE stimulated this parameter, but CEE exerted lesser effects than E2. BZA inhibited CEE-stimulated Ki67 to levels significantly lower than observed in the vehicle control glands. BZA also blocked the effect of E2 on this parameter but not to levels observed in vehicle controls (Fig. 3C). To confirm the effects on cell proliferation, measurement of PCNA, another marker of proliferation, revealed nearly equal stimulation with E2 and CEE, but BZA was less effective in their inhibition (Fig. 3D).
Gene expression
As potentially more sensitive parameters to compare the estrogenic effects of E2 and CEE, we evaluated the expression of several E2-regulated genes (25). At the dose used, E2 stimulated aphiregulin (AREG) by 873-fold and CEE to a lesser extent of approximately 186-fold. BZA blocked the effects of both estrogens but not completely back to vehicle levels (Fig. 4A). E2 and CEE induced effects in similar directions on keratin complex 1, acidic, gene 19 (Krt1-19) and WNT1 inducible signaling pathway protein 2 (WISP2). Notably, CEE exerted greater effects than E2 on both of these genes (Fig. 4, B and C.). Taken together, these experiments demonstrate stimulatory effects of both CEE and E2 on normal mammary tissue and blockade with BZA but differences in degree of stimulation when comparing the two estrogens. It is recognized that full dose-response studies would be required to precisely assess potency vs. efficacy.
Fig. 4.
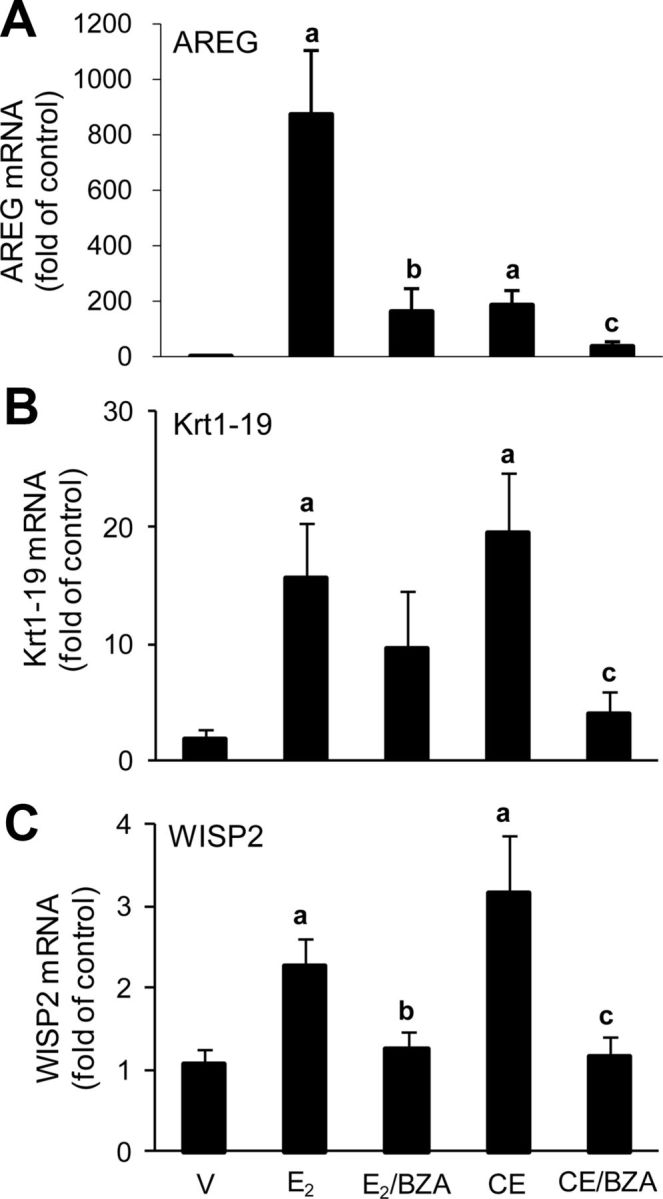
A, AREG gene expression in mammary glands of ovariectomized (OVX) C57BL/6 mice after 4 wk of treatment with vehicle, sc E2 (5 μg/kg) alone and in combination with 2 mg/kg BZA, and CEE (3 mg/kg) alone and in combination with BZA (2 mg/kg). Columns are average relative amount of AREG mRNA ± sem (n = 9); a, P < 0.01 compared with vehicle control (V); b and c, P < 0.05 compared with E2 and CEE group, respectively. B, Krt1-19 gene expression. Columns are average relative amount of Krt1-19 mRNA ± sem (n = 9); a, P < 0.05 compared with vehicle control (V); c, P < 0.05 compared with CEE group. C, WISP2 gene expression. Columns are average relative amount of WISP2 mRNA ± sem (n = 9); a, P < 0.01 compared with vehicle control (V); b and c, P < 0.05 compared with E2 and CEE group, respectively.
Estrogenic effects on MCF-7 xenografts
Tumor growth
The same doses of CEE were used in tumor growth experiments as in evaluation of benign tissue. The dose of E2 was clamped to 80 pg/ml plasma concentrations. This dose of E2 was demonstrated to stimulate growth of MCF-7 tumors in vivo (20). CEE stimulated uterine weight to levels 80% of that induced by E2, confirming the effects observed in animals without tumors (Supplemental Fig. 2). BZA blocked both effects but to a lesser extent in E2-treated animals. After a 7-wk latency period (see Supplemental Fig. 3), tumors in E2 treatment group grew log linearly with a slope of 24.78 and reached the size required for killing at 11 wk (Fig. 5, A and B). In contrast, vehicle-treated tumors grew minimally with a slope of 1.54. BZA completely abrogated E2-induced tumor growth (P < 0.05 vs. E2 alone). Surprisingly, CEE did not stimulate growth with slopes of 2.89, similar to those in animals receiving vehicle alone (Fig. 5, A and B) or BZA (slope 2.96). Tumor weights measured at 11 wk (Fig. 5C) confirmed the findings from slope measurements. E2-treated tumors weighed approximately 170 vs. 10 mg in vehicle-treated animals. BZA markedly reduced tumor weight when combined with E2. CEE exerted no effects on tumor weight compared with CEE/BZA or vehicle. To potentially explain the lack of CEE stimulation, we postulated that the CEE dose was not sufficient to stimulate tumor growth and repeated the study using 10, 30, and 50 mg/kg CEE rather than 3 mg/kg. Neither the wild-type MCF-7 xenografts (Fig. 5D) nor the hypersensitive LTED (19) xenografts (Fig. 5E) grew in response to 10, 30, or 50 mg/kg CEE. To confirm absorption and deconjugation of these high doses of CEE, we measured uterine weight and found substantial stimulation over vehicle control levels (data not shown). These findings demonstrated an unexpected lack of the ability of CEE to stimulate xenograft growth, even at high doses.
Fig. 5.

A, Logarithmic curve of tumor growth. The data are shown as average tumor volume in cubic millimeters ± sem, 20 tumors per group. B, Average slopes of tumor growth curves. The data are average slopes of growth curves of 20 tumors of each group from wk 7 to 11; a, P < 0.05 compared with vehicle control (V); b, P < 0.05 compared with E2 group. C, Weight of MCF-7 tumors. Columns are average tumor weight in milligrams ± sem (20 tumors per group); a, P < 0.001 compared with vehicle control (V); b, P < 0.001 compared with E2 group. Tumor volumes of MCF-7 xenografts (D) and LTED-MCF-7 xenografts (E) in mice receiving various doses of CEE. None of the curves are statistically significantly different.
Gene expression of tumors
Because CEE did not stimulate tumor growth, we examined gene expression as a more sensitive endpoint to determine whether CEE exerted any estrogenic effects on the xenografts (Fig. 6). Compared with the vehicle control, E2 increased progesterone receptor gene expression by 267-fold and CEE by 13-fold. BZA blocked the stimulatory effect of E2 by 89% and that of CEE by 80%, respectively. In contrast, although E2 treatment increased AREG gene expression by 131-fold (vs. vehicle), CEE induced no effects. However, BZA plus CEE reduced AREG levels by 94% below vehicle control levels. WISP 2, cellular myelocytomatosis oncogene (cMyc), and Cyclin D1 exhibited variable effects. Taken together, these experiments demonstrated a weak estrogenic effect of CEE on certain target genes but to a much lesser extent than induced by E2.
Fig. 6.

Estrogen-regulated gene mRNA expression of MCF-7 tumors grown in ovariectomized (OVX) nude mice. Transcription of estrogen-regulated genes, progesterone receptor (PR), AREG, pS2 (trefoil factor 1), cMyc, WISP2, and cyclin D1, was determined by qPCR using SYBR Green method. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene for quantification. Results represent relative mRNA copies compared with vehicle control (n = 3); a, compared with vehicle control (V); b, compared with estrogen alone (E2 or CEE); ***, P < 0.001; **, P < 0.01; *, P < 0.05.
Histological analysis of breast tumors
It was surprising that CEE exerted only weak estrogenic effects on gene expression and did not stimulate tumor growth. To determine whether any other estrogenic effects were induced, we asked whether CEE might induce histological changes in the residual tumor tissue at the time of killing. As shown by H&E staining (Fig. 7), vehicle-treated xenografts contained small tumor cell islands scattered among stromal tissue. CEE-treated tumors contained a greater number of cells than those receiving vehicle, whereas E2 markedly increased tumor cell number. BZA slightly reduced tumor cell densities in both E2 and CEE groups. These data demonstrated a weak but positive response to CEE.
Fig. 7.

H&E staining of MCF-7 breast tumors grown in ovariectomized (OVX) nude mice treated with vehicle (V), E2, CEE, and BZA and killed at the end of the experiment presented in Fig. 5A. Magnification, ×5; inset magnification, ×20.
We then assessed apoptosis by TUNEL staining and quantified this parameter with Image-Pro computer analysis (Fig. 8A). E2 markedly reduced apoptosis compared with vehicle-treated mice. As expected, this reduction was abrogated by the antiestrogen, BZA. Although CEE reduced apoptosis compared with the vehicle control, the percentage of apoptotic cells in CEE group was 13-fold higher than that in E2 group (Fig. 8A and Supplemental Figs. 4 and 5). Paradoxically, BZA exerted antiapoptotic effect when combined with CEE (Fig. 8A). Ki67 as a measure of cell proliferation increased in response to E2 as shown in Fig. 8B, and this effect was partially blocked by BZA. CEE also stimulated Ki67, but again, this effect was not blocked by BZA. To confirm the Ki67 results, we also quantitated another marker of proliferation, PCNA, which paralleled the responses of Ki67 (Fig. 8C).
Fig. 8.
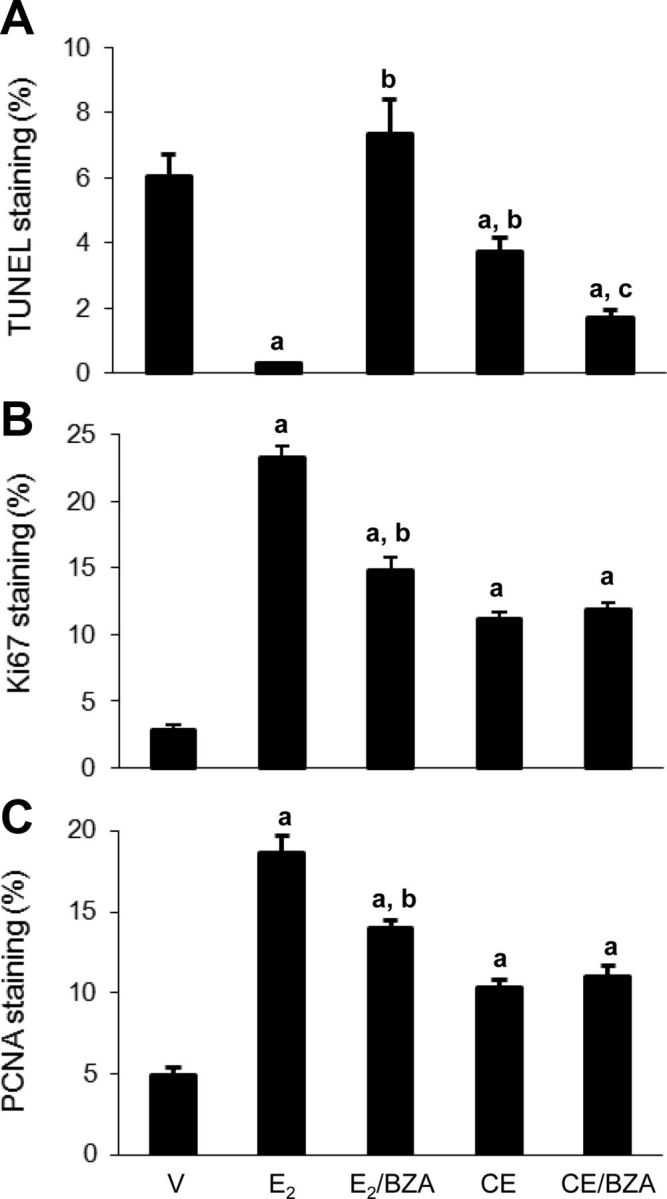
A, Percentage of TUNEL positive tumor cells per field quantified by Image-Pro software. Columns are percentage of TUNEL positive cell ± sem (four tumors per group, nine fields per tumor); a, P < 0.05 compared with vehicle control (V); b, P < 0.001 compared with E2 group; c, P < 0.001 compared with CEE. B, Percentage of Ki67 positive cell per field. Columns are percentage of Ki67 positive cell ± sem (four tumors per group, nine fields per tumor); a, P < 0.001 compared with vehicle control (V); b, P < 0.001 compared with E2 group. C, Percentage of PCNA positive cell per field. Columns are percentage of PCNA positive cell ± sem (four tumors per group, nine fields per tumor). a, P < 0.001 compared with vehicle control (V); b, P < 0.001 compared with E2 group.
Discussion
BZA effectively blocked the estrogenic effects of CEE and E2 on multiple parameters, including ductal length, terminal end bud development, cell proliferation, apoptosis, and gene expression in breast tissue from ovariectomized mice. In human MCF-7 tumor xenografts, BZA also inhibited tumor growth and the tumor weight increase induced by E2 clamped at plasma levels of 80 pg/ml. Taken together, these data provide strong evidence that BZA can effectively block the biologic effects of exogenous estrogens on multiple benign and malignant breast parameters. These results support the concept that the TSEC containing BZA and CEE would exert antiestrogenic effects on breast tissue in postmenopausal women.
The results of this study provide a rationale for testing whether the BZA/CEE TSEC would prevent breast cancer in women. Data critically evaluating the Women's Health Initiative (WHI) results suggest that the CEE + medroxyprogesterone acetate (MPA) regimen did not increase de novo breast cancer but stimulated occult, preexisting tumors to grow more rapidly through a promotional effect (26). Autopsy data indicate that 7% of otherwise healthy women harbor small, undiagnosed breast cancer with estimated doubling times averaging 200 d (1, 27). A recently developed tumor growth model provided insight into the promotional effects of MHT on breast cancer. This model suggests that more than 10 yr are required for de novo breast tumors to reach the mammographic detection threshold in women. Based on doubling times, the model estimates that only 7% of the new tumors diagnosed in the WHI arose de novo and the remaining 93% represented preexisting tumors (26, 27). These models suggested that an effect of CEE + MPA to increase proliferation sufficiently to reduce doublings time from 200 to 150 d would explain the increased incidence of breast cancer in the WHI study. Based on the findings in this study, the BZA/CEE TSEC would be expected to exert an opposite effect and slow growth of occult, estrogen-dependent tumors in women. The result would be to reduce breast cancer incidence in a manner similar to that of the antiestrogenic effects associated with tamoxifen and raloxifene (16).
In this study, we demonstrated that E2 and CEE exert similar effects on certain endpoints and differential ones on others. Both CEE and E2 induced estrogenic effects on uterine weight and on benign breast development in ovariectomized mice. In contrast, these two estrogens induced nonparallel gene expression responses with differential potencies on specific genes. These studies confirmed the conclusions drawn from estrogen receptor cofactor recruitment data and from studies of ductal branching and fat pad invasion in oophorectomized mice (21, 23). In addition, the current studies demonstrated stimulation of proliferation with Ki67 immunohistochemical staining and inhibition of apoptosis by CEE in normal mammary glands. With respect to human breast cancer tissue, E2 stimulated the growth of breast cancer xenografts, whereas CEE, even at very high doses, unexpectedly did not. Lack of stimulation of tumor growth with CEE might result from its less stimulatory (AREG) or even inhibitory (cMyc) effect on proliferation-related gene expression and moderate resistance to suppression of apoptosis. Differential regulation of gene expression by CEE and E2 could also explain why they exerted dissimilar effects on apoptosis in breast cancer xenografts. Chang et al. (25) have reported that in MCF-7 cells, CEE but not E2 specifically down-regulated several apoptosis-related genes. Tumor protein 53 induced nuclear protein 1 (TP53INP1) is one of these genes. TP53INP1 is a major mediator of the antioxidant function of p53. TP53INP1 inactivation induces a persistent accumulation of reactive oxygen species (ROS) and facilitates cell death (28, 29). In addition, it has been reported that equine estrogen can be oxidatively metabolized to 4-hydroxyequilenin and quinone. The redox cycles of this pathway generate more ROS and induce DNA damage (30, 31). Down-regulation of TP53INP1 by CEE would be expected to exacerbate the oxidative stress that drives apoptosis. It should be mentioned that long-term exposure to elevated levels of ROS will cause more DNA damage and mutations that would lead to tumor formation. In postmenopausal women, long-term MHT is associated with increased risk of breast cancer, whereas less than 5-yr MHT with CEE alone reduces breast cancer incidence.
The different effect of CEE on mouse mammary gland and breast xenografts could also be due to the SERM-like components of CEE. Combination of one SERM with another could exert estrogen agonistic, antagonistic, or neutral effects depending on resulting alteration of receptor configuration. Data from monkeys had previously demonstrated that CEE in clinically relevant doses exerted lesser proliferative effects on normal breast than E2 (32). These findings suggested that higher concentrations of CEE might be required to stimulate human tumor growth in our studies. Accordingly, we used 3- to 15-fold higher doses of CEE in a separate experiment but still demonstrated no tumor stimulation. These observations correspond in some respect to human data where CEE alone was not associated with an increase in breast cancer risk in the WHI study (2), whereas E2 and its derivatives have been associated with an increased risk in large observational studies (33, 34). It may be, however, that a concomitant progestogen is required to stimulate breast cancer as in the WHI study (1, 4). To assess this possibility, we have preliminarily added MPA to animals bearing xenografts treated for 12 wk with CEE. Observations over the ensuing 12 wk revealed no tumor growth. Additional experiments are now planned to add MPA to cell cultures before implantation to induce cancer stem cell formation as described by Horwitz and Sartorius (35). These experiments will test whether CEE will stimulate tumor growth when MPA is added before nipple inoculation. It is also possible that even the 50 mg/kg dose of CEE was insufficient to reach a threshold for proliferation, but this seems unlikely.
CEE contains a mixture of more than 100 components, some of which may be antiestrogenic and serve to limit the stimulation of growth of breast cancer xenografts (Komm, B. S., personal communication). If this were the case in women as well, a CEE/progestogen combination might be associated with less of an increase in breast cancer than E2/progestogen combinations. This differential effect was not observed in the large observational study reported by the European Prospective Investigation into Cancer and Nutrition group (36). The intriguing concept that CEE might have lesser effects on breast cancer in women requires further exploration
Although several possibilities exist as discussed above, it remains unresolved why CEE alone did not stimulate MCF-7 or LTED-MCF-7 tumor growth, even at doses 15-fold higher (i.e. 50 mg/kg) than needed to stimulate uterine development (i.e. 3 mg/kg), whereas E2 did markedly enhance tumor growth. CEE was clearly absorbed, because it increased ductal development in ovariectomized mice and uterine weight in tumor bearing animals. To our knowledge, this is the first time that the effects of CEE on human xenografts have been studied, and no previous data are available to explain this finding.
A critical question in assessing the ultimate importance of the current findings is whether the data are potentially applicable to women. Specifically, could a SERM block the proliferative effects of a substantial dose of exogenous estrogen in women such as the 0.45-mg dose of PremarinR in the CEE/BZA TSEC. Previous studies in women suggest the affirmative, because tamoxifen, a classical SERM, prevents breast cancer in premenopausal women (16). A 50% reduction of breast cancer incidence occurs even in the presence of plasma E2 levels of 500-2000 pg/ml, which result from interruption of hypothalamic-pituitary negative feedback (37). It should be noted that BZA blocked the breast tumor stimulatory effect of 80 pg/ml E2 in this study and could then be expected to inhibit the estrogenic effect of the less potent CEE compound.
In summary, our data suggest that BZA blocks the effect of CEE and E2 on multiple parameters and that a CEE/BZA TSEC regimen might then be expected to prevent breast cancer in women. On this basis, our data provide a rationale for a clinical trial in women to test this possibility.
Acknowledgments
Present address for Y.S.: Department of Molecular and Cellular Pharmacology, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China.
This work was supported by Pfizer, Inc.
Disclosure Summary: Y.S., J.-p.W., and W.Y. have nothing to disclose. R.J.S. is a consultant for Pfizer, Inc.
Footnotes
- AREG
- Amphiregulin
- BZA
- Bazedoxifene
- CEE
- conjugated equine estrogen
- cMyc
- cellular myelocytomatosis oncogene
- E2
- estradiol
- H&E
- hematoxylin and eosin
- Krt1-19
- keratin complex 1, acidic, gene 19
- MHT
- menopausal hormone therapy
- MPA
- medroxyprogesteron acetate
- PCNA
- proliferating cell nuclear antigen
- qPCR
- quantitative real time PCR
- ROS
- reactive oxygen species
- SERM
- selective estrogen receptor modulator
- TP53INP1
- tumor protein 53 induced nuclear protein 1
- TSEC
- tissue-selective estrogen complex
- TUNEL
- terminal deoxynucleotidyl transferase 2′-deoxyuridine, 5′-triphosphate nick end labeling
- WHI
- Women's Health Initiative
- WISP2
- WNT1 inducible signaling pathway protein 2.
References
- 1. Santen RJ , Allred DC , Ardoin SP , Archer DF , Boyd N , Braunstein GD , Burger HG , Colditz GA , Davis SR , Gambacciani M , Gower BA , Henderson VW , Jarjour WN , Karas RH , Kleerekoper M , Lobo RA , Manson JE , Marsden J , Martin KA , Martin L , Pinkerton JV , Rubinow DR , Teede H , Thiboutot DM , Utian WH. 2010. Postmenopausal hormone therapy: an Endocrine Society scientific statement. J Clin Endocrinol Metab 95:s1–s66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson GL , Chlebowski RT , Aragaki AK , Kuller LH , Manson JE , Gass M , Bluhm E , Connelly S , Hubbell FA , Lane D , Martin L , Ockene J , Rohan T , Schenken R , Wactawski-Wende J. 2012. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: extended follow-up of the Women's Health Initiative randomised placebo-controlled trial. Lancet Oncol 13:476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson GL , Limacher M , Assaf AR , Bassford T , Beresford SA , Black H , Bonds D , Brunner R , Brzyski R , Caan B , Chlebowski R , Curb D , Gass M , Hays J , Heiss G , Hendrix S , Howard BV , Hsia J , Hubbell A , Jackson R , Johnson KC , Judd H , Kotchen JM , Kuller L , LaCroix AZ , Lane D , Langer RD , Lasser N , Lewis CE , Manson J , Margolis K , Ockene J , O'Sullivan MJ , Phillips L , Prentice RL , Ritenbaugh C , Robbins J , Rossouw JE , Sarto G , Stefanick ML , Van Horn L , Wactawski-Wende J , Wallace R , Wassertheil-Smoller S, Women's Health Initiative Steering Committee 2004. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA 291:1701–1712 [DOI] [PubMed] [Google Scholar]
- 4. Rossouw JE , Anderson GL , Prentice RL , LaCroix AZ , Kooperberg C , Stefanick ML , Jackson RD , Beresford SA , Howard BV , Johnson KC , Kotchen JM , Ockene J, Writing Group for the Women's Health Initiative Investigators 2002. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA 288:321–333 [DOI] [PubMed] [Google Scholar]
- 5. Komm BS. 2008. A new approach to menopausal therapy: the tissue selective estrogen complex. Reprod Sci 15:984–992 [DOI] [PubMed] [Google Scholar]
- 6. Pickar JH , Yeh IT , Bachmann G , Speroff L. 2009. Endometrial effects of a tissue selective estrogen complex containing bazedoxifene/conjugated estrogens as a menopausal therapy. Fertil Steril 92:1018–1024 [DOI] [PubMed] [Google Scholar]
- 7. Archer DF. 2010. Tissue-selective estrogen complexes: a promising option for the comprehensive management of menopausal symptoms. Drugs Aging 27:533–544 [DOI] [PubMed] [Google Scholar]
- 8. Mitwally MF. 2008. Bazedoxifene: a selective estrogen-receptor modulator. Womens Health 4:319–326 [DOI] [PubMed] [Google Scholar]
- 9. Harvey JA , Holm MK , Ranganath R , Guse PA , Trott EA , Helzner E. 2009. The effects of bazedoxifene on mammographic breast density in postmenopausal women with osteoporosis. Menopause 16:1193–1196 [DOI] [PubMed] [Google Scholar]
- 10. Lobo RA , Pinkerton JV , Gass ML , Dorin MH , Ronkin S , Pickar JH , Constantine G. 2009. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic parameters and overall safety profile. Fertil Steril 92:1025–1038 [DOI] [PubMed] [Google Scholar]
- 11. Kagan R , Williams RS , Pan K , Mirkin S , Pickar JH. 2010. A randomized, placebo- and active-controlled trial of bazedoxifene/conjugated estrogens for treatment of moderate to severe vulvar/vaginal atrophy in postmenopausal women. Menopause 17:281–289 [DOI] [PubMed] [Google Scholar]
- 12. Lindsay R. 2011. Preventing osteoporosis with a tissue selective estrogen complex (TSEC) containing bazedoxifene/conjugated estrogens (BZA/CE). Osteoporos Int 22:447–451 [DOI] [PubMed] [Google Scholar]
- 13. Bachmann G , Bobula J , Mirkin S. 2010. Effects of bazedoxifene/conjugated estrogens on quality of life in postmenopausal women with symptoms of vulvar/vaginal atrophy. Climacteric 13:132–140 [DOI] [PubMed] [Google Scholar]
- 14. Pinkerton JV , Stovall DW. 2010. Bazedoxifene when paired with conjugated estrogens is a new paradigm for treatment of postmenopausal women. Expert Opin Investig Drugs 19:1613–1621 [DOI] [PubMed] [Google Scholar]
- 15. Pinkerton JV , Utian WH , Constantine GD , Olivier S , Pickar JH. 2009. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens: a randomized, controlled trial. Menopause 16:1116–1124 [DOI] [PubMed] [Google Scholar]
- 16. Cuzick J , Powles T , Veronesi U , Forbes J , Edwards R , Ashley S , Boyle P. 2003. Overview of the main outcomes in breast-cancer prevention trials. Lancet 361:296–300 [DOI] [PubMed] [Google Scholar]
- 17. Song Y , Santen RJ , Wang J-P , Yue W, Inhibitory effects of a bazedoxifene/conjugated equine estrogen combination on human breast cancer cells in vitro. Proceedings 94th Annual Meeting of The Endocrine Society, Houston, 2012 (Abstract) [Google Scholar]
- 18. Kim T , Park H , Yue W , Wang JP , Atkins KA , Zhang Z , Rogan EG , Cavalieri EL , Mohammad KS , Kim S , Santen RJ , Aiyar SE. 2011. Tetra-methoxystilbene modulates ductal growth of the developing murine mammary gland. Breast Cancer Res Treat 126:779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shim WS , Conaway M , Masamura S , Yue W , Wang JP , Kmar R , Santen RJ. 2000. Estradiol hypersensitivity and mitogen-activated protein kinase expression in long-term estrogen deprived human breast cancer cells in vivo. Endocrinology 141:396–405 [DOI] [PubMed] [Google Scholar]
- 20. Yue W , Wang JP , Hamilton CJ , Demers LM , Santen RJ. 1998. In situ aromatization enhances breast tumor estradiol levels and cellular proliferation. Cancer Res 58:927–932 [PubMed] [Google Scholar]
- 21. Peano BJ , Crabtree JS , Komm BS , Winneker RC , Harris HA. 2009. Effects of various selective estrogen receptor modulators with or without conjugated estrogens on mouse mammary gland. Endocrinology 150:1897–1903 [DOI] [PubMed] [Google Scholar]
- 22. Heitjan DF , Manni A , Santen RJ. 1993. Statistical analysis of in vivo tumor growth experiments. Cancer Res 53:6042–6050 [PubMed] [Google Scholar]
- 23. Berrodin TJ , Chang KC , Komm BS , Freedman LP , Nagpal S. 2009. Differential biochemical and cellular actions of Premarin estrogens: distinct pharmacology of bazedoxifene-conjugated estrogens combination. Mol Endocrinol 23:74–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhavnani BR. 2003. Estrogens and menopause: pharmacology of conjugated equine estrogens and their potential role in the prevention of neurodegenerative diseases such as Alzheimer's. J Steroid Biochem Mol Biol 85:473–482 [DOI] [PubMed] [Google Scholar]
- 25. Chang KC , Wang Y , Bodine PV , Nagpal S , Komm BS. 2010. Gene expression profiling studies of three SERMs and their conjugated estrogen combinations in human breast cancer cells: insights into the unique antagonistic effects of bazedoxifene on conjugated estrogens. J Steroid Biochem Mol Biol 118:117–124 [DOI] [PubMed] [Google Scholar]
- 26. Santen RJ , Yue W , Heitjan DF. 2012. Modeling of the growth kinetics of occult breast tumors: role in interpretation of studies of prevention and menopausal hormone therapy. Cancer Epidemiol Biomarkers Prev 21:1038–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bailey SL , Sigal BM , Plevritis SK. 2010. A simulation model investigating the impact of tumor volume doubling time and mammographic tumor detectability on screening outcomes in women aged 40–49 years. J Natl Cancer Inst 102:1263–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cano CE , Gommeaux J , Pietri S , Culcasi M , Garcia S , Seux M , Barelier S , Vasseur S , Spoto RP , Pébusque MJ , Dusetti NJ , Iovanna JL , Carrier A. 2009. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res 69:219–226 [DOI] [PubMed] [Google Scholar]
- 29. N'guessan P , Pouyet L , Gosset G , Hamlaoui S , Seillier M , Cano CE , Seux M , Stocker P , Culcasi M , Iovanna JL , Dusetti NJ , Pietri S , Carrier A. 2011. Absence of tumor suppressor tumor protein 53-induced nuclear protein 1 (TP53INP1) sensitizes mouse thymocytes and embryonic fibroblasts to redox-driven apoptosis. Antioxid Redox Signal 15:1639–1653 [DOI] [PubMed] [Google Scholar]
- 30. Chen Y , Liu X , Pisha E , Constantinou AI , Hua Y , Shen L , van Breemen RB , Elguindi EC , Blond SY , Zhang F , Bolton JL. 2000. A metabolite of equine estrogens, 4-hydroxyequilenin, induces DNA damage and apoptosis in breast cancer cell lines. Chem Res Toxicol 13:342–350 [DOI] [PubMed] [Google Scholar]
- 31. Li Y , Yao J , Chang M , Cuendet M , Bolton JL. 2004. Altered apoptotic response in MCF-10A cells treated with the equine estrogen metabolite, 4-hydroxyequilenin. Toxicol Lett 154:225–233 [DOI] [PubMed] [Google Scholar]
- 32. Wood CE , Clarkson TB , Chen H , Veenstra TD , Xu X , Scott L , Cline JM. 2008. Comparative effects of oral conjugated equine estrogens and micronized 17β-estradiol on breast proliferation: a retrospective analysis. Menopause 15:890–898 [DOI] [PubMed] [Google Scholar]
- 33. Beral V , Collaborators MWS. 2003. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 362:419–427 [DOI] [PubMed] [Google Scholar]
- 34. Chen WY , Manson JE , Hankinson SE , Rosner B , Holmes MD , Willett WC , Colditz GA. 2006. Unopposed estrogen therapy and the risk of invasive breast cancer. Arch Intern Med 166:1027–1032 [DOI] [PubMed] [Google Scholar]
- 35. Horwitz KB , Sartorius CA. 2008. Progestins in hormone replacement therapies reactivate cancer stem cells in women with preexisting breast cancers: a hypothesis. J Clin Endocrinol Metab 93:3295–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bakken K , Fournier A , Lund E , Waaseth M , Dumeaux V , Clavel-Chapelon F , Fabre A , Hémon B , Rinaldi S , Chajes V , Slimani N , Allen NE , Reeves GK , Bingham S , Khaw KT , Olsen A , Tjønneland A , Rodriguez L , Sánchez MJ , Etxezarreta PA , Ardanaz E , Tormo MJ , Peeters PH , van Gils CH , Steffen A , Schulz M , Chang-Claude J , Kaaks R , Tumino R , Gallo V , Norat T , Riboli E , Panico S , Masala G , González CA , Berrino F. 2011. Menopausal hormone therapy and breast cancer risk: impact of different treatments. The European Prospective Investigation into Cancer and Nutrition. Int J Cancer 128:144–156 [DOI] [PubMed] [Google Scholar]
- 37. Sawka CA , Pritchard KI , Paterson AH , Sutherland DJ , Thomson DB , Shelley WE , Myers RE , Mobbs BG , Malkin A , Meakin JW. 1986. Role and mechanism of action of tamoxifen in premenopausal women with metastatic breast carcinoma. Cancer Res 46:3152–3156 [PubMed] [Google Scholar]