

Abstract
Nonalcoholic fatty liver disease is common in developed countries and is associated with obesity, metabolic syndrome, and type 2 diabetes. T deficiency is a risk factor for developing these metabolic deficiencies, but its role in hepatic steatosis has not been well studied. We investigated the effects of T on the pathogenesis of hepatic steatosis in rats fed a high-fat diet (HFD). Adult male rats were randomly placed into four groups and treated for 15 weeks: intact rats on regular chow diet (RCD), intact rats on liquid HFD (I+HFD), castrated rats on HFD (C+HFD), and castrated rats with T replacement on HFD (C+HFD+T). Fat contributed 71% energy to the HFD but only 16% of energy to the RCD. Serum T level was undetectable in castrated rats, and T replacement led to 2-fold higher mean serum T levels than in intact rats. C+HFD rats gained less weight but had higher percentage body fat than C+HFD+T. Severe micro- and macrovesicular fat accumulated in hepatocytes with multiple inflammatory foci in the livers of C+HFD. I+HFD and C+HFD+T hepatocytes demonstrated only mild to moderate microvesicular steatosis. T replacement attenuated HFD-induced hepatocyte apoptosis in castrated rats. Serum glucose and insulin levels were not increased with HFD in any group. Immunoblots showed that insulin-regulated proteins were not changed in any group. This study demonstrates that T deficiency may contribute to the severity of hepatic steatosis and T may play a protective role in hepatic steatosis and nonalcoholic fatty liver disease development without insulin resistance.
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in the Western world and is strongly associated with obesity and metabolic syndrome (Met S) (1–6). Nonalcoholic steatohepatitis (NASH) is part of the spectrum of NAFLD that may precede hepatic cirrhosis and hepatic carcinoma (7). NAFLD is also associated with peripheral insulin resistance, type 2 diabetes mellitus, and dyslipidemia. The estimated prevalence of NAFLD in the United States is reaching 20%, with NASH present in 3% of the population (8). Worsening of the disease can lead to development of cirrhosis with estimated progression rates of up to 20% over 10 years in patients with NAFLD. The presence of NASH or fibrosis signifies a rate of 30%–60% of progression to cirrhosis within 7 years (9).
Obesity and Met S are associated with an increased risk of hypogonadism. A negative correlation exists between body mass index and total serum T (10–13). Approximately 20%–50% of men with type 2 diabetes and Met S have low T levels. Androgen deficiency is associated with increased visceral adiposity, which is reversed with T treatment (14, 15). Cross-sectional, population-based studies showed that men with low serum T levels have a higher risk of developing hepatic steatosis, and T replacement in hypogonadal men may decrease hepatic steatosis (16–19).
The studies in men are supported by animal studies showing that caponization of male chickens followed by T replacement depressed hepatic lipogenesis and lipid accumulation (20). Androgen actions on adipose tissue and hepatic steatosis have been studied in androgen receptor knockout (ARKO) mice. In some studies, despite demonstration of late-onset obesity with decreased energy expenditure, male ARKO mice exhibited increased adiponectin, normal insulin sensitivity, and increased lipolysis (21–23). In contrast, other studies reported that aging male ARKO mice developed obesity, insulin resistance, glucose intolerance, increased triglycerides in liver and muscle, and elevated leptin and low adiponectin levels (24, 25). When fed a high-fat diet, liver-specific eugonadal male ARKO mice, but not female ARKO mice, developed hepatic steatosis and insulin resistance with reduced peroxisome proliferator-activated receptor (PPAR)-α and hepatic lipid β-oxidation (26). The differences between these studies could be due to differences in genetic background or diet.
Experimental models of steatosis with different histopathological and pathophysiological features include those caused by diet, toxins and drugs, or genetic modifications that result in increased hepatic lipogenesis and uptake or decreased fatty acid oxidation or export (27). To better understand the relationship between low T and hepatic steatosis, we used an adult rodent model of androgen deficiency and diet-induced hepatic steatosis to evaluate the effect of T on NAFLD. We administered to rats an ad libitum high-fat (HFD; low carbohydrates) emulsion diet for 15 weeks. This model has previously been shown to result in abnormalities similar to patients with NAFLD and NASH (28, 29). The effect of T in modulating the development of diet-induced hepatic steatosis was studied in this HFD-induced rat NAFLD model.
Materials and Methods
Animals and experimental design
Young male Sprague Dawley rats (8 wk) were purchased from Charles River Laboratories, Inc and housed individually in temperature- and humidity-controlled rooms and exposed to 12-hour light and 12-hour dark cycles. An adaptation period of 2 weeks preceded the initiation of the experiment, during which time all animals had access to a regular chow diet (RCD) and water. The rats were randomly placed into four treatment groups with eight animals per group. Data from three rats were excluded for the following reasons: one rat from the control group fed RCD had bilateral testicular atrophy, and two rats from the group of animals that were castrated and T replaced and fed a HFD lost their SILASTIC implants (Dow Corning Corp) resulting in very low T levels. The final number of animals in each treatment group that were included in the analysis was as follows: intact rats fed RCD (I+RCD) (n = 6), intact rats fed HFD (I+HFD) (n = 8), castrated rats fed HFD (C+HFD) (n = 8), and castrated rats + T fed HFD (C+T+HFD) (n = 7). The RCD provided 16% energy from fat, 27% from protein, and 56% from carbohydrates (5008 Formulab Diet). The HFD derived 71% energy from fat, 18% energy from protein, and 11% from carbohydrates with 1 mL of HFD being equivalent to 1 kcal (Lieber-Decarli 71% FDC Diet; Dyets Inc). In the HFD, 60.9% fat was from corn oil, 35.7% from olive oil, and 3.4% from safflower oil (44.5% monounsaturated, 42.3% polyunsaturated, and 13.2% saturated fat). The daily HFD intake was recorded, and all animals were weighed twice each week.
After 15 weeks the rats (aged 25 wk) were killed by pentobarbital anesthesia after fasting overnight. The blood was collected from the aorta and centrifuged (4°C, 3200 rpm, 10 min), and serum was frozen at −20°C for future analysis. A small portion of the left liver lobe was fixed in 10% formalin for histological examination, and the remaining liver lobes were snap frozen in liquid nitrogen and stored at −80°C. Animal handling and experimentation were in accordance with the recommendation of the American Veterinary Medical Association and were approved by the Animal Care and Use Review Committee at the Los Angeles Biomedical Research Institute at Harbor-University of California, Los Angeles (Harbor-UCLA) Medical Center.
Castration and T replacement
T SILASTIC implants (Dow Corning) were prepared according to procedures described previously (30). Briefly, polydimethylsilozane tubing, 3 cm in length (outer diameter 3.18 mm; inner diameter 1.98; Dow Corning), was packed with T (Sigma) and sealed with SILASTIC medical adhesive A (Dow Corning). The T-filled implants were implanted subdermally at the back of each rat after castration under pentobarbital anesthesia.
Dual-energy X-ray absorptiometry (DEXA)
Body composition, including total fat and lean mass, of the animals was assessed by DEXA scan (Hologic 4500A, with small animal software) at the beginning and at the end of the experiment. Rats were anesthetized (ketamine 100 mg/kg and xylazine 20 mg/kg) and positioned supine on the scanning table. The DEXA software was optimized for adult rats weighing 200–750 g.
Histopathology
Osmium staining was performed by immersion of formalin-fixed liver tissue into a solution of osmium tetraoxide for 2 hours. Then the tissue was embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Osmium fixation was used to make the fat in the liver insoluble, preventing fat extraction during embedding. The black color of osmium provided better visualization and identification of intracellular lipid droplets in liver specimens (31). Using a modification of a previously published scoring system (32), sections were scored for micro- and macrovesicular fat accumulation, inflammation, necrosis, and fibrosis by an experienced hepatopathologist (S.W.F.), who was blinded to treatment allocation. The total pathology score was calculated by adding the scores of all the aforementioned parameters. Reticulin staining was used to identify presence of fibrosis. In addition, using a Nikon 400 microscope (Nikon Inc) equipped with a morphometric system and MetaVue imaging system (Universal Imaging Corp), five randomly chosen areas from each liver section were analyzed morphometrically for fat and the percentage of total pixels measured as fat indicated the percentage fat in the field by the same blinded observer (S.W.F.).
Serum and liver analyses
Serum levels of leptin, adiponectin, and insulin were measured with rat ELISA kits (ALPCO Diagnostics), and serum free fatty acids were measured using a nonesterified fatty acids (NEFA) test kit (Wako Diagnostics) by the Endocrine and Metabolic Research Laboratory at Los Angeles Biomedical Research Institute. The intraassay and interassay coefficients of variation were less than 6% and 8% for these hormones. The lower limits of quantification were 25 pg/mL for leptin, 0.38 ng/ml for adiponectin, and 0.15 ng/mL for insulin respectively. Serum T and estradiol were measured by liquid chromatography tandem mass spectrometry as previously described (33) by the same laboratory. The intra- and interassay coefficients of variation for T and estradiol were less than 5% and the lower limit of quantification was 0.02 ng/mL for T and 2 pg/mL for estradiol. Serum glucose, triglycerides, cholesterol, alanine aminotransferase, and aspartate aminotransferase were measured by an autoanalyzer by the Clinical Chemistry Laboratory of the Harbor-UCLA Medical Center using approved standardized methods. Homeostatic model assessment of insulin resistance (HOMA-IR) was calculated by using the classic homeostatic model assessment formula [HOMA-IR = serum glucose (milligrams per deciliter) × plasma insulin (microinternational units per milliliter)/405]. The total lipid in the liver was measured by a modified extraction method using methanol/chloroform (34). The extracts were pooled, air dried, and reconstituted with 0.5 mL of PBS with 0.1% Nonidet P-40. The total triglycerides of the extract were measured by the Clinical Chemistry Laboratory of Harbor-UCLA Medical Center using standard methods.
Tissue preparation and Western blotting analysis
Briefly, liver tissues were homogenized in lysis buffer (0.25 M sucrose, 50 mM HEPES, 10 mM NaCl, 10 mM EDTA, 2 mM dithiothreitol) supplemented with protease inhibitors (Complete Protease Inhibitors; Roche). Radioimmunoprecipitation assay buffer (Santa Cruz Biotechnology) was added after homogenization. Western blotting was performed as described previously (35). Proteins were denatured and separated by SDS-PAGE (Invitrogen). After transferring and blocking, the immunoblot polyvinyl difluoride membrane (Bio-Rad Laboratories) was probed using anticleaved polyADP ribose polymerase (PARP; Cell Signaling Technology); antifatty acid synthase (FAS; Santa Cruz Biotechnology); antiadipose triglyceride lipase (ATGL; Cayman Chemical Co); antisterol regulatory element-binding protein (SREBP)-1; anti-SREBP-2; antihormone-sensitive lipase (HSL); anti-PPARα (Abcam Co); anticarnitine palmitotyltransferase 1 (CPT1; Santa Cruz Biotechnology); and antiglyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (EMD Millipore. For detailed information about antibodies used in present study, see Table 1. After washing and incubating with antimouse (for SREBP-1 and GAPDH antibodies; Santa Cruz Biotechnology) or antirabbit (for the other antibodies; Amersham Biosciences) secondary antibody, membranes were exposed to Hyperfilm enhanced chemiluminescence (Denville Scientific Inc) with enhanced chemiluminescence solutions per the manufacturer's specifications (Amersham Biosciences). Band intensities were determined using Quantity One software (Bio-Rad Laboratories). No band was observed using anti-CPT1 antibodies despite repeated attempts.
Table 1.
Description of Antibodies Used for Immunoblots in the Present Study
| Protein Target | Antibody Name | Manufacturer Name | Catalog Number | Species Raised | Dilution Used |
|---|---|---|---|---|---|
| GAPDH | Anti-GAPDH | EMD Millipore | MAB374 | Mouse monoclonal Ab | 1:5000 |
| FAS | Anti-FAS | Santa Cruz Biotechnology, Inc | Sc-20140 | Rabbit polyclonal Ab | 1:500 |
| SREBP-1 | Anti-SREBP-1 | Abcam PLC | Ab3259 | Mouse monoclonal Ab | 1:200 |
| SREBP-2 | Anti-SREBP-2 | Abcam PLC | Ab30682 | Rabbit polyclonal Ab | 1:500 |
| HSL | Anti-HSL | Abcam PLC | Ab45422 | Rabbit polyclonal Ab | 1:500 |
| ATGL | Anti-ATGL | Cayman Chemical | 10006409 | Rabbit polyclonal Ab | 1:200 |
| PPARα | Anti-PPARα | Abcam PLC | Ab24509 | Rabbit polyclonal Ab | 1:500 |
| CPT-1 | Anti-CPT-1 (N-17) | Santa Cruz Biotechnology, Inc | Sc-20514 | Goat polyclonal | 1:200 to 1:1000 |
| CPT-1 | Anti-CPT-1 (H-95) | Santa Cruz Biotechnology, Inc | Sc-20669 | Rabbit polyclonal | 1:200 to 1:1000 |
| PARP | PARP | Cell Signaling Technology, Inc | 9542 | Rabbit polyclonal Ab | 1:500 |
Abbreviation: Ab, antibody.
Assessment of apoptosis
Terminal deoxynucleotidyl transferase-mediated deoxyuridine 5-triphosphate nick end labeling was performed in formalin-fixed, paraffin-embedded liver sections (5 μm) by using an ApopTag-peroxidase kit (Chemicon International) (36). The number of the apoptotic hepatocytes with distinct staining was assessed using an Olympus BH-2 microscope (Olympus) with a ×100 oil immersion objective. For each rat at least 50 mm2 were counted. Cleaved PARP was used as an additional marker of apoptosis by Western blotting analysis.
Statistical analysis
Statistical analyses were performed using the StatPlus 2007 Program (AnalystSoft) and SAS version 9.3 (SAS Institute). Normal data are presented as mean ± SEM and analyzed by one-way ANOVA and post hoc tests by Tukey-Kramer correction for multiple comparisons. Liver pathology scores, including macrovesicular fat score, microvesicular fat score, inflammation score, and total pathology score, were not normally distributed and data are individually presented. These data were assessed by a Kruskal-Wallis test and post hoc tests by Hochberg correction for multiple comparisons. Post hoc tests were confined a priori to three comparisons (I+RCD vs I+HFD, I+HFD vs C+HFD, and C+HFD vs C+HFD+T). Statistical significance was construed at P < .05.
Results
Daily intake, body weight, and body composition
Castrated rats fed a HFD gained less weight than rats in the other groups fed the same diet (Figure 1A, overall group comparison P < .001). With T replacement, the body weights of castrated rats fed HFD were similar to the body weights of intact rats fed RCD. The greatest body weight gain was seen in the intact rats fed HFD (Figure 1A). The castrated rats fed HFD consumed less liquid diet than intact or castrated and T replaced groups on HFD with significant differences (Figure 1B, overall group comparison P < .001). The daily intake to weight ratio demonstrated that the weight gain of rats on HFD was proportional to the daily food intake (Figure 1C). The DEXA results demonstrated that C+HFD rats had a higher percentage body fat than C+HFD+T rats (Figure 2B, P = .026), despite gaining less body weight than other groups (Figure 2A). The I+HFD rats had the lowest lean mass (percentage) compared with other groups (Figure 2C, overall group comparison P < .001). T replacement led to higher lean mass (percentage) compared with the C+HFD groups. (Figure 2C, P = .028).
Figure 1.
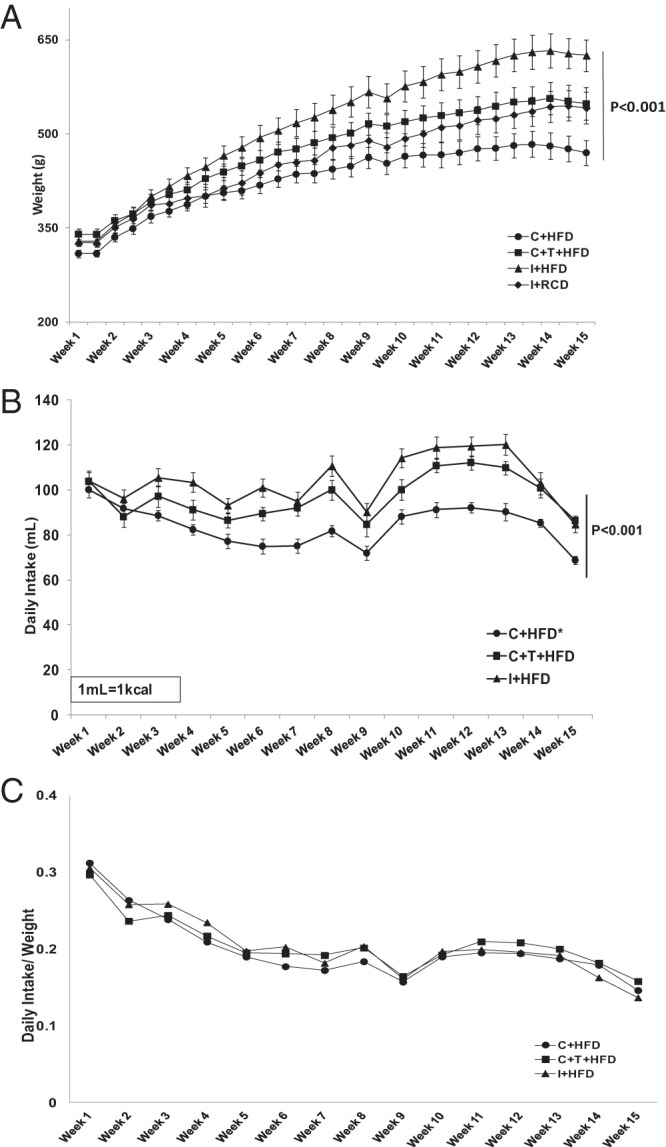
Body weight and daily food intake. A, Body weight was measured twice a week (overall group comparison P < .001). B, Daily intake of liquid HFD in the three groups of rats fed a HFD (overall group comparison P < .05). C, Daily intake to weight ratio demonstrated that the weight gain of each group was proportional to the daily intake. In this and Figures 2–5, the numbers of rats in each group were: I+RCD, n = 6; I+HFD, n = 8; C+HFD, n = 8; and C+HFD+T, n = 7. I+RCD, Intact rat fed RCD; I+HFD, intact rat fed HFD; C+HFD, castrated rat fed HFD; and C+HFD+T, castrated rat fed HFD with T replacement.
Figure 2.
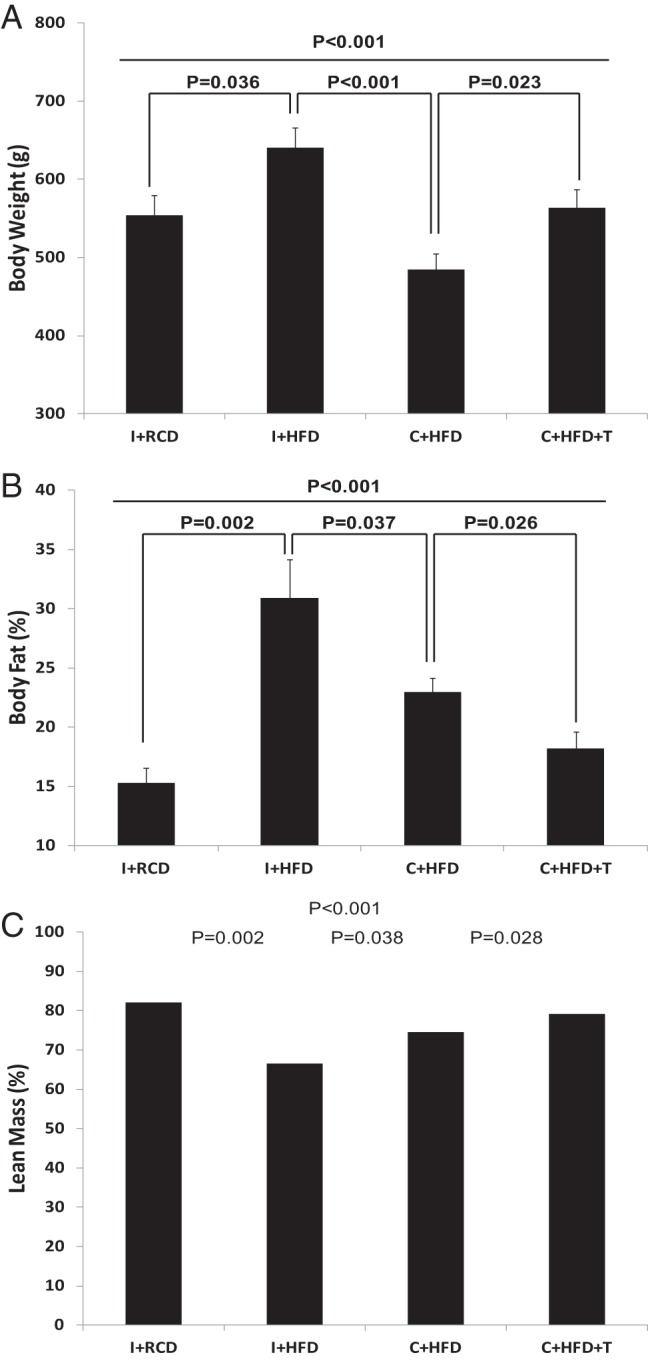
Body composition analyses at the end of the feeding experiment. A, Body weight was different among the groups (overall group comparison P < .001). B, Percentage body fat was different among the groups (overall group comparison P < .001). C, Percentage lean mass was different among the groups (overall group comparison P < .001).
Serum T levels
Serum T levels were 1.49 ± 0.26 ng/mL and 1.40 ± 0.23 ng/mL for intact rats fed HFD or RCD, respectively. As expected, castrated rats had undetectable serum T. Castrated rats with T pellets implanted had significantly higher levels (2.49 ± 0.24 ng/mL, P < .01, figure not shown) than intact rats. Serum estradiol levels were below the detectable range in all animals.
Serum liver enzymes, histopathology, and lipid content of liver tissue
Serum alanine aminotransferase was higher in the C+HFD group (60.6 ± 6.4 IU/L) compared with the I+RCD group (40.7 ± 4.4 IU/L) and the other two groups (C+HFD+T = 44.4 ± 4.5 IU/L and I+HFD = 47.4 ± 3.8 IU/L), but the differences were not significant among groups (P > .05). The aspartate aminotransferase levels were highest in the C+HFD, but the difference did not reach statistical significance because of large variability (C+HFD = 168 ± 48 IU/L, C+HFD+T = 132 ± 31 IU/L, I+HFD = 128 ± 22 IU/L, and I+RCD = 112 ± 32 IU/L; P > .05).
Liver histopathology revealed severe micro- and macro-vesicular accumulation of fat in the hepatocytes of the C+HFD rats (Figure 3A, e and f) as compared with the normal liver architecture of I-RCD (Figure 3A, a and b). The black color staining for fat with osmium confirmed that all rats on HFD had microvesicular fat but on C+HFD had macrovesicular fat (Figure 3B, a–d). Multiple inflammatory foci were seen in the livers of C+HFD rats (Figure 3A, e and f). However, the hepatocytes of the C+HFD+T and I+HFD rats demonstrated only mild to moderate microvesicular steatosis (Figure 3A, c and d, and g and h). No significant fibrosis, evaluated by reticulin staining, or necrosis was found during histological examination in any of the treatment groups. Figure 3C shows the microvesicular and macrovesicular inflammation and total pathology score in each rat. The C+HFD group had the highest macrovesicular, inflammation, and total pathology score compared with I+RCD and I+HFD groups (Figure 3C, a, c, and d). Macrovesicular fat (P = .002), inflammation (P = .003), and total pathology scores (P = .020) in the C+HFD group were ameliorated significantly by T replacement in C+HFD+T group (Figure 3C, a, c, and d). Morphometry analysis also showed the percentage fat was higher in all groups fed HFD (I+HFD: 3.209% ± 0.622%; C+HFD: 7.087% ± 2.350%; C+HFD+T: 2.425% ± 0.628%) compared with intact rats fed RCD (0.149% ± 0.071%; P < .05). This was confirmed by liver triglyceride content, which was higher in all groups fed HFD (I+HFD: 0.071 ± 0.015 mg/mg tissue; C+HFD: 0.073 ± 0.017 mg/mg tissue; C+HFD+T: 0.088 ± 0.012 mg/mg tissue) compared with intact rats fed RCD (0.028 ± 0.018 mg/mg tissue; P < .05).
Figure 3.

Liver histology and pathology scores analysis. A, Liver histology in low (×200, a, c, e, and g) and high (×400, b, d, f, and h) magnifications stained with hematoxylin and eosin. The I+RCD group showed normal hepatocytes (a and b). The I+HFD group developed microvesicular steatosis and minimal macrovesicular fatty accumulation (c and d). The C+HFD group developed significantly more macrovesicular steatosis and inflammation (e and f) with multiple foci of inflammation were seen at low magnification (e, black arrows); one focus of inflammation was shown in the center of the high-magnification picture (f, black arrow). With T replacement, the C+T+HFD group (g) had less microvesicular steatosis and minimal macrovesicular fatty accumulation and fewer inflammation foci compared with the C+HFD group. B, Osmium staining of fat in hepatocytes (×400). Osmium staining demonstrated minimal lipid deposition in the I+RCD group, microvesicular fat accumulation in the I+HFD and C+HFD+T groups, and massive micro- and macrovesicular steatosis in the C+HFD group. C, Liver pathology scores: a, C+HFD group (open diamonds) showed the highest score of macrovesicular fat, whereas T replacement decreased the score (P = .002); b, the I+HFD (open triangles), C+HFD (open diamonds), and C+HFD+T (open circles) groups had higher microvesicular fat score than the I+RCD group (open squares) (overall group comparison P < .001, but no significant difference between three groups); c, C+HFD showed the highest inflammation score, whereas T replacement decreased the score (P = .003); d, highest total pathology score was found in the C+HFD group, which decreased with T replacement (P = .020). The horizontal line across symbols represents the median and statistical significance was calculated using the Kruskal-Wallis test.
Hepatocyte apoptosis in liver
Apoptotic cells were rare in liver sections in the I+RCD group (Figure 4, A and B, black arrows). Intact animals fed HFD showed more liver cell apoptosis (expressed as numbers per square millimeter of liver tissue) than intact animals fed RCD (Figure 4B, P = .003). When fed a HFD, castrated rats had significantly higher liver cell apoptosis compared with intact rats (Figure 4B, P = .003). Hepatocyte apoptosis was reduced by T replacement in castrated rats fed a HFD (Figure 4B, P = .048). The results of the hepatocyte apoptosis were confirmed by changes in levels of cleaved PARP (Figure 4, C and D). In intact animals, a HFD led to higher cleaved PARP levels compared with the RCD group (Figure 4C, P < .05). Among castrated animals, T replacement (in C+HFD+T) resulted in reduction in cleaved PARP levels compared with untreated castrated (C+HFD) animals (Figure 4D, P < .05).
Figure 4.

Hepatocyte apoptosis. A, By terminal deoxynucleotidyl transferase-mediated deoxyuridine 5-triphosphate nick end labeling staining, more apoptotic hepatocytes (black arrows) were detected in the C+HFD group compared with other groups. B, Quantification of the apoptotic cells in liver showed that rats fed a HFD had increased hepatocyte apoptosis, which was further elevated by castration (P = .003) and reduced by T replacement (P = .048). C and D, By Western blotting, HFD led to higher levels of cleaved PARP compared with the RCD group in intact rats (Figure 4C, P < .05), and T replacement attenuated the increased PARP cleavage in hepatocytes induced by a HFD in the castrated group (panel D, P < .05). GAPDH was used as loading control.
Serum metabolic markers and adipokines
Serum glucose and insulin were not elevated and showed no significant difference among the groups (Figure 5, A and B, overall group comparisons P > .05). Calculated from serum glucose and insulin levels, HOMA-IR results also showed no significant difference among the groups (Figure 5H, overall group comparisons P > .05). Leptin levels were highest in I+HFD (4.6 ± 0.9 ng/mL) followed by C+HFD groups (2.4 ± 0.3 ng/mL) and with lower levels of leptin in C+HFD+T (1.8 ± 0.4 ng/mL) and I+RCD (1.1 ± 0.2 ng/mL) (Figure 5C, overall group comparisons P = .001). Adiponectin was significantly higher in C+HFD group compared with the C+HFD+T group (Figure 5D, P = .019). Serum cholesterol was significantly higher in C+HFD rats (38.8 ± 3.28 mg/dL) compared with C+HFD+T animals (29.00 ± 5.00 mg/dL) (Figure 5E, P = .001). There were no significant differences in serum triglycerides or NEFA among the groups (Figure 5, F and G, overall group comparisons P > .05).
Figure 5.

Fasting serum glucose (A), insulin (B), leptin (C), adiponectin (D), cholesterol (E), triglyceride (F), and NEFA (G) levels were measured at the end of the experiment. No significant differences in overall group comparisons were found for fasting glucose and insulin (A and B). Leptin levels were highest in I+HFD (C; overall group comparison P = .001). Adiponectin level was higher in the C+HFD compared with the C+HFD+T group (P = .019) (D). Serum cholesterol showed significant group differences (E; P = .025), and T replacement in the C+HFD+T group led to lower cholesterol levels compared with the C+HFD group (P = .001). No significant differences were observed among the groups in the fasting triglyceride and NEFA levels (F and G).
Protein expression levels of HSL, ATGL, FAS, SREBP-1, SREBP-2, and PPARα in liver tissues
In intact rats, HFD caused an approximately 25% decrease in FAS (P < .05) and increases in SREBP1 (∼25%), SREBP-2 (∼33%), and PPARα (∼15%) (P < .05 for all three proteins) but not in ATGL and HSL level in the liver (Figure 6, A–F, and Figure 7, A–F). After castration, there were no significant differences in liver FAS, HSL, ATGL, SREBP-1, SREBP-2, or PPARα protein levels in the C+HFD animals with or without T supplementation (Figure 6, G–L, and Figure 7, G–L).
Figure 6.

Western blot analyses of PPARα, FAS, SREBP-1, SREBP-2, ATGL, and HSL in rat livers (see quantification in Figure 7). GAPDH was used as loading control. In this and Figure 7, the numbers of rats in each group were: I+RCD, n = 6; I+HFD, n = 8; C+HFD, n = 7; and C+HFD+T, n = 7.
Figure 7.

Protein levels of PPARα, FAS, SREBP-1, SREBP-2, ATGL, and HSL in rat livers. A HFD decreased FAS (Figure 6A, P < .05), increased PPARα (P < .05), SREBP1 (Figure 6C, P < .05), and SREBP-2 (P < .05) but did not change the ATGL and HSL level in rat liver tissues compared with a regular diet (Figure 6, B and D). T replacement in castrated rats did not change FAS, PPARα, SREBP-1, SREBP-2, ATGL, or HSL expression in rat liver compared with castrated rats fed a HFD (Figure 6, E–H). GAPDH was used as loading control.
Discussion
Our study demonstrated that a high-fat, low-carbohydrate liquid diet induced NAFLD in androgen-deficient adult male rats. NAFLD was characterized by mildly elevated liver alanine aminotransferases and high liver histopathology scores with increased macrovesicular fat accumulation, evidence of inflammation, increased hepatocyte apoptosis, and PARP cleavage. These changes were accompanied by increased percentage body fat in the castrated rats fed a HFD despite gaining the least amount of weight when compared with the other treatment groups. This increase in fat mass is consistent with previous studies examining ARKO mice fed RCD, which demonstrated significantly lower body weight gain in the first 20 weeks of the experiment, with a subsequent and progressive increase in weight gain as the animals aged between 20 and 40 weeks of the experiment (24). Another study of liver-specific ARKO mice showed significant differences in weight gain in ARKO compared with control mice only after 14–16 weeks of the experiment when animals were fed RCD for 8 weeks followed by 8 weeks of a HFD (25). Food consumption has been reported to decrease by castration and increased by T treatment both in immature and adult rats (37, 38); the mechanisms of action is not clear but might be related to low T levels that result in suppression of appetite and decreased energy. There are many candidate factor(s) that could mediate the effects of low T levels to appetite and food intake, including ghrelin, cholecystokinin, glucagon, insulin, and leptin (39). In castrated model, the histopathology of liver steatosis is similar to those reported in ARKO and liver-specific ARKO (21–23). Importantly, pathological changes in the liver and hepatocyte apoptosis were significantly improved with T replacement. T replacement also lowered percentage body fat in castrated rats to the percentage found in intact rats fed RCD. The results of our study support the hypothesis that T protects against HFD-induced fat accumulation in the liver and the development of NAFLD and provides evidence that androgen deficiency could be a risk factor for increasing the severity of hepatic steatosis and inflammation.
The molecular mechanisms by which T deficiency is involved in the pathogenesis of NAFLD are poorly understood. Several pathways have been proposed: increased adipose tissue lipolysis, increased hepatic lipogenesis, decreased hepatic fatty acid β-oxidation, and decreased export of lipids from the liver (40, 41). Obesity, hyperglycemia, and insulin resistance often precede increased lipogenesis, hepatic steatosis, and NAFLD. In the current study, both intact and castrated rats fed a high-fat but not a high-carbohydrate diet did not develop hyperglycemia, hyperinsulinemia, or insulin resistance. These results are different from other rodent studies in which animals are fed high-fat and high-carbohydrate diets. In these models the high-carbohydrate diet induces insulin resistance and drives the target genes and proteins to accumulate fat in the liver (41). Our rat steatohepatitis model provided the evidence that NAFLD may develop in the absence of insulin resistance and consistent with recent findings in genetic mouse models and clinical studies demonstrating the ability to dissociate hepatic steatosis from insulin resistance and diabetes (42).
Levels of leptin reflecting body fat of each group showed higher levels in the groups fed a HFD irrespective of their androgen status. Moreover, adiponectin, which is a marker of insulin sensitivity, was higher in the castrated rat fed a HFD compared with T-treated castrated rats. This finding is consistent with the lack of insulin resistance, which is usually associated with low levels of adiponectin. Therefore, the development of hepatic steatosis in our animal model is independent of hyperglycemia or hyperinsulinemia and supports that fat accumulation due to T deficiency may be an independent causal factor for NAFLD. In ARKO mice fed a RCD, the lack of hyperphagia, lower spontaneous activity, and decreased overall oxygen consumption were attributed to decreased expression of thermogenic uncoupling protein 1. The ARKO mice had elevated levels of adiponectin and did not develop insulin resistance despite their obese phenotype (22). Several other genetically modulated animal models, not androgen or androgen receptor specific, described significant hepatic steatosis without development of insulin resistance. These studies include transgenic mice overexpressing diacylglycerol acyltransferase 2 in liver (Liv-dag2 mice) fed a standard diet and mice deficient in long-chain fatty acid elongase (Elovl6/− mice) when fed a high-fat and high-carbohydrate diet (41). Our model of adult androgen deficiency is different from other studies because it uses an intact androgen receptor system with ligand deficiency, which relates to clinical male hypogonadism due to a variety of causes.
Increased adipose tissue lipolysis releases NEFAs into the serum and provides NEFA flux to the liver, contributing to the development of NAFLD. Our study did not show significant differences in serum NEFA levels among groups. The end product of adipose tissue lipolysis NEFA was not elevated in the C+HFD rats and was therefore unlikely to be the causative factor of the fatty liver pathology observed in the androgen-deficient rats.
Western blotting was performed to examine whether protein expression of key transcription factors and enzymes in lipid metabolism contributed to NAFLD. HSL (8) and ATGL (37) induce lipid turnover through lipase activity by hydrolyzing triglycerides in liver and adipose tissues, respectively (43), whereas SREBP-1 (44) and FAS (45) have been shown to be important in liver lipogenesis. SREBP-1 is a lipogenic transcription factor, whereas FAS catalyzes fatty acid synthesis (36). These proteins are regulated by insulin. In our NAFLD rat model, HFD caused very small decreases FAS with small increases in SREBP-1 and -2 and PPAR compared with RCD, indicating that this high-fat, low-carbohydrate diet did not induce changes consistent with a marked increase in fat synthesis. FAS, ATGL, HSL, and SREBP-1 showed no change after castration with or without T treatment in rats fed the HFD. These negative results also suggest that insulin resistance may not be involved in the development of NAFLD in T-deficient rats. We also tried to assess CPT1, which regulated β-oxidation of fatty acid, but immunoblots performed using two antibodies were unable to detect CPT1 protein expression. PPARα mediates multiple aspects of lipid metabolism through targets that include genes related to lipid uptake, trafficking, lipogenesis, and lipid oxidation (46). In the present experiment, no differences in PPARα protein levels were detected in in castrated rats with or without T treatment fed a HFD. Stearoyl-CoA desaturase 1 (SCD-1) is also a key enzyme in fatty acid metabolism especially lipogenesis via regulating the synthesis of unsaturated fatty acids (47). It has been shown that SCD-1 synthesis in rat liver is positively regulated by saturated fatty acids (48) and negatively by unsaturated fatty acids (49). Unfortunately, we were not able to detect SCD-1 changes at the protein level because of nonspecificity of the antibodies available. SREBP2 is a regulator of cholesterol biosynthesis (50). Our data showed that T replacement did not change the protein levels of SREBP-2 in castrated rats fed the HFD, and therefore, perturbations in cholesterol synthesis may not be involved in NAFLD in this model. Although no changes were detected in levels of these six proteins related to lipid metabolism among T-replaced and untreated castrated animals, the modulation effect of T on the enzyme activities such as stimulating phosphorylation cannot be excluded. More studies are needed to explore the molecular pathway for the protective effect of T against HFD-induced hepatic steatosis.
The possible mechanism of action of anabolic steroids on liver may involve hepatic fatty acid oxidation by increasing ketogenesis and activity of hepatic lipase and phospholipase A (51, 52). Evaluation of short-term administration of oxandrolone (17β-hydroxy-17α-methyl-2-oxa-5α-androstan-3-one, an anabolic steroid) to adult men showed marked increase in hepatic ketogenesis demonstrated by increases of 3-hydroxybutyrate levels. This finding was consistent with influx of fatty acids into the liver from increased activity of hepatic lipase on lipoprotein lipolysis (51). Further studies are needed to evaluate the extent of T involvement in liver lipogenesis or β-oxidation to better understand the molecular basis of androgen amelioration of hepatic steatosis.
Prior studies showed that hepatic steatosis occurs both in the ARKO and liver-specific ARKO mice, suggesting that lack of androgens action via the androgen receptor may be the cause of the accumulation of fat in the viscera and in the liver (21–26). T is aromatized to estradiol in the body. Thus, treatment with T may elevate serum estradiol and act through estrogen receptors. Antiestrogens such as tamoxifen have been shown to induce the development of fatty liver and steatohepatitis in rodents and men (53–55). In our study, using a sensitive liquid chromatography and tandem mass spectrometry study, we were not able to detect estradiol levels in rat sera. The role of estrogens in the prevention of the development of NAFLD was not the primary goal of our study and will be investigated in the future by using the concomitant treatment of T with aromatase inhibitors to dissect androgen receptor and estrogen receptor roles in NAFLD. Recently Zhang et al (56) studied whether estradiol and/or a nonaromatizable androgen, dihydrotestosterone (DHT), can mitigate hepatic steatohepatitis in castrated male rats fed a HFD. Their study showed that estradiol decreased lipogenesis by decreasing FAS and the phosphorylation of acetyl coenzyme A. DHT decreased cholesterol synthesis and increased β-oxidation of fatty acids by increasing gene expression of the key enzyme carnitine palmitotyltransferase 1. We could not detect the protein expression of this enzyme in liver using available antibodies. Decreased β-oxidation of fatty acids is usually a minor contributor in the development of hepatic steatosis (41). It should be noted that the composition of the diet and the duration of treatment is different. In the study by Zhang et al (56), rats were treated for about 11 weeks using a high-fat (36.9% of calories) and high-carbohydrate (42.7% of calories) diet, which probably led to insulin resistance, whereas in our study we treated the rats for a longer period with a high-fat (75% of calories) but low-carbohydrate (11% of calories) diet without inducing insulin resistance. Both studies used castration model in which the serum androgen and estrogens levels should be very low. In our study serum T and estradiol were not detected in the rats after castration, but in the study reported by Zhang et al (56), both estradiol and DHT were measurable, suggesting that these hormones were derived from another source, which was unlikely or that the assays used to measure these hormones were not specific. It should be recognized that in both studies the levels of androgen achieved with replacement were at least 2-fold higher than control rats, and future experiments are needed to clarify the dose response of the protective effects of T in NAFLD.
In summary, our study demonstrates that androgen deficiency exacerbated HFD-induced hepatic steatosis and apoptosis and that T replacement decreases the overall body fat accumulation and ameliorates the pathophysiological features of NAFLD and liver cell apoptosis. Given that insulin resistance was not observed in our experiments, this animal model of androgen deficiency and diet-induced NAFLD represents a useful tool for further analysis of mechanisms by which androgen may provide a protective action against hepatic steatosis and NAFLD.
Acknowledgments
We thank Vince Atienza and Andrew Leung for their technical expertise and support for the study.
This work was supported by Grant MO1 RR00425 from the General Clinical Research Center (to L.N.); Grant 1UL1TR000124 (to the UCLA Clinical and Translational Science Institute) at Los Angeles Biomedical Research Institute (LA BioMed) at Harbor-UCLA Medical Center; and the Endocrine, Metabolism, and Nutrition Training Grant (T32 DK007571) and the Summer High School Student Program at LA BioMed.
The results from this work were presented in part at the 92nd Annual meeting of The Endocrine Society in San Diego, California, June 2010.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by Grant MO1 RR00425 from the General Clinical Research Center (to L.N.); Grant 1UL1TR000124 (to the UCLA Clinical and Translational Science Institute) at Los Angeles Biomedical Research Institute (LA BioMed) at Harbor-UCLA Medical Center; and the Endocrine, Metabolism, and Nutrition Training Grant (T32 DK007571) and the Summer High School Student Program at LA BioMed.
Footnotes
- ARKO
- androgen receptor knockout
- ATGL
- adipose triglyceride lipase
- CPT1
- carnitine palmitotyltransferase 1
- DEXA
- dual-energy X-ray absorptiometry
- DHT
- dihydrotestosterone
- FAS
- fatty acid synthase
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- HFD
- high-fat diet
- HOMA-IR
- homeostatic model assessment of insulin resistance
- HSL
- hormone-sensitive lipase
- Met S
- metabolic syndrome
- NAFLD
- nonalcoholic fatty liver disease
- NASH
- nonalcoholic steatohepatitis
- PARP
- polyADP ribose polymerase
- PPAR
- peroxisome proliferator-activated receptor
- RCD
- regular chow diet
- SCD-1
- stearoyl-CoA desaturase 1
- SREBP
- sterol regulatory element-binding protein.
References
- 1. Bellentani S, Marino M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Ann Hepatol. 2009;8(suppl 1):S4–S8. [PubMed] [Google Scholar]
- 2. Adams LA, Waters OR, Knuiman MW, Elliott RR, Olynyk JK. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up study. Am J Gastroenterol. 2009;104(4):861–867. [DOI] [PubMed] [Google Scholar]
- 3. Almeda-Valdes P, Cuevas-Ramos D, Aguilar-Salinas CA. Metabolic syndrome and non-alcoholic fatty liver disease. Ann Hepatol. 2009;8(suppl 1):S18–S24. [PubMed] [Google Scholar]
- 4. Boppidi H, Daram SR. Nonalcoholic fatty liver disease: hepatic manifestation of obesity and the metabolic syndrome. Postgrad Med. 2008;120(2):E01–E07. [DOI] [PubMed] [Google Scholar]
- 5. Khashab MA, Liangpunsakul S, Chalasani N. Nonalcoholic fatty liver disease as a component of the metabolic syndrome. Curr Gastroenterol Rep. 2008;10(1):73–80. [DOI] [PubMed] [Google Scholar]
- 6. van der Poorten D, Milner KL, Hui J, et al. Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology. 2008;48(2):449–457. [DOI] [PubMed] [Google Scholar]
- 7. Adams LA, Lymp JF, Sanderson SO, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129(1):113–121. [DOI] [PubMed] [Google Scholar]
- 8. Botion LM, Green A. Long-term regulation of lipolysis and hormone-sensitive lipase by insulin and glucose. Diabetes. 1999;48(9):1691–1697. [DOI] [PubMed] [Google Scholar]
- 9. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2 suppl 1):S99–S112. [DOI] [PubMed] [Google Scholar]
- 10. Allan CA, Strauss BJ, Burger HG, Forbes EA, McLachlan RI. The association between obesity and the diagnosis of androgen deficiency in symptomatic ageing men. Med J Aust. 2006;185(8):424–427. [DOI] [PubMed] [Google Scholar]
- 11. Jensen TK, Andersson AM, Jorgensen N, et al. Body mass index in relation to semen quality and reproductive hormones among 1,558 Danish men. Ferti Steril. 2004;82(4):863–870. [DOI] [PubMed] [Google Scholar]
- 12. Allen NE, Appleby PN, Davey GK, Key TJ. Lifestyle and nutritional determinants of bioavailable androgens and related hormones in British men. Cancer Causes Control. 2002;13(4):353–363. [DOI] [PubMed] [Google Scholar]
- 13. Allan CA, Strauss BJ, McLachlan RI. Body composition, metabolic syndrome and testosterone in ageing men. Int J Impot Res. 2007;19(5):448–457. [DOI] [PubMed] [Google Scholar]
- 14. Marin P, Arver S. Androgens and abdominal obesity. Baillieres Clin Endocrinol Metab. 1998;12(3):441–451. [DOI] [PubMed] [Google Scholar]
- 15. Allan CA, Strauss BJ, Burger HG, Forbes EA, McLachlan RI. Testosterone therapy prevents gain in visceral adipose tissue and loss of skeletal muscle in nonobese aging men. J Clin Endocrinol Metab. 2008;93(1):139–146. [DOI] [PubMed] [Google Scholar]
- 16. Hoyos CM, Yee BJ, Phillips CL, Machan EA, Grunstein RR, Liu PY. Body compositional and cardiometabolic effects of testosterone therapy in obese men with severe obstructive sleep apnoea: a randomised placebo-controlled trial. Eur J Endocrinol. 2012;167(4):531–541. [DOI] [PubMed] [Google Scholar]
- 17. Kim S, Kwon H, Park JH, et al. A low level of serum total testosterone is independently associated with nonalcoholic fatty liver disease. BMC Gastroenterol. 2012;12:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adams LA, Feldstein A, Lindor KD, Angulo P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology. 2004;39(4):909–914. [DOI] [PubMed] [Google Scholar]
- 19. Volzke H, Aumann N, Krebs A, et al. Hepatic steatosis is associated with low serum testosterone and high serum DHEAS levels in men. Int J Androl. 2010;33(1):45–53. [DOI] [PubMed] [Google Scholar]
- 20. Chen KL, Chi WT, Chu C, Chen RS, Chiou PW. Effect of caponization and testosterone implantation on hepatic lipids and lipogenic enzymes in male chickens. Poult Sci. 2007;86(8):1754–1759. [DOI] [PubMed] [Google Scholar]
- 21. Yanase T, Fan W, Kyoya K, et al. Androgens and metabolic syndrome: lessons from androgen receptor knock out (ARKO) mice. J Steroid Biochem Mol Biol. 2008;109(3–5):254–257. [DOI] [PubMed] [Google Scholar]
- 22. Fan W, Yanase T, Nomura M, et al. Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion. Diabetes. 2005;54(4):1000–1008. [DOI] [PubMed] [Google Scholar]
- 23. Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun. 2003;300(1):167–171. [DOI] [PubMed] [Google Scholar]
- 24. Lin HY, Xu Q, Yeh S, Wang RS, Sparks JD, Chang C. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes. 2005;54(6):1717–1725. [DOI] [PubMed] [Google Scholar]
- 25. Lin HY, Yu IC, Wang RS, et al. Increased hepatic steatosis and insulin resistance in mice lacking hepatic androgen receptor. Hepatology. 2008;47(6):1924–1935. [DOI] [PubMed] [Google Scholar]
- 26. Yu IC, Lin HY, Liu NC, et al. Hyperleptinemia without obesity in male mice lacking androgen receptor in adipose tissue. Endocrinology. 2008;149(5):2361–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. London RM, George J. Pathogenesis of NASH: animal models. Clin Liver Dis. 2007;11(1):55–74, viii. [DOI] [PubMed] [Google Scholar]
- 28. Lieber CS, Leo MA, Mak KM, et al. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79(3):502–509. [DOI] [PubMed] [Google Scholar]
- 29. Wang Y, Ausman LM, Russell RM, Greenberg AS, Wang XD. Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J Nutr. 2008;138(10):1866–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lue Y, Hikim AP, Wang C, Im M, Leung A, Swerdloff RS. Testicular heat exposure enhances the suppression of spermatogenesis by testosterone in rats: the “two-hit” approach to male contraceptive development. Endocrinology. 2000;141(4):1414–1424. [DOI] [PubMed] [Google Scholar]
- 31. Deng QG, She H, Cheng JH, et al. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology. 2005;42(4):905–914. [DOI] [PubMed] [Google Scholar]
- 32. Morgan K, Uyuni A, Nandgiri G, et al. Altered expression of transcription factors and genes regulating lipogenesis in liver and adipose tissue of mice with high fat diet-induced obesity and nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2008;20(9):843–854. [DOI] [PubMed] [Google Scholar]
- 33. Shiraishi S, Lee PW, Leung A, Goh VH, Swerdloff RS, Wang C. Simultaneous measurement of serum testosterone and dihydrotestosterone by liquid chromatography-tandem mass spectrometry. Clin Chem. 2008;54(11):1855–1863. [DOI] [PubMed] [Google Scholar]
- 34. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. [DOI] [PubMed] [Google Scholar]
- 35. Jia Y, Hikim AP, Lue YH, et al. Signaling pathways for germ cell death in adult cynomolgus monkeys (Macaca fascicularis) induced by mild testicular hyperthermia and exogenous testosterone treatment. Biol Reprod. 2007;77(1):83–92. [DOI] [PubMed] [Google Scholar]
- 36. Sinha Hikim AP, Rajavashisth TB, Sinha HI, et al. Significance of apoptosis in the temporal and stage-specific loss of germ cells in the adult rat after gonadotropin deprivation. Biol Reprod. 1997;57(5):1193–1201. [DOI] [PubMed] [Google Scholar]
- 37. Nunez AA. Dose-dependent effects of testosterone on feeding and body weight in male rats. Behav Neural Biol. 1982;34(4):445–449. [DOI] [PubMed] [Google Scholar]
- 38. Nunez AA, Grundman M. Testosterone affects food intake and body weight of weanling male rats. Pharmacol Biochem Behav. 1982;16(6):933–936. [DOI] [PubMed] [Google Scholar]
- 39. Asarian L, Geary N. Modulation of appetite by gonadal steroid hormones. Philos Trans R Soc Lond B Biol Sci. 2006;361(1471):1251–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Postic C, Girard J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008;34(6 Pt 2):643–648. [DOI] [PubMed] [Google Scholar]
- 41. Postic C, Girard J, Tsochatzis EA, Papatheodoridis GV, Archimandritis AJ. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice adipokines in nonalcoholic steatohepatitis: from pathogenesis to implications in diagnosis and therapy. J Clin Invest. 2008;118(3):829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun Z, Lazar MA. Dissociating fatty liver and diabetes. Trends Endocrinol Metab. 2013;24(1):4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology. 2011;53(1):116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yellaturu CR, Deng X, Cagen LM, et al. Posttranslational processing of SREBP-1 in rat hepatocytes is regulated by insulin and cAMP. Biochem Biophys Res Commun. 2005;332(1):174–180. [DOI] [PubMed] [Google Scholar]
- 45. Radenne A, Akpa M, Martel C, Sawadogo S, Mauvoisin D, Mounier C. Hepatic regulation of fatty acid synthase by insulin and T3: evidence for T3 genomic and nongenomic actions. Am J Physiol Endocrinol Metab. 2008;295(4):E884–E894. [DOI] [PubMed] [Google Scholar]
- 46. Rakhshandehroo M, Knoch B, Muller M, Kersten S. Peroxisome proliferator-activated receptor α target genes. PPAR Res. 2010;2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ntambi JM. Dietary regulation of stearoyl-CoA desaturase 1 gene expression in mouse liver. J Biol Chem. 1992;267(15):10925–10930. [PubMed] [Google Scholar]
- 48. Enser M, Roberts JL. The regulation of hepatic stearoyl-coenzyme A desaturase in obese-hyperglycaemic (ob/ob) mice by food intake and the fatty acid composition of the diet. Biochem J. 1982;206(3):561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jeffcoat R, Roberts PA, Ormesher J, James AT. Stearolyl-CoA desaturase: a control enzyme in hepatic lipogenesis. Eur J Biochem. 1979;101(2):439–445. [DOI] [PubMed] [Google Scholar]
- 50. Caballero F, Fernandez A, De Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol. 2009;50(4):789–796. [DOI] [PubMed] [Google Scholar]
- 51. Hasuwa H, Muro Y, Ikawa M, Kato N, Tsujimoto Y, Okabe M. Transgenic mouse sperm that have green acrosome and red mitochondria allow visualization of sperm and their acrosome reaction in vivo. Exp Anim. 2010;59(1):105–107. [DOI] [PubMed] [Google Scholar]
- 52. Ikawa M, Inoue N, Benham AM, Okabe M. Fertilization: a sperm's journey to and interaction with the oocyte. J Clin Invest. 2010;120(4):984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cole LK, Jacobs RL, Vance DE. Tamoxifen induces triacylglycerol accumulation in the mouse liver by activation of fatty acid synthesis. Hepatology. 2010;52(4):1258–1265. [DOI] [PubMed] [Google Scholar]
- 54. Lelliott CJ, Lopez M, Curtis RK, et al. Transcript and metabolite analysis of the effects of tamoxifen in rat liver reveals inhibition of fatty acid synthesis in the presence of hepatic steatosis. FASEB J. 2005;19(9):1108–1119. [DOI] [PubMed] [Google Scholar]
- 55. Osman KA, Osman MM, Ahmed MH. Tamoxifen-induced non-alcoholic steatohepatitis: where are we now and where are we going? Expert Opin Drug Saf. 2007;6(1):1–4. [DOI] [PubMed] [Google Scholar]
- 56. Zhang H, Liu Y, Wang L, et al. Differential effects of estrogen/androgen on the prevention of nonalcoholic fatty liver disease in the male rat. J Lipid Res. 2013;54(2):345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]