

Abstract
Na+/I− symporter (NIS) mediates iodide (I−) uptake in the thyroid gland, the first and rate-limiting step in the biosynthesis of the thyroid hormones. The expression and function of NIS in thyroid cells is mainly regulated by TSH and by the intracellular concentration of I−. High doses of I− for 1 or 2 days inhibit the synthesis of thyroid hormones, a process known as the Wolff-Chaikoff effect. The cellular mechanisms responsible for this physiological response are mediated in part by the inhibition of I− uptake through a reduction of NIS expression. Here we show that inhibition of I− uptake occurs as early as 2 hours or 5 hours after exposure to excess I− in FRTL-5 cells and the rat thyroid gland, respectively. Inhibition of I− uptake was not due to reduced NIS expression or altered localization in thyroid cells. We observed that incubation of FRTL-5 cells with excess I− for 2 hours increased H2O2 generation. Furthermore, the inhibitory effect of excess I− on NIS-mediated I− transport could be recapitulated by H2O2 and reverted by reactive derived oxygen species scavengers. The data shown here support the notion that excess I− inhibits NIS at the cell surface at early times by means of a posttranslational mechanism that involves reactive derived oxygen species.
The Na+/I− symporter (NIS) translocates iodide (I−) from the bloodstream into the thyrocyte against its electrochemical gradient to promote the synthesis of the thyroid hormones (T3 and T4) (1, 2). NIS is a plasma membrane glycoprotein with 13 transmembrane segments facing the COOH terminal to the intracellular and the NH2 terminal the extracellular (3). The expression and function of NIS is under hormonal regulation, mainly TSH, and regulates NIS biosynthesis and targeting to the plasma membrane (4). The excess of I− is an important factor that regulates NIS function (5–7).
More than 60 years ago, Wolff and Chaikoff reported that I− organification in the rat thyroid was inhibited when plasma concentrations of I− reached a high threshold level, a phenomenon known as the Wolff-Chaikoff effect (8). Wolff et al showed that after 2 days of this inhibitory effect, an adaptation or escape takes place, and organification and thyroid hormone biosynthesis return to a normal state (9). Socolow et al (10) showed that rats fed with high doses of I− displayed a decrease in 125I uptake by the thyroid gland, a process that is independent of TSH. The mechanisms triggered by excess I− over I− uptake begun to be elucidated after NIS was cloned (11). Studies using animal models and thyroid cell lines showed that after 24 hours of exposure to excess I−, a reduction on NIS protein and mRNA was observed (5–7). However, 30 years before, Socolow et al demonstrated that the inhibition of I− uptake by excess I− in the thyroid gland could occur much earlier than 24 hours. These authors showed that inhibition took place as early as 2 hours after exposure to an excess I−, and they named this phenomenon as acute inhibition (10). This acute regulation by excess I− was also observed by Grollman et al (12) in the highly functional rat thyroid-derived FRTL-5 cell line. Aiming to understand the mechanism behind acute NIS inhibition after excess I−, Serrano-Nascimento et al (13) showed a reduction in the levels of NIS mRNA and shortened the length of its poly-A in rats exposed for 30 minutes to excess I−. Eng et al (5) showed a significant reduction of NIS mRNA but not NIS protein in rats exposed 6 hours to excess I−. Later, Leoni et al (7), by working with PCCI3 cell line and thyroid gland, found that at the 6-hour time point, NIS protein but not NIS mRNA was reduced. Even though this latter observation suggests that a posttranscriptional mechanism could contribute to the regulation of NIS activity by excess I−, how excess I− acutely inhibits I− uptake still remains under debate.
One of the most important posttranscriptional mechanisms for NIS activity is the targeting of this symporter to the plasmatic cellular membrane (4). The contribution of this mechanism to NIS regulation by excess I− has not been yet evaluated. Furthermore, it has been suggested that the mechanisms responsible of the Wolff-Chaikoff inhibitory effect could be an alteration in the production of reactive derived oxygen species (ROS) molecules, such as H2O2 (14). This notion is supported by the observation made by Leoni et al (7), that high doses of I− increase the levels of ROS in PCCI3 cells and induce a reduction in the expression of thioredoxin reductase. One type of ROS molecule that can be responsible for this regulation is H2O2, which has been shown to be increased in thyroid slices of pork, sheep, and dog by high I− levels (15). Although H2O2 has been described as a signaling molecule that can modulate several cellular processes (16–19), such as I− uptake and organification (20–22), the contribution of ROS to NIS inhibition by high I− levels remains unknown. Here we approached this question by using a mixture of three ROS scavengers (ROSsc) to reduce ROS production in FRTL-5 cells.
Importantly, previous studies about I− uptake inhibition in rats used doses of I− 2000 times higher than the recommended for euthyroid condition (5, 13, 23), suggesting that the conclusions obtained from these reports are difficult to extrapolate to humans. Thus, in this study we evaluated the acute inhibition of NIS at doses only 20 times higher than the recommended doses of I− for rats, being closest to what could happen in humans with high I− intake (24).
In summary in this work, first, we show that the effect of acute excess I− occurs with doses only 20 times higher of the recommended dose. Second, we show that the acute inhibition of I− uptake is not due to changes in the content and localization of NIS in both the thyroid gland and FRTL-5 cells, suggesting that NIS is being inactivated at the plasma membrane. Third, we show that the changes induced by acute excess I− are mediated by ROS produced in response to high I− levels.
Materials and Methods
Animals and excess I− administration
For in vivo experiments, male Sprague Dawley rats with a weight of 250 g were used. They were kept at a constant temperature of 22°C and a 12-hour light, 12-hour dark photoperiod. All animal work was performed according to institutional and CONICYT bioethical guidelines and was supervised by a veterinarian. Three groups (5 h, 1 d, or 6 d) were established for excess I− treatment. The treatment with excess I− for 5 hours was carried out in male rats via a single ip injection of 100 μg of I− (Sigma; catalog number 221945) in 500 μL of distilled water. Rats that received excess I− for 1 or 6 days drank tap water with 4 mg/L of I− over the time period. Rats drank approximately 30 mL of water per day. The control group was given tap water without the addition of I−.
In vivo 125I uptake assays
After the treatment with excess I−, rats were ip injected with 75 μCi Na125I (PerkinElmer; catalog number NEZ033A), as previously described by Ferreira et al (23). After 15 minutes of 125I, rats were anesthetized with an ip injection of a mixture of 0.05 mg of ketamine (Agrovet) and 0.01 mg xylazine (Agroland) per gram of rat body weight. Then the thyroid glands were removed and weighed, and the amount of 125I in the thyroid gland was measured in a γ-counter (PerkinElmer; model Wizard). The counts per minute in the thyroid gland were used to calculate the percentage of 125I in the thyroid gland, having in account that 100% corresponded to the counts per minute injected I− into the rat.
Immunofluorescence analyses in thyroid glands
After the thyroid glands were removed, they were immersed in medium for tissue freezing (Tissue-Tek OCT compound). Then the thyroid glands were frozen in liquid nitrogen-cold and isopentane and cut in slices of 16 μm with cryostat (Leica; model CM1510S). The slices were fixed with 90% ethanol for 1 hour at −20°C and then with 100% ethanol for 30 minutes at room temperature. The thyroid slices were rehydrated with 95% ethanol for 30 minutes and 75% ethanol for 5 minutes. Then the thyroid sections were permeabilized with PBS/0.4% Triton X-100 for 5 minutes and washed two times with PBS. Endogenous fluorescence was quenched with 50 mM NH4Cl in PBS for 10 minutes at room temperature, and the unspecific binding was blocked with a solution containing fish gelatin solution two times (5 mM EDTA, 1% gelatin, 1% IgG free BSA, and 1% horse serum). After blocking, thyroid sections were incubated with 4.6 nM of rabbit anti-NIS polyclonal antibody diluted in PBS/0.4% Triton X-100/fish gelatin solution one time (solution A) for 24 hours at 4°C (25). Na+/K+ ATPase-α1 was detected with 5 ng/mL of mouse monoclonal antibody (Upstate; catalog number 05-369). Tissue sections were washed with solution A and then incubated with the secondary antibodies, antirabbit Alexa Fluor 488 (Invitrogen; catalog number A11008) for anti-NIS and 3.3 ng/mL of antimouse coupled to Alexa Fluor 594 (Invitrogen; catalog number A11032) for anti-Na+/K+ ATPase-α1 for 1 hour at room temperature. Finally, tissues were washed with 0.4% PBS/Triton X-100 and PBS before being mounted with the Dabco medium (Sigma; catalog number 290734). The fluorescence was visualized using an Olympus FV-1000 confocal microscope.
Treatments of FRTL-5 cells
The FRTL-5 rat thyroid cell line was used (26) and cultured in a F12 medium (Gibco; catalog number 21700-075) with 5% of newborn calf serum (Gibco; catalog number 16010-159). The medium was supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco; catalog number 15140-122), and the following hormones: 1.3 μM insulin (Sigma; catalog number I6634), 60 pM transferrin (Sigma; catalog number T8158), 2.5 μM gly-his-lys acetate salt (Sigma; catalog number G7387), 6.1 nM somatostatin (Sigma; catalog number S1763), and 1 mU/mL TSH (Sigma; catalog number T8931). Cells were maintained for growth in a humid atmosphere with 5% CO2 and 37°C. When FRTL-5 cells reached 70% confluence, the cells were subjected to different treatments. First, for experiments using excess I−, FRTL-5 cells were incubated for 2 hours with 100 μM I−. This I− was supplemented by potassium iodide (Sigma; catalog number 221945), and during this period control cells were kept in Hanks' balanced salt solution (HBSS) medium. Second, for experiments using ROSsc, FRTL-5 cells were first incubated with 500 μg/L catalase (Sigma; catalog number C9322), 180 μM 3-methyl-1-phenyl-2-pyrazolin-5-one (MCI-186) (Calbiochem; catalog number 443300), and 500 μM 4,5-dihydroxy-1,3-benzenedisulfonic acid disodium salt (Tiron) (Sigma; catalog number 17255-3) or 3 nM rotenone (Sigma; catalog number R88755G) or 1 mM diphenyliodonium (DPI) (Calbiochem; catalog number 300260) for 3 hours. Third, for those cells that also received excess I− and ROSsc or DPI or rotenone, the drugs were added for 1 hour, and then I− was added for 2 hours more. Fourth, for experiments using H2O2, FRTL-5 cells were incubated for 1 hour with 100 μM H2O2 (Merck; catalog number 1.97210). And finally, for the experiments using H2O2 and ROSsc, FRTL-5 cells were incubated for 1 hour with both reagents added at the same time. These treatments were performed in growth medium when I− uptake was analyzed and in HBSS when carboxy-2′, 7′-dichlorofluorescein diacetate (DCF) fluorescence was measured.
125I uptake in FRTL-5 cells
I− uptake in FRTL-5 cells was measured at steady state as previously described (4, 12). Briefly, FRTL-5 cells in 24-well plates at 90% confluence were washed two times with a modified HBSS. Then FRTL-5 cells were incubated with the HBSS buffer containing 100 μM I− and Na125I (PerkinElmer) with a specific activity of 100 mCi/mmol for 40 minutes at 37°C in a humidified atmosphere with 5% CO2. Eighty micromoles of KClO4 was added to some FRTL-5 cells to block the NIS iodide uptake. Reactions were terminated by aspirating the radioactive solution and by performing three washes with cold HBSS. Intracellular 125I was released by permeabilizing the cells with 500 μL cold ethanol. The released 125I was quantified in a γ-counter (PerkinElmer; model Wizard). The counts per minute value obtained for each well was normalized by the amount of DNA of this respective well. The results of I− uptake are shown as a percentage being the 100% of the uptake of those FRTL-5 cells that were not subjected to any treatment. Quantification of the DNA was performed by the colorimetric assay of the diphenylamine method (27). The absorbance was measured at 570 nm, and the standard curve was performed with salmon sperm DNA (Invitrogen; catalog number 15632011).
Immunofluorescence analyses in FRTL-5 cells
The localization of NIS in FRTL-5 cells was performed by immunofluorescence and confocal microscopy as previously described by Riedel et al (4). Briefly, cells were seeded into 24-well plates with coverslips. At 90% confluence and after treatments, cells were washed with PBS supplemented with 0.1 mM CaCl2 and 1 mM MgCl2 (PBS/CM) three times before being fixed in 2% paraformaldehyde in PBS for 30 minutes at room temperature. The cells were then permeabilized with solution B (0.1% Triton X-100, 0.2% BSA in PBS/CM) for 10 minutes at room temperature. Endogenous fluorescence was quenched with 50 mM NH4Cl in PBS/CM at room temperature for 10 minutes and then rinsed with solution B. Cells were incubated with the primary antibodies rabbit antirat NIS (25), at a concentration of 4.5 nM, and anti-Na+/K+ ATPase-α1 (Millipore; catalog number 05-369), at a concentration of 5 ng/mL, in solution B for 1 hour at room temperature. Next, cells were washed three times with solution B for 10 minutes before being incubated with the secondary antibodies, antirabbit Alexa Fluor 488 (5 μg/mL) for anti-NIS and antimouse Alexa Fluor 594 (4 μg/mL) for anti-Na+/K+ ATPase-α1, for 1 hour at room temperature. The cells were washed with solution B and then with PBS and were finally mounted on slides with the Dabco mounting medium (Sigma). Fluorescent visualization was carried out with an Olympus FV-1000, a laser-scanning confocal microscope at the Universidad Andrés Bello.
Western blot assays
Membrane fractions were prepared from thyroid gland or FRTL-5 cells as previously described by Riedel et al (4). Briefly, 20 μg of total protein from membrane fractions was diluted to a 1:2 ratio with loading buffer and heated at 37°C for 30 minutes prior to electrophoresis. Then the protein samples were loaded in 8% sodium dodecyl sulfate (SDS)-polyacrylamide gel. The Western blot analysis of NIS was performed with 930 pM of polyclonal antirat NIS antibody and incubated with 1:10 000 dilution of antirabbit secondary antibody coupled to horseradish peroxidase (Jackson; catalog number 111-035-003) as previously described (25). The detection of E-cadherin by Western blot was performed by using 125 ng/mL of primary antibody against E-cadherin (BD Bioscience; catalog number 610182). Then the membranes were washed and the proteins were visualized by an enhanced chemiluminescent Western blot detection system (Amersham; catalog number RPN2106). The peptides and antibodies are listed in Table 1.
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|---|
| NIS | Peptide sequence AETHPLYLGHDVETNL | Primary rabbit antirat NIS | Nancy Carrasco Laboratory | Rabbit; polyclonal | 930 pM |
| Antirabbit IgG (H+L) | Antirabbit secondary antibody coupled to HRP | Jackson, catalog number 111-035-003 | Goat; polyclonal | 1:10 000 | |
| E-cadherin | Primary E-cadherin | BD Biosciences, catalog number 610182 | Mouse; monoclonal | 125 ng/mL | |
| Na+/K+ ATPase-α1 | Primary mouse anti-Na+/K+ ATPase-α1 | Upstate, catalog number 05-369 | Mouse; monoclonal | 5 ng/mL | |
| Antirabbit IgG (H+L) | Secondary antirabbit Alexa Fluor 488 | Invitrogen, catalog number A11008 | Goat | 3.3 μg/mL | |
| Antimouse IgG (H+L) | Secondary antimouse Alexa Fluor 594 | Invitrogen, catalog number A11032 | Goat | 3.3 μg/mL |
Abbreviations: HRP, horseradish peroxidase; (H+L): Heavy + Light Chains.
Cell surface biotinylation assays in FRTL-5 cells
To quantify the expression of NIS at the plasma membrane in FRTL-5 cells, cell surface proteins were labeled with biotin as previously described by Riedel et al (4). Cells were washed with cold PBS/CM and then incubated two times with 1 mg/mL sulfo-NHS-SS-biotin (Pierce; catalog number 21328) in a biotin buffer (20 mM HEPES, pH 8.5; 2 mM CaCl2; and 150 mM NaCl) for 20 minutes with gentle agitation at 4°C. To stop the binding reaction, cells were incubated in a glycine buffer (PBS/CM and 100 mM glycine) for 20 minutes and then lysed with a lysis buffer (50 mM Tris-HCl, pH 7.5; 1% Triton X-100; 150 mM NaCl; and 5 mM EDTA) supplemented with 1% SDS and protease inhibitors (100 mM phenylmethylsulfonyl fluoride, 1 mM leupeptin, and 20 mg/mL aprotinin) for 30 minutes with gentle shaking at 4°C. Samples were diluted 10 times by adding 450 μL of lysis buffer, without SDS or protease inhibitors, and collected in microcentrifuge tubes. NeutrAvidin beads of 100 μL (Pierce; catalog number 29200) were added to the lysate and incubated overnight at 4°C. The lysate was centrifuged at 5000 × g for 2 minutes to separate the beads from the supernatant. Beads were washed three times with a lysis buffer, without SDS, and two times with a high ionic strength buffer (500 mM NaCl; 0.1% Triton X-100; 5 mM EDTA; and 50 mM Tris-HCl, pH 7.5). The final wash was done with 50 mM Tris-HCl (pH 7.5). Beads were resuspended in sample buffer containing 100 mM β-mercaptoethanol (Sigma; catalog number M6250) and heated for 5 minutes at 75°C. Finally, these samples were loaded onto a polyacrylamide gel and analyzed by immunoblot as detailed previously (1, 4).
Measurement of ROS in FRTL-5 cells
The fluorescence emitted by the DCF (Invitrogen; catalog number C400) probe was used to analyze ROS levels in FRTL-5 cells exposed to I− excess and to ROS scavengers as described previously (3, 28–30). Cells were seeded on 96-well plates and after reaching 80% confluence were loaded with 7 mM DCF in HBSS/HEPES (pH 7.4) for 30 minutes at 37°C. Then cells were washed and the different treatments were applied in HBSS/HEPES solution. Cells were maintained at 37°C inside the microplate reader (Biotek; model Synegy MX), and fluorescence readings were taken every 30 minutes for two wavelengths of 490/525 nm (em/ex). Fluorescence intensity results were normalized with respect to the control group, which was defined as 100% for each time point analyzed.
Cell viability quantification
Viability of FRTL-5 cells exposed to excess I−, H2O2, and ROSsc was determined as an indirect measurement of mitochondrial activity using 3-(4,5-dimethylthiazol-2-yl)-2,5-dipheniltetrazolium (MTT). Cells were cultured in 96-well plates and after exposure to treatments were incubated with 10 mL MTT (5 mg/mL) for 4 hours at 37°C in the same medium. Then the cells were incubated with 100 mL of isopropanol with 5% (wt/vol) of dimethylsulfoxide for a few minutes to dissolve the formazan cristal produced. The absorbance was measured at 540 nm.
Statistical analyses
The mean values for each experiment were calculated and expressed using the SE as a measure of dispersion. Significant differences between the mean values of different treatments were analyzed by a parametric ANOVA. Posttest analyses to determine the specific differences between the groups were done using the Student-Newman-Keuls method. Significant differences between groups were established at P < .05.
Supplemental methodology
Detection of thyroid hormones and TSH in serum
Thyroid hormones and the TSH were analyzed to check thyroid status after excess I− treatment. Blood samples were obtained after the decapitation of experimental animals. Serum was separated from the blood and used to measure total T3 and total T4 by a RIA using Siemens Healthcare Coat-A-Count diagnostic kits (catalog number TKT41 for total T3 and catalog number TKT31 for total T4). TSH was analyzed by using a rat ultrasensitive TSH ELISA kit (Mybiosource; catalog number MBS731244) according to the manufacturer's instructions (4, 31).
Determination of I− and selenium concentrations in rat serum
The measurement of I− and selenium in rat serum was performed by Vetlab Laboratories using a PerkinElmer series 200 HPLC with C18 5μ 8-mm cartridge in a RCM-100.
TdT-mediated dUTP Nick End Labeling (TUNEL) staining
FRTL-5 cell death was analyzed using TUNEL staining. Staining was performed according to the manufacturer's instructions using the in situ cell death detection kit (Roche Applied Science; catalog number 11684795910). Briefly, FRTL-5 cells fixed in 4% paraformaldehyde were incubated with the TUNEL reaction mixture kit. Nuclei were counterstained with 4′6-diamidino-2-phenylindole, and positive control was performed incubating FRTL-5 cells with deoxyribonuclease I. Immunofluorescence was analyzed using a laser-scanning confocal microscope (Fluoview FV1000; Olympus) with a ×20 objective.
Results
Acute excess I− reduces I− uptake without altering NIS expression or localization in thyroid glands
The effect of excess I− on I− uptake and NIS regulation has been studied in rat thyroids using 0.05% of NaI in water, which could potentially represents an amount that are about 1500–2000 times higher than the recommended dose for euthyroid condition in rats (5 μg/d) (5–7, 23) Therefore, we first analyzed in rat thyroid glands the effect of exposure during 6 days to various doses of excess I− (Figure 1A). A significant reduction of I− uptake was observed in both rats that ingested either 20 times or 2000 times the amount of recommended daily I−. Because the dose equal to 100 mg of I− per day is closer to the I− concentration found in some human populations (8, 32–34), it was used for the following in vivo experiments. To test the acute regulation of I− uptake by excess I−, rats were exposed to this condition for 5 hours. To compare the effect of acute excess I− (5 hours) with chronic treatment, 1 and 6 days were added as controls of the experiments (Figure 1B). Significant reduction in I− uptake was observed after 5 hours, 1 day, and 6 days of treatment with excess I−. Thyroid hormones and TSH were similar for the three conditions, supporting that the inhibition was induced by excess I− and is independent of TSH (Supplemental Figure 1). The concentration of I− in the serum has a tendency to increase after rats were treated with excess I−; however, these increments were not significantly different after 5 hours, 1 day, and 6 days of excess I− (Supplemental Figure 2A).
Figure 1. Acute excess of I− inhibits I− uptake in rat thyroid glands.
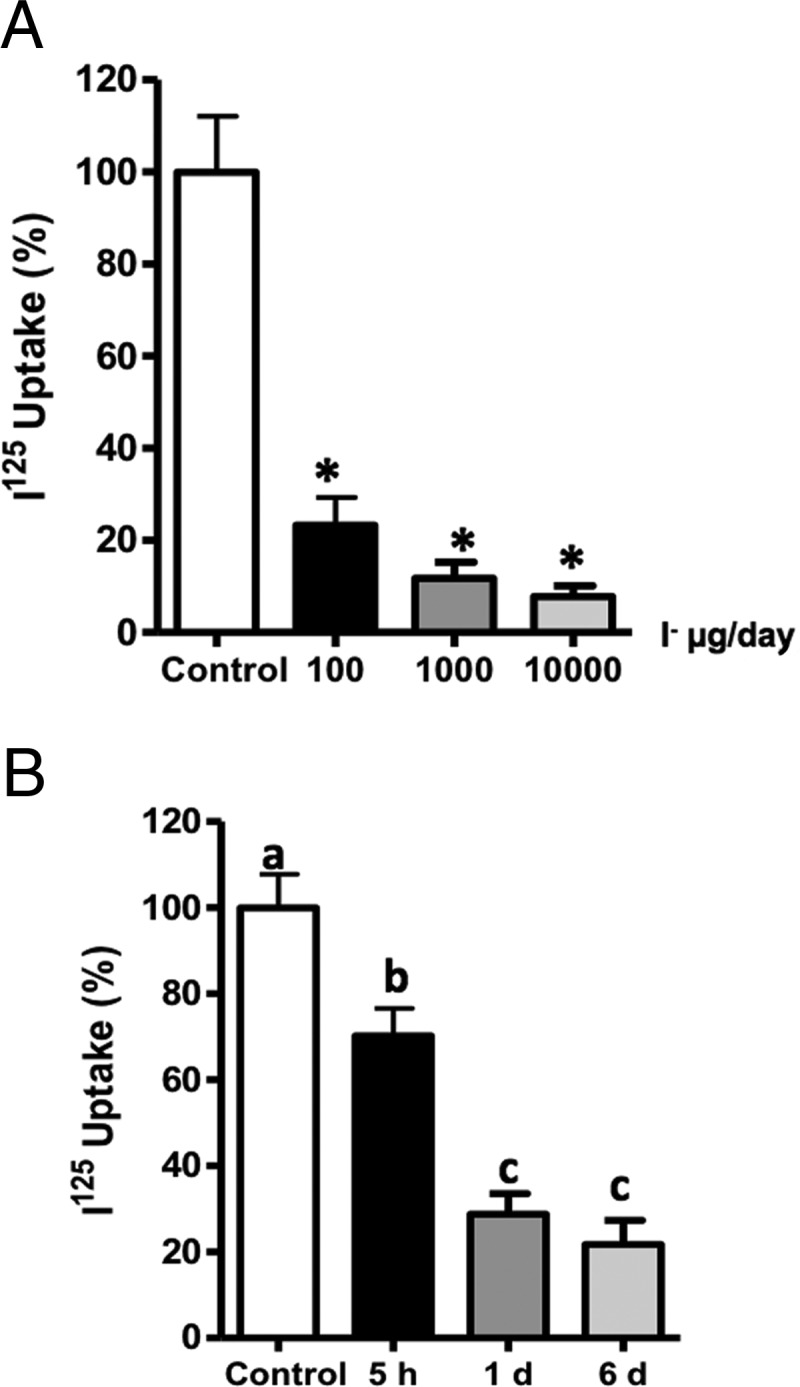
A, The percentage of I− uptake by the thyroid gland as a function of treatment, with a range of three high doses (100, 1000, and 10 000 μg/d) of I− over a period of 6 days. The percentage of I− uptake was expressed as a mean ± SEM (n ≥ 3) (P < .05). B, I− uptake in the thyroid glands of rats that received excess I− (100 μg) for 5 hours, 1 day, or 6 days. This was measured and plotted as the percentage of I− uptake as compared with thyroid glands from rats without excess I−. The data were expressed as the mean ± SEM (n ≥ 3).
Similar concentrations of selenium were found in the serum of rats treated or not with excess I− (Supplemental Figure 2B) and were comparable with previous publications (35–38). Western blot analyses, to measure NIS expression, were performed to evaluate whether acute excess I− alters NIS content. Figure 2A shows a representative immunoblot of membrane fractions from the thyroid glands. The 95-kDa band corresponds to the mature form of NIS, and the 68-kDa band corresponds to the NIS precursor (9, 39). Densitometric analyses for the mature form of NIS indicated a reduction in NIS content in the thyroid glands of rats treated with excess I− for 1 or 6 days (Figure 2B). Thus, there is a correlation between NIS content and I− uptake after 1 or 6 days with high I− doses, suggesting that the decrease in I− uptake could be due to a reduction in the expression of NIS. However, 5 hours after exposure to excess I−, the content of NIS was equivalent between treated and control cells, even though in this condition there was a 25% decrease in I− uptake, suggesting that other mechanisms are responsible for the reduction in I− uptake after acute treatment with excess I−.
Figure 2. NIS expression was unaltered after acute excess I−.

A, Representative immunoblot of the thyroid NIS and E-cadherin from rats treated with excess I− (100 μg/d) for 5 hours, 1 day, or 6 days. The NIS protein is visualized as an 83-kDa band and E-cadherin as a 120-kDa band. B, The graph represents the relative quantification of NIS content as performed by densitometry analysis of the 83-kDa band, with the normalization value obtained from densitometry analysis of the E-cadherin band. The data were expressed as the mean ± SEM of the control (n = 3). Different letters denote significant differences between control and experimental rats (P < .05).
To evaluate whether excess I− has an effect on the subcellular localization of NIS in thyrocytes, immunofluorescence analyses were performed to measure NIS location in thyroid glands of rats that received excess I− for 5 hours, 1 day, and 6 days (Figure 3). In these experiments Na+/K+ ATPase was used as a cell surface marker (Figure 3). Both types of staining, NIS (green fluorescence) and Na+/K+ ATPase (red fluorescence), delineated the cell surface of thyrocytes from control cells as well as those exposed to excess I− for 5 hours, 1 day and 6 days. Thus, excess I− did not alter the location of NIS at the cell surface in any condition.
Figure 3. NIS localization was unaltered after excess I−.

Representative photographs of confocal immunofluorescence microscopy analysis of NIS and Na+/K+ ATPase localization in the thyroid glands of male rats (n = 3) that received excess I− (100 μg/d) for 5 hours, 1 day, or 6 days are shown. NIS is visualized in green, whereas the α1-subunit of Na+/K+ ATPase, a plasma membrane marker, is visualized in red. Colocalization of the NIS with the α1-subunit of Na+/K+ ATPase is visualized in yellow. The white bar corresponds to 20 μm. The negative control corresponded to immunofluorescence of only the secondary antibodies used for NIS and the α1-subunit of the Na+/K+ ATPase. A picture with hematoxylin-eosin stain was included as a reference to show thyroid follicle structure in a control animal.
Acute inhibition of I− uptake due to excess I− is mediated by increased ROS levels
The inhibition of I− uptake and the localization of NIS at the cell surface after acute treatment with excess I− suggest that NIS activity can be regulated at the plasma membrane. The ROS level is a plausible candidate for the regulation of NIS function because these molecules play an essential role at regulating the function of several cellular proteins (10, 17, 28, 29), including thyroid cells (21, 40). Thus, FRTL-5 cells were used as a thyroid cell model because they endogenously express thyroid proteins and have been extensively used to study NIS regulation (4–7, 12). ROS levels were analyzed by using DCF probes after exposure to acute excess I−. As shown in Figure 4A, an increase in DCF-derived fluorescence was observed when cells were incubated with excess I− for 2 hours. A study of different time points of incubation with excess I− showed that after 1 hour the fluorescence of DCF increased (Supplemental Figure 3). This increase in DCF fluorescence induced by excess I− was reverted after the cells were incubated in the presence of a mixture of catalase, Tiron, and MCI-186 (Figure 4A).
Figure 4. ROS increased after acute excess I− and it is required for I− uptake inhibition in FRTL-5 cells.

A, ROS levels were analyzed by DCF fluorescence in FRTL-5 cells incubated with 100 μM I− in the presence or absence of ROSsc for 2 hours and H2O2 (100 μM) for 1 hour. The ROSsc used were a combination of 500 μg/L catalase, 180 μM MCI-186, and 500 μM Tiron. The percentage of DCF fluorescence was plotted for FRLT-5 cells incubated with or without I− excess. The data were expressed as the mean ± SEM. Asterisks denote significant differences between control and experimental cells (P < .05). B, I− uptake was measured after FRTL-5 cells were incubated with or without 100 μM I− for 2 hours in the presence or absence of ROSsc, with 100 μM H2O2 with or with ROSsc and with KClO4 for 1 hour. Asterisks denote significant differences between control and experimental cells (P < .05). C, The effects of I−, ROSsc, and H2O2 on FRTL-5 cell viability were analyzed by an MTT assay. The percentage for each experimental group is expressed as the mean ± SEM (n = 3).
These three molecules among others are considered ROSsc thought to inhibit different sources of ROS in the cell. DCF fluorescence remained high when one or two ROSsc were used in the presence of excess I− (data not shown), which would suggest that various sources of ROS in FRTL-5 cells contribute to the response triggered by excess I−. Before, to assay the role of ROS in the mechanism of acute I− uptake inhibition by excess I−, FRTL-5 cells were incubated at different time points of exposure to excess I− (Supplemental Figure 4). These results showed that the inhibition of I− uptake was observed from 1 hour of exposure to excess I−.Then FRTL-5 cells were incubated in the presence of excess I− (2 h) and/or ROSsc (3 h), and I− uptake was measured (Figure 4B). KClO4, a specific inhibitor of NIS, inhibited 90% of the I− uptake, indicating that I− uptake in these experiments is mediated by NIS (Figure 4B). As observed in Figure 4B, the inhibition of I− uptake by excess I− was reverted in the presence of ROSsc (catalase, Tiron, and MCI-186) (Figure 4B). However, the inhibition of I− uptake by excess I− was still observed when each of the ROSsc was separately incubated with FRTL-5 cells (Supplemental Figure 5) or with rotenone (Supplemental Figure 5) or when the dual oxidase system was inhibited with DPI (Supplemental Figure 6). To evaluate the effect of ROS on I− uptake, FRTL-5 cells were incubated in the presence of H2O2, which caused a significant reduction on I− uptake (Figure 4B). The level of inhibition on I− uptake by H2O2 was similar to the inhibition observed by excess I− and was reverted by coincubation with ROSsc (Figure 4B). Furthermore, as shown in Figure 4C, no significant reduction of FRTL-5 cell viability was observed after treatment with H2O2 both by MTT assay (Figure 4C) and by TUNEL (Supplemental Figure 7).
No changes on NIS expression or localization are observed in response to excess I− and ROSsc
A representative Western blot analysis is shown in Figure 5A for NIS from cells exposed to excess I− and/or ROSsc. The densitometric analyses of the mature form of NIS indicated that none of the conditions altered NIS expression in FRTL-5 cells (Figure 5B). Next, experiments were performed to determine whether NIS localization was altered in the presence of I− with or without ROSsc. First, NIS content at the plasma membrane was analyzed by the biotin labeling of plasma membrane proteins facing to the extracellular milieu. Figure 5C shows a representative Western blot for NIS from a cell surface biotinylation experiments. As expected, one single band at 85 kDa was detected at the plasma membrane because only the mature form of NIS is found at the cell surface (Figure 5C) (4, 10). Densitometric analyses indicated that NIS content showed no significant modifications at the plasma membrane (Figure 5D). These results were consistent by NIS immunofluorescence analyses (Figure 6), which show that NIS (Figure 6, green) and the Na+/K+ ATPase (Figure 6, red) colocalized similarly in all experimental conditions tested. As shown in Figure 6, NIS location delineated the plasma membrane and colocalized with the α-subunit of Na+/K+ ATPase in all conditions. These data suggest that no significant alterations on NIS content or localization were caused by excess I− or ROS activity.
Figure 5. NIS expression in FRTL-5 cells remained unchanged after acute excess I−.

A, Representative picture of immunoblots of NIS expression in FRTL-5 cells treated with 100 μM I− for 2 hours in the presence or absence of ROSsc. E-cadherin was probed as a loading control in the same nitrocellulose. B, The graph depicts densitometry analysis of the 86-kDa band of NIS normalized with respect to the value obtained from the densitometry analysis of the E-cadherin band. The data were plotted as the mean ± SEM (n = 3). The value corresponding to 100% was the control group. C, Representative immunoblot of a cell surface biotinylation experiment analyzing NIS content at the plasma membrane in FRTL-5 cells incubated with excess I− in the presence or absence of ROSsc. D, The graph depicts the NIS content quantified by a densitometry analysis of the 85-kDa bands. Results were expressed as the mean ± SEM (n = 3), where the control group was set at 100%.
Figure 6. NIS localization at the cell surface remained unchanged after excess I− in FRTL-5 cells.

Representative confocal immunofluorescence microscopy image of NIS localization in FRTL-5 cells treated with 100 μM I− for 2 hours in the presence or absence of ROSsc. NIS and α1-subunit of Na+/K+ ATPase were visualized in green and red, respectively. Yellow fluorescence corresponds to the colocalization of the NIS with α1-subunit of the Na+/K+ ATPase. The negative control corresponds to immunofluorescence of only secondary antibodies used for NIS and the α1-subunit of the Na+/K+ ATPase. The white bar corresponds to 20 μm.
Discussion
Here we provide evidence supporting the notion that the acute inhibition of I− uptake by excess I− occurs without altering the content and localization of NIS, and its mechanism of inhibition of I− uptake is dependent on ROS production and activity. This work is the first to show the recovery of I− uptake after the inhibition exerted by excess I− and that this recovery was accomplished by the inhibition of different ROS sources in the cell (Figure 4B). Moreover, in this work we report for the first time that the regulation of I− uptake due to excess I− occurs with a dose 20 times higher the recommended for rats (100 μg vs 5 μg of I−), and we observed this effect at chronic and acute I− exposure (Figure 1A and B). Thus, these results support that the acute consumption of I− of at least 1 order of magnitude higher than the recommended dose for humans can prompt the inactivation of NIS at the plasma membrane.
The temporality of I− uptake inhibition by excess I− in the thyroid gland has been evaluated by several groups, which have shown inhibition after 2 hours (10, 12), 5 hours (9, 13), 6 hours (5), and longer than 24 hours until 6 days of exposure to excess I− (5, 7). The mechanisms behind this inhibitory effect over I− uptake had been elucidated for periods of 24 hours or longer (4–6, 23). These studies concluded that NIS inhibition required transcriptional and/or translational regulation (4–6, 14). However, the mechanism behind the acute inhibitory effect of excess I− over NIS function still remains unknown.
There are two studies that focus the inhibition in thyroid gland at the time point of 6 hours and follow NIS protein (5, 7). Leoni et al (7) found a slightly reduction in NIS content, whereas Eng et al (5) found no differences. The authors discussed that the slight reduction of NIS cannot account for the inhibition of I− uptake. Therefore, other mechanisms like the inactivation of NIS at the plasma membrane can be responsible for this phenotype (7, 16–19). In agreement with Eng et al (5), we found at 5 hours with excess I− the content of NIS protein in the cell remained similar (Figure 2), and this result was supported by the immunofluorescence analysis in the thyroid gland (Figure 3). Moreover, two in vitro studies that used FRTL-5 cells to analyze the mechanisms behind acute inhibition of NIS by excess I− had shown a reduction of NIS mRNA (42).
The third study performed by Leoni et al (7) found a reduction on NIS protein but not in NIS mRNA after 6 hours of I− excess. We have shown, in FRTL-5 cells, by two different approaches that at 2 hours of excess I−, the content of NIS protein remained similar in the cell and at the plasma membrane (Figures 5 and 6). The concentration of excess I− used in FRTL-5 cell experiments were approximately 6 times higher than the concentration of I− in the serum of in vivo experiments (Supplemental Figure 2A). Despite that, the findings from FRTL-5 cells correlated with the in vivo experiments; thus, both approaches in vivo and in vitro strongly suggest that the acute inhibition of I− uptake by excess I− must be mediated by the inactivation of NIS at the plasma membrane. Our results are also consistent with previous studies (7, 23) that observed no significant changes on the levels of NIS at the plasma membrane in response to excess I−, suggesting that inhibition of I− uptake could be due to modifications of NIS molecules located at the cell surface. A posttranslational mechanism of I− uptake inhibition seems to apply after thyroid cells are exposed to excess I−. Thus, we propose that excess I− initiate a cascade of mechanisms in thyroid cells to inhibit NIS activity. Our data support that one of the first and fast mechanism is the inactivation of NIS at the plasma membrane.
Even though the specific posttranslational mechanism responsible for the inhibition has not been elucidated, here we provide evidence for an involvement of ROS as key molecules contributing to this regulation process. Consistent with this notion, excess I− has been shown to induce an increase of ROS amounts over basal toxicity levels in thyroid cells (25, 42). Thus, after 6 hours of excess I−, ROS and seleno proteins were significantly induced both in the thyroid gland and PCCl3 cells (7). Those studies found an increase in ROS after exposing either rat or PCCl3 thyroid cells to an excess I− for 6–72 hours. Our data support the notion that excess I− increased ROS levels in FRTL-5 cells (Figure 4A), and for the first time, it is shown that the inhibition of I− uptake by excess I− was overcome by using a combination of three ROSsc (Figure 4B), indicating that ROS molecules are responsible for mediating the inhibition of NIS function by excess I−.
Previous findings have shown that excess I− increased H2O2 levels in slices of human thyroid glands, which was also observed for dogs, horses, and sheep (15). Although our data provide further support to the notion that H2O2 contributes to the inhibition of I− uptake induced by excess I− (Figure 4B), the involvement of ROS other than H2O2 cannot be ruled out. In fact, by using DPI, an inhibitor of the Duox system, the main H2O2 producer in thyroid cells (43), a reduction of I− uptake after an acute exposure to excess I− is still observed (Supplemental Figure 6). Tiron, MCI-186, catalase, rotenone, or DPI by themselves failed to restore I− uptake and is suggesting the involvement of different sources of ROS (Supplemental Figures 5 and 6). In the absence of excess I−, Tiron, rotenone, and DPI each one by themselves showed a tendency to reduce I− uptake. Although such a reduction was not statistically significant as compared with controls, it was not observed for MCI-186 and catalase (Supplemental Figure 5). Although Tiron reduced the levels of H2O2 by acting as scavenger (44), DPI achieved the same result by inhibiting the production of this molecule (45). On the other hand, rotenone is an inhibitor of mitochondrial electron chain (46). MCI-186 inhibits the generation of hydroxyl (47). These results support that ROS participates in a complex manner regulating NIS activity (41–44).
Recently Yao et al (48) showed that excess I− induces mitochondrial superoxide anion production; thus, it is probable that more than one ROS species could be simultaneously produced. It is possible that other reactive molecules, such as nitric oxide (NO) can contribute to the inhibition of NIS function caused by an excess I− (49). Although it is unclear whether excess I− can stimulate NO production, it has been shown that NO inhibits I− uptake in thyroid cells. Currently it is difficult to determine what reactive species may be directly responsible for the biological effects of excess I− because many ROS coexist at the same time in thyroid cells.
The mechanisms by which ROS inhibits NIS remain unknown. It has been reported that ROS can directly or indirectly regulate the activity of several proteins (28–30, 50). Direct inhibitory mechanisms of ROS molecules could be the oxidation of cysteine residues on the target protein by H2O2, thus inducing conformational changes that could alter protein function (49, 50). Along these lines, it has been proposed that cysteine 272 on NIS could be specifically targeted by ROS as part of the regulatory mechanism (7). On the other hand, an indirect mechanism could involve the modification of kinases and/or phosphatases by ROS molecules, which in turn would modify the activity of target proteins (52). It has been previously shown that NIS is a phosphoprotein whose phosphorylation pattern can be modified by the signaling pathway triggered by TSH (4). NIS phosphorylation at Ser-43 and Ser-581 modifies the kinetic properties of I− transport. In particular, S43A and S581A mutation residues in rat NIS protein decreased the maximal velocity of the I− transport by more than 60% and 40%, respectively, without affecting the Michaelis constant parameter (53). Another possible mechanism for the indirect inhibition of NIS through ROS could be through the iodination of organic compounds, such as arachidonic acid to produce 6-iodine d-lactone (54). This reaction is also mediated by thyroperoxidase and requires ROS (55). It has been described that 6-iodine d-lactone is capable of exerting several of the inhibitory effects of I− excess, such as inhibiting I− uptake and decreasing NIS content (54). Interestingly, methimazole, an inhibitor of thyroperoxidase that also plays an antioxidant effect (56), has been shown to revert and protect NIS activity from the effects of excess I− (12, 13, 23), supporting that the generation of ROS could be involved in NIS inactivation.
Finally, in this study we have shown that it was not necessary to expose thyroid cells to such high doses of I− to observe the inhibitory effect of excess I− over I− uptake (Figure 1A). In fact, the inhibition occurred under a chronic or acute treatment with an I− dose 100 times lower than the doses previously published (5, 23). We believe that it is important for thyroid cells to have an acute mechanism of I− uptake inhibition after excess I− that will not only protect and defend the thyroid cells against high levels of ROS and the oxidative power generated during the incorporation and organification of I− (41) but also will help to prevent thyroid diseases like hypothyroidism, hyperthyroidism, and autoimmune thyroid diseases (24).
Acknowledgments
This work was supported by Proyecto de Inicio Grant UNAB DI 06-08, Fondecyt Grant 1130996, Fondecyt Grant 1121078, Millennium Institute on Immunology and Immunotherapy Grant P09/016-F, Proyecto Cochilco-Fondecyt 1100995, and Proyecto Núcleo Grant UNAB DI-209-12/N.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by Proyecto de Inicio Grant UNAB DI 06-08, Fondecyt Grant 1130996, Fondecyt Grant 1121078, Millennium Institute on Immunology and Immunotherapy Grant P09/016-F, Proyecto Cochilco-Fondecyt 1100995, and Proyecto Núcleo Grant UNAB DI-209-12/N.
Footnotes
- DCF
- carboxy-2′, 7′-dichlorofluorescein diacetate
- DPI
- diphenyliodonium
- HBSS
- Hanks' balanced salt solution
- I−
- iodide
- MCI-186
- 3-methyl-1-phenyl-2-pyrazolin-5-one
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-dipheniltetrazolium
- NIS
- Na+/I− symporter
- NO
- nitric oxide
- PBS/CM
- PBS supplemented with CaCl2 and MgCl2
- ROS
- reactive derived oxygen species
- ROSsc
- ROS scavengers
- SDS
- sodium dodecyl sulfate
- Tiron
- 4,5-dihydroxy-1,3-benzenedisulfonic acid disodium salt
- TUNEL
- TdT-mediated dUTP Nick End Labeling.
References
- 1. Carrasco N. In: Braverman MD, Robert D, Utizer MD, Ingbar SH, Wegner SC, eds. Werner Ingbar's Thyroid: A Fundamental Clinical Text. 8th ed Philadelphia: Williams & Wilkins; 2000. [Google Scholar]
- 2. Dohan O, Carrasco N. Advances in Na(+)/I(−) symporter (NIS) research in the thyroid and beyond. Mol Cell Endocrinol. 2003;213:59–70. [DOI] [PubMed] [Google Scholar]
- 3. Dohan O, De la Vieja A, Paroder V, et al. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr Rev. 2003;24:48–77. [DOI] [PubMed] [Google Scholar]
- 4. Riedel C, Levy O, Carrasco N. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem. 2001;276:21458–21463. [DOI] [PubMed] [Google Scholar]
- 5. Eng PH, Cardona GR, Fang SL, et al. Escape from the acute Wolff-Chaikoff effect is associated with a decrease in thyroid sodium/iodide symporter messenger ribonucleic acid and protein. Endocrinology. 1999;140:3404–3410. [DOI] [PubMed] [Google Scholar]
- 6. Eng PH, Cardona GR, Previti MC, Chin WW, Braverman LE. Regulation of the sodium iodide symporter by iodide in FRTL-5 cells. Eur J Endocrinol. 2001;144:139–144. [DOI] [PubMed] [Google Scholar]
- 7. Leoni SG, Kimura ET, Santisteban P, De la Vieja A. Regulation of thyroid oxidative state by thioredoxin reductase has a crucial role in thyroid responses to iodide excess. Mol Endocrinol (Baltimore, Md). 2011;25:1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wolff J, Chaikoff IL. Plasma inorganic iodide as a homeostatic regulator of thyroid function. J Biol Chem. 1948;174:555–564. [PubMed] [Google Scholar]
- 9. Wolff J, Chaikoff IL, Goldberg RC, Meier JR. The temporary nature of the inhibitory action of excess iodide on organic iodine synthesis in the normal thyroid. Endocrinology. 1949;45:504–513. [DOI] [PubMed] [Google Scholar]
- 10. Socolow EL, Dunlap D, Sobel RA, Ingbar SH. A correlative study of the effect of iodide administration in the rat on thyroidal iodide transport and organic iodine content. Endocrinology. 1968;83:737–743. [DOI] [PubMed] [Google Scholar]
- 11. Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature. 1996;379:458–460. [DOI] [PubMed] [Google Scholar]
- 12. Grollman EF, Smolar A, Ommaya A, Tombaccini D, Santisteban P. Iodine suppression of iodide uptake in FRTL-5 thyroid cells. Endocrinology. 1986;118:2477–2482. [DOI] [PubMed] [Google Scholar]
- 13. Serrano-Nascimento C, Calil-Silveira J, Nunes MT. Posttranscriptional regulation of sodium-iodide symporter mRNA expression in the rat thyroid gland by acute iodide administration. Am J Physiol Cell Physiol. 2010;298:C893–C899. [DOI] [PubMed] [Google Scholar]
- 14. Corvilain B, Van Sande J, Dumont JE. Inhibition by iodide of iodide binding to proteins: the “Wolff-Chaikoff” effect is caused by inhibition of H2O2 generation. Biochem Biophys Res Commun. 1988;154:1287–1292. [DOI] [PubMed] [Google Scholar]
- 15. Corvilain B, Collyn L, Van Sande J, Dumont JE. Stimulation by iodide of H(2)O(2) generation in thyroid slices from several species. Am J Physiol Endocrinol Metab. 2000;278:E692–E699. [DOI] [PubMed] [Google Scholar]
- 16. D'Autreaux BIT, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. [DOI] [PubMed] [Google Scholar]
- 17. Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. [DOI] [PubMed] [Google Scholar]
- 18. Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science (New York, NY). 2006;312:1882–1883. [DOI] [PubMed] [Google Scholar]
- 19. Foreman J, Demidchik V, Bothwell JHF, et al. Reactive oxygen species produced by NADPH oxidase regulate plant cell growth. Nature. 2003;422:442–446. [DOI] [PubMed] [Google Scholar]
- 20. Fukayama H, Murakami S, Nasu M, Sugawara M. Hydrogen peroxide inhibits iodide uptake and iodine organification in cultured porcine thyroid follicles. Thyroid. 1991;1:267–271. [DOI] [PubMed] [Google Scholar]
- 21. Nadolnik LI, Niatsetskaya ZV, Lupachyk SV. Effect of oxidative stress on rat thyrocyte iodide metabolism. Cell Biochem Funct. 2008;26:366–373. [DOI] [PubMed] [Google Scholar]
- 22. Sugawara M, Yamaguchi DT, Lee HY, et al. Hydrogen peroxide inhibits iodide influx and enhances iodide efflux in cultured FRTL-5 rat thyroid cells. Eur J Endocrinol. 1990;122:610–616. [DOI] [PubMed] [Google Scholar]
- 23. Ferreira ACF, Lima LIVP, Araujo RL, et al. Rapid regulation of thyroid sodium-iodide symporter activity by thyrotrophin and iodine. J Endocrinol. 2005;184:69–76. [DOI] [PubMed] [Google Scholar]
- 24. Bürgi H. Iodine excess. Best Pract Res Clin Endocrinol. 2010;24(1):107–115. [DOI] [PubMed] [Google Scholar]
- 25. Levy O, Dai G, Riedel C, et al. Characterization of the thyroid Na+/I− symporter with an anti-COOH terminus antibody. Proc Natl Acad Sci USA. 1997;94:5568–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ambesi-Impiombato FS, inventor. Living, fast-growing thyroid cell strain, FRTL-5. US Patent US4608341A 1983.
- 27. Kaminsky SM, Levy O, Salvador C, Dai G, Carrasco N. Na(+)-I− symport activity is present in membrane vesicles from thyrotropin-deprived non-I(−)-transporting cultured thyroid cells. Proc Natl Acad Sci USA. 1994;91:3789–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nunez-Villena F, Becerra A, Echeverria C, et al. Increased expression of the transient receptor potential melastatin 7 channel is critically involved in lipopolysaccharide-induced reactive oxygen species-mediated neuronal death. Antioxid Redox Signal. 2011;15:2425–2438. [DOI] [PubMed] [Google Scholar]
- 29. Becerra A, Echeverra C, Varela D, et al. Transient receptor potential melastatin 4 inhibition prevents lipopolysaccharide-induced endothelial cell death. Cardiovasc Res. 2011;91:677–684. [DOI] [PubMed] [Google Scholar]
- 30. Simon F, Fernandez R. Early lipopolysaccharide-induced reactive oxygen species production evokes necrotic cell death in human umbilical vein endothelial cells. J Hypertens. 2009;27:1202–1216. [DOI] [PubMed] [Google Scholar]
- 31. Albornoz EA, Carreño LJ, Cortés C, et al. Gestational hypothyroidism increases the severity of experimental autoimmune encephalomyelitis in adult offspring. Thyroid. 2013;23:1627–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hetzel BS, Maberly GF. Iodine. In: Mertz W, ed. Trace Elements in Human and Animal Nutrition. 5th ed New York: Academic Press; 1986:499. [Google Scholar]
- 33. Muzzo S, Ramírez I, Carvajal F, Biolley E, Leiva L. Iodine nutrition in school children of four areas of Chile during the year 2001. Rev Med Chil. 2003;131:1391–1398. [PubMed] [Google Scholar]
- 34. Mussig K. Iodine-induced toxic effects due to seaweed consumption. In: Comprehensive Handbook of Iodine. New York: Academic Press; 2009:897–908. [Google Scholar]
- 35. Su L, Wang M, Yin S-T, et al. The interaction of selenium and mercury in the accumulations and oxidative stress of rat tissues. Ecotoxicol Environ Saf. 2008;70:483–489. [DOI] [PubMed] [Google Scholar]
- 36. Chiu-Ugalde J, Theilig F, Behrends T, et al. Mutation of megalin leads to urinary loss of selenoprotein P and selenium deficiency in serum, liver, kidneys and brain. Biochem J. 2010;431:103–111. [DOI] [PubMed] [Google Scholar]
- 37. Zhou X, Smith AM, Failla ML, Hill KE, Yu Z. Estrogen status alters tissue distribution and metabolism of selenium in female rats. J Nutr Biochem. 2012;23:532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hill KE, Zhou J, McMahan WJ, Motley AK. Deletion of selenoprotein P alters distribution of selenium in the mouse. J Biol Chem. 2003;278:13640–13646. [DOI] [PubMed] [Google Scholar]
- 39. Levy O, De la Vieja A, Ginter CS, Riedel C, Dai G, Carrasco N. N-linked glycosylation of the thyroid Na+/I− symporter (NIS). Implications for its secondary structure model. J Biol Chem. 1998;273:22657–22663. [DOI] [PubMed] [Google Scholar]
- 40. Song Y, Driessens N, Costa M, et al. Review: roles of hydrogen peroxide in thyroid physiology and disease. J Clin Endocrinol Metab. 2007;92:3764–3773. [DOI] [PubMed] [Google Scholar]
- 41. Suzuki K, Kimura H, Wu H, et al. Excess iodide decreases transcription of NIS and VEGF genes in rat FRTL-5 thyroid cells. Biochem Biophys Res Commun. 2010;393:286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vitale M, Di Matola TD, D'Ascoli F, et al. Iodide excess induces apoptosis in thyroid cells through a p53-independent mechanism involving oxidative stress. Endocrinology. 2000;141:598–605. [DOI] [PubMed] [Google Scholar]
- 43. Dupuy C, Virion A, Ohayon R, Kaniewski J, Deme D, Pommier J. Mechanism of hydrogen peroxide formation catalyzed by NADPH oxidase in thyroid plasma membrane. J Biol Chem. 1991;266, 3739–3743. [PubMed] [Google Scholar]
- 44. Taiwo FA. Mechanism of tiron as scavenger of superoxide ions and free electrons. Spectroscopy. 2008;22:491–498. [Google Scholar]
- 45. Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–299. [DOI] [PubMed] [Google Scholar]
- 46. Dröse S, Hanley PJ, Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim Biophys Acta. 2009;1790(6):558–565. [DOI] [PubMed] [Google Scholar]
- 47. Perez-Gonzalez A, Galano A. OH radical scavenging activity of Edaravone: mechanism and kinetics. J Phys Chem B. 2011;115:1306–1314. [DOI] [PubMed] [Google Scholar]
- 48. Yao X, Li M, He J, et al. Effect of early acute high concentrations of iodide exposure on mitochondrial superoxide production in FRTL cells. Free Radic Biol Med. 2012;52:1343–1352. [DOI] [PubMed] [Google Scholar]
- 49. Bocanera LV, Krawiec L, Silberschmidt D, et al. Role of cyclic 3“5” guanosine monophosphate and nitric oxide in the regulation of iodide uptake in calf thyroid cells. J Endocrinol. 1997;155:451–457. [DOI] [PubMed] [Google Scholar]
- 50. Simon F, Stutzin A. Protein kinase C-mediated phosphorylation of p47 phox modulates platelet-derived growth factor-induced H2O2 generation and cell proliferation in human umbilical vein endothelial cells. Endothelium. 2008;15:175–188. [DOI] [PubMed] [Google Scholar]
- 51. Simon F, Leiva-Salcedo E, Armisén R, et al. Hydrogen peroxide removes TRPM4 current desensitization conferring increased vulnerability to necrotic cell death. J Biol Chem. 2010;285:37150–37158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poole LB, Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol. 2008;12:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vadysirisack DD, Chen ESW, Zhang Z, Tsai MD, Chang GD, Jhiang SM. Identification of in vivo phosphorylation sites and their functional significance in the sodium iodide symporter. J Biol Chem. 2007;282:36820–36828. [DOI] [PubMed] [Google Scholar]
- 54. Thomasz L, Oglio R, Dagrosa MIAA, Krawiec LON, Pisarev MA, Juvenal GJ. 6 Iodo-δ-lactone reproduces many but not all the effects of iodide. Mol Cell Endocrinol. 2010;323:161–166. [DOI] [PubMed] [Google Scholar]
- 55. Dugrillon A. Iodolactones and iodoaldehydes—mediators of iodine in thyroid autoregulation. Exp Clin Endocrinol Diabetes. 1996;104(suppl 4):41–45. [DOI] [PubMed] [Google Scholar]
- 56. Kim H, Lee TH, Hwang YS, et al. Methimazole as an antioxidant and immunomodulator in thyroid cells: mechanisms involving interferon-γ signaling and H(2)O(2) scavenging. Mol Pharmacol. 2001;60:972–980. [DOI] [PubMed] [Google Scholar]