

Abstract
Neuroglobin (Ngb) is a recently discovered globin with preferential localization to neurons. Growing evidence indicates that Ngb has distinct physiological functions separate from the oxygen storage and transport roles of other globins, such as hemoglobin and myoglobin. We found increased ATP production and decreased glycolysis in Ngb-overexpressing immortalized murine hippocampal cell line (HT-22), in parallel with inhibition of AMPK signaling and activation of acetyl-CoA carboxylase (ACC). In addition, lipid and glycogen content was increased in Ngb-overexpressing HT-22 cells. AMPK signaling was also inhibited in brain and heart from Ngb-overexpressing transgenic mice. Although Ngb overexpression did not change glycogen content in whole brain, glycogen synthase was activated in cortical neurons of Ngb overexpressing mouse brain and Ngb overexpression primary neurons. Moreover, lipid and glycogen content was increased in hearts derived from Ngb-overexpressing mice. These findings suggest that Ngb functions as a metabolic regulator and enhances cellular anabolism through the inhibition of AMPK signaling.
Introduction
Globins are O2-binding heme proteins present in bacteria, fungi, plants and animals, which have diverged widely in evolution, and which function by binding, transporting, scavenging, detoxifying and sensing of gases like O2 and NO [1]. Four types of globin have been discovered in vertebrates: erythrocyte-specific hemoglobin, muscles expressed myoglobin [2], ubiquitously expressed cytoglobin [3], and neurons expressed neuroglobin (Ngb) [4]. Although Ngb fulfills the basic requirements of a respiratory protein employed in O2 supply, increasing evidence suggests that Ngb has a distinct evolutionary history and physiological functions [4,5]. Numerous studies indicate that Ngb has neuroprotective effects against various insults, although the exact mechanisms that underlie protection are uncertain [6-11].
Accumulating evidence points to a role for Ngb in cell metabolism. Ngb preferentially localizes in metabolically active cells and subcellular compartments [4,12,5]. The concentration of Ngb is tightly correlated with the distribution of mitochondria [5]. Ngb has been found to interact with many mitochondrial proteins including VDAC and Cyc1 and plays important roles in mitochondrial functions such as ATP production, ROS generation, and apoptosis signaling [13]. AMP-activated protein kinase (AMPK) is positioned at the crossroad of multiple metabolic pathways and its activation stimulates catabolism and, concomitantly, inhibits anabolism [14]. In the adult mammalian brain, AMPK is predominantly expressed in neurons [15,16]. Given the well-established function of AMPK signaling in cellular metabolism, the high affinity of Ngb to oxygen, and the fact that both Ngb and AMPK are preferentially localized to neurons in the CNS, it is plausible that Ngb might interact with AMPK signaling and play a critical role in neuronal metabolism. In the current study, we investigated the function of Ngb in cell metabolism using both in vitro and in vivo approaches. Our study suggests that Ngb inhibits AMPK signaling and enhances cell anabolism.
Materials and Methods
Animals and reagents
All animal experiments were approved by the Buck Institute for Research on Aging and University of North Texas Health Science Center at Fort Worth Animal Care and Use Committees and conducted according to National Institutes of Health guidelines. Transgenic mice that overexpress murine neuroglobin under the control of chicken β-actin promoter were created as described previously [8]. Ngb-Tg mice and wild type (WT) mice (23–25 g; approximately 8 weeks old) were housed individually with controlled temperature (22–25 °C) and humidity (55%). A 12-h light-dark cycle was maintained, with lights on between 7 a.m. and 7 p.m. The mice were euthanized and brains and hearts collected for further analysis. All regents were purchased from Sigma-Aldrich except otherwise indicated in the methods section.
Establishment of neuroglobin-overexpressing cell lines
HT22 cells (Murine hippocampal cell line, passages 10–25) were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum (Life technologies, Grand Island, NY), 50 IU/ml penicillin, and 50 μg/ml streptomycin (Invitrogen, Carlsbad, CA), and incubated at 37 °C under 5% CO2. Medium was changed three times weekly and cultures were split at confluence. Recombinant plasmid (pTRUF12d-GFP-Ngb and pTRUF12d-GFP blank vectors) [6] was amplified and its sequence verified. HT22 wild type (HT22-WT) cells were plated at 1 × 105 cells per well on six-well plates and transfected with recombinant plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), Following transfection, cells were selected for neomycin (G418) (Invitrogen, Carlsbad, CA) resistance (2 mg/ml) for 2 weeks. As HT22 cells are resistant to neomycin [17], resistant cells were further sorted for GFP green fluorescence using a BD LSR-II flow cytometer. Then the transfected cells were seeded in 96-well culture plates at 1 cell per well. After culture for 2 wk, each colony with GFP green fluorescence was harvested for determination of Ngb protein level by immunocytochemistry and Western blotting. Ngb-expressing HEK 293 and vector cell lines were established using the same method except for selection by neomycin without sorting. Recombinant human Ngb (Prosepc, East Brunswick, NJ) was loaded as control for Western blot of Ngb.
Neuroprotection analysis
HT-22 cells were seeded at a density of 3,000 cells/well and were incubated overnight in 96-well plates in 100 μl of DMEM (high glucose with 1 mM pyruvate and 10% FBS) at 37 °C with 5% CO2. For oxygen glucose deprivation (OGD), HT22 cells were washed with PBS to remove residual glucose and FBS, switched to DMEM without glucose, pyruvate, and FBS, and placed in auto-controlled hypoxia chambers (BioSpherix, Lacona, NY, USA) with 0.5% O2 at 37 °C for 3 hr followed by 24 hr reoxygenation in normoxic O2 at 37 °C. For glutamate cytotoxicity, HT22 cells were treated with 5 mM glutamate. At 24 hr after treatment, cell viability was determined by Calcein AM assay (Invitrogen, Carlsbad, CA). Media was removed and replaced with a 1 μM solution of Calcein AM in PBS. Cells were incubated for 5 minutes at 37 °C and fluorescence was measured using a Tecan Infinite F200 plate reader (excitation 485 emission 530).
MitoSox Red (Invitrogen, Carlsbad, CA) was used to detect mitochondrial superoxide. After 16 hrs glutamate treatment, cells were stained with MitoSox Red according to the protocols provided by the manufacturers. The cells were incubated in pre-warmed PBS containing 0.5% BSA and MitoSox (5 μM) for 15 min at 37 °C, followed by washing with PBS. The fluorescent signal was visualized and photographed on a Zeiss fluorescence microscope (Axio Observer Z1).
Mitochondrial membrane potential was analyzed by TMRE (tetramethylrhodamine, ethyl ester) (Invitrogen, Carlsbad, CA). HT22 cells were incubated with glutamate for 16 hr. The media was then removed and the cells were washed once with PBS, then incubated in PBS containing 1 μM TMRE for 30 min at 37 °C. Cells were washed twice in PBS. The fluorescent signal was visualized and photographed on a Zeiss fluorescence microscope.
Primary neuron culture
Primary hippocampal neurons were cultured as described previously with minor modifications [18]. Briefly, hippocampus was dissected from postnatal day 0 mouse pups. Tissue was incubated with trypsin for 20 min at 37 °C and dissociated to single cells using fire-polished glass pipettes. Cells were resuspended in Neurobasal medium with B27 supplement and passed through 40-μm cell strainer, then counted and seeded on poly-L-lysine-coated cover glasses. Half of the medium was replaced with pre-warmed fresh medium every other day. Two days after seeding, cytosine arabinofuranoside (AraC) were added to the medium to a final concentration of 5 μM and cells were cultured for 2 days. Primary neurons were used for experiments at 7 days in vitro.
Cellular bioenergetics assay
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured by a Seahorse XF-24 Metabolic Flux Analyzer (Seahorse Bioscience, North Billerica MA). HT22 cells were plated at 8000/well and cultured on Seahorse XF-24 plates for 24 hr. On the day of analysis, cells were changed to unbuffered DMEM (DMEM base medium supplemented with 25 mM glucose, 10 mM sodium pyruvate, 31 mM NaCl, 2 mM glutamine, pH 7.4) and incubated at 37 °C in a non-CO2 incubator for 1 hr. All medium and injection reagents were adjusted to pH 7.4 on the day of assay. The initial 35 min established a baseline reading. Three subsequent injections followed, consisting of 1 μg/mL oligomycin (complex V inhibitor), 1 μM carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP, proton gradient uncoupler), and 0.1 μM rotenone (complex I inhibitor). After each injection, 4 time points were recorded with about 35 min between each injection. OCR and ECAR were automatically recorded and calculated by the Seahorse XF-24 software. Plates were saved and protein readings were measured for each well to confirm the presence of equal cell numbers per well. The percentage of change compared with the basal rates was calculated as the value of change divided by the average value of baseline readings.
For ATP assay, wild type, GFP- and Ngb-expressing HT22 cells were plated at 250,000 cells/well in 6-well plates and cultured overnight. ATP concentrations in lysates were quantified using an ATP determination kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Cells were rinsed with PBS and lysed with ATP-releasing buffer, and 10 μl of the lysate and ATP standard was added to a white Nunc 96-well plate. Then 100 μl of Luciferase Buffer was added to each well and immediately the luminescence was determined using an Infinite M200 Microplate reader (Tecan). Total ATP levels were normalized by protein amount and shown as a ratio to levels in HT22-WT cells.
Lipid analysis
Lipid content was analyzed as described by Pedrini et al. [19] with minor modifications. Cells were fixed in 4% formaldehyde for 60 min then rinsed with water. Subsequently, oil red O staining was performed as previously described [20]. Cells were washed with 60% isopropanol (isopropyl alcohol) for 5 min, then, incubated with oil red O working solution for 10 min. Microscopic images were obtained after washing the cells with double-distilled H2O. For quantitative lipid content analysis, HT-22 cells were stained with oil red O and the dye was eluted with 100% isopropanol. The OD of the elution solutions were measured at 500 nm and using 100% isopropanol as blank. The measurement was normalized to the protein concentration of each sample.
For brain and heart oil red O staining, frozen heart and brain sections were cut by a cryostat at 10 μm and mounted on slides. The sections were air dried for 30 min at room temperature, fixed in ice cold 10% formalin for 10 min, air dried again, and incubated with 60% isopropanol for 5 min. Then, the sections were stained with oil red O working solution at room temperature for 10 min, washed thoroughly in running tap water for 3 min, and examined by microscopy.
Glycogen analysis
Tissue was isolated and homogenized at 10 mg/100 μl in water. The homogenate was boiled for 5 min and then centrifuged at 13,000 × g for 10 min. The supernatant was collected for glycogen assay using a Glycogen Assay Kit (Sigma-Aldrich, St. Louis MO) and following the manufacturer's manual. Glycogen level was normalized to protein concentration of the tissue. Glycogen in cultured cells was isolated by ethanol precipitation to reduce glucose background for the assay. Briefly, the cell pellet was boiled in 200 μl of 30% KOH for 2 hr. After cooling on ice for 30 min, 400 μl of 100% ethanol was added to the sample and glycogen was precipitated by centrifugation at 12,000 × g for 15 min. The glycogen pellet was washed with cold 70% ethanol and, after air drying for 5 min, dissolved in water for glycogen assay analysis.
Immunohistochemistry and Western blot analysis
Cerebral hemispheres were incubated in 30% sucrose overnight, then frozen in OCT (Optimal Cutting Temperature) compound (VWR, San Francisco CA), and floating sections (20 μm) were obtained using a cryostat. For single-label immunohistochemistry, sections were blocked with 5% normal goat serum and incubated in primary antibodies at 4 °C overnight. Sections were then immunostained with Picture Plus immunohistochemistry kits (Invitrogen, Carlsbad, CA). 3,3′-diaminobenzidine tetrachloride (DAB) was used to visualize immunoreactivity. Primary antibodies against p-AMPKα, p-AMPKβ, and p-ACC (Cell Signaling, 1:100) were used. For controls, primary antibodies were omitted. Fluorescence microscopic images were captured using a Zeiss microscope with selective filter sets to visualize FITC, rhodamine, and DAPI. DAB-immunostained sections were visualized with a bright-field microscope.
For Western blots, cells were plated at a density of 200,000 cells/well in a 6-well plate. Cells were grown confluence and lysed in radioimmunoprecipitation assay (RIPA) buffer with protease and phosphatase inhibitors. Protein lysate was run in 6% or 10% polyacrylamide gels and transferred onto nitrocellulose. After blocking with 5% milk for 1 hr, nitrocellulose membranes were incubated with primary antibody overnight at 4 °C at the indicated concentrations (AMPKα, Cell Signaling, 1:500; p- AMPKα, Cell Signaling, 1:500; AMPKβ, Cell Signaling, 1:500; p-AMPKβ, Cell Signaling, 1:500; ACC, 1:500, Cell Signaling; p-ACC, Cell Signaling, 1:1000; β-Actin, Santa Cruz Biotechnology, 1:1000). The membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch) for 2 hr at room temperature (1:2000). Chemiluminescence was detected and analyzed with a UVP Biospectrum 500.
Statistical analysis
All data are presented as mean +/- S.E.M. The significance of differences among groups with one independent variable was determined by one-way ANOVA with a Tukey's multiple-comparisons test for planned comparisons between groups when significance was detected. The significance of differences among groups with two independent variables was determined by two-way ANOVA with a Bonferronni post-test for planned comparisons between groups when significance was detected. For all tests, p < 0.05 was considered significant.
Results
Establishment of Ngb-overexpressing HT-22 and HEK293 cell lines
We stably transfected pTRUF12d-GFP-Ngb [8] to HT-22 and HEK293 cells and overexpression of Ngb was confirmed by Western blots and immunocytochemistry. Ngb immunoreactivity with the same molecular weight as recombinant Ngb protein (Ngb control) was detected in HT22-Ngb transfected cells, but not in wild type (HT22-WT) or green fluorescent protein-transfected (HT22-GFP) cells (Figure 1A, left panel) and in HEK293-Ngb transfected but not in the HEK293-WT or and HEK293-GFP cells (Figure 1A, right panel). Immunohistochemistry confirmed that Ngb immunoreactivity was observed in HT22-Ngb but not HT22-WT or HT22-GFP cells (Figure 1B).
Fig 1. Establishment of stable Ngb expression cell lines.
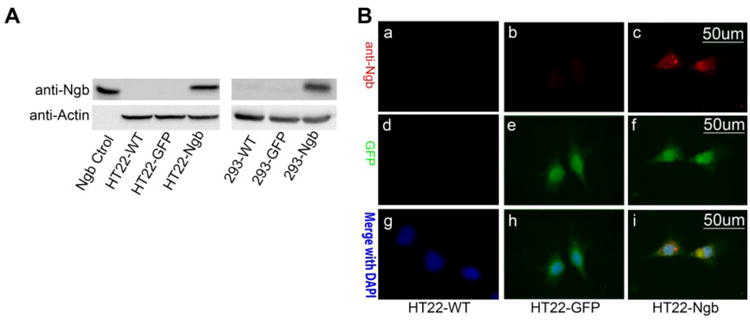
A. HT22 wide type cells were stably transfected with pTRUF12d-GFP-Ngb or pTRUF12d-GFP vector to create the Ngb-expressing HT22 cells (HT22-Ngb) and the control GFP-fluorescence HT22 cell lines (HT22-GFP). Western blot of Ngb was conducted to verify the overexpression of Ngb in HT22-Ngb cell line. Recombinant Ngb protein was used as the positive control (A, left panel). The Ngb-expressing HEK 293 cells were established and Ngb expression was verified by Western blot (A, right panel). B. Immunocytochemistry staining of Ngb and GFP was conducted to confirm the expression of Ngb in HT22-Ngb cells. HT22 wide type (HT22-WT) and vector transfected cell (HT22-GFP) were used as controls. Depicted are representative fluorescent microscopic images of Ngb, GFP and merge with DAPI staining.
A neuroprotective effect of Ngb has been identified in a wide range of models of neurological disorders in vitro and in vivo [6] [21,22,10,11,23-25]. We confirmed that Ngb overexpression protected HT-22 cells against cytotoxicity induced by oxygen and glucose deprivation (OGD) (Figure 2A, B) and glutamate (Figure 2C, D). We also determined the effect of Ngb overexpression on glutamate-induced mitochondrial superoxide production using MitoSox™ probe. A dramatic increase in MitoSox™ red fluorescence was observed in HT22-WT and HT22-GFP cells at 16 hrs after glutamate treatment, but not in HT22-Ngb cells (Figure 2E). Glutamate also induced mitochondrial membrane potential depolarization, which was attenuated by Ngb overexpression (Figure 2F).
Fig. 2. Ngb overexpression protects HT22 cells from OGD or glutamate injury.
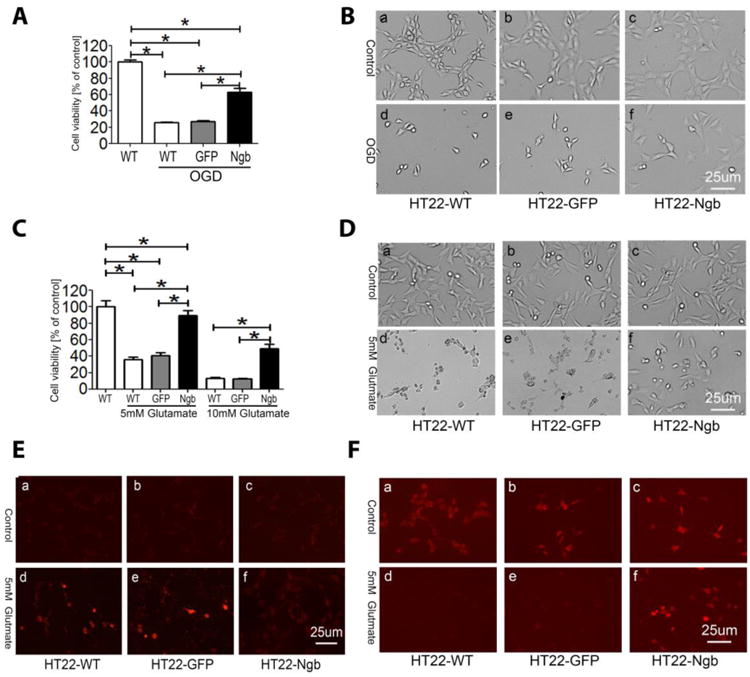
HT-22 cells (including HT22-WT, HT22-GFP, HT22-Ngb) were subjected to OGD (A, B) or glutamate at 5 or 10 mM (C, D) and cell viability was determined by Calcein AM assay (n=8). The experiment was repeated more than three times and similar results were observed. * P < 0.05. E-F. HT22 cells were treated with 5 mM glutamate for 16 hr and stained with MitoSox Red (E) or TMRE (F), then were visualized using a fluorescence microscope.
Ngb increases cellular ATP production and inhibits glycolysis
Next we determined the effect of Ngb on energy metabolism. Our cellular bioenergentics analysis demonstrated a significant increase in ATP concentration in HT22-Ngb compared with HT22-WT and HT22-GFP cells (Figure 3A). We investigated the effect of Ngb overexpression on cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) using a Seahorse XF24 Flux Analyzer. No significant difference in OCR was observed between HT22-WT, HT22-GFP, and HT22-Ngb cells under normal culture conditions, nor after complex V inhibition (oligomycin), uncoupling (FCCP), and complex I inhibition (rotenone) (Figure 3B). On the other hand, a significant decrease in basal ECAR was found in HT22-Ngb compared with HT22-WT and HT22-GFP cells (Figure 3C, D).
Fig. 3. Ngb expression enhances ATP production and decreases basal extracellular acidification rate in HT22 cells.
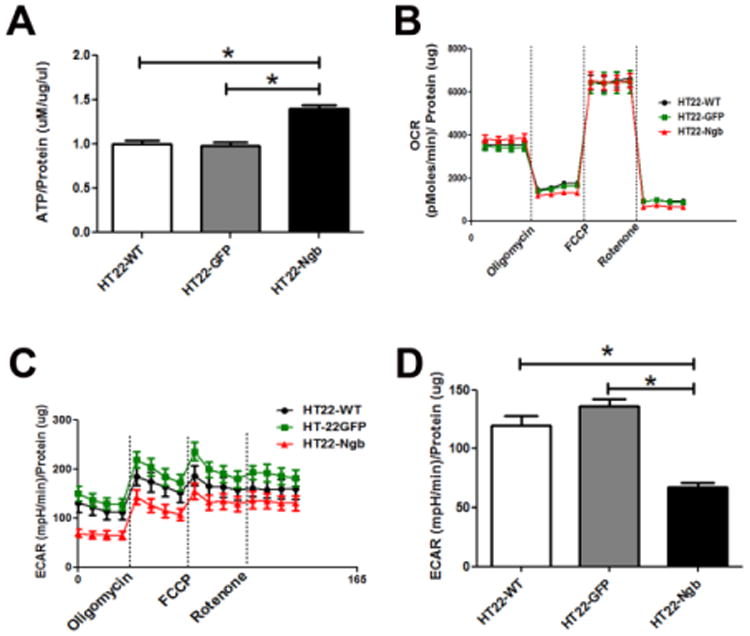
A. ATP content in HT22 cells was analyzed using an ATP determination kit. The ATP content of HT22-Ngb cells was significantly higher than that of HT22-WT and HT22-GFP cells. B. Oxygen consumption rate (OCR) recording at baseline and after treatment with oligomycin, FCCP, and rotenone. Oligomycin decreased OCR under all conditions. Injection of FCCP resulted in maximum OCR. Rotenone inhibited complex I causing a decrease in OCR. No difference in OCR was observed between HT22-Ngb, vector and wild type HT22 cells. C. Extracellular cellular acidification rate (ECAR) recording at baseline and after addition of oligomycin, FCCP, and rotenone. The ECAR baseline of HT22-Ngb cells was reduced compared to that of vector and wild type HT22 cells. D. Quantitative analysis of basal ECAR demonstrated reduction of ECAR in HT22-Ngb compared with HT22-WT and HT22-GFP cells. * p<0.05 vs. control groups.
Ngb inhibits AMPK signaling and increases lipid and glycogen contents in HT22 cells
We investigated the effect of Ngb expression on AMPK signaling by Western blot analysis in HT22 cells. Ngb overexpression significantly decreased the expression and phosphorylation of AMPKβ. Ngb overexpression also showed a trend toward decreasing the expression and phosphorylation of AMPKα, although no statistical significance was reached (Figure 4A, B). AMPK phosphorylates and inactivates acetyl-CoA-carboxylase (ACC), a key enzyme for fatty acid biosynthesis. Consistent with the inhibitory effect of Ngb overexpression on AMPK, a significant reduction of ACC phosphorylation was observed in Ngb-overexpressing HT22 compared with HT22-WT and HT22-GFP cells, suggesting that Ngb overexpression inhibits AMPK signaling (Figure 4A, B).
Fig. 4. Ngb expression inhibits AMPK signaling in HT22 cells.
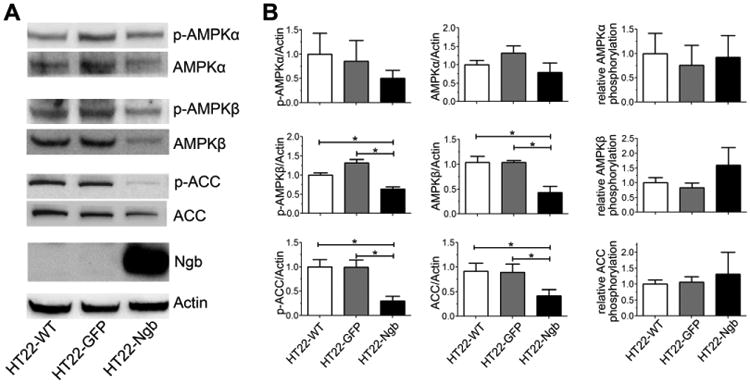
A. Representative western blots depict decreased AMPKβ, p-AMPKβ, ACC, and p-ACC in HT22-Ngb compared with HT22-WT and HT22-GFP cells. B. Quantitative western blot analysis showed that Ngb overexpression decreased protein levels of AMPKβ, p-AMPKβ, ACC, and p-ACC in HT22 cells. *, p < 0.05.
We further explored the effect of Ngb overexpression on lipid and glycogen metabolism in HT22 cells. Oil red O staining demonstrated that Ngb overexpression markedly increased lipid content in HT22-Ngb compared with HT22-WT and HT22-GFP cells (Figure 5A). Quantitative lipid assay also indicated a significant increase of lipid content in HT22-Ngb compared with HT22-WT and HT22-GFP cells (Figure 5B). In addition, quantitative glycogen analysis indicated that Ngb overexpression increased glycogen content in HT22 cells (Figure 5C).
Fig. 5. Ngb overexpression increases lipid and glycogen content in HT-22 cells.
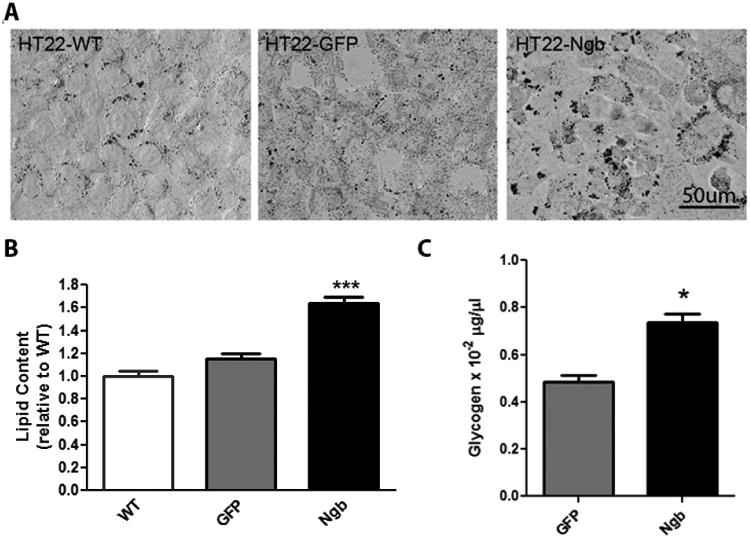
A. Representative oil red O staining showed that Ngb overexpression increased lipid content in HT22-Ngb compared with HT22-WT and HT22-GFP cells (controls). B. Quantitative lipid analysis demonstrated that lipid content in HT22-Ngb cells was higher than that in HT22-WT and HT22-GFP cells. C. Quantitative glycogen analysis demonstrated that glycogen content was increased in HT22-Ngb cells. * p < 0.05.
Ngb overexpression inhibits AMPK signaling and induces glycogen synthase activation in mouse cortical neurons
We determined the effect of Ngb overexpression on the AMPK pathway in brain using Ngb-overexpressing transgenic (Ngb-Tg) mice. Immunohistochemistry demonstrated a substantial reduction of phospho- (p-) AMPKα, p-AMPKβ, and p-ACC in Ngb-Tg mouse brain cortex (Figure 6A). Western blot analysis indicated that Ngb overexpression significantly decreased the expression and phosphorylation of AMPKα and ACC (Figure 6B-C).
Fig. 6. Ngb overexpression inhibits AMPK signaling in mouse brain.
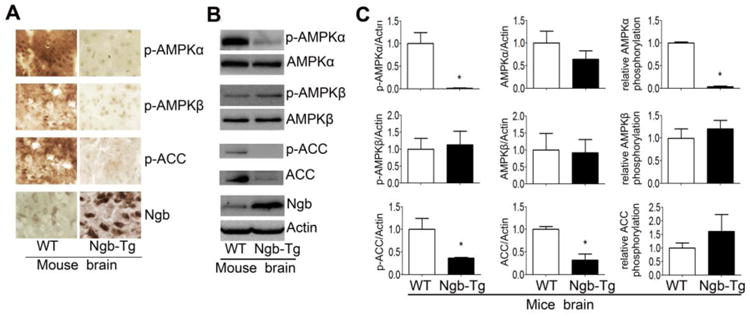
A. Representative immunohistochemical staining of AMPK signaling mediators and Ngb expression in brain indicated decreased p-AMPKα, p-AMPKβ and p-ACC in Ngb-Tg mouse brain. B. Representative western blots of AMPK pathway mediators and Ngb demonstrated reduction of p-AMPKα, ACC, p-ACC in Ngb-Tg mouse brain. C. Quantitative western blot analysis showed that Ngb overexpression decreased p-AMPKα, ACC, p-ACC, and the phosphorylation ratio of AMPKα in Ngb-Tg mouse brain. * p < 0.05 vs. WT.
We investigated the effect of Ngb overexpression on brain glycogen synthesis. Our quantitative glycogen analysis indicated no significant difference in whole brain glycogen content between wild type and Ngb-Tg mice. Immunohistochemistry showed that glycogen synthase (GS) was mainly expressed in astrocytes, while p-GS localized to the nucleus of neurons. In addition, a remarkable decrease in nuclear p-GS and increase of cytosolic GS were found in cortical neurons of Ngb overexpressing mouse brain (Figure 7A, B). We further determined the effect of Ngb expression and glycogenesis in primary neuron cultures. Consistent with findings in whole brain, overexpression of Ngb in primary neurons was associated with a decrease in p-GS (Figure 7C).
Fig. 7. Ngb overexpression inhibits phosphorylation of glycogen synthase in mouse cortical neurons.
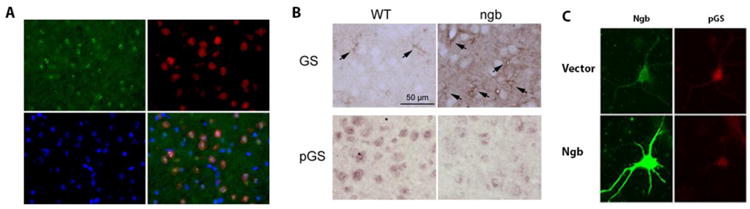
A. Representative immunohistochemistry of phospho-glycogen synthase (pGS, red) and NeuN (green) and merged image with DAPI (blue) demonstrated nuclear localization of pGS in mouse cortical neurons. B. Representative immunohistochemistry of GS and pGS demonstrated predominant localization of GS in astrocyte (arrows). Cortical neurons from Ngb-overexpressing mice showed increased cytosolic (active) GS and reduced nuclear (inactive) pGS compared with neurons from wild type (WT) mice. C. Representative immunocytochemistry of Ngb and pGS demonstrated that Ngb overexpression decreased and activated pGS in primary neuron cultures.
Ngb overexpression inhibits AMPK signaling and increases lipid and glycogen content in mouse heart
The transgenic Ngb mouse was generated under the control of a chicken β-actin promoter, resulting in enhanced Ngb expression in both brain and heart [7,8]. Therefore, we determined the effect of Ngb overexpression on AMPK signaling in mouse heart. Western blot analysis showed significant reductions in heart p-AMPKα and p-ACC (Figure 8A, B).
Fig. 8. Ngb overexpression inhibits AMPK pathway and increases lipid and glycogen content in mouse heart.
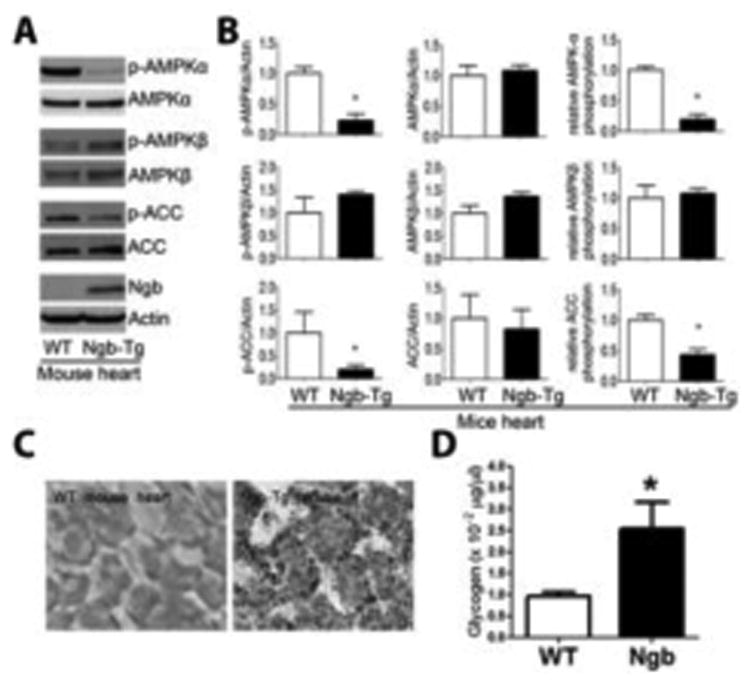
A. Representative Western blots of AMPK pathway mediators and Ngb demonstrated decreased p-AMPKα and p-ACC in Ngb-Tg compared with WT mouse heart. B. Quantitative western blot analysis indicated reduction of p-AMPKα, p-ACC, and phosphorylation ratio of AMPKα and ACC in Ngb-Tg mouse heart. C. Representative oil red O staining of mouse hearts demonstrated that Ngb overexpression increases lipid content in mouse heart. D. Quantitative glycogen analysis demonstrated that Ngb overexpression increases glycogen content in mouse heart. *, p < 0.05.
Finally, we investigated the effect of Ngb overexpression on heart lipid and glycogen content. Oil red O staining demonstrated that Ngb overexpression increased heart lipid content (Figure 6C). Quantitative glycogen analysis indicated that Ngb overexpression also increased heart glycogen content (Figure 8D).
Discussion
Ngb shares little amino-acid sequence similarity with vertebrate myoglobin (<21% identity) and hemoglobin (< 25% identity). It has an oxygen affinity (1.9-2.6 torr) 10 fold higher than that of hemoglobin (∼26 torr), but lower than that of myoglobin (∼1 torr) [4]. Given the lack of sequence homology and distinctive oxygen affinity, neuroglobin may exert unique functions other than the O2 delivery role of hemoglobin and myoglobin. In addition, the relatively low concentration of Ngb in brain compared to myoglobin in muscle suggests that Ngb is unlikely to be a primary O2 reservoir in neurons. Consistent with this notion, both a previous study [6] and our current data indicated that Ngb expression does not affect cellular O2 consumption rate. Our current study provides the first evidence that Ngb inhibits AMPK signaling, and thus regulates neuronal energy metabolism.
Brain is characterized by high metabolic activity with tight regulatory mechanisms to ensure adequate energy substrate delivery in accordance with neuronal activity. AMPK, a heterotrimeric protein comprising catalytic α (α1, α2), and regulatory β (β1, β2) and γ (γ1, γ2, γ3) subunits, is strategically located at the crossroads of multiple metabolic pathways [26]. AMPK is activated by events that compromise cellular ATP production or increase ATP consumption [27,26]. Activation is stimulated by binding of AMP to the γ-subunit, which is antagonized by high concentrations of ATP [28]. In the brain, AMPK regulates energy metabolism in both physiological and pathophysiological conditions.
There is increasing evidence that Ngb may play a role in ATP production [13]. Knockdown of Ngb has been found to reduce the activity of mitochondrial oxidative phosphorylation complex I and III [29]. Our study demonstrates that Ngb overexpression increases intracellular ATP content, thus inhibiting AMPK signaling. Cells generate ATP through a combination of oxidative and glycolytic metabolism. Interestingly, our cellular bioenergetic analysis indicates that Ngb overexpression significantly inhibits extracellular acidification rate, suggesting Ngb may inhibit glycolysis and play a critical role in glucose metabolism. Furthermore, the inhibitory effect of Ngb on glycolysis suggests that the increase in intracellular ATP concentration upon Ngb overexpression might be due to increased mitochondrial efficiency, and hence ATP production.
Activated AMPK stimulates catabolism (glycolysis and fatty acid oxidation) and concomitantly inhibits anabolic reactions (glycogenesis and lipogenesis) [14]. AMPK inhibits glycogen and fatty acid synthesis by phosphorylating and inactivating GS and ACC, respectively [26]. On the other hand, loss of AMPK signaling increases ATP levels and promotes anabolic metabolism [30]. Consistent with the inhibition of AMPK signaling, we observed an increase of glycogenesis and lipogenesis upon Ngb overexpression both in vitro and in vivo.
Brain energy homeostasis is mainly maintained by oxidative metabolism of glucose, the obligatory energy substrate of brain in normal circumstances. Moreover, neurons have poor nutrient storage capacity with no glycogen and lipid synthesis and are particularly vulnerable to ischemic insult. Nonetheless, glycogen has been found in both embryonic neural and glial tissue [31,32]. In the adult brain, glycogen is found predominantly in astrocytes and can be metabolized to lactate for transfer to neurons [33]. Recent studies indicate that neurons have the enzymatic machinery for glycogenesis but in less or inactivated state [34-37]. Accordingly, our study demonstrated that glycogen synthase was indeed expressed in neurons, but at the inactive phosphorylation state in the nucleus. Overexpression of Ngb activated glycogen synthase and increased glycogen content. Interestingly, neuronal glycogen metabolism has been indicated to contribute to tolerance to hypoxia [37]. Together with the well-established neuroprotective action of Ngb, we speculated that activation of glycogen synthesis might partially underlie the neuroprotection of Ngb. In addition, our data demonstrated that Ngb overexpression decrease glycolysis, which might also contribute to the increase of cellular glycogen level in the Ngb overexpression cells. However, it is unclear which contributes more to the outcome. Future studies on the effect of Ngb overexpression on the activation of key enzymes might provide further insight in the action of Ngb in glucose metabolism.
Consistent with increased Ngb expression in the heart of Ngb-overexpressing mice [7,8], we observed increased glycogen content. However, whole brain glycogen content was not increased. This may not be surprising given that AMPK and Ngb are predominantly expressed in neurons whereas glycogenesis is most active in astrocytes. AMPK inhibits glycogen synthesis by phosphorylating and inactivating glycogen synthase [38-40]. Our immunohistochemistry demonstrated that, while glycogen synthase is mainly localizes to astrocytes, inactive glycogen synthase is predominately present in the neuronal nucleus, which is consistent with the nuclear localization of p-AMPKα [41]. In addition, a substantial increase of cytosolic (active) GS and reduction of phosphorylated (inactive) GS were found in cortical neurons of Ngb transgenic compared with wild type mouse brains. These data suggest that Ngb overexpression enhances glycogen synthesis via inhibiting AMPK signaling.
Brain is one of the most lipid-rich organs and primarily relies on local synthesis because the blood-brain barrier prevents uptake of lipid from the circulation. Neurons have a particularly high demand for lipid to form and maintain axons, dendrites and synaptic connections. In adult brain, neurons cannot produce energetically expensive lipid efficiently and rely on astrocytes for lipid synthesis and shuttling. Indeed, lipid synthetic pathways are highly enriched in astrocytes [42]. The lack of lipid synthesis in neurons is also consistent with the exclusive neuronal localization of AMPK. Consistent with the inhibitory action of Ngb in vitro, we found that lipid content was increased in heart of Ngb-overexpressing compared with wild type mice.
Neurons are characterized by high metabolic activity and execute fast-paced energy-demanding processes. Our study suggests that Ngb overexpression inhibits AMPK signaling and enhances anabolism in neurons. Although the expression level of Ngb in adult brain is rather low with the concentration of ∼100-fold less than that of myoglobin in skeletal muscle [4], Ngb expression is at the highest in developing and neonatal brain [5,43]. This is coincident with the peak of brain lipogenesis [44,45] and with the presence of glycogen in neurons at neonatal brain [32]. AMPK expression is also highest in developing neurons [15,46]. Thus, neuronal metabolism might be orchestrated by the fine tuning between the Ngb expression level and AMPK activation. Ngb overexpression has been indicated to be neuroprotective both in vitro and in vivo. Paradoxically, Ngb-deficiency has also been found to enhance the expression of a few hypoxia-dependent response genes and reduce infarct size in an ischemic stroke model [47,23]. Therefore, Ngb expression level might play a critical role in neuron metabolism through AMPK pathway. Further studies using Ngb overexpression, knockdown, and knockout approaches may provide additional insight for the role of Ngb in neuron metabolism and its effect on ischemic stroke outcome.
In summary, our current study indicates that Ngb plays important role in energy metabolism. Overexpression of Ngb in vitro and in vivo promotes cellular anabolism evidenced by increased glycogenesis and lipogenesis. The enhancement of anabolism upon Ngb overexpression is associated with inhibition of AMPK signaling. Given the well-established role of AMPK signaling in energy metabolism, we predict that Ngb overexpression inhibits AMPK activation, hence enhancing anabolism (Figure 9).
Fig. 9. Proposed mechanism.
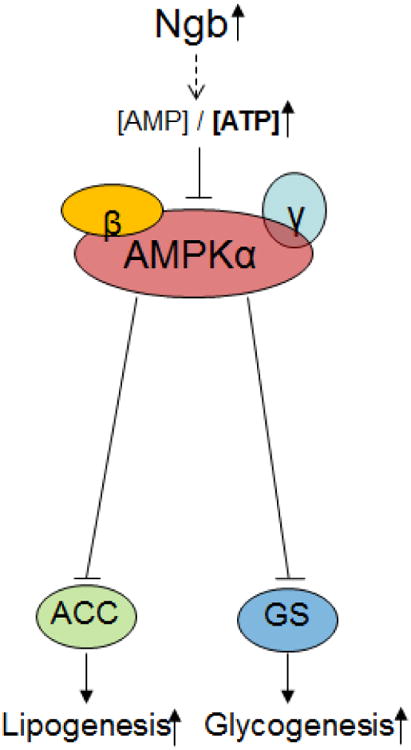
Ngb increases lipogenesis and glycogenesis through AMPK inhibition.
Acknowledgments
This work was partly supported by National Institutes of Health grants R01NS054651 (SY), R01NS088596 (SY), and R01AG021980 (KJ); American Heart Association SDG 16960084 (RL); The National Key Clinical Specialty Discipline Construction program, China (NW); Grant No. 81171114 of Natural Science Foundation of China (BC), Grant No. 2011J01158 Fujian Provincial Natural Science Foundation (BC) and Grant No. 2011-CXB-12 of Fujian Provincial medical innovation Project (BC); and the Buck Institute for Research on Aging (DAG).
Abbreviations
- ACC
acetyl-CoA carboxylase
- AMPK
AMP-activated protein kinase
- ECAR
extracellular acidification rate
- GS
glycogen synthase
- Ngb
neuroglobin
- OCR
oxygen consumption rate
- OGD
oxygen glucose deprivation
References
- 1.Weber RE, Vinogradov SN. Nonvertebrate hemoglobins: functions and molecular adaptations. Physiol Rev. 2001;81(2):569–628. doi: 10.1152/physrev.2001.81.2.569. [DOI] [PubMed] [Google Scholar]
- 2.Pesce A, Bolognesi M, Bocedi A, Ascenzi P, Dewilde S, Moens L, Hankeln T, Burmester T. Neuroglobin and cytoglobin. Fresh blood for the vertebrate globin family. EMBO Rep. 2002;3(12):1146–1151. doi: 10.1093/embo-reports/kvf248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burmester T, Ebner B, Weich B, Hankeln T. Cytoglobin: a novel globin type ubiquitously expressed in vertebrate tissues. Mol Biol Evol. 2002;19(4):416–421. doi: 10.1093/oxfordjournals.molbev.a004096. [DOI] [PubMed] [Google Scholar]
- 4.Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407(6803):520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- 5.Burmester T, Hankeln T. What is the function of neuroglobin? The Journal of experimental biology. 2009;212(Pt 10):1423–1428. doi: 10.1242/jeb.000729. [DOI] [PubMed] [Google Scholar]
- 6.Sun Y, Jin K, Mao XO, Zhu Y, Greenberg DA. Neuroglobin is upregulated by and protects neurons from hypoxic-ischaemic injury. Proc Natl Acad Sci U S A. 2001 doi: 10.1073/pnas.251466698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan AA, Wang Y, Sun Y, Mao XO, Xie L, Miles E, Graboski J, Chen S, Ellerby LM, Jin K, Greenberg DA. Neuroglobin-overexpressing transgenic mice are resistant to cerebral and myocardial ischemia. Proc Natl Acad Sci U S A. 2006;103(47):17944–17948. doi: 10.1073/pnas.0607497103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan AA, Sun Y, Jin K, Mao XO, Chen S, Ellerby LM, Greenberg DA. A neuroglobin-overexpressing transgenic mouse. Gene. 2007;398(1-2):172–176. doi: 10.1016/j.gene.2007.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin K, Mao XO, Xie L, Khan AA, Greenberg DA. Neuroglobin protects against nitric oxide toxicity. Neurosci Lett. 2008;430(2):135–137. doi: 10.1016/j.neulet.2007.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Z, Fan X, Lo EH, Wang X. Neuroprotective roles and mechanisms of neuroglobin. Neurol Res. 2009;31(2):122–127. doi: 10.1179/174313209X389866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Yu Z, Guo S, Lee SR, Xing C, Zhang C, Gao Y, Nicholls DG, Lo EH, Wang X. Effects of neuroglobin overexpression on mitochondrial function and oxidative stress following hypoxia/reoxygenation in cultured neurons. Journal of neuroscience research. 2009;87(1):164–170. doi: 10.1002/jnr.21826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt M, Giessl A, Laufs T, Hankeln T, Wolfrum U, Burmester T. How does the eye breathe? Evidence for neuroglobin-mediated oxygen supply in the mammalian retina. J Biol Chem. 2003;278(3):1932–1935. doi: 10.1074/jbc.M209909200. [DOI] [PubMed] [Google Scholar]
- 13.Yu Z, Poppe JL, Wang X. Mitochondrial mechanisms of neuroglobin's neuroprotection. Oxidative medicine and cellular longevity. 2013;2013:756989. doi: 10.1155/2013/756989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes & development. 2011;25(18):1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17(1):45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 16.Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer's disease and other tauopathies. Acta neuropathologica. 2011;121(3):337–349. doi: 10.1007/s00401-010-0759-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukui M, Choi HJ, Zhu BT. Rapid generation of mitochondrial superoxide induces mitochondrion-dependent but caspase-independent cell death in hippocampal neuronal cells that morphologically resembles necroptosis. Toxicol Appl Pharmacol. 2012;262(2):156–166. doi: 10.1016/j.taap.2012.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beaudoin GM, 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7(9):1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- 19.Pedrini MT, Kranebitter M, Niederwanger A, Kaser S, Engl J, Debbage P, Huber LA, Patsch JR. Human triglyceride-rich lipoproteins impair glucose metabolism and insulin signalling in L6 skeletal muscle cells independently of non-esterified fatty acid levels. Diabetologia. 2005;48(4):756–766. doi: 10.1007/s00125-005-1684-8. [DOI] [PubMed] [Google Scholar]
- 20.Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc. 2013;8(6):1149–1154. doi: 10.1038/nprot.2013.055. [DOI] [PubMed] [Google Scholar]
- 21.Sun Y, Jin K, Peel A, Mao XO, Xie L, Greenberg DA. Neuroglobin protects the brain from experimental stroke in vivo. Proc Natl Acad Sci U S A. 2003;100(6):3497–3500. doi: 10.1073/pnas.0637726100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Liu J, Zhu H, Tejima E, Tsuji K, Murata Y, Atochin DN, Huang PL, Zhang C, Lo EH. Effects of neuroglobin overexpression on acute brain injury and long-term outcomes after focal cerebral ischemia. Stroke; a journal of cerebral circulation. 2008;39(6):1869–1874. doi: 10.1161/STROKEAHA.107.506022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hundahl CA, Luuk H, Ilmjarv S, Falktoft B, Raida Z, Vikesaa J, Friis-Hansen L, Hay-Schmidt A. Neuroglobin-deficiency exacerbates Hif1A and c-FOS response, but does not affect neuronal survival during severe hypoxia in vivo. PloS one. 2011;6(12):e28160. doi: 10.1371/journal.pone.0028160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe S, Takahashi N, Uchida H, Wakasugi K. Human neuroglobin functions as an oxidative stress-responsive sensor for neuroprotection. J Biol Chem. 2012;287(36):30128–30138. doi: 10.1074/jbc.M112.373381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shang A, Feng X, Wang H, Wang J, Hang X, Yang Y, Wang Z, Zhou D. Neuroglobin upregulation offers neuroprotection in traumatic brain injury. Neurol Res. 2012;34(6):588–594. doi: 10.1179/1743132812Y.0000000052. [DOI] [PubMed] [Google Scholar]
- 26.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8(10):774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 27.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. The Journal of clinical investigation. 2006;116(7):1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403(1):139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lechauve C, Augustin S, Cwerman-Thibault H, Bouaita A, Forster V, Celier C, Rustin P, Marden MC, Sahel JA, Corral-Debrinski M. Neuroglobin involvement in respiratory chain function and retinal ganglion cell integrity. Biochim Biophys Acta. 2012;1823(12):2261–2273. doi: 10.1016/j.bbamcr.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 30.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, Mamer OA, Avizonis D, DeBerardinis RJ, Siegel PM, Jones RG. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17(1):113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oz G, Seaquist ER, Kumar A, Criego AB, Benedict LE, Rao JP, Henry PG, Van De Moortele PF, Gruetter R. Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. American journal of physiology. 2007;292(3):E946–951. doi: 10.1152/ajpendo.00424.2006. [DOI] [PubMed] [Google Scholar]
- 32.Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. 2007;55(12):1263–1271. doi: 10.1002/glia.20557. [DOI] [PubMed] [Google Scholar]
- 33.DiNuzzo M, Maraviglia B, Giove F. Why does the brain (not) have glycogen? Bioessays. 2011;33(5):319–326. doi: 10.1002/bies.201000151. [DOI] [PubMed] [Google Scholar]
- 34.Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B, Criado-Garcia O, Fernandez-Sanchez E, Medrano-Fernandez I, Dominguez J, Garcia-Rocha M, Soriano E, Rodriguez de Cordoba S, Guinovart JJ. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nature neuroscience. 2007;10(11):1407–1413. doi: 10.1038/nn1998. [DOI] [PubMed] [Google Scholar]
- 35.Magistretti PJ, Allaman I. Glycogen: a Trojan horse for neurons. Nature neuroscience. 2007;10(11):1341–1342. doi: 10.1038/nn1107-1341. [DOI] [PubMed] [Google Scholar]
- 36.Duran J, Tevy MF, Garcia-Rocha M, Calbo J, Milan M, Guinovart JJ. Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO molecular medicine. 2012;4(8):719–729. doi: 10.1002/emmm.201200241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, Martinez-Pons C, Milan M, Guinovart JJ. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab. 2014 doi: 10.1038/jcbfm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009;9(1):23–34. doi: 10.1016/j.cmet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McBride A, Hardie DG. AMP-activated protein kinase--a sensor of glycogen as well as AMP and ATP? Acta Physiol (Oxf) 2009;196(1):99–113. doi: 10.1111/j.1748-1716.2009.01975.x. [DOI] [PubMed] [Google Scholar]
- 40.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89(3):1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 41.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280(21):20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- 42.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28(1):264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hummler N, Schneider C, Giessl A, Bauer R, Walkinshaw G, Gassmann M, Rascher W, Trollmann R. Acute hypoxia modifies regulation of neuroglobin in the neonatal mouse brain. Exp Neurol. 2012;236(1):112–121. doi: 10.1016/j.expneurol.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Quan G, Xie C, Dietschy JM, Turley SD. Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Brain Res Dev Brain Res. 2003;146(1-2):87–98. doi: 10.1016/j.devbrainres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 45.Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45(8):1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Ramamurthy S, Chang E, Cao Y, Zhu J, Ronnett GV. AMPK activation regulates neuronal structure in developing hippocampal neurons. Neuroscience. 2014;259:13–24. doi: 10.1016/j.neuroscience.2013.11.048. [DOI] [PubMed] [Google Scholar]
- 47.Raida Z, Hundahl CA, Kelsen J, Nyengaard JR, Hay-Schmidt A. Reduced infarct size in neuroglobin-null mice after experimental stroke in vivo. Exp Transl Stroke Med. 2012;4(1):15. doi: 10.1186/2040-7378-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]