

Abstract
Context: The typically indolent behavior of pituitary tumors is juxtaposed with high rates of tumor cell invasion into adjacent dural structures, and occasional aggressive behavior. Although clinically significant invasion and malignant transformation remain uncommon, there are limited treatment options available for the management of these aggressive tumors. Recently, case reports have described efficacy of temozolomide for the treatment of aggressive pituitary tumors.
Design: Seven patients with aggressive pituitary tumors have been treated with temozolomide. We compared O6-methylguanine methyltransferase (MGMT) promoter methylation and MGMT expression in 14 surgical specimens from these seven patients and correlated these molecular features with the clinical response to temozolomide.
Results: Significant tumor regression was seen in two patients (29%), a 20% reduction in tumor volume with subsequent stable tumor size was noted in one patient, arrest of tumor growth occurred in three patients, and progressive metastatic disease developed during treatment in one patient. The DNA promoter site for MGMT was unmethylated in all 14 adequate specimens, and variable MGMT expression was seen in all 14 cases. There was no correlation between MGMT expression and clinical outcomes.
Conclusions: We conclude that medical therapy with temozolomide can be helpful in the management of life-threatening pituitary tumors that have failed to respond to conventional treatments. The optimal duration of treatment in patients with stabilization or reduction of tumor size has not been established, and long-term follow up studies are needed.
Adjuvant medical therapy with temozolomide can be helpful for the management of life-threatening invasive pituitary tumors that have failed to respond to conventional treatments.
The natural history of pituitary adenomas varies widely. The typically indolent growth rate of these tumors is juxtaposed with high rates of tumor cell invasion into adjacent dural structures (1). Clinically significant invasion and malignant transformation remain uncommon, but morbidity and mortality are high when aggressive tumor behavior occurs (2). With the exception of dopamine agonist therapy for prolactinomas, surgery has long been the first line treatment for pituitary tumors. Options for medical treatment are limited, and none of the current therapies is tumoricidal (3, 4, 5, 6). When invasive tumors occur, repeat surgery and radiation therapy are second-line therapies that have variable efficacy depending on the characteristics of the tumor. Extrasellar tumor extension, proximity to cranial nerves and critical blood vessels, and the delayed therapeutic effects of radiotherapy can all limit the efficacy of these treatment modalities (7, 8). These therapeutic shortfalls are most critical in the management of highly invasive adenomas and pituitary carcinomas.
Therapeutic efforts have been made with conventional chemotherapeutic agents that cross the blood-brain barrier to treat invasive and malignant pituitary tumors, but efficacy has been poor, and partial responses are generally short-lived (9, 10, 11, 12, 13). Temozolomide is an oral alkylating agent that readily crosses the blood-brain barrier and has been shown to be efficacious in malignant gliomas. Several recent case reports have reported efficacy of temozolomide for aggressive pituitary tumors (14, 15, 16, 17, 18). The mechanism by which temozolomide acts on pituitary tumors has not been established, but in general, the alkylating agents disrupt gene transcription by inducing DNA damage by attaching a methyl group to the guanine base. Rapidly dividing tumor cells often lack competent DNA repair mechanisms, and thus are more prone to cytotoxic DNA disruption (19, 20). Epigenetic inactivation of the gene for the DNA repair enzyme O6-methylguanine methyltransferase (MGMT) via promoter hypermethylation has been show to predict therapeutic response to temozolomide in glioblastomas (21, 22), and retrospective analyses of pituitary tumors have reported MGMT methylation and expression by immunohistochemistry (17, 23).
We report a series of seven patients with aggressive pituitary tumors that have been treated with temozolomide. We compare MGMT promoter methylation status, as established by methylation-specific PCR (MSP), and nuclear MGMT expression by immunohistochemistry (IHC) and correlate these molecular features with the clinical response.
Patients and Methods
From the records of the University of Virginia Pituitary Clinic, we identified all of the patients with invasive pituitary tumors who received temozolomide as adjuvant medical therapy. Medical records were reviewed to establish duration of treatment, tumor response, tolerability of temozolomide treatment, and adverse events. Tumor response was established by review of serial magnetic resonance imaging (MRI) studies, and in the case of hormonally active tumors, the hormone response to treatment. The chemotherapeutic dose, the calendar regimen employed, and the number of cycles administered were established by review of medical records. Tumor volume was calculated as elliptical volume—the product of the largest diameter in three planes from coronal and sagittal postgadolinium T1 MRI images, multiplied by 0.5. The study was approved by the University of Virginia Institutional Review Board.
The presence of tumor within selected specimen blocks and confirmation of the cell lineage of each tumor were made by review of the standard histological, IHC, and ultrastructural studies. MGMT methylation was established by the MSP. The intrapatient stability of MGMT promoter methylation was determined in surgical specimens obtained before and after radiation therapy in patients who underwent multiple operations during the course of multimodality treatment; this includes pre- and postradiation specimens in case 3. IHC was used to determine nuclear expression of the MGMT DNA repair enzyme.
Methylation-specific PCR
Genomic DNA was isolated from three to five 10-μm paraffin tissue sections containing at least 70% tumor (QIAamp DNA Mini Kit; QIAGEN, Valencia, CA). DNA methylation patterns in the CpG island of the MGMT gene (GenBank accession number AL355531, nucleotides 46931–47011) was determined by chemical (bisulfite) modification of unmethylated, but not methylated, cytosine to uracil (CpGenome Fast DNA Modification Kit; from Chemicon, now part of Millipore, Billerica, MA). The bisulfite-treated DNA was precipitated, rehydrated, and amplified by MSP using primers specific for either the methylated or the modified unmethylated DNA (18). PCR amplification with Taq-Gold (Applied Biosystems, Foster City, CA) was performed, and PCR products were analyzed in duplicate parallel runs by capillary gel electrophoresis (ABI 3130xl; Applied Biosystems) with an expected product of 93 bp for methylated DNA and 80 bp for unmethylated DNA. Bisulfite-treated CPGenome Universal Unmethylated DNA (Millipore) and SssI methyltransferase (New England Biolabs, Ipswich, MA)-treated human DNA were used as negative and positive methylation controls, respectively. No template controls were performed. The sensitivity of the assay based on DNA dilution studies is at least 1:1000. A dichotomous result of methylated or unmethylated MGMT promoter was reported for each case.
Immunohistochemistry
IHC for MGMT was performed in formalin-fixed, paraffin-embedded tissue using the avidin-biotin-peroxidase technique (24). Antigen retrieval by boiling in a citrate solution (Target Retrieval Solution, pH 6.1; Dako, Carpinteria, CA) was performed using a pressure cooker (25). Tissue sections were incubated with anti-MGMT monoclonal antibody (clone MT3.1; 1:50 dilution; Chemicon) using the automated staining system (DakoCytomation Autostainer Plus; Dako, Glostrup, Denmark). Positive and negative controls were run concomitantly in all analyses. Tissue sections from human glioblastomas in which MGMT methylation status was known were used to establish positive IHC control. In addition, normal tissues adjacent to the adenoma specimens including blood vessels were used as positive internal controls.
IHC scoring and statistical analysis
IHC specimens were analyzed independently by three reviewers using a semiquantitative method to determine both the proportion of MGMT-positive cells and the intensity of the immunoreactivity. Reviewers were blinded to clinical outcome. Positivity was quantified as negative or 1 for tumors with less than 10% of cells positive for MGMT; 2 for tumors with 10–50% positive cells; or 3 for tumors with more than 50% positive cells. Intensity was scored on a scale where 1 = faint, 2 = moderate, and 3 = distinct immunoreactivity equivalent to the intensity seen in internal controls. Marginals were not known to the reviewers a priori; thus, interoperator variability was determined using Randolph’s multirater variation on Fleiss’ free-marginal κ equation (26, 27, 28, 29). The free-marginal κ statistic for the semiquantitative IHC analysis was determined for interoperator agreement for both the whole integer score (IHC positivity score 0 vs. 1 vs. 2, etc.) and the dichotomous outcome measure of positive (scores of 1, 2, or 3) vs. negative.
Results
We identified seven cases of invasive pituitary tumors that had been treated with temozolomide between 2005 and 2009. All seven patients were initially treated with surgery. Six of the seven patients had repeat surgery before receiving adjuvant treatment. A total of 19 operations were performed on the seven patients during multimodality management. We eliminated redundant surgical specimens obtained in rapid sequence within the same treatment phase (i.e. before or after radiation). Fifteen specimens from the seven patients were analyzed by IHC and MSP. One specimen was too necrotic to yield results in our molecular studies.
Five of the seven patients were treated with conventional radiation or GammaKnife stereotactic radiosurgery (GKR) before temozolomide treatment. The interval between the last radiation dose and temozolomide treatment ranged from 5 months to 17 yr (Table 1). The duration of temozolomide treatment ranged from 2 to 18 months. The initial treatment regimen for all seven patients was a 28-d cycle of temozolomide 75 mg/m2·d for 21 d followed by 7 d off therapy. One patient had a dose adjustment after 12 standard cycles to 75 mg/m2·d for 18 d, 10 d off therapy in an effort to reduce side effects of fatigue and headache. One patient with a null cell tumor received octreotide concomitant with seven cycles of temozolomide; progressive metastatic tumor burden in this patient resulted in progressive clinical decline and death. One patient with an undifferentiated pituitary carcinoma was treated with thalidomide concomitant with temozolomide; the latter drug was stopped after two cycles because of debilitating dizziness and headaches. There was no evidence of tumor growth during 2 months of treatment.
TABLE 1.
Temozolomide treatment cohort
| Case no. | Surgery | Cell lineage | Tumor features | Radiation therapy | MGMT score | Temozolomide | Response | Side effects | Outcome | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Biochemical | Imaging | ||||||||||
| 1 | TSR: 10/08 | Null-cell | Ki-67 < 3%; prominent nucleoli | None | 3 | 10 of 12 cycles completed | N/A | 20% reduction in tumor volume; subsequent stable tumor volume | Fatigue, headaches | Clinically stable | |
| 2 | TSR: 07/04; craniectomies: 07/07; 06/09 | Corticotroph-cell with clinical Cushing’s disease | Ki-67 = 18%; p53 +; large, multinucleated cells | GKRT 10/18/07; 5/02/08 | 1 | 11 completed | ACTH: 221 pg/ml decreased to 18 ng/ml; adrenally insufficient at cycle 8/11 | >80% tumor reduction | Sensory neural hearing loss, fatigue, headaches | Clinically improved during treatment | |
| 3 | TSR: 01/92; 06/03; 12/06 | FSH/TSH/α-SU positive; clinically nonfunctional | Ki-67 < 3%; no atypical features | Conventional fractionated radiation 1992 | 2 | 13 completed, still on tx; 12 cycles of 21/7, now on 18/10 | N/A | Stable tumor size | Fatigue and headaches, lessened with dose reduction | Clinically stable | |
| 4 | TSR: 07/94; 02/00 | Null-cell | Ki-67 = 6%; mitotic figures present | None | 3 | 10/12 completed | N/A | Stable tumor size | Fatigue, dry mouth | Clinically stable | |
| 5 | TSR: 08/06; 12/06 | Prolactinoma | Ki-67 > 20%; prominent nucleoli | GKRT 02/2007 | 1 | 11 completed still on tx 21/7 | Prolactin: 5702 ng/ml decreased to 121 ng/ml | >80% tumor reduction | Well tolerated | Clinically improved during treatment | |
| 6 | TSR: 06/94; 10/01; 04/03 | Pituitary carcinoma; null-cell | Ki-67 > 20%; brisk mitotic activity | Conventional fractionated radiation 1994; GKRT 02/05 | 3 | 2 cycles, Apr/May 2005 temodar/ thalidomide | N/A | Stable tumor size during 2 months of treatment | Fatigue, severe dizziness | Deceased; cause of death unrelated to pituitary carcinoma | |
| 7 | TSR: 01/05; 03/05 | Pituitary carcinoma; null-cell | Ki-67 > 20%; rare mitotic figures | IMRT 04/05 | 1 | 7 cycles | N/A | Progressive metastatic disease on treatment | Fatigue, headaches | Deceased; death attributed to complications of pituitary carcinoma | |
TSR, Transsphenoidal surgery; GKRT, GammaKnife radiation treatment; IMRT, intensity modulated radiation therapy; N/A, not available; tx, treatment; α-SU, α-subunit of glycoproteins. Median MGMT IHC score reported for each case (1 = <10% MGMT positive cells; 2 = 10%–50% MGMT positive cells; 3 = >50% MGMT positive cells).
Clinical response
Tumor regression of more than 80% tumor volume resulted in rapid clinical improvement in two patients (29%; Fig. 1), a 20% reduction in tumor size with subsequent stable tumor volume was noted in one patient, arrest of tumor growth was seen in three patients (Fig. 2), and progressive metastatic disease while on temozolomide occurred in one patient with a null cell carcinoma that resulted in death (Table 1).
Fig. 1.

Serial T1, Postcontrast MRI images before and after temozolomide treatment. A and B, Case 2, an invasive corticotroph tumor pretreatment (A) and after 11 cycles (B) (arrows highlight tumor extension). C and D, Case 5, an invasive prolactinoma pretreatment (C) and after 8 cycles (D) (arrows highlight substantial reduction of tumor).
Fig. 2.

Serial MRI images from case 4. A–E, Progressive tumor growth in a patient with no history of radiation treatment. E, Immediately before temozolomide; F, arrest of tumor growth during eight cycles of temozolomide.
In two patients, the aggressive pituitary tumor—one prolactinoma (case 5, Table 1) and one corticotroph tumor (case 2, Table 1)—had caused compression of cranial nerves and the brain stem. Both tumors had more than 80% reduction in tumor volume with chronic temozolomide therapy, and clinical improvement was evident within the first four cycles; serial MRI studies demonstrated marked reduction in tumor size, but after 11 cycles of treatment some residual tumor remained in both cases (Fig. 1). Biochemically, the prolactin levels in case 5 decreased from 5702 ng/ml to 121 ng/ml over 11 cycles, but have not fully normalized. Symptoms of cranial nerve (CN) dysfunction (partial 6th CN palsy and a painful 5th CN neuropathy) improved during the first 6 wk of treatment and resolved within four cycles of temozolomide. This patient received GKR treatment 26 months before temozolomide, with evidence of rapidly progressive disease up to the point of initiating temozolomide treatment. We attribute her treatment response to temozolomide. Case 2 had an ACTH-secreting macroadenoma with clinical Cushing’s disease. The marked reduction in tumor volume led to a cerebrospinal fluid (CSF) leak during cycle 11 of temozolomide (a phenomenon seen when prolactinomas rapidly respond to dopamine agonist therapy). The ACTH level pretreatment was 221 pg/ml, which steadily declined during 13 cycles, ultimately normalizing at 18 pg/ml, with clinical adrenal insufficiency developing near the end of the treatment period. Tissue specimens obtained during transsphenoidal surgery to repair the CSF leak revealed histological evidence of invasive corticotroph tumor cells in dural structures; nonetheless, the patient has been off treatment for 6 months without signs or symptoms of tumor regrowth or recurrence of Cushing’s disease, and there has been no MRI evidence of growth in the residual tumor. Importantly, this patient had a second GKR treatment 5 months before starting temozolomide, so it is possible that the treatment response in this patient is in part the result of radiosurgery (therapeutic response in Cushing’s patients has been shown to be delayed 3–48 months after GKR) (30).
Side effects and adverse events
Side effects were common in our cohort. Fatigue was experienced by six patients and prompted dose reduction after 12 cycles in one patient with a stable tumor burden (case 3, Table 1). Case 2 experienced fatigue throughout treatment, and once biochemical remission was achieved temozolomide was discontinued to relieve the persistent side effect, given the unknown benefit of continued therapy. Headache occurred in four patients, but was not dose-limiting. Severe dizziness occurred in one patient (case 6) who was on concomitant thalidomide; the temozolomide was stopped after two cycles, and symptoms were relieved (thalidomide is also know to cause dizziness and may have contributed to the severity of this side effect). One adverse event occurred during the first four cycles of temozolomide treatment in case 2; the patient developed progressive bilateral sensorineural hearing loss, which necessitated hearing aids that were unable to correct the sensory deficit. We have not seen hearing loss in the hundreds of patients receiving temozolomide monotherapy for malignant gliomas at our center, but we cannot exclude temozolomide as the causative agent in this case. Despite the adverse event, the patient and treating physicians elected to continue treatment because of clinical and radiological evidence of tumor response. There has been no improvement in the patient’s hearing loss over the 6 months since temozolomide was stopped.
MSP and IHC
The MGMT promoter was unmethylated in all 14 adequate specimens. The unmethylated state was stable over time in case 3 in which both pre- and postradiation surgical specimens were available (Table 2). The IHC analysis demonstrated variable immunoreactivity for MGMT in a nuclear pattern in all 14 adequate specimens (Table 2 and Fig. 3). Four specimens were scored as negative for MGMT immunoreactivity by at least one reviewer, but there was not a consensus for negative MGMT immunoreactivity in any of the 14 specimens (Table 2). Median MGMT scores for each case are reported in Table 1. Three cases had less than 10% of cells immunoreactive for MGMT; one case had 10–50% MGMT immunoreactive cells; three cases had MGMT immunoreactivity in more than 50% of cells. Therefore, methylation status of the MGMT gene promoter was not predictive of relative MGMT expression by IHC in any of the cases. Marked heterogeneity in MGMT expression was noted within many of the tumors, some with expression predominantly in single regions of the tumor, some with predominant expression in the periphery of the tumor. Likewise, there was significant heterogeneity of the intensity of MGMT expression as demonstrated in Table 2 and Fig. 3.
TABLE 2.
Fifteen surgical specimens from seven patients with invasive or metastatic pituitary tumors
| Patient no. | Specimen | PCR: MGMT promoter | MGMT IHC | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reviewer 1 | Reviewer 2 | Reviewer 3 | |||||||||||||||||||
| Methylated | Unmethylated | MGMT positive | Intensity | Total | MGMT positive | Intensity | Total | MGMT positive | Intensity | Total | |||||||||||
| 1 | A | X | 3 | 1 | 4 | 3 | 1 | 4 | 3 | 2 | 5 | ||||||||||
| 2 | A | X | 1 | 2 | 3 | Negative | 0 | 0 | 1 | 1 | 2 | ||||||||||
| B | X | 1 | 1 | 2 | 2 | 3 | 5 | Negative | 0 | 0 | |||||||||||
| 3 | A | X | 2 | 2 | 4 | 2 | 2 | 4 | 3 | 2 | 5 | ||||||||||
| B1 | X | 2 | 2 | 4 | 2 | 1 | 3 | 1 | 1 | 2 | |||||||||||
| C1 | X | 2 | 1 | 3 | 1 | 3 | 4 | 2 | 1 | 3 | |||||||||||
| 4 | A | X | 3 | 2 | 5 | 3 | 1 | 4 | 3 | 2 | 5 | ||||||||||
| B | X | 2 | 1 | 3 | 2 | 3 | 5 | 3 | 2 | 5 | |||||||||||
| 5 | A | X | 1 | 1 | 2 | Negative | 0 | 0 | 1 | 1 | 2 | ||||||||||
| B | X | 1 | 1 | 2 | 2 | 2 | 4 | 1 | 1 | 2 | |||||||||||
| 6 | A1 | X | 3 | 1 | 4 | 3 | 1 | 4 | 3 | 2 | 5 | ||||||||||
| B1 | N/A; necrotic specimen | N/A; necrotic specimen | N/A | N/A | Necrotic | N/A | N/A | Necrotic | N/A | N/A | Necrotic | ||||||||||
| C1 | X | 3 | 2 | 5 | 3 | 1 | 4 | 3 | 2 | 5 | |||||||||||
| 7 | A | X | 1 | 1 | 2 | 1 | 2 | 3 | Negative | 0 | 0 | ||||||||||
| B | X | 1 | 1 | 2 | 1 | 2 | 3 | 1 | 1 | 2 | |||||||||||
Methylation status of the MGMT promoter by MSP and MGMT immunoreactivity are shown. MGMT positive: 1 = <10% of cells positive in tumor; 2 = 10%–50%; 3 = > 50%. MGMT intensity: 1, faint; 2, moderate; 3, equivalent to positive controls. N/A, Not available.
Postradiation surgical specimen.
Fig. 3.
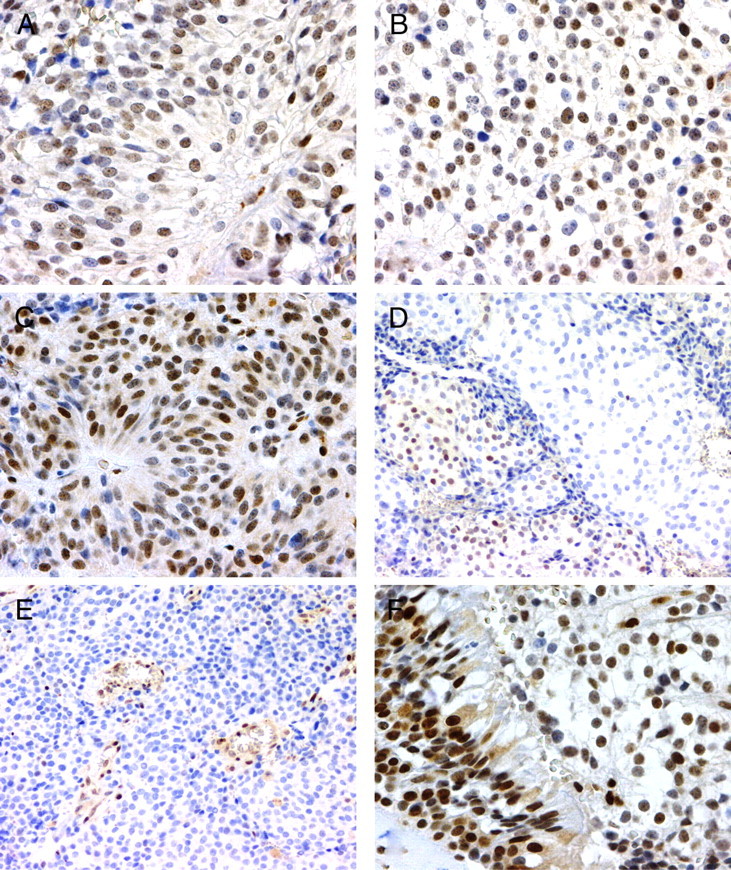
IHC for MGMT. A and B, Diffuse pattern of stain (score 2); C, strong stain (score 3); D, example of heterogeneous stain in a single focus; E, internal positive control endothelial cells; F, internal positive control sinus mucosa.
IHC statistical analysis
The free-marginal κ statistic for the semiquantitative IHC analysis of MGMT positivity was 0.40, which suggests poor to moderate agreement by three reviewers on the proportion of cells with MGMT immunoreactivity within any given specimen (κ > 0.70 indicates good interoperator reliability). With a dichotomous outcome measure of positive (scores of 1, 2, or 3) vs. negative, the free-marginal κ score is 0.62, suggesting moderate interoperator agreement.
Correlation of clinical outcomes and molecular studies
The MGMT promoter regions in the two postradiation surgical specimens from the most aggressive tumor (case 7, Table 1) in our cohort remained unmethylated, but IHC immunoreactivity for MGMT was less than 10% of cells in both specimens. The lack of MGMT expression would be expected to predict impaired DNA repair capacity in this tumor and thus predict temozolomide responsiveness; however, the patient developed spinal metastasis and increasing parasellar tumor burden during 7 months of temozolomide treatment, which was discontinued 2 months before her death to alleviate the side effects of severe fatigue and headaches. The two tumors with the most significant response to temozolomide therapy were cases 2 and 5 described in Clinical response, both of which were locally invasive with encroachment on cranial nerves, optic apparatus, and midbrain structures. Like case 7, these tumors were MGMT positive in less than 10% of cells. Thus, clinical response was not reliably predicted in our cohort by either MGMT promoter methylation or MGMT IHC immunoreactivity.
Discussion
To our knowledge, this is the largest published cohort of patients treated with temozolomide for aggressive pituitary tumors (invasive adenomas and carcinomas). The lack of clinical guidelines for chemotherapy management of life-threatening pituitary tumors has led to significant heterogeneity in the clinical use of temozolomide for these rare tumors. The single-center aspect of this cohort has allowed for some uniformity in regard to the multimodality treatment algorithm and temozolomide dosing. Nonetheless, our data underscore the limitations of our current understanding of aggressive pituitary tumors and the underlying molecular features that determine the heterogeneity in tumor behavior and response to multimodality therapy. We believe that our data raise more questions than are answered, and we caution against any conclusions regarding what type of aggressive pituitary tumors may or may not respond to temozolomide based on this limited data set. Previously published case reports demonstrate at least a partial response to temozolomide in some aggressive tumors arising from all anterior pituitary cell types including functioning and nonfunctioning pituitary adenomas and pituitary carcinomas.
Most patients receiving treatment for aggressive pituitary tumors in our center undergo at least two operations; the second procedure is routinely undertaken to decompress compromised neural structures, reduce symptoms, or create adequate space from the optic chiasm to allow for focused radiation therapy. Radiation therapy with GammaKnife is routinely performed for treating residual tumor seen either intraoperatively or by MRI after a second surgical procedure. In those cases in which there is tumor adjacent to optic nerves or other barriers to GKR treatment, conventional fractioned radiation therapy is recommended. In cases 1 and 4, the patients refused radiation. Temozolomide is rarely used to treat pituitary tumors at our center and is only used as last-line treatment for life-threatening pituitary tumors that have been refractory to standard treatment modalities. We use the 21 d on, 7 d off temozolomide regimen—a “dose-dense” schedule—that delivers a higher dose of temozolomide per unit time (75 mg delivered 21/28 d vs. 150 or 200 mg delivered 5/28 d). There is a theoretical advantage to the dose-dense schedule in that it may deplete tumor cells of their MGMT and thereby circumvent an important mechanism of resistance in tumors that lack promoter methylation. This regimen is associated with a greater risk of lymphopenia than standard 5/28-d regimens. However, the association with higher risk of other side effects or secondary malignancies is unclear.
Our clinical results suggest that temozolomide induces clinically significant tumor volume regression in a minority of invasive pituitary tumors [two of seven patients (29%)], but stabilization of tumor growth occurred in four additional cases [four of seven patients (57%)]. In contrast to other published findings (31, 32), our data suggest that MGMT promoter methylation and MGMT expression by IHC are not clinically useful in predicting tumor response to temozolomide therapy. The intraoperator heterogeneity in MGMT IHC scores in our series is in line with recent large studies that have evaluated MGMT expression in gliomas; the intraoperator variability seen across the various scoring methods used in these studies reflects the significant intratumor variability of immunoreactivity patterns (multiple regions within a single tumor can vastly differ in regard to the frequency and the relative intensity of the immunoreactivity) (33, 34, 35). Furthermore, our MSP data suggest that MGMT promoter methylation is rare among invasive pituitary tumors and does not explain the wide variability of IHC expression seen in our cohort of tumors.
Clonality studies employing X-chromosome inactivation analyses have demonstrated that the majority of sporadic human pituitary adenomas are monoclonal (36). This implies that these tumors arise from de novo somatic genetic changes in a single pituitary cell. Our DNA methylation data suggest that the heterogeneity of MGMT expression within these monoclonal tumor cell populations may result not from epigenetic modification of the MGMT promoter but instead from a unique transcriptional microenvironment dictated by nuclear receptors and their corepressors and coactivators or at the level of mRNA translation.
The alkylating action of temozolomide can cause epigenetic modification of DNA by methylation of gene promoter sites, which disrupts protein synthesis. In vitro studies suggest that temozolomide arrests tumor growth through cell cycle arrest rather than direct cytotoxic mechanisms. As such, complete remission is not an expected outcome of temozolomide treatment; instead, growth arrest, diminished tumor volume, and biochemical control are the goals of temozolomide treatment. Tumor cells undergoing cell replication are most vulnerable to alkylating agents. This was demonstrated in the recent publication from Kovacs et al. (17) in which Ki-67 staining of posttemozolomide treatment specimen demonstrated a greater proportion of cells in G0 (fewer Ki-67-positive cells) than the pretreatment specimen without any evidence of apoptosis or necrotic cell death. Our own pre- and posttemozolomide surgical specimens from case 2 confirm these findings, with Ki-67 labeling index in the pretreatment specimen being very high at 18% and subsequently undetectable in the posttreatment specimen (Fig. 4). A population of apparently viable corticotroph tumor cells is evident in both pre- and posttemozolomide specimens.
Fig. 4.
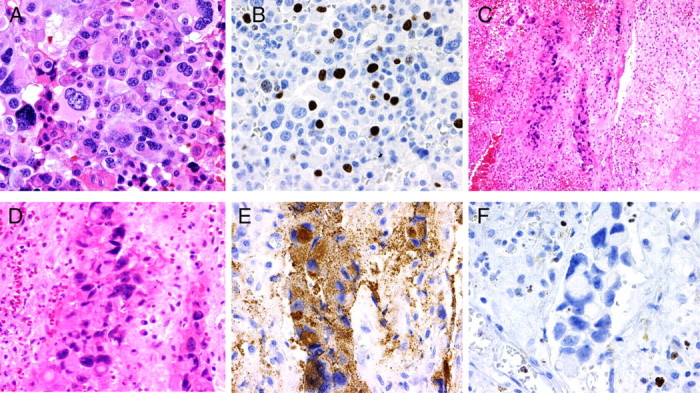
Pre- and posttemozolomide treatment specimens from case 2. A and B, Surgical specimen before GammaKnife radiation and temozolomide treatment showing extensive cellular atypia (A) and high Ki-67 labeling index (18%) (B). C–F, Surgical specimen after treatments and collected during CSF leak correction showed focal islands of tumor cells entrapped on fibrous connective tissue with intense inflammatory reaction (C). Extensive cellular pleomorphism was present (D), but adenoma cells were still immunoreactive for ACTH (E). Ki-67 labeling was practically absent on tumor cells, but present in inflammatory cells seen on the specimen (F).
Radiation therapy is known to exert some antitumor effects via DNA methylation (37, 38); however, the MGMT promoter in our five postradiation specimens remained unmethylated (prior conventional radiation in two specimens from case 3, and prior conventional and GKR treatment in three specimens from case 6, Table 2), and MGMT IHC does not suggest significant impact of radiation on MGMT expression. This suggests that pretemozolomide radiation treatment may not have the same therapeutic synergy with temozolomide that has been observed in glioblastomas (22, 39, 40). A recent publication by Su et al. (41) that evaluated MGMT promoter methylation in sputum samples of uranium miners showed increasing promoter methylation with cumulative low-dose radiation exposure. Multicenter investigation will be necessary to establish the treatment effect of the combined effects of radiation and temozolomide. Evaluation of differential tumor response depending on interval between radiation and temozolomide along with the study of the molecular effects of single-dose radiation treatment vs. chronic or intermittent low-dose radiation will be necessary to establish definitive treatment guidelines.
We conclude that adjuvant medical therapy with temozolomide may be considered for the management of life-threatening invasive pituitary tumors that have failed to respond to conventional treatments. Based on our cumulative experience and previous reported data, we do not believe that there are any molecular or clinical features of these tumors that adequately predict tumor response; therefore, we do not recommend that tumor cell lineage or MGMT molecular analysis influence the clinical decision for treatment with temozolomide in individual cases. Case 5 in our series, a prolactinoma, was one of the two tumors with significant tumor regression. There is a preponderance of prolactinomas among those tumors reported in the literature to be temozolomide-responsive (14, 15, 16, 17, 31, 42). However, we believe this is indicative of the overrepresentation of prolactinomas among functional tumors that progress to a more malignant phenotype (13) rather than a true molecular predisposition to temozolomide responsiveness.
Complete remission is not an expected outcome of temozolomide therapy, but tumor stabilization or reduction of tumor volume can improve clinical outcomes. Any benefit of long-term therapy with a tumorstatic agent such as temozolomide is typically limited by the increased likelihood of adverse effects with cumulative doses. Cytopenias are the most likely to occur, but myelodysplasia syndrome and secondary malignancies including myeloid leukemia have been observed with temozolomide (43). Thus, in our opinion, temozolomide remains a last-line of defense for life-threatening pituitary tumors, and the duration of therapy must be determined on a patient-by-patient basis.
The lack of prospective clinical trial data for these rare aggressive tumors necessitates multidisciplinary patient care teams to provide patients with the most informed treatment options and demands multicenter cooperation to facilitate the future clinical studies that are needed to establish treatment guidelines for aggressive pituitary tumors.
Footnotes
Disclosure Summary: All authors have nothing to declare.
First Published Online July 28, 2010
Abbreviations: CN, Cranial nerve; CSF, cerebrospinal fluid; GKR, GammaKnife stereotactic radiosurgery; IHC, immunohistochemistry; MGMT, O6-methylguanine methyltransferase; MRI, magnetic resonance imaging; MSP, methylation-specific PCR.
References
- 1.Meij BP, Lopes MB, Ellegala DB, Alden TD, Laws Jr ER 2002. The long-term significance of microscopic dural invasion in 354 patients with pituitary adenomas treated by transsphenoidal surgery. J Neurosurg 96:195–208 [DOI] [PubMed] [Google Scholar]
- 2.Pernicone PJ, Scheithauer BW, Sebo TJ, Kovacs KT, Horvath E, Young Jr WF, Lloyd RV, Davis DH, Guthrie BL, Schoene WC 1997. Pituitary carcinoma: a clinicopathologic study of 15 cases. Cancer 79:804–812 [DOI] [PubMed] [Google Scholar]
- 3.Melmed S, Sternberg R, Cook D, Klibanski A, Chanson P, Bonert V, Vance ML, Rhew D, Kleinberg D, Barkan A 2005. A critical analysis of tumor shrinkage during primary medical therapy in acromegaly. J Clin Endocrinol Metab 90:4405–4410 [DOI] [PubMed] [Google Scholar]
- 4.Buchfelder M, Weigel D, Droste M, Mann K, Saller B, Brübach K, Stalla GK, Bidlingmaier M, Strasburger CJ 2009. Investigators of German Pegvisomant Observational Study. Pituitary tumor size in acromegaly during pegvisomant treatment: experience from MR re-evaluations of the German Pegvisomant Observational Study. Eur J Endocrinol 161:27–35 [DOI] [PubMed] [Google Scholar]
- 5.Batista DL, Zhang X, Gejman R, Ansell PJ, Zhou Y, Johnson SA, Swearingen B, Hedley-Whyte ET, Stratakis CA, Klibanski A 2006. The effects of SOM230 on cell proliferation and adrenocorticotropin secretion in human corticotroph pituitary adenomas. J Clin Endocrinol Metab 91:4482–4488 [DOI] [PubMed] [Google Scholar]
- 6.Boscaro M, Ludlam WH, Atkinson B, Glusman JE, Petersenn S, Reincke M, Snyder P, Tabarin A, Biller BM, Findling J, Melmed S, Darby CH, Hu K, Wang Y, Freda PU, Grossman AB, Frohman LA, Bertherat J 2009. Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. J Clin Endocrinol Metab 94:115–122 [DOI] [PubMed] [Google Scholar]
- 7.Yildiz F, Zorlu F, Erba° T, Atahan L 1999. Radiotherapy in the management of giant pituitary adenomas. Radiother Oncol 52:233–237 [DOI] [PubMed] [Google Scholar]
- 8.Knosp E, Steiner E, Kitz K, Matula C 1993. Pituitary Adenomas with invasion of the cavernous sinus space: a magnetic resonance imaging classification compared with surgical findings. Neurosurgery 33:610–618 [DOI] [PubMed] [Google Scholar]
- 9.Petterson T, MacFarlane IA, MacKenzie JM, Shaw MD 1992. Prolactin secreting pituitary carcinoma. J Neurol Neurosurg Psychiatry 55:1205–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan NJ, Laroche CM, Goodman I, Davies MJ, Jenkins JS 1985. Pituitary Cushing’s disease arising from a previously nonfunctional corticotrophic chromophobe adenoma. Clin Endocrinol (Oxf) 22:147–153 [DOI] [PubMed] [Google Scholar]
- 11.Mixson AJ, Friedman TC, Katz DA, Feuerstein IM, Taubenberger JK, Colandrea JM, Doppman JL, Oldfield EH, Weintraub BD 1993. Thyrotropin-secreting pituitary carcinoma. J Clin Endocrinol Metab 76:529–533 [DOI] [PubMed] [Google Scholar]
- 12.Kasperlik-Zaluska AA, Wislawski J, Kaniewska J, Zborzil J, Frankiewicz E, Zgliczyñski S 1987. Cytostatics for acromegaly. Marked improvement in a patient with an invasive pituitary tumour. Acta Endocrinol 116:347–349 [PubMed] [Google Scholar]
- 13.Lopes MB, Scheithauer BW, Schiff D 2005. Pituitary carcinoma: diagnosis and treatment. Endocrine 28:115–121 [DOI] [PubMed] [Google Scholar]
- 14.Neff LM, Weil M, Cole A, Hedges TR, Shucart W, Lawrence D, Zhu JJ, Tischler AS, Lechan RM 2007. Temozolomide in the treatment of an invasive prolactinoma resistant to dopamine agonists. Pituitary 10:81–86 [DOI] [PubMed] [Google Scholar]
- 15.Fadul CE, Kominsky AL, Meyer LP, Kingman LS, Kinlaw WB, Rhodes CH, Eskey CJ, Simmons NE 2006. Long-term response of pituitary carcinoma to temozolomide. J Neurosurg 105:621–626 [DOI] [PubMed] [Google Scholar]
- 16.Lim S, Shahinian H, Maya MM, Yong W, Heaney AP 2006. Temozolomide: a novel treatment for pituitary carcinoma. Lancet Oncol 7:518–520 [DOI] [PubMed] [Google Scholar]
- 17.Kovacs K, Horvath E, Syro LV, Uribe H, Penagos LC, Ortiz LD, Fadul CE 2007. Temozolomide therapy in a man with an aggressive prolactin-secreting pituitary neoplasm: morphological findings. Hum Pathol 38:185–189 [DOI] [PubMed] [Google Scholar]
- 18.Thearle MS, Freda PU, Bruce JN, Isaacson SR, Lee Y, Fine RL 4 December 2009. Temozolomide and capecitabine treatment of an aggressive corticotroph pituitary tumor. Pituitary doi:10.1007/s11102-009-0211-1 [DOI] [PMC free article] [PubMed]
- 19.Roos WP, Batista LF, Naumann SC, Wick W, Weller M, Menck CF, Kaina B 2007. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 26:186–197 [DOI] [PubMed] [Google Scholar]
- 20.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG 1999. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59:793–797 [PubMed] [Google Scholar]
- 21.Hegi ME, Diserens AC, Godard S, Dietrich PY, Regli L, Ostermann S, Otten P, Van Melle G, de Tribolet N, Stupp R 2004. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res 10:1871–1874 [DOI] [PubMed] [Google Scholar]
- 22.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R 2005. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003 [DOI] [PubMed] [Google Scholar]
- 23.Widhalm G, Wolfsberger S, Preusser M, Woehrer A, Kotter MR, Czech T, Marosi C, Knosp E 2009. O(6)-methylguanine DNA methyltransferase immunoexpression in nonfunctioning pituitary adenomas: are progressive tumors potential candidates for temozolomide treatment? Cancer 115:1070–1080 [DOI] [PubMed] [Google Scholar]
- 24.Hsu SM, Raine L, Fanger H 1981. The use of antiavidin antibody and avidin-biotin-peroxidase complex in immunoperoxidase techniques. Am J Clin Pathol 75:816–821 [DOI] [PubMed] [Google Scholar]
- 25.Norton AJ, Jordan S, Yeomans P 1994. Brief, high-temperature heat denaturation (pressure cooking): a simple and effective method of antigen retrieval for routinely processed tissues. J Pathol 173:371–379 [DOI] [PubMed] [Google Scholar]
- 26.Brennan RL, Prediger DJ 1981. Coefficient kappa: Some uses, misuses, and alternatives. Educ Psychol Meas 41:687–699 [Google Scholar]
- 27.Fleiss JL 1971. Measuring nominal scale agreement among many raters. Psychol Bull 76:378–382 [Google Scholar]
- 28.Randolph JJ Free-marginal multirater κ: an alternative to Fleiss’ fixed-marginal multirater κ. Proc of Joensuu University Learning and Instruction Symposium, Joensuu, Finland, 2005
- 29.Jackson TJ, Michel JL, Roberts R, Shepheard J, Cheng D, Rust J, Perry C 2009. Development of a validation algorithm for ‘present on admission’ flagging. BMC Med Inform Decis Mak 9:48 [DOI] [PMC free article] [PubMed]
- 30.Sheehan JM, Vance ML, Sheehan JP, Ellegala DB, Laws Jr ER 2000. Radiosurgery for Cushing’s disease after failed transsphenoidal surgery. J Neurosurg 93:738–742 [DOI] [PubMed] [Google Scholar]
- 31.Hagen C, Schroeder HD, Hansen S, Hagen C, Andersen M 2009. Temozolomide treatment of a pituitary carcinoma and two pituitary macroadenomas resistant to conventional therapy. Eur J Endocrinol 161:631–637 [DOI] [PubMed] [Google Scholar]
- 32.Mohammed S, Cusimano MD, Scheithauer BW, Rotondo F, Horvath E, Kovacs K 2010. O-methylguanine-DNA methyltransferase immunoexpression in a double pituitary adenoma: case report. Neurosurgery 66:E421–E422; discussion E422 [DOI] [PubMed]
- 33.Christmann M, Nagel G, Horn S, Krahn U, Wiewrodt D, Sommer C, Kaina B 3 February 2010. MGMT activity, promoter methylation and immunohistochemistry of pre-treatment and recurrent malignant gliomas: a comparative study on astrocytoma and glioblastoma. Int J Cancer doi: 10.1002/ijc.25229 [DOI] [PubMed]
- 34.Preusser M, Charles Janzer R, Felsberg J, Reifenberger G, Hamou MF, Diserens AC, Stupp R, Gorlia T, Marosi C, Heinzl H, Hainfellner JA, Hegi M 2008. Anti-O6-methylguanine-methyltransferase (MGMT) immunohistochemistry in glioblastoma multiforme: observer variability and lack of association with patient survival impede its use as clinical biomarker. Brain Pathol 18:520–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mellai M, Caldera V, Annovazzi L, Chiò A, Lanotte M, Cassoni P, Finocchiaro G, Schiffer D 2009. MGMT promoter hypermethylation in a series of 104 glioblastomas. Cancer Genomics Proteomics 6:219–227 [PubMed] [Google Scholar]
- 36.Clayton RN, Farrell WE 2004. Pituitary tumour clonality revisited. Front Horm Res 32:186–204 [DOI] [PubMed] [Google Scholar]
- 37.Koturbash I, Pogribny I, Kovalchuk O 2005. Stable loss of global DNA methylation in the radiation-target tissue: a possible mechanism contributing to radiation carcinogenesis. Biochem Biophys Res Commun 337:526–533 [DOI] [PubMed] [Google Scholar]
- 38.Kaup S, Grandjean V, Mukherjee R, Kapoor A, Keyes E, Seymour CB, Mothersill CE, Schofield PN 2006. Radiation-induced genomic instability is associated with DNA methylation changes in cultured human keratinocytes. Mutat Res 597:87–97 [DOI] [PubMed] [Google Scholar]
- 39.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO 2005. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996 [DOI] [PubMed] [Google Scholar]
- 40.Stupp R, Dietrich PY, Ostermann Kraljevic S, Pica A, Maillard I, Maeder P, Meuli R, Janzer R, Pizzolato G, Miralbell R, Porchet F, Regli L, de Tribolet N, Mirimanoff RO, Leyvraz S 2002. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol 20:1375–1382 [DOI] [PubMed] [Google Scholar]
- 41.Su S, Jin Y, Zhang W, Yang L, Shen Y, Cao Y, Tong J 2006. Aberrant promoter methylation of P16-INK4a and O6-methylguanine-DNA methyltransferase in workers at a Chinese uranium mine. J Occup Health 48:261–266 [DOI] [PubMed] [Google Scholar]
- 42.McCormack AI, McDonald KL, Gill AJ, Clark SJ, Burt MG, Campbell KA, Braund WJ, Little NS, Cook RJ, Grossman AB, Robinson BG, Clifton-Bligh RJ 2009. Low O6-methylguanine-DNA methyltransferase (MGMT) expression and response to temozolomide in aggressive pituitary tumours. Clin Endocrinol (Oxf) 71:226–233 [DOI] [PubMed] [Google Scholar]
- 43.Su YW, Chang MC, Chiang MF, Hsieh RK 2005. Treatment-related myelodysplastic syndrome after temozolomide for recurrent high-grade glioma. J Neurooncol 71:315–318 [DOI] [PubMed] [Google Scholar]