

Abstract
The interferon-inducible protein X (IFIX), a member of the PYHIN family, was recently recognized as an antiviral factor against infection with herpes simplex virus 1 (HSV-1). IFIX binds viral DNA upon infection and promotes expression of antiviral cytokines. How IFIX exerts its host defense functions and whether it is inhibited by the virus remain unknown. Here, we integrated live cell microscopy, proteomics, IFIX domain characterization, and molecular virology to investigate IFIX regulation and antiviral functions during HSV-1 infection. We find that IFIX has a dynamic localization during infection that changes from diffuse nuclear and nucleoli distribution in uninfected cells to discrete nuclear puncta early in infection. This is rapidly followed by a reduction in IFIX protein levels. Indeed, using immunoaffinity purification and mass spectrometry, we define IFIX interactions during HSV-1 infection, finding an association with a proteasome subunit and proteins involved in ubiquitin-proteasome processes. Using synchronized HSV-1 infection, microscopy, and proteasome-inhibition experiments, we demonstrate that IFIX co-localizes with nuclear proteasome puncta shortly after 3 h of infection and that its pyrin domain is rapidly degraded in a proteasome-dependent manner. We further demonstrate that, in contrast to several other host defense factors, IFIX degradation is not dependent on the E3 ubiquitin ligase activity of the viral protein ICP0. However, we show IFIX degradation requires immediate-early viral gene expression, suggesting a viral host suppression mechanism. The IFIX interactome also demonstrated its association with transcriptional regulatory proteins, including the 5FMC complex. We validate this interaction using microscopy and reciprocal isolations and determine it is mediated by the IFIX HIN domain. Finally, we show IFIX suppresses immediate-early and early viral gene expression during infection. Altogether, our study demonstrates that IFIX antiviral functions work in part via viral transcriptional suppression and that HSV-1 has acquired mechanisms to block its functions via proteasome-dependent degradation.
The initiation of human innate immune responses is one of the early host defenses against DNA viruses. These antiviral mechanisms rely on proteins termed pattern recognition receptors (PRRs)1 that have the ability to identify molecular signatures of DNA viruses, subsequently triggering a cascade of events that leads to the induction of antiviral cytokines, such as interferons (IFN) (1). DNA sensors are PRRs that can specifically sense the presence of foreign DNA to initiate host defense processes. A PRR protein family that has gained recognition in recent years is that of PYHIN proteins (2). Four PYHIN proteins are known in humans as follows: absent in melanoma 2 (AIM2); the interferon-inducible protein 16 (IFI16); the interferon-inducible protein X (IFIX), and myeloid cell nuclear differentiation antigen (MNDA). The name of this family derives from the two common features of these proteins, a pyrin (PY) domain and at least one HIN200 (HIN) domain. The pyrin domain (also known as PAAD or DAPIN) is a relative of the evolutionarily conserved Death domain that mediates homotypic interactions (3–5). The HIN200 domain(s) confers to these proteins their ability to bind viral DNA. Oligonucleotide/oligosaccharide binding folds present within the HIN200 domain interact with DNA in a sequence-independent manner, thereby allowing these proteins to bind DNA from diverse viruses (6, 7). The ability of PYHIN proteins to act as DNA sensors was first discovered for AIM2. AIM2 is a cytoplasmic DNA sensor that assembles the inflammasome complex in macrophages, which leads to the activation of proinflammatory cytokines IL-1 and IL-18 (8, 9). The interest in this protein family was further elevated when two PYHIN proteins were shown to be able to bind viral DNA in the nuclei of infected cells (10). Given the similarities between human and viral DNA molecules, DNA sensing was thought for a long time to only take place in subcellular compartments that do not contain host DNA. However, as the majority of known DNA viruses that are human pathogens deposit their DNA and replicate in the nucleus, the ability of a human cell to recognize this foreign molecule in the nucleus and to defend itself remained an open question. We and others have found that IFI16 binds nuclear viral DNA upon infection with several herpesviruses, including herpes simplex virus 1 (HSV-1), aiding the induction of antiviral cytokines (5, 11–13). More recently, we discovered that IFIX can also bind nuclear viral DNA during HSV-1 infection (2). Furthermore, this IFIX-DNA binding was sequence-independent, similar to AIM2 and IFI16.
IFIX is the newest member of the PYHIN family (14). Its functions have been linked to tumor suppression, as evidenced by its anti-tumor and anti-proliferation activities and by its down-regulation in human breast tumors and breast cancer cell lines (14). IFIX was shown to destabilize human double minute homolog (HDM2), which in turn leads to increased p53 levels (15). This effect of IFIX on p53 stabilization may delineate the mechanism by which IFIX exerts its tumor-suppressing function. Our recent finding that IFIX binds viral DNA upon HSV-1 infection expands its range of functions, pointing to a role in host antiviral defense. This was supported by our observation that IFIX overexpression decreased HSV-1 progeny titers, whereas IFIX knockdown had the opposite effect (2). However, the pathways through which IFIX exerts its defense functions remain unknown.
Another important aspect that has to be considered when studying viral DNA sensors is the constant tug of war that occurs in mammalian cells during DNA virus infection. Just as host cells try to limit the spread of infection and promote host survival via immune responses and other antiviral activities, viruses have evolved mechanisms to inhibit these defense strategies (16). Upon infection of a host cell, the HSV-1 life cycle relies on a temporal cascade of viral gene expression, with the sequential expression of immediate-early (IE), delayed-early (DE), and late viral genes. During its subcellular life cycle, HSV-1 uses multiple strategies to evade host defenses. For example, the viral E3 ligase ICP0 of HSV-1 targets the intrinsic immune factor promyelocytic leukemia (PML) and the DNA sensor DNA-PKcs for proteasomal degradation, and it was also shown to play a role in the degradation of the nuclear DNA sensor IFI16 (12, 17–22). The ability of host cells to induce immune responses was rescued upon infection with HSV-1 mutant virus strains that lack a functional viral ICP0 protein (12, 22). One such HSV-1 virus has a mutated RING finger domain in the viral ICP0 protein, here abbreviated as RF, where two cysteine residues have point mutations (C116G/C156A) that abolish the E3 ligase activity (23). The fact that antiviral responses mediated by IFI16 are detected only during infection with mutant virus strains highlights the ability of HSV-1 to overcome host defenses mediated by a PYHIN protein. Whether HSV-1 can also inhibit IFIX and how this antiviral protein is regulated during infection remain unknown.
Here, we used an integrative virology-proteomics approach to explore IFIX protein regulation during infection and to gain insights into its antiviral functions upon HSV-1 infection in two different cells types, primary human fibroblasts and HEK293 cells. Using live cell time-lapse microscopy, we define the localization of IFIX during the early time points post-infection with a fine spatial and temporal resolution. We find a significant change in IFIX distribution following infection, observing its recruitment to subnuclear puncta followed by its degradation. We show that the IFIX domain targeted for degradation is the pyrin domain. Using immunoaffinity purification and mass spectrometry, we identify IFIX protein interactions during infection with either wild type or RF HSV-1 strains, finding associations linked to both IFIX degradation and its antiviral functions. We determine that IFIX degradation is a virus-induced phenomenon, requiring immediate-early viral gene expression, and is mediated by the proteasome, although not dependent on the E3 ligase activity of ICP0. Additionally, we demonstrate that IFIX associates with transcriptional regulatory proteins and discovered that it functions to suppress the expression of immediate-early and early viral genes. Altogether, this work provides the first evidence that IFIX works to transcriptionally suppress viral gene expression and that HSV-1 has acquired mechanisms to induce its proteasome-dependent degradation.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
All Western blottings and immunofluorescence microscopy experiments were performed in biological duplicates or triplicates in both primary human fibroblasts stably expressing EGFP or IFIX-GFP and inducible HEK293s. Virus titers were performed in biological duplicates, followed by technical duplicates in human fibroblasts. Gene expression analyses by quantitative PCR were performed in biological triplicate in human fibroblasts stably expressing EGFP or IFIX-GFP. Significance for virus titers and quantitative PCR was determined by Student's t test. p values of p ≤ 0.05 were considered significant. IFIX-GFP and EGFP immunoaffinity purifications followed by mass spectrometry (IP-MS) analyses were performed in biological duplicates for each of the following three tested conditions: uninfected cells (mock-infected); cells infected with wild-type (WT) HSV-1; and cells infected with RF HSV-1 strain. Specificity assessments were performed by SAINT analysis (24). IFIX-GFP WT sample spectral counts were normalized to IFIX-GFP in the mock samples. Fold change of IFIX-GFP-specific interacting proteins was calculated by dividing the average of the total number of spectral counts found during WT infection by the average of the total number of spectral counts found in mock samples and expressed as a log2 transformation for an even distribution.
Reagents
The antibodies used for immunoaffinity purifications, Western blotting, and immunofluorescence were as follows: an in-house-generated α-green fluorescent protein (GFP) for IPs (25); α-GFP (Roche Applied Science) for Western blottings; α-PYHIN1 (Sigma-Aldrich); α-IFI16 antibodies (ab50004 and ab55328, Abcam, Cambridge, MA) used at a 1:1 mixture as described previously (22); α-ICP0 (H1A027-100, Virusys Corp., Taneytown, MD); α-ICP4 (sc-69809, Santa Cruz Biotechnology, Dallas, TX); α-ICP27 (sc-69806, Santa Cruz Biotechnology); α-ICP8 (sc-53329, Santa Cruz Biotechnology); α-tubulin (T6199, Sigma-Aldrich); α-PML (sc-9862, Santa Cruz Biotechnology); α-proteasome 20S core subunit (PW8155-0100, Enzo Life Sciences, Farmingdale, NY); α-HUWE1 (NB100-652, Novus, Littleton, CO); α-SENP3 (sc-67076, Santa Cruz Biotechnology); and α-LAS1L (ab140656) and α-PELP1 (sc-393534, Santa Cruz Biotechnology). Secondary antibodies were as follows: goat anti-mouse IgG conjugated with HRP (Jackson ImmunoResearch, West Grove, PA); mouse anti-rabbit IgG conjugated to HRP, and goat anti-mouse and anti-rabbit IgG conjugated with Alexa 568 or Alexa 628 (Life Technologies, Inc., Carlsbad, CA). The proteasome inhibitor (S)-MG132 (Cayman Chemical catalog no. 10012628, Ann Arbor, MI) was used at 10 μm final concentration; M-270 Epoxy magnetic beads (Invitrogen) and Lipofectamine 2000 (Life Technologies, Inc.) were used to transfect Phoenix cells to generate stable cell lines. Oligofectamine (Life Technologies, Inc.) was used to transfect siRNA.
Cell Culture and Viruses
T-Rex HEK293 cells (a gift from Dr. Loren W. Runnels, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School), primary human foreskin fibroblast (HFF) cells, and U2OS cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma) supplemented with 10% (v/v) fetal bovine serum (Atlantic Biologicals, Flowery Branch, GA) and 1% (v/v) penicillin/streptomycin (Gibco) at 37 °C in 5% CO2. Viruses used were strain 17+ wild-type HSV-1 (WT HSV-1), and strain 17+ ICP0-RING finger mutant, gifts from Dr. Saul Silverstein, Columbia University, and Dr. Bernard Roizman, University of Chicago. The blue fluorescent protein (BFP)-tagged WT HSV-1 was previously generated in our laboratory (26). Viruses were grown in U2OS cells and collected when cells exhibited 100% cytopathic effect. The d109 mutant virus and the complementing FO6 cells used to propagate the virus were gifts from Dr. Neal DeLuca, University of Pittsburgh. To harvest virus, both culture supernatant and cell-associated virus were collected, and the cell-associated virus samples were sonicated and centrifuged to pellet cell debris. Supernatants were then combined and subjected to ultracentrifugation over a sorbitol cushion to purify virus. Virus pellets were resuspended in MNT buffer (200 mm MES, 30 mm Tris-HCl, 100 mm NaCl, pH 7.4) and titered by plaque assay on U2OS cells or FO6 cells for the d109 virus. For infections, virus (or no virus for mock infection) was diluted in DMEM containing 2% (v/v) FBS and incubated on cells at the indicated multiplicity of infection (m.o.i.) for 1 h at 37 °C, 5% CO2 with gentle rocking every 15 min to allow for virus attachment. Cells were then washed once with phosphate-buffered saline (PBS), overlaid with DMEM containing 10% (v/v) FBS, and incubated at 37 °C for the indicated lengths of time. Ultraviolet (UV)-induced inactivation of viral gene expression was achieved by a Stratalinker UV cross-linker (Agilent Technologies, Santa Clara, CA) using an energy of 300 mJ/cm2.
Construction of Stable Cell Lines
Tetracycline-inducible EGFP and IFIX-GFP Flp-Ins as well as pLXSN (Clontech, catalog no. 631509) plasmids containing EGFP or mGFP were previously generated in our laboratory (2). Primary human fibroblasts stably expressing GFP, mGFP, IFIX-GFP, or IFIX-mGFP were generated for this study by retrovirus transduction. IFIX was cloned from pEGFP (2), PCR-amplified, and subcloned into pLXSN-EGFP or pLXSN-mGFP using restriction enzymes SalI and AgeI (New England Biolabs). To generate retrovirus, Phoenix cells were transfected using Lipofectamine 2000 (Invitrogen) with the pLXSN constructs, and supernatant was collected at 48 and 72 h post-transfection, filtered (0.45-μm membrane, Millipore), and concentrated using Retro-X concentrator (Clontech) as per the manufacturer's instructions. HFFs were transduced with the retroviruses. Selection began 3 days post-transduction using 400 μg/ml G418 in 10% FBS DMEM. Cells were in selection for 7 days, and expression was confirmed by microscopy and Western blottings.
Immunofluorescence Staining and Microscopy
Cells were seeded in a 35-mm glass bottom culture dish and infected with the indicated viruses. The cells were then fixed with 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.1% Triton X-100 in PBS for 15 min. Three washing steps with PBS containing 0.2% (v/v) Tween 20 (PBS-T) were performed after fixation and again after permeabilization. For indirect fluorescence, the cells were blocked with 3% (w/v) BSA and 3% (v/v) human serum in PBS-T for 1 h at room temperature or overnight. The cells were then stained sequentially for 1 h with the indicated primary and secondary antibodies in blocking solution. After incubation with secondary antibody, cells were stained with 1 μg/ml DAPI in PBS-T for 10 min. Cells were washed three times with PBS-T after each incubation, stored, and imaged in DPBS.
Direct imaging and immunofluorescence imaging of fixed cells were performed using a Nikon A1 or Nikon A1-RS confocal microscope and Nikon elements software. All images were taken at ×60 oil objective, exported as original .ND2 files, and analyzed in ImageJ with an .ND2 reader plugin. Live cell imaging was performed on a Nikon Ti-E with a Yokogawa spinning disc (CSU-21) using a Hamamatsu ORCA-Flash4.0 camera and the Perfect Focus System at ×60 oil objective.
siRNA-mediated Knockdown
ON-TARGETplus SMARTPool siRNA against human HUWE1, SENP3, and PELP1, as well as the ON-TARGETplus non-targeting control pool (Dharmacon by GE Healthcare), was used to knock down (KD) the indicated genes or serve as an siRNA control. HFFs or 293 Flp-Ins were seeded at a density of 2E5 cells/well in a 12-well plate for virus titer analysis and KD efficiency analysis. siRNA transfection was performed at a final concentration of 50 nm siRNA pool, 2 μl of Oligofectamine (Invitrogen) per well, in 200 μl of OptiMEM added to 800 μl of 10% FBS DMEM in each well. At 24 h post-transfection, cells were washed one time with DPBS and either infected for virus titer or HUWE1 Western blotting analysis, or overlaid with 10% FBS DMEM to determine KD efficiency by Western blotting. KD efficiency samples were collected when virus titer samples were collected (24 h post-infection (hpi)).
Measuring Virus Titers
EGFP or IFIX-GFP HFF cells were seeded at a density of 2E5 in 12-well plates. 24 h following seeding or transfection, the cells were infected with WT HSV-1 at an m.o.i. of 10 or RF (to determine the m.o.i. at which RF virus produces similar progeny particles as the WT virus) at an m.o.i. of 20. At 95% cytopathic effect, which occurred at ∼24 hpi, cells and supernatants were collected and frozen at −80 °C. To determine virus titers, samples were thawed on ice and bath-sonicated at 60% duty cycle for 10× 1-s pulses. Samples were then centrifuged to pellet cell debris, and supernatants were taken for serial dilution and titer determination. Samples were titered on U2OS cells using 10% FBS DMEM in methylcellulose until plaque formation (3 days), fixed, and stained with 70% methanol, 0.5% methylene blue.
Immunoblotting
Cells were collected for Western blotting analysis in SDS sample buffer with 100 mm dithiothreitol (DTT) and boiled at 95 °C for 5 min. All samples were resolved by 10% SDS-PAGE. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes and blocked in 5% milk in Tris-buffered saline with 0.2% Tween (TBST) at room temperature for 1 h. Membranes were incubated with primary antibody in blocking solution overnight at 4 °C. Secondary antibody incubations were performed at room temperature for 1 h. Membranes were washed three times in TBST before and after secondary antibody incubations. ECL Western blotting detection reagents (GE Healthcare) were used to detect proteins.
Isolation of IFIX Protein Complexes
Determination of IFIX protein interactions was performed in biological duplicate using immunoaffinity purification followed by nano-LC-MS/MS analysis. Five 15-cm plates per sample of EGFP Flp-In 293 cells or IFIX-GFP Flp-In 293 cells were induced with tetracycline (1 μg/ml) and were either mock-infected or infected 18 h later with wild-type HSV-1 or ICP0-RF mutant for 4 h at an m.o.i. of 5. Cells were then washed and scraped in PBS, resuspended in freezing buffer (20 mm Na-HEPES, 1.2% polyvinylpyrrolidone (w/v), pH 7.4), and flash-frozen as cell pellets in liquid nitrogen, as described previously (27). Frozen cell pellets were ground with a Retch MM301 Mixer Mill (Retch, Newtown, PA) for 1.5 min at 30.0 Hz for 8 rounds with re-cooling in liquid nitrogen between rounds, as described (28). Each replicate was resuspended in 5 ml (1 ml per 15-cm plate) of optimized lysis buffer (20 mm K-HEPES, pH 7.4, 0.11 m KOAc, 2 mm MgCl2, 0.1% Tween 20 (v/v), 1 μm ZnCl2, 1 μm CaCl2, 0.6% Triton X-100, 200 mm NaCl, 1:100 protease inhibitor mixture (Sigma), and 100 units/ml Benzonase (Pierce) for DNA and RNA digestion). Lysates were incubated at room temperature for 10 min to activate Benzonase. Lysates were then homogenized using a PT 10-35 GT Polytron (Kinematica, Bohemia, NY) for 20 s at 20,000 rpm and then subjected to centrifugation at 8,000 × g for 10 min at 4 °C. The clarified supernatants were decanted into new conical tubes, and immunopurifications were performed by mixing each cell lysate sample with 6 mg of M-270 epoxy magnetic beads (Life Technologies, Inc.) that were conjugated with our in-house-generated polyclonal GFP antibody as described in Ref. 27. The mixing was allowed to proceed for 1 h at 4 °C, after which the beads were washed five times with lysis buffer and twice with PBS. The co-isolated proteins were eluted in 1× lithium dodecyl sulfate sample buffer (Life Technologies, Inc.) by incubating the beads in a thermomixer at 70 °C with agitation (1500 rpm) for 10 min, followed by an additional 10-min agitation on a TOMY shaker at room temperature. 10% of the eluates, 2% of the inputs, and 2% of the unbound flow-through fractions were resuspended in SDS sample buffer to assess the efficiency of isolation by Western blotting. To analyze protein solubilization efficiency by Western blotting, the pellet fraction was resuspended in 1 ml of water, and 10% was taken for solubilization in SDS sample buffer.
Sample Preparation and Mass Spectrometry Analysis
To prepare the co-isolated proteins from uninfected and WT HSV-1-infected cells for mass spectrometry analysis, the proteins were reduced and alkylated with 0.05 m final concentrations of each tris(2-carboxyethyl) phosphine (Pierce) and chloroacetamide by heating at 70 °C for 20 min. 1-D SDS-PAGE (4–12% BisTris NuPAGE gel) was used to partially resolve proteins. The gel was stained with SimplyBlue Coomassie Safe Stain (Life Technologies, Inc.) overnight and destained by washing in water until the background became clear. Proteins were processed through in-gel protein digestion by cutting gel lanes into 1-mm-thick cubes, separated into eight total fractions per sample, and processed using an in-gel digestion protocol, as described (22). Samples were digested with 12.5 ng/μl trypsin (Promega, Madison, WI) overnight at 37 °C. Peptides were extracted by incubating the gel pieces in 0.5% formic acid for 1 h at room temperature with agitation, followed by 1 h at room temperature without agitation. A second extraction was performed by incubating the gel pieces in 0.5% formic acid, 50% acetonitrile for 2 h at room temperature and pooled with the first extraction. Each biological sample was pooled from eight fractions to four fractions. The acetonitrile was removed from the extracted peptides by vacuum centrifugation for 30 min. Peptides were acidified to 1% TFA and desalted using StageTips assembled from low retention plastic tips (Eppendorf) and SDB-RPS Empore Discs (Sigma-Aldrich). StageTip membranes were washed with 0.2% TFA and eluted into autosampler vials with 5% ammonium hydroxide, 80% acetonitrile. Samples were vacuum-centrifuged to 1 μl and resuspended in 8 μl of 1% formic acid, 4% acetonitrile. The proteins co-isolated with IFIX upon HSV-1 RF infection were analyzed similar to those in uninfected and WT infected cells (see above), with the exception that the samples were reduced with 100 mm DTT at 37 °C for 30 min and alkylated with iodoacetamide at room temperature for 30 min, followed by in-solution trypsin digestion using a filter-aided sample preparation method (29) as described (30). Four μl of each sample were used for analysis using nano-liquid chromatography tandem mass spectrometry (MS/MS) using a Dionex Ultimate 3000 nanoRSLC (Dionex Corp., Sunnyvale, CA) coupled on line to an EASY-Spray source and an LTQ-Orbitrap Velos (Thermo Fisher Scientific, Waltham, MA). The EASY-Spray column for the reverse-phase chromatography separation of peptides was 50 cm × 75 μm inner diameter, PepMap RSLC C18, 2 μm (Thermo Fisher Scientific), and used at a flow rate of 250 nl/min over a 90-min gradient of acetonitrile consisting of 4–40% B (mobile phase A, 0.1% formic acid in water; mobile phase B, 0.1% formic acid in 97% acetonitrile). Peptide precursors were subjected to collision-induced dissociation (CID) MS/MS fragmentation in the ion trap for the 15 most abundant precursor ions (data-dependent acquisition) with the following parameters: ion trap MS/MS target value of 1E4 (100-ms maximum ion injection time), enabled Fourier transform MS predictive automatic gain control with a target value of 1E6 (500-ms maximum ion injection time), enabled dynamic exclusion (repeat count of 1 and exclusion duration of 70 s), and enabled lock mass (mass list, 371.101233). The m/z range of an MS scan was 350–1700, and the resolution was set to 60,000; CID fragmentation used an isolation width of 2.0 Th, normalized collision energy of 30%, and 10-ms activation time. The mass spectrometry proteomics datasets have been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org/) via the PRIDE partner repository with the dataset identifier PXD005152.
Database Searching and Protein Assignments
Raw data from MS analyses of immunoisolations were extracted and searched against UniProt Swiss-Prot sequence database (22,630 entries, including human, herpesvirus, and common contaminants, and was downloaded August, 2013) in Proteome Discoverer (version 1.4.0.288, Thermo Fisher Scientific) using the Sequest HT algorithm (version 1.3, Thermo Fisher Scientific). The following criteria were used as search parameters: full trypsin specificity, maximum of two missed cleavage sites, precursor and fragment ion mass tolerance of 10 ppm and 0.5 Da, respectively; dynamic modifications: oxidation (+15.995 Da (Met)), phospho (+79.966 Da (Ser, Thr, and Tyr)); static modifications: carbamidomethyl (+57.021 Da (Cys)). Percolator in Proteome Discoverer was used to calculate peptide spectral match probabilities against decoy database. Protein identification validation was performed in Scaffold (version 4.4.8; Proteome Software, Inc., Portland, OR) using X!Tandem (31) algorithm. Scaffold searched the additional variable modifications: Glu → pyro-Glu (−18.01), ammonia loss (−17.03), Gln → pyro-Glu (−17.03), deamidation (+0.98, Asn and Gln), and carbamylation (+43.01, Lys and n). Probabilistic validation of peptide identifications was performed using the Bayesian model local false discovery rate (FDR) algorithm in Scaffold. Probability filters were set to 1% FDR at the protein and peptide levels, which resulted in statistical confidence scores for all proteins of 98% or greater, and protein identification required at least two unique peptides in at least one biological replicate. Common contaminant entries were removed, and spectral counts were exported for further analysis. The supplemental Table S1 presents the number of unique peptides and the sequence coverage for each identified protein. SAINT algorithm (24) was used to filter out nonspecific associations based on spectral counts using n-burn = 2000, n-iter = 4000, LowMode = 0, minFold = 0, normalize = 1 settings. Spectral count matrices were prepared for IFIX-GFP IPs relative to each respective GFP control IPs (mock IPs, wild-type IPs, and RF IPs). Interacting partners passing the stringent probability cutoff score of 0.90 were used for further analysis (supplemental Table S1).
Assembling IFIX Protein Interaction Networks
To generate functional interaction networks, the protein that passed the SAINT 0.9 score filter was first submitted to the STRING database (32) using default parameters except for the interaction confidence threshold, which was set to 0.9 STRING networks. The IFIX interacting proteins were then imported into Cytoscape (version 3.1.1) (33). Overlapping proteins during WT and RF infection are depicted by diamond node shapes to distinguish them from those found in uninfected or WT only samples (circles). A color gradient was used to indicate the log2-transformed fold enrichment of normalized spectral counts between uninfected and WT HSV-1-infected cells, and the color orange indicates interactions unique to infection. Edges represent functional relationships as determined by STRING. The ClueGO plug-in within Cytoscape was used to cluster proteins according to their biological or molecular gene ontology (GO) terms.
Generation and Transfection of IFIX Domain Constructs
IFIX PY was subcloned into pEGFP-N1 plasmid as described previously (5). The HIN200 domain of IFIX (HIN) was amplified from FL IFIX in pEGFP-N1 (2) and subcloned into pEGFP-N1 using XhoI and BamHI restriction sites (supplemental Table S2). Plasmids were transfected into 293T cells using Lipofectamine 2000 and 0.3 μg of DNA per well in a 12-well plate for Western blotting or 6 μg of DNA in a 10-cm plate for IPs.
Validation of IFIX-SENP3 Interaction by Reciprocal Isolation
Two 10-cm dishes of induced EGFP or IFIX-GFP 293 cells were harvested after infection with WT HSV-1 at 4 hpi and lysed in 1 ml of lysis buffer per plate. Preclearing was performed by incubating protein A/G-agarose beads in the lysates for 30 min at 4 °C. Subsequently, cell lysates were incubated with α-SENP3 antibody for 1 h at 4 °C, at which time 20 μl of precleared protein A/G beads was added for an additional 1 h at 4 °C. The beads were washed three times with lysis buffer and two times with PBS, resuspended in 40 μl of SDS sample buffer, and eluted by heating at 95 °C for 5 min. Eluates were then centrifuged to pellet the beads/insoluble fraction and analyzed by Western blotting.
Determination of Cell Line Doubling Times
EGFP or IFIX-GFP-stable HFF cells were seeded at a density of 1E5 in a 12-well dish. Cells were lifted with trypsin and counted by trypan blue exclusion method on a hemocytometer at 48 and 72 h after seeding. Doubling time was calculated by using a standard formula: doubling time = duration(h)·log(2)/log(final cell density) − log(initial cell density). Doubling times were calculated for each time point and averaged.
cDNA Generation and Quantitative PCR
RNA extraction and generation of cDNA via reverse transcription PCR (RT-PCR) were performed using Cell-to-Ct kit (Ambion) according to the manufacturer's instructions. For quantitative PCR, amplification was performed using SYBR Green PCR Master Mix (Life Technologies, Inc.) and gene-specific primers (supplemental Table S2) with an ABI 7900HT Fast Real-Time PCR System (Applied Biosystems). Relative quantification was assessed by the ΔΔCT method using 18S rRNA as the reference gene. 100,000 cells were used per sample; 40% of the lysis was used for RT-PCR, and 0.5% of cDNA was used for quantitative analysis.
RESULTS
Protein Levels and Subnuclear Localization of IFIX Change during Infection with HSV-1
We have previously shown that, upon HSV-1 infection, IFIX localization is predominantly nuclear, where it can bind to viral DNA and promote antiviral cytokine expression (2). However, how IFIX is regulated or how it exerts its defense functions during HSV-1 infection remained unknown. To better understand the regulation of IFIX during the early stages of HSV-1 infection, we first used microscopy to monitor its localization in HEK293 cells over the course of 8 hpi (Fig. 1A). We started by using this cell model system, as we previously demonstrated that IFIX tagged with GFP and expressed in HEK293 cells has an antiviral function, inhibiting HSV-1 titers (2). The viral protein ICP27 was used as a marker of infection. Our microscopy analyses showed that IFIX localization is quickly altered upon infection, changing from a predominantly diffuse nuclear and nucleolar distribution to a concentrated localization within discrete nuclear puncta (Fig. 1A, 2 and 4 hpi). Additionally, IFIX protein levels appeared to be diminished as the infection progressed, with a reduction in both diffuse and punctate IFIX (Fig. 1A, 6 and 8 hpi). To confirm this finding, we next performed analyses in primary human fibroblasts, a commonly used and biologically relevant system for studying HSV-1 infection. We generated primary human fibroblasts stably expressing IFIX-GFP and monitored IFIX levels upon HSV-1 infection using microscopy (Fig. 1B). The observed phenotype was even more striking in these cells, as IFIX-GFP became virtually undetectable by microscopy at 4 hpi (Fig. 1B, compare infected and uninfected cells). These results indicate that the localization and protein levels of IFIX-GFP are dynamically regulated during WT HSV-1 infection.
Fig. 1.

IFIX displays altered nuclear localization and protein levels during infection with WT HSV-1. A, time course showing IFIX-GFP localization in Flp-In 293s at the indicated hpi. m.o.i.: 5, ICP27 is marker of infection. B, IFIX-GFP localization upon infection of primary HFFs stably expressing IFIX-GFP. m.o.i.: 3 was taken at 4 hpi; ICP8 is marker of infection. A and B, bar, 5 μm.
To further confirm the changes in IFIX protein levels in fibroblasts, we performed Western blotting analyses. Indeed, an 8-h time course demonstrated reduction in IFIX-GFP levels during infection (Fig. 2A). Comparison with EGFP control cells showed that this decrease in signal was not driven by the GFP tag, as we did not observe reduction in the levels of control GFP protein. To determine whether IFIX levels return at late time points of infection, we also monitored IFIX levels every 6 h up to 24 hpi, which covers the duration of the life cycle of active HSV-1 infection in fibroblasts. IFIX-GFP levels were not rescued later in infection (Fig. 2B). Altogether, these data demonstrate that IFIX-GFP protein levels diminish by 4 hpi and do not return at late time points post-infection.
Fig. 2.

Protein levels of IFIX-GFP decrease by 4 h post-infection and remain diminished over the course of HSV-1 infection in primary fibroblasts. A, time course of EGFP or IFIX-GFP HFF cells during infection with BFP-HSV-1 analyzed by Western blotting. The blot reveals a decrease in IFIX-GFP levels over time that is not due to GFP being targeted or cleaved. B, late times of WT HSV-1 infection in IFIX-GFP stable HFFs analyzed by Western blotting showing the lack of IFIX-GFP signal late in infection. C, still images extracted from live cell microscopy movies, taken with ×60 objective, monitoring IFIX-GFP in uninfected (mock) and BFP-tagged HSV-1 infected stable HFF cells (see also supplemental Movie S1). Bar, 5 μm. D, schematic of the changes in IFIX sub-nuclear localization and levels during HSV-1 infection. Small green dots represent diffuse IFIX recruitment to distinct puncta that are dispersed over time.
When considering the Western blotting results, something to take into account is that although we optimized the multiplicity of infection to obtain a large percentage of infected cells, the cells lysates may still contain a small percentage of uninfected cells. Therefore, to determine the temporality of this IFIX regulation, we next used time-lapse microscopy to monitor IFIX-GFP in single infected live cells (Fig. 2, C and D). We focused on the first 5 h of infection, given our finding that the detection of IFIX was already significantly reduced in infected fibroblasts by 4 hpi (Fig. 1B). During mock infection (i.e. uninfected cells), IFIX-GFP remained diffuse throughout the nucleus with an enrichment in nucleoli, and its overall levels did not change (Fig. 2C, top row). However, during HSV-1 infection, the diffuse nucleoplasmic localization appeared diminished, concomitant with the appearance of discrete puncta between 130 and 220 min post-infection (Fig. 2C, middle and bottom rows; and supplemental Movie S1). Altogether, our results demonstrate that upon HSV-1 infection, IFIX localized to discrete puncta at the nuclear periphery and within the nucleoplasm, followed by a rapid reduction in its protein levels (Fig. 2D).
IFIX Interacts with Ubiquitin-Proteasome Components and Transcriptional Regulators during HSV-1 Infection
To identify factors that could contribute to IFIX antiviral functions, as well as to the observed modulation of IFIX levels, we performed IP-MS experiments to define IFIX protein interactions during HSV-1 infection. Given the absence of suitable antibodies for isolating endogenous IFIX, and as we previously established that IFIX-GFP can bind viral DNA and induce an interferon response in inducible Flp-In 293 cells (2), we selected this cell system for our interaction study. IFIX-GFP-inducible Flp-In 293 cells or control inducible GFP cells were mock-infected or infected at an m.o.i. of 5 and harvested at 4 hpi to capture IFIX interactions at the time when its levels are modulated. Affinity purifications were carried out on magnetic beads conjugated to anti-GFP antibodies and processed for mass spectrometry (Fig. 3A). The lysis buffer was optimized for the efficient isolation of IFIX, as validated by comparing the pellet, input, flow-through, and elution fractions by Western blotting in mock and infected cells (Fig. 3B).
Fig. 3.

IFIX protein interactions early in HSV-1 infection. A, experimental workflow. B, immunoblot showing the efficiency of IFIX isolations in uninfected and WT infected cells. FT, flow-through. C, IFIX interactions during HSV-1 infection. Diamond-shaped nodes indicate proteins found to be specific in both WT and RF infections. The blue-to-yellow gradient depicts the fold change of WT infected/mock relative quantification of spectral counts. Orange nodes represent proteins found to be unique in infected cells. Proteins are grouped according to their GO annotations assigned by ClueGO.
The specificity of the identified IFIX interactions was assessed by comparing biological replicates of IFIX-GFP and GFP isolations using the SAINT algorithm (24). Proteins that passed a stringent threshold of >0.9 SAINT scores were visualized within a functional interaction network that takes into account known biological or molecular GO terms (Fig. 3C, clusters), as well as prior evidence of interactions from the STRING database (Fig. 3C, lines) (32). The node colors in the network illustrate the fold change in IFIX interaction identified in both mock and WT infection (Fig. 3C, blue-to-yellow gradient). Additionally, proteins found to associate with IFIX only upon infection, and not observed in uninfected cells, are depicted as orange nodes in Fig. 3C. Overall, the identified IFIX interactions fell within functional categories that likely represent both its antiviral functions and the regulation of its levels. For example, we found IFIX interactions with proteins involved in ubiquitin-proteasome processes, including E3 ubiquitin ligases HUWE1, RBX1, and UBR5, as well as with proteins within E3 ubiquitin ligase complexes, including BTBD2, KEAP1, and CUL4. The interactions between IFIX and these ubiquitin ligase complex components suggest that a proteasome degradation of IFIX may trigger the decrease in IFIX levels during infection. Supporting this idea, we observe IFIX to interact with the proteasome subunit PSMB7.
Other observed associations, such as those with transcriptional regulatory proteins and immune factors, may contribute to IFIX antiviral functions. In fact, the majority of IFIX interactions were with proteins involved in transcriptional regulation and the epigenetic regulation by heterochromatin remodeling (Fig. 3C). We previously reported that host cells can more effectively elicit cytokine responses upon infection with an HSV-1 RF strain than upon WT infection (22), as this strain has a mutated RING finger domain in ICP0 that diminishes its E3 ligase activity and its ability to inhibit host immune effectors (23). Therefore, to determine which of these interactions are maintained or enhanced in an environment where cells can trigger immune responses, we also performed AP-MS studies upon HSV-1 RF infection. The specific IFIX interacting partners that were shared between WT and RF infections are depicted as diamond-shaped nodes (Fig. 3C). Among the most prominent associations retained upon HSV-1 RF infection were proteins forming the five friends of methylated chromatin target of Prmt1 (CHTOP) (5FMC) complex. The 5FMC components include PELP1, SENP3, LAS1L, TEX10, and WDR18 (34). These proteins were observed as specific interacting partners of IFIX in both uninfected and HSV-1 WT infected cells (Fig. 3C, gradient nodes), and three components of the 5FMC complex (LAS1L, SENP3, and WDR18) were also found during HSV-1 RF infection. The 5FMC complex and related methyltransferase proteins are known to function in transcription-related processes (34) and, given its dependence on CHTOP, may be a bridge between the GO functional groups' epigenetic and transcriptional regulation. Their association with IFIX is in agreement with the prior thinking that one of the housekeeping functions of IFIX is in transcriptional regulation. In the context of infection, the maintenance of this interaction may provide IFIX the means to act in defense by possibly regulating viral gene expression. Therefore, to further investigate both the regulation and the antiviral roles of IFIX, we chose to further analyze the significance of IFIX interactions with proteins involved proteasome-dependent processes and in transcriptional regulation.
IFIX Pyrin Domain Is Targeted for Degradation in a Proteasome-dependent Manner during HSV-1 Infection
Given the IFIX association with E3 ubiquitin ligases and a proteasome subunit, we considered the possibility that IFIX may be degraded through the proteasome. Therefore, we sought to distinguish between this mechanism of degradation and other possibilities, such as the degradation of IFIX via an autophagy mechanism or its cleavage, leading to the inability to visualize GFP. We first visualized the localization of IFIX puncta relative to proteasomes early in infection. We aimed to capture the 3.5 hpi time point, which was shown by our live cell microscopy study to provide a high number of cells displaying IFIX puncta (Fig. 2C). To achieve the necessary level of synchronization, we adapted a protocol shown to be effective at synchronizing the entry of virus particles during the early stages of HSV-1 infection (35). Specifically, fibroblasts were infected with WT HSV-1 at 4 °C in CO2-independent media, and the co-localization of IFIX-GFP with the 20S core proteasomal subunit was monitored by microscopy at 3.5 hpi. We observed that nuclear proteasomes were formed during HSV-1 infection, appearing at sites of IFIX-GFP puncta (Fig. 4A). Interestingly, we did not see the presence of discrete nuclear proteasomes in uninfected cells, when IFIX-GFP also remained diffusely distributed throughout the nucleus. To confirm the proteasome-mediated degradation of IFIX-GFP during WT infection, we inhibited the proteasome by treating cells with the compound MG132 and analyzed IFIX levels by Western blotting (Fig. 4B). As expected, we observed diminishing levels of IFIX-GFP during WT infection in cells not treated with MG132 and the rescue of IFIX-GFP to uninfected (i.e. mock-infected) levels in cells treated with MG132. Next, to determine the domain of IFIX that is being degraded, we monitored the protein levels of FL, PY, and HIN of IFIX-GFP in infected or uninfected cells (Fig. 4C, and full blots shown in supplemental Fig. S1). Reduction in IFIX levels was only observed for the FL and PY constructs but not for the HIN domain. Altogether, these microscopy and Western blotting results demonstrate that the pyrin domain of IFIX-GFP is targeted for proteasome-mediated degradation in the nucleus during WT HSV-1 infection, and it is in agreement with our proteomic interaction study that showed the IFIX association with the proteasome subunit PSMB7 only upon HSV-1 infection (Fig. 3C).
Fig. 4.
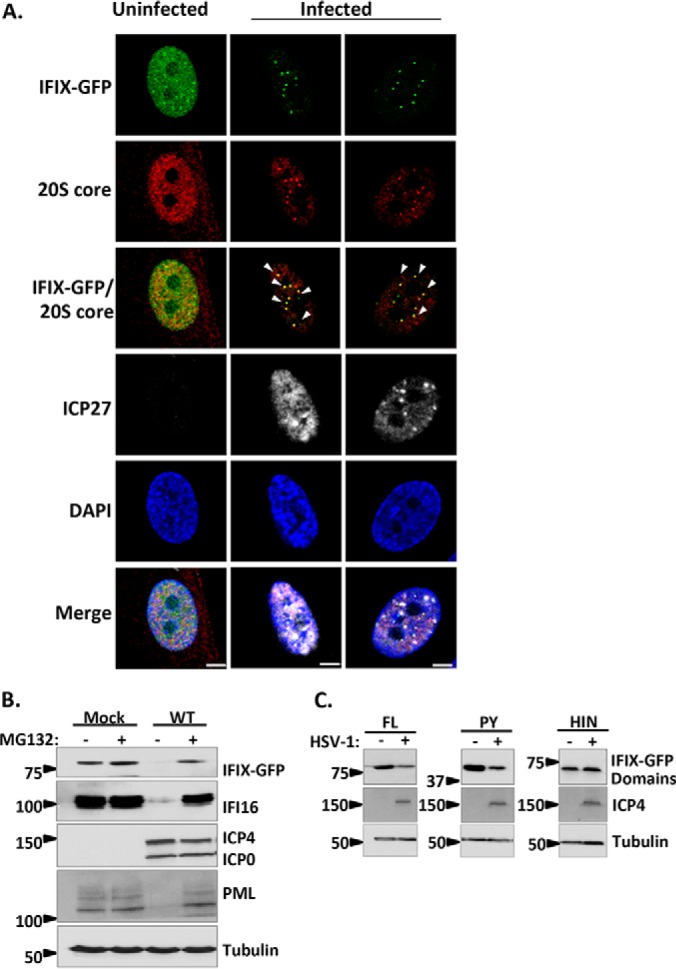
IFIX is targeted for proteasome-dependent degradation during infection with HSV-1. A, direct- and immunofluorescence microscopy during WT HSV-1 infection displaying IFIX-GFP puncta localizing to nuclear proteasomes. The 20S core subunit of the proteasome is in the red channel, and ICP27 as a marker for infection is in white. m.o.i.: 10, 3.5 hpi. Bar, 5 μm. B, Western blot of IFIX-GFP stable HFFs during WT HSV-1 infection with and without proteasome inhibitor MG132 treatment. ICP4 and ICP0 are markers for infection. IFI16 and PML are positive controls. IFIX levels decrease during infection but are rescued by MG132 treatment. m.o.i.: 10, 6 hpi. C, Western blot of 293Ts transfected with IFIX-GFP domain constructs reveals the pyrin domain of IFIX is the target for degradation. FL, full length; PY, pyrin domain; HIN, HIN200 domain. ICP4 is marker for infection. m.o.i.: 10, 6 hpi. See supplemental Fig. S1 for exposures of the intact Western blot membranes.
Decrease in IFIX Levels Requires Immediate-Early Viral Gene Expression but Is Not Dependent on the E3 Ligase Activity of the Viral Protein ICP0
We next investigated which host or viral protein initiates the degradation of IFIX. Our protein interaction network revealed IFIX associations with both host and viral E3 ubiquitin ligases. We first focused on the cellular E3 ubiquitin ligase HUWE1, which interacted with IFIX exclusively in infected cells. Using siRNA-mediated KD of HUWE1, we determined that HUWE1 is not responsible for the degradation of IFIX (supplemental Fig. S2). Our proteomic study also showed that IFIX interacts with the viral E3 ubiquitin ligase ICP0 upon infection with HSV-1 RF mutant strain (Fig. 3C). Although ICP0 was also detected in the IFIX IP in cells infected with WT HSV-1, it did not pass our SAINT threshold due to its low levels. It is possible that this difference is driven by the more transient nature of this interaction when ICP0 is active, but this remains to be determined. As mentioned above, the viral E3 ubiquitin ligase ICP0 was shown to play a role in the degradation of several host defense proteins, including PML and another PYHIN protein, IFI16 (12, 17, 18, 22). Therefore, we asked whether ICP0 may also be responsible for the observed reduction in IFIX protein levels during HSV-1 infection. First, comparison of IFIX-GFP levels upon infection with either WT or UV-inactivated virus showed that although we observed the expected decrease in IFIX levels upon WT infection, the levels are rescued upon infection with the UV-treated virus (Fig. 5A). This demonstrates that viral gene expression is required for the reduction in IFIX protein levels. Next, we tested the specific contribution of ICP0. We compared infections in fibroblasts with either WT HSV-1 or the RF mutant virus, which lacks ICP0 E3 ligase activity. However, the difference in the replication kinetics of these two viruses is important to consider when comparing these infections. At low m.o.i., the HSV-1 RF virus was shown to have delayed replication kinetics, thought to be associated with reduced levels in the viral immediate-early protein ICP27 (23), an important regulator of viral gene transcription (36, 37). Slower replication has also been reported for an ICP0-null virus and was shown to be rescued when cells are infected at a higher m.o.i. (38). Therefore, we reasoned that we could rescue the delayed replication kinetics during RF infection so that we could effectively compare WT to RF infection at the same time post-infection. In primary human fibroblasts, we observed that an m.o.i. of 20 in RF infection yields an equivalent number of HSV-1 progeny as WT infection at an m.o.i. of 10 as determined by t test (Fig. 5B). Therefore, we used these infection conditions to compare IFIX protein levels at 6 hpi with WT or RF virus strains (Fig. 5C). By monitoring PML, a well known substrate of ICP0, we confirmed that the RF mutation inactivates the ICP0 E3 ligase activity. Indeed, PML was not degraded during RF infection. However, we observed that IFIX protein levels were still decreased following RF infection, at a similar extent to that seen during WT infection. Microscopy confirmed that IFIX levels were diminished during infection with the RF mutant (Fig. 5D). Therefore, IFIX degradation is not dependent on the E3 ligase activity of ICP0. However, the time when IFIX levels are observed to decrease (i.e. 4 hpi) coincides with viral immediate-early processes. Thus, to gain further insight into the viral process that is triggering the decrease in IFIX levels, we infected IFIX-GFP-expressing fibroblasts with a mutant virus that has the ability to enter a host cell, but lacks all immediate-early genes, the d109 virus (39). We compared IFIX levels during infection with WT, RF, and d109 viruses (Fig. 5E). Although, as expected, we see the reduction in IFIX levels during WT and RF infections, its levels were not reduced upon infection with the d109 virus. This observation indicates that viral tegument proteins contained in the virion are not sufficient to induce IFIX degradation, and that IE gene expression is likely necessary for this degradation. The IE-mediated degradation of IFIX can occur via either direct targeting by a viral IE protein or as a by-product of an immediate-early process. However, these results do not preclude the possibility that the reduction in IFIX levels is due to a delayed-early viral protein or process, because the delayed-early expression is controlled by immediate-early proteins. To further investigate this, we used treatment with an inhibitor of protein translation, cycloheximide (CHX), to control the expression of viral genes. Our results suggest that IE proteins are required for IFIX degradation (Fig. 5F). When IE proteins are inhibited by CHX (i.e. treatment at 0 hpi and collection of samples at 5 hpi), IFIX is still visible. However, when IE proteins are present, but DE proteins are inhibited (i.e. treatment at 3 hpi and collection of samples at 7 hpi), IFIX is degraded. The presence or absence of IE and DE proteins was monitored by blotting for the IE protein ICP27 and DE protein ICP8. Altogether, our results indicate that (i) IFIX degradation is observed by 4 h post-infection, (ii) viral tegument proteins are not sufficient to induce degradation, (iii) viral gene expression is required for IFIX degradation, as a UV-treated virus cannot induce degradation, (iv) IE proteins are necessary for degradation, and (v) DE proteins do not seem sufficient to fully degrade IFIX suggesting that viral immediate-early proteins are primary contributors to IFIX degradation.
Fig. 5.

Decrease in IFIX levels requires viral gene expression, yet is not dependent on the E3 ligase activity of the viral protein ICP0. A, UV-induced inactivation of viral genes rescues IFIX protein levels. Cells were mock-infected (M) or infected with WT or UV-treated HSV-1 at m.o.i. 10, harvested at 6 hpi, and analyzed by Western blotting. B, testing equivalence of infection progression of wild type HSV-1 and ICP0 RING finger mutant (RF) HSV-1 by comparing HSV-1 progeny titers in HFF cells stably expressing EGFP. Cells were infected with WT at m.o.i. 10 or RF at m.o.i. 20. Cells and supernatants were collected at 22 hpi (95% cytopathic effect) and titered on U2OS cells. n.s., not significant. Error bars represent S.D. of two biological replicates run in technical duplicates. C, Western blot in HFFs stably expressing EGFP or IFIX-GFP comparing the indicated proteins during mock, WT (m.o.i. 10), or RF (m.o.i. 20) infection was at 6 hpi. IFIX levels decrease during infection with WT and RF viruses. PML is blotted to show ICP0 ligase activity is inhibited in the RF virus. ICP0 is marker for infection; ICP27 displays equal levels in both viruses. D, microscopy during RF infection displays decrease in IFIX-GFP in primary fibroblasts stably expressing IFIX-GFP. m.o.i.: 3, 6hpi; ICP27 is marker of infection. Bar, 5 μm. E, Western blot in HFFs stably expressing IFIX-GFP demonstrate IFIX levels do not decrease during d109 virus infection. Mock (M), WT (m.o.i. 10), RF (m.o.i. 20), or d109 (m.o.i. 20) infection was at 6 hpi. ICP4 and ICP0 are markers for infection and a validation for d109, which lacks all immediate-early genes. F, time course analyzed by Western blotting comparing IFIX-GFP levels in fibroblasts treated with cycloheximide (CHX) at 10 μg/ml to inhibit immediate-early viral protein expression (0–5 hpi) or delayed-early viral protein expression (3–7 hpi) and IFIX-GFP levels in fibroblasts not treated with CHX during WT HSV-1 infection.
Our findings demonstrate that HSV-1 has acquired mechanisms to inhibit IFIX functions by inducing its degradation, further emphasizing the importance of IFIX in host defense. However, this virus-mediated suppression of IFIX is only one side of the intertwined mechanisms that regulate the progression and spread of infection. To develop antiviral strategies and rescue host defenses, a better understanding of the mechanisms regulating host response is needed. Although we previously demonstrated that IFIX attenuates HSV-1 titers (2), the mechanisms regulating its antiviral functions remain unknown. Aside from ubiquitin-proteasome processes, our interaction study also revealed prominent IFIX interactions with epigenetic and transcription regulatory proteins. Given the efficiency of WT HSV-1 in suppressing host innate immune responses (12, 22), these IFIX-interacting functional groups could point to a mechanism separate from cytokine induction through which IFIX can act in antiviral response. Therefore, we next explored the role of IFIX and transcription during infection.
HIN Domain of IFIX Mediates Its Interaction with the 5FMC Transcription Complex
As noted above, in addition to pointing to the IFIX association with the proteasome, our protein interaction study also highlighted prominent interactions with transcriptional regulatory proteins. Of interest, IFIX retained its interaction with members of the 5FMC complex (PELP1, SENP3, LAS1L, TEX10, and WDR18 (34)) across infections, as well as in uninfected cells (Fig. 3C). We validated the association with different 5FMC complex members by reciprocal isolation and microscopy. By immunoaffinity purification of endogenous SENP3, we confirmed the co-purification of IFIX-GFP (Fig. 6A). Having validated this association, we next investigated which IFIX domain mediates the interaction with the 5FMC complex. We transfected the IFIX-GFP FL, PY, HIN, or GFP control constructs into 293T cells, and IPs were performed using GFP antibodies. We found that the 5FMC components SENP3 and LAS1L interacted with the FL or the HIN (Fig. 6B). Therefore, the interaction of IFIX with the 5FMC complex is mediated by its HIN domain. It is possible that without the pyrin domain the HIN200 domain, which we know is responsible for the binding of IFIX to dsDNA, can enrich for the associations of IFIX with transcriptional regulatory proteins, leading to increased HIN association with 5FMC members when compared with FL. By microscopy, we observed the co-localization of IFIX-GFP with LAS1L and PELP1 within the nucleoli of uninfected cells (Fig. 6C and supplemental Fig. S3A). Interestingly, the diminished IFIX levels during infection are accompanied by a redistribution of these 5FMC proteins as exhibited by the loss of their nucleolar localization (Fig. 6C and supplemental Fig. S3B; white arrows indicate infected cells). This appears to be independent of IFIX, because we observe the same loss of nucleolar localization in GFP control cells (Fig. 6C and supplemental Fig. S3B). We further investigated whether this redistribution is also connected to a reduction in protein levels (similar to IFIX). However, our results show that the 5FMC LAS1L and PELP1 protein levels remain similar in uninfected and infected cells (Fig. 6D). Altogether, our results show a virus-induced redistribution of 5FMC proteins during infection, which is not representative of a degradation event. This may either represent a virus-induced recruitment of this complex to aid viral gene expression or a host-induced process in defense against infection.
Fig. 6.
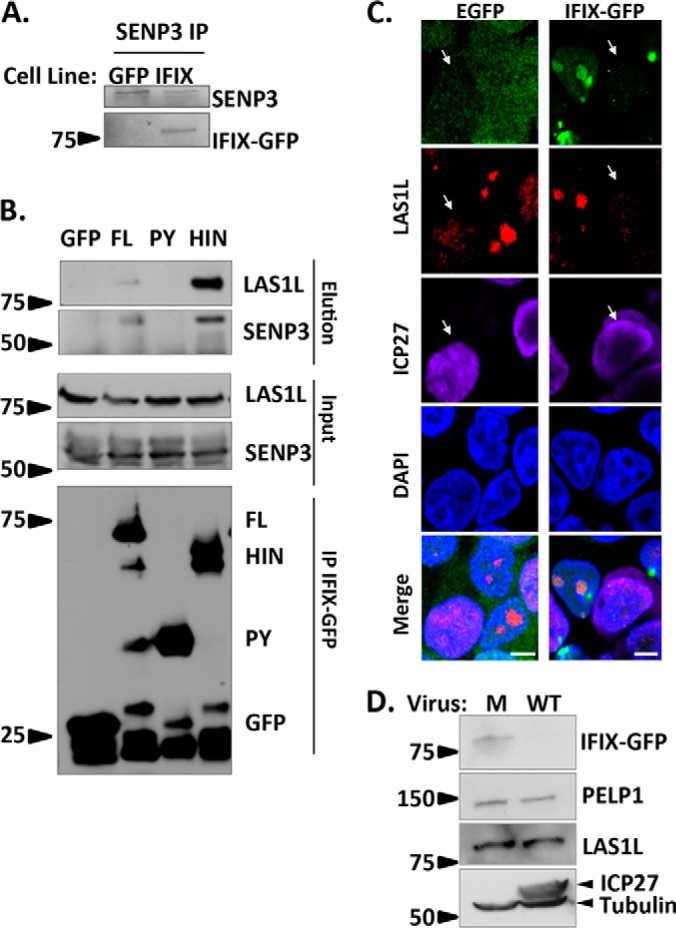
HIN domain of IFIX mediates its interaction with the 5FMC complex. A, validating IFIX co-interaction with SENP3 by reciprocal IP. B, IFIX interacts with 5FMC components LAS1L and SENP3 through its HIN domain. Forward IPs using GFP antibody were performed in cells transfected with IFIX constructs full-length (FL), IFIX Pyrin domain (PY), and IFIX HIN200 domain (HIN) in pEGFP. Inputs (1.5%), elutions (20%), and isolated IFIX constructs (IP, 20%) were blotted for LAS1L, SENP3, and GFP. C, IF microscopy in IFIX-GFP and EGFP control 293 cells showing a redistribution of 5FMC protein LAS1L in infected cells (white arrows), m.o.i.: 5, 4 hpi. Co-localization of IFIX and LAS1L is pronounced in uninfected cells (for PELP1, see supplemental Fig. S3). D, Levels of PELP1 and LAS1L are not reduced during HSV-1 infection. PELP1 and LAS1L levels were monitored by western blotting at 6hpi. ICP27 is marker for infection. M, mock. Microscopy images were taken at ×60 oil objective. Bar, 5 μm.
IFIX Inhibits Viral Gene Transcription Possibly by Sequestering the 5FMC Complex
Our finding that IFIX interacts with numerous transcriptional regulatory proteins (Fig. 3C) and our validation and characterization of its association with the 5FMC complex (Fig. 6) led us to propose that IFIX may exert its antiviral function in part by transcriptional repression of viral genes. To test this, we assessed the impact of IFIX overexpression on viral genes. First, we confirmed the overexpression of IFIX in our primary human fibroblasts stably expressing IFIX-GFP (Fig. 7A). Next, we compared the relative levels of viral genes after infection of either IFIX-GFP or EGFP control cells. Given the known temporal cascade of viral gene expression upon HSV-1 infection (40, 41), we monitored immediate-early and early viral genes. We found IFIX-dependent decreased levels of both the immediate-early viral gene ICP27 and the early viral gene ICP8 (Fig. 7B). To further investigate how IFIX can impact viral gene expression, we monitored its localization relative to viral genomes when IFIX degradation is inhibited (Fig. 7C). As ICP4 protein is localized to viral DNA throughout the early time points of infection, it is commonly used as a marker for viral genomes (42–44). Our results demonstrate that IFIX localizes to viral genomes at 3 and 6 hpi in primary human fibroblasts. This IFIX puncta seems more distinct than the puncta co-localizing with the proteasome. When treating cells with MG132 and monitoring IFIX localization relative to the 20S core, we observed a loss of co-localization of IFIX with the proteasome (supplemental Fig. S4). However, as expected, IFIX was still able to form nuclear aggregates. Therefore, IFIX has two distinct puncta localizations, one at proteasomes (prior to its degradation by HSV-1) and one at viral genomes (when its degradation is inhibited). To assess the impact of IFIX levels on virus titers, we first confirmed that the growth rates of the EGFP and IFIX-GFP primary fibroblasts are equivalent, as shown by the measured cell doubling times (Fig. 7D). This was monitored to ensure that any observed differences in virus titers are not due to differences in cell numbers, i.e. fewer cells yielding fewer virus particles. As expected, we saw a significant decrease in virus titers upon overexpression of IFIX when comparing IFIX-GFP cells to EGFP control cells (Fig. 7E, left). These results support a model in which IFIX works in antiviral response at least in part by suppressing viral gene expression. Additionally, this finding prompted us to determine whether the interaction between IFIX and 5FMC components contributes to this transcriptional inhibition of viral genes. To test this possibility, we knocked down the 5FMC components PELP1 and SENP3 by siRNA and monitored virus titers during infection in IFIX-GFP or control EGFP fibroblasts. The knockdown efficiencies were confirmed by Western blotting (Fig. 7, F and G). Interestingly, the antiviral effect of IFIX overexpression was less prominent upon knockdown of the 5FMC components SENP3 and PELP1 (Fig. 7E). Furthermore, the virus titers even in the control EGFP cells were decreased when SENP3 and PELP1 were knocked down. It is possible that the 5FMC components positively regulate viral gene expression, and IFIX may sequester the complex from its gene targets.
Fig. 7.
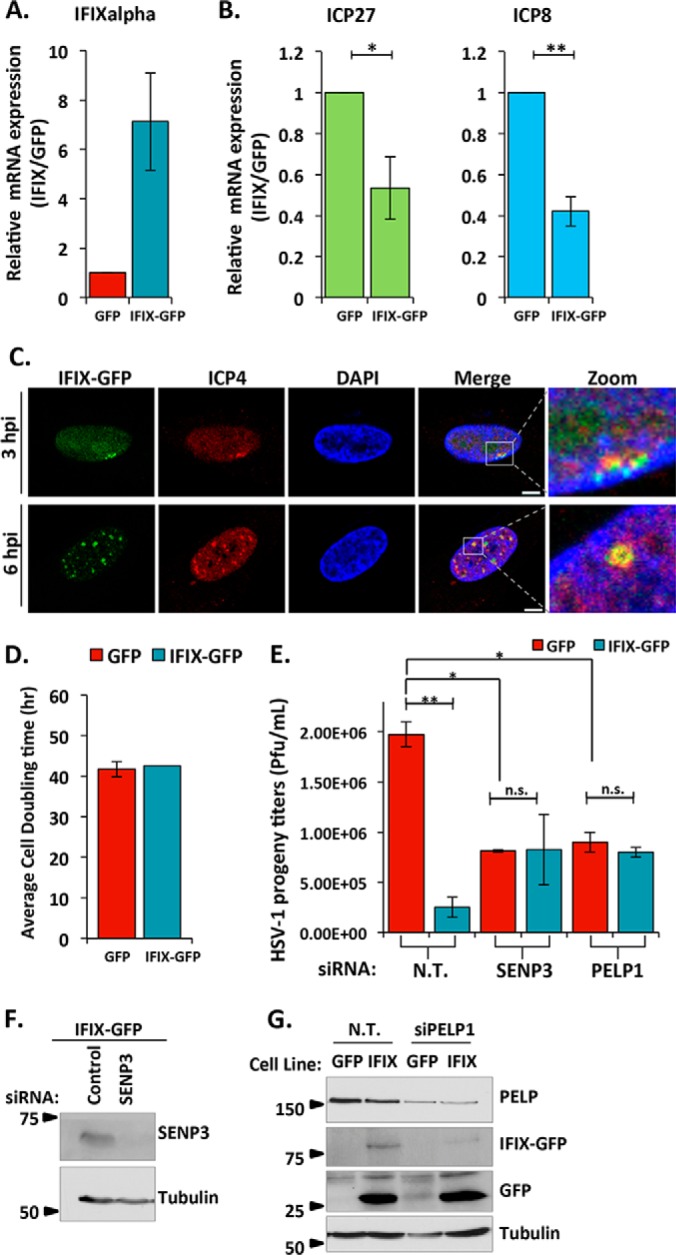
IFIX inhibits viral gene transcription. A, confirmation of IFIX is overexpressed in stable HFFs. B, relative mRNA levels of viral genes ICP27 and ICP8 determined by quantitative PCR. Cells were infected with WT HSV-1 at an m.o.i. of 10 and collected at 6 hpi. Error bars represent S.D. of three biological replicates. Significance was determined by t test. *, p ≤ 0.05; **, p ≤ 0.005. C, IFIX-GFP localizes to viral genomes when its degradation is inhibited. Cells were treated with MG132 to block the proteasome during the course of 3- or 6-h infections. ICP4 is marker for viral genomes. Size bars, 5 μm. D, doubling times of HFFs stably expressing EGFP or IFIX-GFP. E, siRNA-mediated knockdown of 5FMC components PELP1 and SENP3 impact HSV-1 progeny titers in human fibroblasts. EGFP or IFIX-GFP HFFs were transfected with the indicated siRNAs. Cells were infected with WT HSV-1 at an m.o.i. of 10 at 24 h post-transfection. N.T., non-targeted. Error bars indicate S.D. of two biological replicates in technical duplicate; significance was determined by t test. *, p ≤ 0.05; **, p ≤ 0.005. n.s., not significant. F and G, validation of knockdown efficiency of PELP1 and SENP3 by Western blotting.
DISCUSSION
DNA virus infection can trigger a multitude of host defense mechanisms, including the sensing of pathogen-associated molecular patterns that stimulate immune cytokine expression (45), DNA damage response associated defenses (46), and inhibition of viral gene expression at the chromatin regulation levels (47, 48). We have recently identified IFIX as a DNA sensor that is able to bind viral DNA either in the cytoplasm, upon transfection of dsDNA fragments from the cytoplasmic replicating vaccinia virus, or in the nucleus, upon infection with the nuclearly replicating HSV-1 (2). We also found IFIX to promote IFNβ expression and that increased IFIX levels result in decreased virus progeny production (2). Here, we show that one antiviral function of IFIX is to inhibit the expression of viral genes, which suggests that IFIX-mediated host defense is not limited to its DNA-sensing ability. Increased IFIX levels already impacted the first step in the temporal cascade of viral gene expression during HSV-1 infection, which is the expression of viral immediate-early genes. This IFIX-mediated inhibition was also maintained for the next temporal step, i.e. early viral gene expression. As early gene expression depends on immediate-early gene expression, whether the initial inhibitory effect of IFIX causes propagation of viral gene suppression or whether these are separate mechanisms remains to be seen. Noteworthy, increased IFIX levels result in a substantial decrease in HSV-1 titers. Therefore, the cumulative functions of IFIX are effective at inhibiting the spread of infection.
The mechanism by which IFIX inhibits viral gene expression may be at the transcriptional level, where IFIX can bind to the viral genome and effectively block the transcriptional machinery. This would be in agreement with our previous observation that IFIX can directly bind DNA through its HIN domain and co-precipitate with HSV-1 DNA (2). This is also consistent with the data presented here, where we demonstrate that IFIX localizes to viral genomes early in infection. Additionally, in this study we show that the majority of IFIX interactions are with proteins that have roles in transcription, chromatin, or mRNA regulation, further supporting a transcriptional regulatory role for IFIX during HSV-1 infection. A prominent interaction was with the 5FMC complex, as we found IFIX to associate with all five known components of this complex, PELP1, LAS1L, TEX10, SENP3, and WDR18 (34). The IFIX association with 5FMC complex members was observed in all tested conditions, i.e. uninfected HSV-1 WT and HSV-1 RF-infected cells. The 5FMC complex was previously reported to act as a transcriptional activator of ZNF148 target genes (34). Interestingly, we found that knockdown of either PELP1 or SENP3 results in decreased virus titers. This decrease did not seem to be dependent on IFIX levels, as we observed similar virus titers in control and IFIX-overexpressing cells. We also noted a redistribution of 5FMC components LAS1L and PELP1 upon infection. Although it remains to be determined, perhaps the relocalization of these proteins represents the recruitment of the transcription complex to viral genomes. Therefore, it is possible that the 5FMC complex works as a transcriptional activator of viral genes. In turn, IFIX may act as a molecular switch, turning the 5FMC complex from transcriptional activation to repression. In fact, PELP1, the core component of 5FMC, was reported to have either transcriptional activating or repressive functions depending on its co-factors (49, 50). Alternatively, IFIX may simply sequester the complex away from its gene targets or effectively block transcription by binding the viral genome at random genomic loci, and HSV-1 may have acquired the mechanism to induce IFIX degradation as a means to release 5FMC.
Because of the constant co-evolution of viruses, such as HSV-1, with their hosts, many host defense pathways are adequately inhibited by viral factors during infection. For example, WT HSV-1 was shown to inhibit cellular cytokine responses in human fibroblasts (22). WT HSV-1 is also effective in inducing the degradation of cellular defense proteins, such as PML and the PYHIN protein IFI16, in part by means of the viral E3 ubiquitin ligase ICP0 (17, 18, 51). Here, we show HSV-1 also promotes the proteasomal degradation of the antiviral factor IFIX. The homeostatic regulation of IFIX does not seem to be a main driver of its rapid degradation upon infection, as IFIX is already degraded by 4 h of HSV-1 infection when compared with the maintenance of relatively high levels of IFIX after 48 h of removal of its tetracycline inducer (supplemental Fig. S5). Interestingly, we found that the distinct IFIX puncta formed during HSV-1 infection within the nucleus is the location of IFIX degradation. We also found IE viral gene expression to be required for the decrease in IFIX levels; however, although we observed ICP0 to interact with IFIX during RF infection, its E3 ligase activity was not necessary for inducing IFIX degradation. Further studies are needed to determine what IE viral factor(s) or processes are promoting the proteasomal degradation of IFIX. Interestingly, our interaction study also showed IFIX to interact with the ubiquitin carboxyl-terminal hydrolases USP7 and USP11. It is tempting to speculate that this may act to reverse the function of E3 ubiquitin ligases in targeting IFIX for degradation, which could represent a mechanism used by the cell to counter IFIX degradation during infection. This hypothesis could reconcile two observations. 1) We often still detect low levels of IFIX even after its degradation (Figs. 2A and 5C). 2) We have not yet detected IFIX ubiquitination following IP-Western blotting analysis (data not shown). It is possible that IFIX could be degraded through a proteasome-dependent but ubiquitination-independent mechanism, as reported previously for other host and viral proteins (52–54). Furthermore, IFIX interactions with deubiquitinating enzymes may also contribute to our observation that increased IFIX levels result in decreased HSV-1 progeny titers even though the virus induces IFIX degradation. However, determining whether this represents such a back-and-forth between the cell host and the virus to adapt cellular processes for either host defense or viral replication, respectively, requires further investigation.
Altogether, we discovered that IFIX exerts its antiviral function during HSV-1 infection in part by suppressing viral gene expression. IFIX may either restrict viral gene transcription directly or by sequestering the 5FMC transcriptional activating complex. We also found that HSV-1 has acquired a mechanism to attempt to inhibit IFIX function by targeting it for proteasome-mediated degradation. It will be of interest to understand whether this antiviral function for IFIX is used as a general mechanism against multiple DNA viruses, or if IFIX has other antiviral strategies yet to be uncovered.
DATA AVAILABILITY
The mass spectrometry proteomics datasets have been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org/) via the PRIDE partner repository with the dataset identifier PXD005152.
Supplementary Material
Acknowledgments
We thank Dr. Todd Greco and Dr. Joel Federspiel from the Cristea laboratory for technical assistance with the mass spectrometry analyses, Dr. Benjamin Diner and Michelle-Ann Tan for technical assistance with IPs and cloning, and Gary Laevsky (Confocal Microscopy Core Facility, Princeton University) for technical support.
Footnotes
Author contributions: I. M. C. designed the research; M. S. C. performed the research; I. M. C. contributed new reagents or analytic tools; M. S. C. and I. M. C. analyzed the data; and M. S. C. and I. M. C. wrote the paper.
* This work was supported by National Institutes of Health Grant GM114141 from NIGMS and Grant HL127640 from NHBLI (to I.M.C.), from the Edward Mallinckrodt Foundation (to I.M.C.), and by National Institutes of Health Graduate Research Fellowship F31 AI114240 (to M.S.C.) from NIAID. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PRR
- pattern recognition receptor
- EGFP
- enhanced green fluorescent protein
- FL
- full length
- HIN
- HIN200 domain
- hpi
- hours post-infection
- HUWE1
- HECT, UBA, and WWE domain-containing protein 1
- ICP0
- infected cell protein 0
- IFIX
- interferon-inducible protein X
- IFI16
- interferon-inducible protein 16
- IP
- immunoaffinity purification
- PELP1
- proline-, glutamic acid-, and leucine-rich protein 1
- PML
- promyelocytic leukemia
- PY
- pyrin domain RF, ICP0 RING finger mutant virus
- SAINT
- significance analysis of interactome computational tool
- SENP3
- sentrin-specific protease 3
- m.o.i.
- multiplicity of infection
- HFF
- human foreskin fibroblast
- IE
- immediate-early
- DE
- delayed-early
- BFP
- blue fluorescent protein
- mGFP
- monomeric GFP
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CID
- collision-induced dissociation
- CHX
- cycloheximide.
REFERENCES
- 1. Brubaker S. W., Bonham K. S., Zanoni I., and Kagan J. C. (2015) Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 33, 257–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diner B. A., Li T., Greco T. M., Crow M. S., Fuesler J. A., Wang J., and Cristea I. M. (2015) The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol. Syst. Biol. 11, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fairbrother W. J., Gordon N. C., Humke E. W., O'Rourke K. M., Starovasnik M. A., Yin J.-P., and Dixit V. M. (2001) The PYRIN domain: a member of the death domain-fold superfamily. Protein Sci. 10, 1911–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin T., Perry A., Smith P., Jiang J., and Xiao T. S. (2013) Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J. Biol. Chem. 288, 13225–13235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li T., Chen J., and Cristea I. M. (2013) Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jin T., Perry A., Jiang J., Smith P., Curry J. A., Unterholzner L., Jiang Z., Horvath G., Rathinam V. A., Johnstone R. W., Hornung V., Latz E., Bowie A. G., Fitzgerald K. A., and Xiao T. S. (2012) Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shaw N., and Liu Z.-J. (2014) Role of the HIN domain in regulation of innate immune responses. Mol. Cell. Biol. 34, 2–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernandes-Alnemri T., Yu J. W., Datta P., Wu J., and Alnemri E. S. (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D. R., Latz E., and Fitzgerald K. A. (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diner B. A., Lum K. K., and Cristea I. M. (2015) The emerging role of nuclear viral DNA sensors. J. Biol. Chem. 290, 26412–26421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li T., Diner B. A., Chen J., and Cristea I. M. (2012) Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. U.S.A. 109, 10558–10563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Orzalli M. H., DeLuca N. A., and Knipe D. M. (2012) Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U.S.A. 109, SE3008–SE3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kerur N., Veettil M. V., Sharma-Walia N., Bottero V., Sadagopan S., Otageri P., and Chandran B. (2011) IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding Y., Wang L., Su L. K., Frey J. A., Shao R., Hunt K. K., and Yan D. H. (2004) Antitumor activity of IFIX, a novel interferon-inducible HIN-200 gene, in breast cancer. Oncogene 23, 4556–4566 [DOI] [PubMed] [Google Scholar]
- 15. Ding Y., Lee J. F., Lu H., Lee M. H., and Yan D. H. (2006) Interferon-inducible protein IFIXα1 functions as a negative regulator of HDM2. Mol. Cell. Biol. 26, 1979–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crow M. S., Lum K. K., Sheng X., Song B., and Cristea I. M. (2016) Diverse mechanisms evolved by DNA viruses to inhibit early host defenses. Crit. Rev. Biochem. Mol. Biol. 2016, 1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boutell C., Orr A., and Everett R. D. (2003) PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 77, 8686–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuchet-Lourenço D., Vanni E., Glass M., Orr A., and Everett R. D. (2012) Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J. Virol. 86, 11209–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lees-Miller S. P., Long M. C., Kilvert M. A., Lam V., Rice S. A., and Spencer C. A. (1996) Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 70, 7471–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parkinson J., Lees-Miller S. P., and Everett R. D. (1999) Herpes simplex virus type 1 immediate-early protein Vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73, 650–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peters N. E., Ferguson B. J., Mazzon M., Fahy A. S., Krysztofinska E., Arribas-Bosacoma R., Pearl L. H., Ren H., and Smith G. L. (2013) A mechanism for the inhibition of DNA-PK-mediated DNA sensing by a virus. PLoS Pathog. 9, e1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diner B. A., Lum K. K., Javitt A., and Cristea I. M. (2015) Interactions of the antiviral factor interferon γ-inducible protein 16 (IFI16) mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol. Cell. Proteomics 14, 2341–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lium E. K., and Silverstein S. (1997) Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential α27 gene. J. Virol. 71, 8602–8614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi H., Larsen B., Lin Z.-Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A.-C., and Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification–mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cristea I. M., Williams R., Chait B. T., and Rout M. P. (2005) Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics 4, 1933–1941 [DOI] [PubMed] [Google Scholar]
- 26. Diner B. A., Lum K. K., Toettcher J. E., and Cristea I. M. (2016) Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. mBio 7, e01553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cristea I. M., Carroll J. W., Rout M. P., Rice C. M., Chait B. T., and MacDonald M. R. (2006) Tracking and elucidating alphavirus-host protein interactions. J. Biol. Chem. 281, 30269–30278 [DOI] [PubMed] [Google Scholar]
- 28. Cristea I. M., and Chait B. T. (2011) Affinity purification of protein complexes. Cold Spring Harb. Protoc. 2011, pdb.prot5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wiśniewski J. R., Zougman A., Nagaraj N., and Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 30. Joshi P., Greco T. M., Guise A. J., Luo Y., Yu F., Nesvizhskii A. I., and Cristea I. M. (2013) The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Craig R., and Beavis R. C. (2003) A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 17, 2310–2316 [DOI] [PubMed] [Google Scholar]
- 32. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., and von Mering C. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, SD561–SD568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fanis P., Gillemans N., Aghajanirefah A., Pourfarzad F., Demmers J., Esteghamat F., Vadlamudi R. K., Grosveld F., Philipsen S., and van Dijk T. B. (2012) Five friends of methylated chromatin target of protein-arginine-methyltransferase[Prmt]-1 (Chtop), a complex linking arginine methylation to desumoylation. Mol. Cell. Proteomics 11, 1263–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sandbaumhüter M., Döhner K., Schipke J., Binz A., Pohlmann A., Sodeik B., and Bauerfeind R. (2013) Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 15, 248–269 [DOI] [PubMed] [Google Scholar]
- 36. Dai-Ju J. Q., Li L., Johnson L. A., and Sandri-Goldin R. M. (2006) ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. J. Virol. 80, 3567–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rojas S., Corbin-Lickfett K. A., Escudero-Paunetto L., and Sandri-Goldin R. M. (2010) ICP27 phosphorylation site mutants are defective in herpes simplex virus 1 replication and gene expression. J. Virol. 84, 2200–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cuchet-Lourenço D., Anderson G., Sloan E., Orr A., and Everett R. D. (2013) The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 87, 13422–13432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Samaniego L. A., Neiderhiser L., and DeLuca N. A. (1998) Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72, 3307–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weir J. P. (2001) Regulation of herpes simplex virus gene expression. Gene 271, 117–130 [DOI] [PubMed] [Google Scholar]
- 41. Everett R. D. (2014) in Herpes Simplex Virus: Methods and Protocols (Diefenbach J. R., and Fraefel C., eds) pp. 1–17, Springer, New York [Google Scholar]
- 42. Everett R. D., and Murray J. (2005) ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 79, 5078–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Everett R. D., Murray J., Orr A., and Preston C. M. (2007) Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81, 10991–11004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Komatsu T., Nagata K., and Wodrich H. (2016) The role of nuclear antiviral factors against invading DNA viruses: the immediate fate of incoming viral genomes. Viruses 8, eE290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Crow M. S., Javitt A., and Cristea I. M. (2015) A proteomics perspective on viral DNA sensors in host defense and viral immune evasion mechanisms. J. Mol. Biol. 427, 1995–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luftig M. A. (2014) Viruses and the DNA damage response: activation and antagonism. Annu. Rev. Virol. 1, 605–625 [DOI] [PubMed] [Google Scholar]
- 47. Orzalli M. H., Conwell S. E., Berrios C., DeCaprio J. A., and Knipe D. M. (2013) Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. U.S.A. 110, SE4492–SE4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kristie T. M. (2016) Chromatin modulation of herpesvirus lytic gene expression: managing nucleosome density and heterochromatic histone modifications. mBio 7, Se00098–Se00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Choi Y. B., Ko J. K., and Shin J. (2004) The transcriptional corepressor, PELP1, recruits HDAC2 and masks histones using two separate domains. J. Biol. Chem. 279, 50930–50941 [DOI] [PubMed] [Google Scholar]
- 50. Girard B. J., Daniel A. R., Lange C. A., and Ostrander J. H. (2014) PELP1: a review of PELP1 interactions, signaling, and biology. Mol. Cell. Endocrinol. 382, 642–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Everett R. D., Rechter S., Papior P., Tavalai N., Stamminger T., and Orr A. (2006) PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80, 7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kalejta R. F., and Shenk T. (2003) Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. U.S.A. 100, 3263–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jariel-Encontre I., Bossis G., and Piechaczyk M. (2008) Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta 1786, 153–177 [DOI] [PubMed] [Google Scholar]
- 54. Hwang J., Winkler L., and Kalejta R. F. (2011) Ubiquitin-independent proteasomal degradation during oncogenic viral infections. Biochim. Biophys. Acta 1816, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics datasets have been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org/) via the PRIDE partner repository with the dataset identifier PXD005152.