

Abstract
Cellular responses to stimuli involve dynamic and localized changes in protein kinases and phosphatases. Here, we report a generalized functional assay for high-throughput profiling of multiple protein phosphatases with subcellular resolution and apply it to analyze coxsackievirus B3 (CVB3) infection counteracted by interferon signaling. Using on-plate cell fractionation optimized for adherent cells, we isolate protein extracts containing active endogenous phosphatases from cell membranes, the cytoplasm, and the nucleus. The extracts contain all major classes of protein phosphatases and catalyze dephosphorylation of plate-bound phosphosubstrates in a microtiter format, with cellular activity quantified at the end point by phosphospecific ELISA. The platform is optimized for six phosphosubstrates (ERK2, JNK1, p38α, MK2, CREB, and STAT1) and measures specific activities from extracts of fewer than 50,000 cells. The assay was exploited to examine viral and antiviral signaling in AC16 cardiomyocytes, which we show can be engineered to serve as susceptible and permissive hosts for CVB3. Phosphatase responses were profiled in these cells by completing a full-factorial experiment for CVB3 infection and type I/II interferon signaling. Over 850 functional measurements revealed several independent, subcellular changes in specific phosphatase activities. During CVB3 infection, we found that type I interferon signaling increases subcellular JNK1 phosphatase activity, inhibiting nuclear JNK1 activity that otherwise promotes viral protein synthesis in the infected host cell. Our assay provides a high-throughput way to capture perturbations in important negative regulators of intracellular signal-transduction networks.
Protein phosphorylation is a critical component of cellular signal transduction (1, 2). In response to extracellular stimulation by cytokines, hormones, and environmental stresses, protein kinases catalyze phosphorylation events that alter substrate activity, protein localization, gene expression, and cell phenotype (Fig. 1). To reverse these events and return the cell to a resting state, protein phosphatases dephosphorylate many phosphoprotein substrates (3–5). Phosphatase abundance and activity determine the extent of constitutive signaling (6) as well as the magnitude and duration of pathway stimulation (7). Accordingly, misregulated protein phosphatases have been implicated in many diseases, including cardiomyopathy, cancer, and inflammatory conditions (8–11).
Fig. 1.
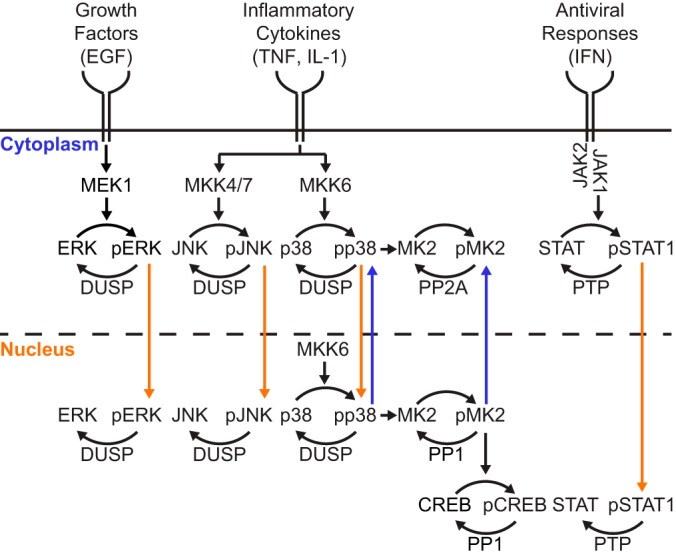
Subcellular phosphatase activities reset intracellular signaling triggered by growth factors, proinflammatory cytokines, and pathogenic stresses. Hierarchical signaling cascades initiated by extracellular stimuli cause downstream protein phosphorylation. Upon phosphorylation, some signaling proteins are shuttled into (orange arrows) or out of (blue arrows) the nucleus. Compartment- and substrate-specific phosphatases dephosphorylate activated proteins thereby returning proteins to their resting compartment.
There are ∼500 protein kinases and ∼180 protein phosphatases in the human genome, indicating that phosphatases must target a larger breadth of substrates (12). The catalytic subunits of the protein phosphatases PP1 and PP2A dephosphorylate most phospho-Ser/Thr-containing proteins, with selectivity conferred by regulatory subunits and subcellular localization (13). In contrast, dual-specificity phosphatases (DUSPs)1 hydrolyze phospho-Tyr residues paired with phospho-Ser/Thr sites, narrowly targeting bisphosphorylated MAP kinases (MAPKs) ERK, JNK, and p38 through kinase-interaction motifs (14) (Fig. 1). DUSP targeting is further refined by subcellular localization and the nucleocytoplasmic shuttling characteristics of each MAPK (5, 15–19). DUSPs comprise part of a larger family of protein tyrosine phosphatases (PTPs) that dephosphorylate phospho-Tyr exclusively (3). Receptor-like PTPs have access to substrates near cell membranes, whereas nontransmembrane PTPs act elsewhere within the cell (Fig. 1). Phosphatases can dephosphorylate a variety of substrates, but multiple phosphatases may also converge upon the same substrate. For example, the bisphosphorylated site in MAPKs is deactivated by DUSPs but also by the coordinate action of Ser/Thr phosphatases and PTPs (20). The extent of targeting is dictated by the abundance of protein phosphatase and phosphosubstrate along with their respective proximity in the cell (4, 5, 21, 22). The redundancy, promiscuity, and multi-layered regulation of protein phosphatases make it challenging to define their specific roles in intracellular signaling (23).
Monitoring cellular protein dephosphorylation events would be greatly aided by high-throughput methods that capture multiple mechanisms of phosphatase regulation. In typical activity assays, phosphatases are purified from extracts and measured using a synthetic phosphopeptide substrate (24–27). This strategy captures changes in protein phosphatase abundance, but the enzyme may lose endogenous regulators during the purification, and subcellular localization is usually homogenized. It is also doubtful that short, unstructured phosphopeptides accurately reflect phosphatase activity in the same way as full-length phosphoproteins. Endogenous phosphatase activity measurements are possible by incubating total cell extracts with 32P-radiolabeled phosphoproteins (28). However, robust protein phosphatase activities or heavily labeled substrates are required; thus, the approach does not scale well to dozens or hundreds of samples. We previously developed a substrate-focused protein phosphatase activity assay using phosphorylated MAPKs and homogenized cellular extracts in a phospho-ELISA format (29). Phosphatase activity in the extract was measured as the decrease in phosphorylated full-length recombinant MAPK substrates adsorbed to a 96-well plate. Although this approach captured substrate-phosphatase interactions, it could not characterize subcellular regulation of protein phosphatase activity and only included MAPKs. A true multi-pathway protein phosphatase assay with subcellular resolution would provide a better systems-level view of how signal transduction is negatively regulated.
Here, we introduce a high-throughput assay that now measures substrate dephosphorylation by all major classes of protein phosphatases in different biochemically defined subcellular compartments. We begin with a high-throughput, scalable lysis procedure that collects paired saponin- and detergent-soluble extracts containing active protein phosphatases from adherent cells. The activity of subcellular phosphatases is then quantified by phospho-ELISA using a panel of recognized full-length phosphoproteins. Building upon our past success with phosphorylated MAPKs (29), we add three new phosphosubstrates—phospho-MK2 (Thr334), phospho-CREB (Ser133), and phospho-STAT1 (Tyr701)—each with distinct patterns of localization and targeting by protein phosphatase enzymes (Fig. 1). Together, these substrates provide a subcellular phosphatase activity signature for the cellular response to growth factors, cytokines, and environmental stress.
As a prototypical cellular stress that engages several host-cell signaling pathways, we investigated changes in protein phosphatase activities during acute viral infection. Coxsackievirus B3 (CVB3) is a cardiotropic picornavirus that causes myocarditis in infants and young children (30, 31). The CVB3 genome encodes neither protein kinases nor phosphatases but widely alters the phosphorylation state of the infected host cell (32–36). For example, CVB3 infection cleaves a negative regulator of Ras, which gives rise to ERK phosphorylation that is important for viral replication (37–40). Various protein phosphatases are required for early CVB3 infection (41), and CVB3-encoded proteins can also modify host phosphatase activity directly. For example, viral protein 2C forms a complex with PP1 to inhibit IKKβ phosphorylation and NF-κB signaling (42). Furthermore, CVB3 infection induces expression of proinflammatory and antiviral cytokines, such as TNF, IL-1, and interferons, in both cardiomyocytes and infiltrating immune cells (43, 44) (Fig. 1). Type I and Type II interferons activate STATs resulting in partially overlapping antiviral transcriptional responses that combat RNA viruses such as CVB3 (45, 46). Understanding the degree to which CVB3 infection intersects with interferons is important for more systematic profiling of protein phosphatase cross-regulation by combinations of cytokines and viral pathogens.
EXPERIMENTAL PROCEDURES
Plasmids
For phosphosubstrate preparation and purification, pGEX-4T-1 3×FLAG-ERK2, pGEX-4T-1 3×FLAG-JNK1, pGEX-4T-1 3×FLAG-p38α, pGEX-4T-1 3×HA-MEK-DD, pGEX-4T-1 3×HA-MKK4-EE, pGEX-4T-1 3×HA-MKK7a1-EE, and pGEX-4T-1 3×HA-MKK6-EE were described previously (29). Human MKK6-EE (Addgene plasmid #13518) (47) was cloned into the BamHI and SalI sites of pCDFDuet-1 (Novagen, Madison, WI) by PCR with the primers gcgcagatctatgtctcagtcgaaaggcaag (forward) and gcgcgtcgacttagtctccaagaatcagttttac (reverse) followed by digestion with BglII and SalI to yield pCDFDuet-1 MKK6-EE. Murine p38α (Addgene plasmid #20351) (48) was cloned into the BglII and XhoI sites of pCDFDuet-1 MKK6-EE by PCR with the primers gcgcggatccatgtcgcaggagaggccc (forward) and gcgcctcgagtcaggactccatttcttcttgg (reverse) followed by digestion with BamHI and XhoI to yield pCDFDuet-1 MKK6-EE p38α. Human MAPKAPK2 (MK2) (hORFeome V5.1 #10384) (49) was cloned into the BamHI and EcoRI sites of pGEX-4T-1 (3×FLAG) by PCR with the primers gcgcggatccatgctgtccaactcccaggg (forward) and gcgcctcgagtcagtgggccagagccg (reverse) followed by digestion with BamHI and EcoRI to yield pGEX-4T-1 3×FLAG-MK2. Human CREB (hORFeome V5.1 #3038) (49) was cloned into the BamHI and EcoRI sites of pGEX-4T-1 (3×FLAG) by PCR with the primers gcgcggatccatgaccatggaatctggagc (forward) and gcgcgaattcttaatctgatttgtggcagtaaag (reverse) followed by digestion with BamHI and EcoRI to yield pGEX-4T-1 3×FLAG-CREB. Human STAT1 (hORFeome V5.1 #4126) (49) was cloned into the BamHI and EcoRI sites of pGEX-4T-1 (3×FLAG) by PCR with the primers gcgcggatccatgtctcagtggtacgaact (forward) and gcgcgaattcttacacttcagacacagaaatca (reverse) followed by digestion with BamHI and EcoRI to yield pGEX-4T-1 3×FLAG-STAT1.
For constitutive lentiviral overexpression, human CXADR/CAR (hORFeome V5.1 #356) (49) was recombined with pLX304 (Addgene plasmid #25890) (49) using Gateway LR clonase (Invitrogen, Carlsbad, CA) to yield pLX304 CAR-V5. Human DUSP10/MKP5 (hORFeome V5.1 #8448), DUSP16/MKP7 (hORFeome V5.1 #11351), DUSP19 (hORFeome V5.1 #2039), DUSP22/JSP1 (hORFeome V5.1 #8622), PTPRR/PTP-SL (hORFeome V5.1 #56450), and PPP1R8/NIPP1 (hORFeome V5.1 #6578) were recombined with pLX302 (Addgene plasmid #25896) using Gateway LR clonase (Invitrogen) to yield pLX302 MKP5-V5, pLX302 MKP7-V5, pLX302 DUSP19-V5, pLX302 JSP1-V5, pLX302 PTP-SL-V5, and pLX302 NIPP1-V5.
For inducible lentiviral expression, human MKK4-EE was cloned into the SpeI and MfeI sites of pEN_TTmiRc2 3×FLAG (50) by PCR with the primers gcgcactagtggatccatgcagggtaaacg (forward) and gcgccaattgctacactttacgttttttcttcggaccagaaccaccatcgacatacatgg (reverse, adding a short linker and the monopartite NLS of SV40 large T antigen) followed by digestion with SpeI and MfeI to yield pEN_TTmiRc2 3×FLAG-MKK4-EE-NLS. Human MKK7a1-EE was cloned into the SpeI and MfeI sites of pEN_TTmiRc2 3×FLAG by PCR with the primers gcgctctagaggatccatgctggggctc (forward) and gcgccaattgctacactttacgttttttcttcggaccagaaccacccctgaagaagggca (reverse, adding a short linker and the monopartite NLS of SV40 large T antigen) followed by digestion with XbaI and MfeI to yield pEN_TTmiRc2 3×FLAG-MKK7-EE-NLS. EGFP was cloned into the SpeI and MfeI sites of pEN_TTmiRc2 3×FLAG by PCR with the primers gcgcactagtgtgagcaagggcgaggagct (forward) and gcgccaattgctacactttacgttttttcttcggaccagaaccaccgtcggcgcgcccac (reverse, adding a short linker and the monopartite NLS of SV40 large T antigen) followed by digestion with SpeI and MfeI to yield pEN_TTmiRc2 3×FLAG-EGFP-NLS. The pEN_TTmiRc2 donor vectors were recombined with pSLIK hygro (50) using Gateway LR clonase (Invitrogen) to yield pSLIK 3×FLAG-MKK4-EE-NLS, pSLIK 3×FLAG-MKK7-EE-NLS, and pSLIK 3×FLAG-EGFP-NLS.
All PCR-cloned constructs and donor vectors were verified by restriction digest and sequencing, and all plasmids can be obtained through Addgene (plasmids #82718 - 82723 and #87770 - 87781).
Protein Induction and Purification
ERK2 and MEK-DD purifications were performed as described previously (29). BL21-CodonPlus (DE3)-RIPL competent cells (#230280, Stratagene, San Diego, CA) were transformed with pGEX-4T-1 or pCDFDuet-1 and pGEX-4T-1 plasmids, and liquid cultures were grown at 37 °C until OD 0.6–1. Cultures were cooled to 12 °C for 30–45 min and protein expression was induced at 12 °C overnight with one of the following isopropyl-d-1-thiogalactopyranoside (IPTG) concentrations: 0.4 mm (CREB, MKK4), 1 mm (p38α, JNK1, STAT1, MKK7), 2 mm (MK2). Bacterial cultures were collected by centrifugation and resuspended in 7.5 ml RIPL TNE buffer (54 mm Tris [pH 7.4], 160 mm NaCl, 1 mm EDTA, 2 μg/ml leupeptin, 5 μg/ml aprotinin, 1 μg/ml pepstatin, 32 mm sodium fluoride, 100 μm Na3VO4, 10 mm sodium pyrophosphate, 2 mm ATP, 1.2 mg/ml MgSO4, 13 mm MgCl2, 7 μg/ml DNase, and 1 mm DTT) per 250 ml culture. Bacteria were mechanically lysed using an EmulsiFlex B15 (Avestin) at 80 psi. Lysates were clarified by centrifugation and flowed over a 5-ml GSTrap column (#17–5131-02, GE Healthcare, Chicago, IL) using an ÄKTAprime Plus chromatography system at 0.1 ml/min (GE Healthcare, Chicago, IL). The column was washed at 0.1 ml/min with 20–25 ml of Buffer A (25 mm sodium phosphate [pH 7.2], 150 mm NaCl), and proteins were eluted in 2 ml fractions with glutathione elution buffer (6 mg/ml glutathione in Buffer A plus 2 mm DTT). For MKK4-EE and MKK7-EE purifications, clarified lysates were incubated with glutathione-agarose beads (Sigma, St. Louis, MO) for 4 h at 4 °C. Beads were then spun down and washed with PBS twice before use.
Phosphorylation of Protein Substrates
ERK2 phosphorylation was performed in vitro with purified MEK-DD as described previously (29). In vivo phosphorylation of p38 and MK2 was achieved by co-transformation of RIPL cells with pCDFDuet-1 MKK6-EE and pCDFDuet-1 MKK6-EE p38α respectively. Phosphorylation of JNK1 was performed in vitro with purified 3×HA-MKK4-EE and 3×HA-MKK7a1-EE bound to glutathione-agarose beads (Sigma, St. Louis, MO) by incubating for 1 h at 37 °C in kinase assay buffer (30 mm Tris [pH 7.5], 3 mm ATP, 45 mm MgCl2, 7.5 mm β-glycerophosphate, 1.5 mm Na3VO4, 1.5 mm EGTA, 0.6 mm DTT). The supernatant containing phosphorylated JNK1 was collected after centrifugation of the kinase assay reactions. CREB and STAT1 were phosphorylated in vitro with phospho-MK2 or active JAK1 fragment (#14–918, Millipore, Dundee, UK) respectively by incubation at 37 °C in kinase assay buffer for 24 h.
Phospho-stoichiometry of substrates was assessed in the phosphoprotein preparations relative to unstimulated or stimulated RIPA lysates from AC16-CAR cells by immunoblotting with antibodies specific to the phosphoprotein and the total protein as described below. Phosphoprotein bands were normalized to the total protein immunoreactivity and phospho-to-total ratio was assessed relative to unstimulated cell lysate.
Cell Lines and Culture
MCF10A-5E cells were obtained and cultured as previously reported (51, 52). CVB3-permissive HeLa cells were provided by Dr. Bruce McManus (University of British Columbia, Vancouver BC, Canada) and were cultured in DMEM (#11965092, Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (#15140–122, Gibco). HT-29 cells (ATCC, Manassas, VA) were cultured according to the distributor's recommendations. AC16 cells were purchased from Dr. Mercy Davidson (Columbia University, NY, New York) (53) and cultured in DMEM/F12 (#11330–032, Gibco) supplemented with 12.5% fetal bovine serum and 1% penicillin-streptomycin. AC16-CAR cells were prepared by transducing parental AC16 cells with pLX304 CAR-V5 lentiviruses as described previously (54) and selecting for stable expression with 10 μg/ml blasticidin (#46–1120, Invitrogen) until control plates had cleared. AC16-CAR cells overexpressing pLX302 MKP7-V5, pLX302 JSP1-V5, pLX302 NIPP1-V5, or pLX302 PTP-SL-V5 were prepared by transducing AC16-CAR cells with lentiviruses as described previously (46). Two days after transduction, cells were either fractionated as described below or selected in 2 μg/ml puromycin (#100552, MP Biomedicals, Santa Ana, CA) plus 10 μg/ml blasticidin (#46–1120, Invitrogen) until control plates had cleared. AC16-CAR cells dually expressing 3×FLAG-MKK4-EE-NLS and 3×FLAG-MKK7-EE-NLS (3×FLAG-MKK4/7-EE-NLS) or 3×FLAG-EGFP-NLS were prepared by transducing AC16-CAR cells with single or pooled lentiviruses as described previously (54). Two days after transduction, cells were selected for stable integration with 100 μg/ml hygromycin (#ant-hg-5, Invivogen, Toulouse, France) and 10 μg/ml blasticidin (#46–1120, Invitrogen) until control plates had cleared.
On-plate Subcellular Fractionation
Cells were washed briefly with ice-cold PBS and then incubated with saponin extract (SE) buffer (50 mm HEPES [pH 7.5], 0.05% [w/v] saponin, 20 μg/ml aprotinin, 20 μg/ml leupeptin, 1 μg/ml pepstatin, 2 mm MgCl2, 1 mm DTT, 50 mm 2-mercaptoethanol, 5 mm d-glucose, and 15 μg/ml hexokinase [#H5000, Sigma]) on a platform rocker for 30 min at 4 °C. The hexokinase stock for the extraction buffer was prepared at 5 mg/ml with 15 μm phenylmethylsulfonyl fluoride (PMSF). After incubation, the SE fraction was collected and cells were washed three times with 0.05% (w/v) saponin in ice-cold PBS for 5 min on ice with rocking. After the third wash, permeabilized cells were incubated with Nonidet P-40 extract (NE) buffer (50 mm HEPES [pH 7.5], 0.1% [w/v] Nonidet P-40 [NP40], 150 mm NaCl, 20 μg/ml aprotinin, 20 μg/ml leupeptin, 1 μg/ml pepstatin, 2 mm MgCl2, 1 mm DTT, 50 mm 2-mercaptoethanol, 5 mm d-glucose, and 15 μg/ml hexokinase) for 5 min on ice with rocking. After incubation, the NE fraction was collected and cells were washed with 0.05% (w/v) saponin in ice-cold PBS for 5 min on ice with rocking. Then, the insoluble fraction was collected by addition of 1× sample buffer (62.5 mm Tris pH 6.8, 2% [w/v] sodium dodecyl sulfate [SDS], 10% [v/v] glycerol, 0.01% [w/v] bromphenol blue) and cell scraping. EGF-stimulated subcellular fractions for immunoblotting were lysed in SE and NE buffers supplemented with phosphatase inhibitors (200 μm Na3VO4, 10 mm sodium pyrophosphate, and 1 μg/ml microcystin-LR) to preserve phosphoproteins.
Subcellular Protein Phosphatase Assays
High protein-binding 96-well plates (#9018, Corning Costar, Lowell, MA) were coated overnight with recombinant phosphoprotein diluted in 100 μl PBS (see supplemental Table S1 for details). The next morning, plates were washed three times with Tris-buffered saline + 0.1% (v/v) Tween-80 (TBS-T) and blocked for 1 h at room temperature on platform rocker with 5% (w/v) BSA in TBS-T (blocking buffer) and washed three times with TBS-T before use.
SE and NE fractions were diluted to cellular equivalents (an amount of cell extract estimated from the cellular density at the time of fractionation) that fall within the optimal dynamic range of each assay (see supplemental Tables S2 and S3 for equivalents used in each figure). SE fractions were diluted to 85 μl in SE buffer, whereas NE fractions were diluted to 85 μl in 10% (v/v) NE buffer + 90% (v/v) NP40- and NaCl-free NE buffer (for phospho-ERK2, phospho-p38α, phospho-JNK, and phospho-STAT1 assays) or 30% (v/v) NE buffer + 70% (v/v) NP40- and NaCl-free NE buffer (for phospho-MK2 and phospho-CREB assays). Diluted extracts were added to the phosphosubstrate-coated plate and incubated in a Jitterbug Microplate Incubator-Shaker (Thomas Scientific, Swedesboro, NJ) at 575 RPM and 30 °C for 30–90 min depending on the kinetics of the assay (see supplemental Table S1 for details). Dephosphorylation reactions were terminated with 85 μl of 2× phosphatase inhibitor solution (20 mm sodium pyrophosphate, 60 mm sodium fluoride, and 400 μm Na3VO4 in TBS) followed by three washes with 1× phosphatase inhibitor solution diluted in TBS-T.
After washing once with TBS-T lacking phosphatase inhibitors, each well was incubated for one hour at room temperature on a platform rocker with 50 μl of one of the following primary antibodies diluted in blocking buffer: phospho-ERK2 (Thr202/Tyr204) (1:1000, #4370, Cell Signaling Technology, Beverly, MA), phospho-p38 (Thr180/Tyr182) (1:1000, #4511, Cell Signaling Technology), phospho-JNK (Thr183/Tyr185) (1:100, #9251, Cell Signaling Technology), phospho-MK2 (Thr334) (1:1000, #3007, Cell Signaling Technology), phospho-CREB (Ser133) (1:1000, #9198, Cell Signaling Technology), phospho-STAT1 (Tyr709) (1:5000, #9167, Cell Signaling Technology). Primary antibody solutions were removed and plates washed three times with TBS-T. Each well was then incubated for 1 h at room temperature on a platform rocker with 50 μl of biotinylated goat anti-rabbit secondary antibody (1:10,000, #111–065-045, Jackson Immunoresearch, West Grove, PA) diluted in blocking buffer. The secondary antibody solution was removed and wells were washed three times with TBS-T followed by incubation for 1 h at room temperature on a platform rocker with 50 μl of streptavidin-HRP diluted in blocking buffer (1:200, #DY998, R&D Systems, Minneapolis, MN). After washing three times with TBS-T, wells were incubated at room temperature on a platform rocker with 100 μl of 1:1 mix of ELISA Reagent A (stabilized hydrogen peroxide) and Reagent B (stabilized tetramethylbenzidine) (#DY999, R&D Systems). ELISA reactions were stopped with 50 μl of 1 m sulfuric acid after 10 min, with the exception of phospho-p38α assays that were allowed to proceed for 15 min. Well absorbance of the phospho-ELISA end point was measured at 450 nm with background correction at 540 nm on an Optima plate reader.
Phosphatase activity was calculated as the decrease in phospho-ELISA signal relative to buffer-only controls. For calibration of relative activities, a 2-fold serial dilution of pooled SE or NE fraction was used to prepare a standard alongside the unknown samples. Standards were regressed against input material by using a four-parameter logistic curve, which was inverted to calculate relative phosphatase activity from unknown samples. Last, activity measurements were adjusted for relative total protein concentration as quantified by o-pthalaldehyde assay (see below). For inhibitor studies, concentrated inhibitors were spiked into diluted extracts to achieve a final concentration of 10 mm sodium pyrophosphate, 200 μm activated Na3VO4 (55), or 1 μg/ml microcystin-LR before the start of the assay.
Cell Lysis
Total cell extracts were prepared in radioimmunoprecipitation assay (RIPA) buffer (25 mm Tris-HCl [pH 7.6], 150 mm NaCl, 1% NP40, 1% sodium deoxycholate, 0.1% SDS, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μg/ml pepstatin, 200 μm Na3VO4, 1 μg/ml microcystin-LR and 30 μm phenylmethylsulfonyl fluoride [PMSF]) as described previously (56).
Cell Infection and Stimulation
For assessment of the stoichiometry of recombinant phosphosubstrates, AC16-CAR cells were seeded at 50,000 cells/cm2 for 24 h and stimulated with EGF (#AF-100–15, Peprotech, Rocky Hill, NJ), TNF (#300–01A, Peprotech, Rocky Hill, NJ), or IFNβ (#300–02BC, Peprotech) by spiking in a 20× concentrated stock for the indicated times before lysis in RIPA buffer.
For the EGF time course, AC16-CAR cells were seeded at 50,000 cells/cm2 for 24 h on a 12-well plate precoated for 2 h with 0.02% [w/v] gelatin (#G9391, Sigma) at 37 °C. Cells were either unstimulated or stimulated for 120, 60, 30, 15, or 5 min with 100 ng/ml EGF before simultaneous extraction using the on-plate subcellular fractionation technique described above.
For CVB3 infection of AC16 and AC16-CAR cells, culture plates were coated with 0.02% [w/v] gelatin as described above before plating at ∼25,000 cells/cm2 for 24 h. Before infection, 75% of the culture medium volume was removed, and virus stock was spiked in at a multiplicity of infection (MOI) of 10 virions per cell. Cells were infected for one hour with gentle rocking every 10–15 min, and then cells were washed with PBS and refed with growth medium lacking selection antibiotics until lysis. In the full-factorial experiment with type I/II interferons, CVB3-infected cells were stimulated with 30 ng/ml IFNβ, 50 U/ml IFNγ (#11040596001, Roche, Mannheim, Germany), or both by spiking in cytokine stocks at 3 h after the start of CVB3 infection (two hours after refeeding). After 4 h of cytokine stimulation, SE and NE fractions were prepared as described above.
For experiments involving JNK inhibition, AC16-CAR cells were seeded ∼25,000 cells/cm2 for 21 h and treated with 1 μm JNK-IN-8 (#sc-364745, Santa Cruz Biotechnology, Santa Cruz, CA) prepared as a 1 mm stock in DMSO. Three hours after JNK-IN-8 addition, cells were infected and stimulated with 30 ng/ml IFNβ as described above. Cells were lysed in RIPA buffer 20 h after the start of CVB3 infection.
For the genetic perturbation of JNK activity, AC16-CAR cells stably expressing inducible 3×FLAG-MKK4/7-EE-NLS or 3×FLAG-EGFP-NLS control were seeded at ∼25,000 cells/cm2. To induce expression, 1 μg/ml doxycycline hyclate (#D9891, Sigma, St. Louis, MO) was added two hours before the start of CVB3 infection (MOI = 10), and 30 ng/ml IFNβ was added 3 h after the start of CVB3 infection as before. Cells were lysed in RIPA buffer 20 h after the start of CVB3 infection.
Protein Quantification
RIPA lysates were quantified for total protein as previously described (56). Subcellular extracts were quantified by o-pthalaldehyde assay with incomplete phthaldialdehyde reagent (#P7914, Sigma) in 96-well black-walled, clear-bottom microtiter plates (#3720, Corning, Lowell, MA). Phthaldialdehyde reagent was activated with 1/500th volume of 2-mercaptoethanol and then 50 μl of activated reagent was incubated with 10 μl of sample for 2 min on an orbital shaker at room temperature. Fluorescence (λex = 355 nm,λem = 440 nm) was detected using an Optima plate reader. All subcellular extracts were regressed against a bovine serum albumin standard run on the same plate under the same conditions.
ATP Quantification
Subcellular SE and NE fractions were collected with or without hexokinase in the extraction buffer. ATP concentration in the extracts was quantified using the Kinase-Glo Assay (#V6701, Promega, Madison, WI). Extracts (25 μl) or ATP standards were incubated with 25 μl of Kinase-Glo Reagent for 10 min at room temperature in black-walled, solid-bottom microtiter plates (#655209, Greiner Bio-One, Kremsmünster, Austria). Luminescence was detected using an Optima Plate reader. ATP standards were diluted in either SE or NE buffer lacking glucose and saponin, which act as substrates for hexokinase and cause loss of luminescence in the ATP standards.
Plaque Assay
CVB3-permissive HeLa cells were plated at 1 million cells/well in a 6-well dish overnight, washed, and incubated with 200 μl of diluted conditioned medium from AC16 or AC16-CAR cells infected with CVB3 for 24 h. HeLa cells were infected for 40 min, washed with serum-free DMEM, and overlaid with 2 ml of a 1:1 mix of 1.5% (w/v) agar and 2× DMEM (#12100–046, Gibco) for 72 h. Wells were fixed with 2 ml of Carnoy's fixative (75% [v/v] ethanol, 25% [v/v] acetic acid) for 30 min at room temperature. Fixative was decanted and agar plugs removed with a pliable weighing spatula. Viral plaques were counterstained with 0.5% (w/v) crystal violet. Plates were scanned on a LI-COR Odyssey scanner in the 700 channel.
Immunofluorescence
Glass coverslips were coated with 2 μg/cm2 poly-d-lysine (#P6407, Sigma) in a 6-well dish for 5 min, washed briefly with cell culture grade water, and allowed to dry for at least 2 h. AC16 or AC16-CAR cells were plated at 20,000 cells/cm2 for 24 h and then infected as described above. Cells were LIVE/DEAD stained and processed for immunofluorescence as described (39, 54) with the following primary antibodies: mouse anti-VP1 (1:1000, #M7064, Dako, Carpinteria, CA) and rabbit anti-cleaved caspase 3 antibody (1:200, #9661, Cell Signaling Technology). For MKK4/7-EE-NLS and EGFP-NLS lines, cells were plated in medium containing 1 μg/ml doxycycline to induce expression. Cells were processed for immunofluorescence as described (39, 54) with the following primary antibodies: rabbit anti-p-cJun (1:100, #9164, Cell Signaling Technology), mouse anti-FLAG (1:200, #F3165, Sigma).
Immunoblotting
Quantitative immunoblotting of RIPA lysates and subcellular extracts was performed as described (56) with the following primary antibodies: caspase 3 (1:1000, #9662, Cell Signaling Technology), eIF4G (1:1000, #2498, Cell Signaling Technology), FLAG (1:5000, #F3165, Sigma), HSP90 (1:2000, #sc-7947, Santa Cruz Biotechnology), IκBα (1:1000, #4814, Cell Signaling Technology), JunD (1:1000, #sc-74, Santa Cruz Biotechnology), MEK1/2 (1:1000, #4694, Cell Signaling Technology), MKP3 (1:1000, #2138, Epitomics, Burlingame, CA), PARP (1:1000, #9532, Cell Signaling Technology), PP1c (1:1000, custom polyclonal from D.L.B.), PP2Ac (1:1000, custom polyclonal from D.L.B.), tubulin (#ab89984, Abcam, Cambridge, MA), V5 (1:5000, #46–0705, Invitrogen), VP1 (1:1000, #M7064, Dako), phospho-ERK2 (Thr202/Tyr204) (1:1000, #4370, Cell Signaling Technology), total ERK (1:1000, #4695, Cell Signaling Technologies), phospho-p38 (Thr180/Tyr182) (1:1000, #4511, Cell Signaling Technology), total p38 (1:5000, #sc-535, Santa Cruz Biotechnology), phospho-JNK (Thr183/Tyr185) (1:100, #9251, Cell Signaling Technology), total JNK (1:1000, #9252, Cell Signaling Technology), phospho-MK2 (Thr334) (1:1000, #3007, Cell Signaling Technology), total MK2 (1:500, #ADI-KAP-MA015-F, Enzo Life Sciences, Farmingdale, NY), phospho-CREB (Ser133) (1:1000, #9198, Cell Signaling Technology), total CREB (1:1000, #9197, Cell Signaling Technology), phospho-STAT1 (Tyr709) (1:1000, #9167, Cell Signaling Technology), and total STAT1 (1:1000, #9172, Cell Signaling Technology).
MKP1, DUSP19-V5 and MKP5-V5 phosphatases were immunoblotted by chemiluminescence as described (56) using the following primary antibodies: MKP1 (1:200, #sc-1102, Santa Cruz Biotechnology), V5 (1:5000, #46–0705, Invitrogen), and MKP5 (1:1000, #3483, Cell Signaling Technology). Blots were then stripped for 30 min at 50 °C in high stringency stripping buffer (2% [w/v] SDS, 62.5 mm Tris pH 6.8, 100 mm 2-mercaptoethanol in water) and reprobed for vinculin (1:10,000, #05–386, EMD Millipore, Dramstadt, Germany) and GAPDH (1:20,000, #AM4300, Thermo Fisher Scientific, Waltham, MA) as loading controls.
shRNA Cloning and Validation
The following shRNA sequences were obtained from the RNAi Consortium and cloned into tet-pLKO-puro (57) as previously described (58): shMKP1 (#1: TRCN0000356127 and #2: TRCN0000367631), shMKP5 (#1: TRCN0000220147 and #2: TRCN0000314618), shMKP7 (#1: TRCN0000052013 and #2: TRCN0000052017), and shDUSP19 (TRCN0000356162). These plasmids can be obtained through Addgene (plasmid #87790 - 87796). Lentiviruses were packaged, transduced into AC16-CAR cells, and selected with 2 μg/ml puromycin and 100 μg/ml blasticidin as previously described (54). shRNAs in AC16-CAR stable lines were induced with 1 μg/ml doxycycline for 72 h before the start of the experiment. shMKP5 and shDUSP19 sequences were validated by transient co-transfection with pLX302 MKP5-V5 and pLX302 DUSP19-V5, respectively, using Lipofectamine 2000 (#11668019, Invitrogen) in the presence of 1 μg/ml doxycycline for 48 h. shMKP1 sequences were verified in AC16-CAR stable lines induced with 1 μg/ml doxycycline 24 h prior to stimulation with 100 ng/ml EGF for 1 h.
Experimental Design and Statistical Rationale
The number of samples analyzed per experiment is indicated by “n = ” at the bottom of each figure legend, where the distinction is made between assay replicates (same extract, different microtiter wells) and biological replicates (different extracts). All quantitative data were collected with n ≥ 3 replicates, providing ∼82% minimum power to detect a 1.5-fold difference in unpaired means according to noncentral t statistics assuming a coefficient of variation (CV) of 20%, which conservatively models error in the assay. Among biological replicates, the error of some assays was much higher (CV = 30–50%), prompting a paired design with n = 9 samples to maintain ∼75% minimum power to detect a 1.5-fold difference when CV = 50%. Negative controls in the phosphatase assays (n = 4–14) were phosphosubstrate-coated wells incubated with the appropriate extraction buffer instead of cellular extract. Negative controls in the biological studies (n = 9) were cells treated with viral or cytokine diluent lacking CVB3 or cytokines. No randomization of plates or wells within plates was performed, but lack of spatial bias or artifacts was confirmed retrospectively.
Statistical Analysis
Standard statistical analyses are described in the figure subpanel legend where used. For the full-factorial experiment involving CVB3, IFNβ, and IFNγ, the assumption of normally distributed biological replicates was assessed by Lilliefors test. Given their empirical cumulative distribution functions, the null hypothesis was rejected for the following phosphatase assays: phospho-JNK1 (SE fraction), phospho-MK2 (SE fraction), phospho-CREB (SE and NE fractions), and phospho-STAT1 (SE and NE fractions). For these six assays, differences in relative ranks were assessed by rank-sum test with Bonferroni correction for multiple-hypothesis testing, and main effects or two-factor interactions were assessed by the three-way extension of the Scheirer-Ray-Hare test at a 10% false-discovery rate. For the remaining six assays where the normality assumption could be retained, differences in means were assessed by two-tailed Student's t test with Bonferroni correction for multiple-hypothesis testing, and main effects or two-factor interactions were assessed by three-way ANOVA at a 10% false-discovery rate.
RESULTS
Reliable Subcellular Extraction for Protein Phosphatase Activity Profiling
Preserving endogenous protein phosphatase activity from different subcellular fractions is technically challenging. Detergents rapidly extract proteins but lyse subcellular compartments indiscriminately (59). Cell lysis itself can inactivate protein phosphatases by oxidation, and phosphatase activity in lysates may be offset by constitutive kinase activities that co-extract (60, 61). We surmounted all of these hurdles by heavily modifying our whole-cell extraction procedure (29) originally developed to preserve the activity of protein phosphatases targeting MAPKs (Fig. 2A, Experimental Procedures). Adherent cells are gently permeabilized without mechanical disruption by using a saponin extraction buffer to permeabilize cells by displacing cholesterol selectively in cell membranes (62). The nuclear envelope has negligible cholesterol and thus only small nuclear proteins (< 40 kDa) that freely diffuse through nuclear pore complexes will be released with saponin extraction (59, 63). Cells are washed with saponin-containing PBS and then incubated briefly with phosphate-free Nonidet P-40 (NP40) extraction buffer to solubilize lipid bilayers, including the nuclear envelope. The NP40 buffer also contains isotonic NaCl to partially disrupt electrostatic interactions between nuclear protein complexes and DNA. Both extraction buffers were supplemented with hexokinase and glucose to consume ATP and therefore prevent kinase-catalyzed phosphorylation in the extract (see below). Multiple reducing agents were included in both buffers to preserve the active site Cys of extracted protein phosphatases. Collectively, we reasoned that these modifications to the lysis procedure should yield two matched biochemical fractions—a saponin extract (SE) and an NP40 extract (NE)—which each retain the activity of endogenous protein phosphatases.
Fig. 2.
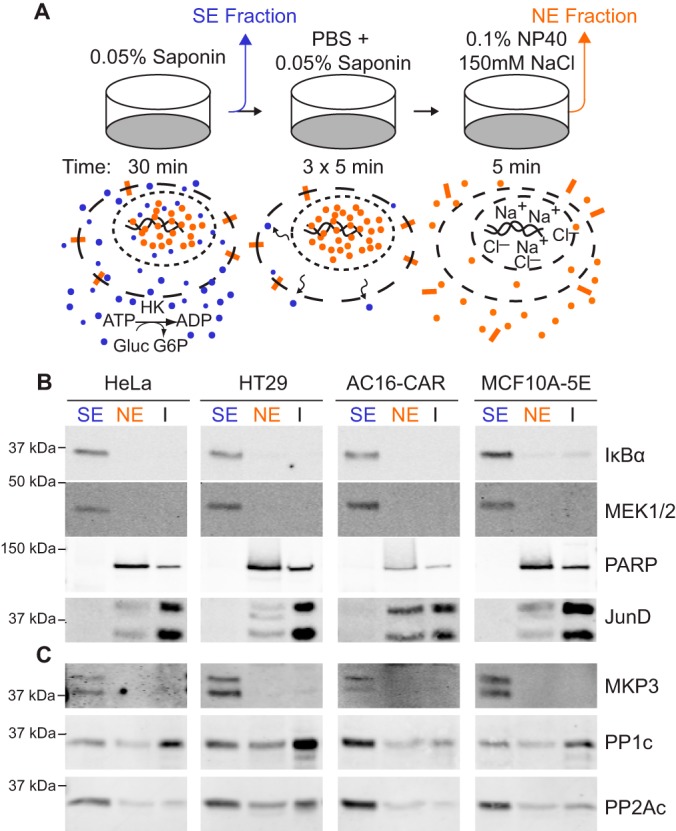
On-plate subcellular fractionation of adherent human cells. A, Extraction procedure schematic for collection of cytoplasmic proteins (blue circles), nuclear proteins (orange and small blue circles), and transmembrane proteins (orange rectangles) from adherent cell cultures. HK, hexokinase. Gluc, glucose. G6P, glucose-6-phosphate. B–C, Immunoblot comparison of saponin extracts (SE), Nonidet P-40 extracts (NE) and insoluble proteins (I) for cytoplasmic (MEK1/2 and IκBα) and nuclear (PARP and JunD) proteins (B) as well as endogenous MKP3 phosphatase, the catalytic subunit of PP1 (PP1c) phosphatase, and the catalytic subunit of PP2A (PP2Ac) phosphatase (C) in the indicated human cell lines.
We first evaluated whether the on-plate extraction procedure accurately fractionated subcellular proteins in multiple biological settings. Using cell lines of breast (MCF10A-5E), cervical (HeLa), colonic (HT29) and cardiac (AC16-CAR) origin, we immunoblotted SE and NE fractions for multiple proteins that strongly localize to the cytoplasm or the nucleus (Fig. 2B). We found that the dual-specificity kinase MEK and the NF-κB inhibitor IκBα were restricted to the S.E. fraction, as expected (64, 65). The lack of MEK and IκBα immunoreactivity in the NE fractions confirmed that the intermediate washing steps completely removed residual SE proteins from the plate. To assess the overall fractionation efficiency, we extracted the remaining insoluble (I) material on the cell culture plate with Laemmli sample buffer (66) and noted that pure SE proteins were almost completely removed. Reciprocally, the DNA-repair enzyme PARP and the transcription factor JunD were only detectable in the NE and I fractions (Fig. 2B). These results indicated that the preceding permeabilization and washing steps retained the proper localization of nuclear proteins within the size restriction of the nuclear pore complex (67), while recognizing that proteins bound tightly to DNA and chromatin-associated factors would not be completely removed by NE fractionation.
The extraction characteristics of SE and NE proteins extended to endogenous protein phosphatases (Fig. 2C). The cytoplasmic DUSP MKP3 resided entirely in the SE fraction, whereas the Ser/Thr phosphatases PP1 and PP2A were distributed in both SE and NE fractions with overall efficiencies of ∼53 ± 17% and ∼81 ± 11% respectively. The extraction of marker proteins and protein phosphatases varied across cell lines but did not change when cells were stimulated with epidermal growth factor (EGF) (supplemental Fig. S1). Replicated fractionation of resting and EGF-stimulated cells further showed that partitioning of endogenous protein phosphatases into SE and NE fractions was highly reproducible (supplemental Fig. S2). We concluded that the biochemical fractionation strategy (Fig. 2A) robustly isolates proteins from distinct subcellular compartments.
To assess protein separation more broadly, we ectopically expressed various V5-tagged constructs in AC16-CAR cells and repeated the fractionation. The DUSP MKP7 predominantly resided in the SE fraction (supplemental Fig. S3A), consistent with the cytoplasmic localization reported in previous overexpression studies (68). The nuclear regulatory subunit of PP1, NIPP1 (69), was also mostly extracted in the S.E. fraction probably because of its small size and passive diffusion through nuclear pores (supplemental Fig. S3B). However, ∼17% was still detectable in the NE fraction, including a faster migrating form that was exclusively NE resident. We observed a similarly interesting fractionation of the myristoylated JNK-stimulatory phosphatase, JSP1, whose localization has been reported to be perinuclear (70). A slower migrating form of JSP1 resided in the SE fraction, whereas a doublet was apparent in the NE fraction, which contained a faster migrating JSP1 that was also partially insoluble (supplemental Fig. S3C). Curiously, the transmembrane phosphatase PTP-SL, which localizes to intracellular vesicles (71–74), was completely solubilized in the SE fraction (supplemental Fig. S3D), suggesting that it may reside in a population of vesicles that is especially cholesterol rich. By contrast, the tight junction-associated transmembrane receptor CAR was negligibly extracted by saponin but solubilized efficiently in the NE fraction as expected (supplemental Fig. S3E). The V5-tagging experiments together indicated that the SE and NE fractions access many compartments within cells.
The precision of subcellular extraction was determined in different culture formats by measuring total extracted protein content with o-pthalaldehyde, a fluorogenic reagent that is compatible with strong reducing conditions (75). We found that on-plate SE and NE extraction was consistent from day to day and compatible with 10-cm, 6-well, 12-well, and 24-well formats (supplemental Fig. S4A–S4B). Total protein extraction decreased in the smaller formats, likely because of reduced shear forces and mixing during incubations on the platform rocker (see Experimental Procedures). However, the impact was equivalent for the SE and NE extraction steps, such that the ratio of the two fractions was roughly equal across all formats (supplemental Fig. S4C). The overall generality of the on-plate extraction procedure ensured that the method could be rapidly adapted to different biological applications.
Multiplex Quantification of Subcellular Protein Phosphatase Activity
To capture a range of endogenous protein phosphatase activities, we generated six recombinant phosphosubstrates, which are compartmentalized in different subcellular locales and regulated by various stimuli (Fig. 1). Phosphorylated MAPKs were previously produced using mutated, constitutively active dual-specificity kinases in vitro with their cognate MAPK substrate: MEK1-DD with ERK2, MKK4-EE and MKK7-EE with JNK1, and MKK6-EE with p38α (29). The efficiency of phosphorylated p38α (phospho-p38α) generation was increased considerably by coexpressing the constitutively active MKK6-EE in bacteria together with GST-tagged p38α. We built upon the success of in vivo p38α phosphorylation and produced phosphorylated MK2 (phospho-MK2) by triple coexpression of MKK6-EE, p38α, and GST-tagged MK2 in bacteria. To expand beyond phosphosubstrates that were themselves kinases, we purified two transcription factors, CREB and STAT1. Like ERK2 and JNK1, CREB and STAT1 were sufficiently phosphorylated in vitro: CREB with phospho-MK2 (described above) and STAT1 with a recombinant JAK1 fragment purified commercially (see Experimental Procedures). Using quantitative immunoblotting (56), we found that the phospho-stoichiometry of all substrates was well below that observed in stimulated cells (Table I), excluding the possibility of off-target dephosphorylation arising from excess phosphosubstrate. For each target, we identified adsorption conditions and phosphospecific antibody titers that yielded an extended linear dynamic range of the ELISA end point (supplemental Fig. S5, arrows). These conditions provided a starting point for each protein phosphatase assay, where loss of phosphoryl groups on the microtiter well caused a proportional loss of ELISA signal.
Table I. Phospho-stoichiometry of Recombinant Phosphosubstrates.
| Substrate | Phosphosite | Recombinanta | In vivoa (stimulationb) |
|---|---|---|---|
| ERK2 | Thr202/Tyr204 | 0.10 | 1.9 (EGF) |
| p38α | Thr180/Tyr182 | 0.040 | 3.5 (TNF) |
| JNK1 | Thr183/Tyr185 | 0.046 | 3.1 (TNF) |
| MK2 | Thr334 | 0.69 | 4.8 (TNF) |
| CREB | Ser133 | 0.033 | 2.2 (TNF) |
| STAT1 | Tyr709 | 6.4 | 36 (IFNβ) |
a Values are reported relative to unstimulated AC16-CAR cells.
b EGF, 100 ng/ml EGF for 5 min; TNF, 20 ng/ml TNF for 15 min; IFNβ, 50 ng/ml IFNβ for 30 min.
The expanded panel of phosphosubstrates presented new challenges in measuring protein phosphatase activity. In SE fractions prepared with earlier versions of extraction buffers (29), we found that the phospho-ELISA end point increased rather than decreased for substrates such as CREB, suggesting that uninhibited kinase activity was overwhelming phosphatase activity (Fig. 3A, columns 1 and 2). This phenomenon was not observed in NE fractions, which as a second-step extraction would contain much lower concentrations of residual ATP (Fig. 3A, columns 5 and 6, supplemental Fig. S6A–S6B). We depleted ATP from the SE fractions by adding recombinant hexokinase together with its substrate glucose in the extraction buffer. To irreversibly deactivate serine proteases that copurify with hexokinase (76) and destroy phosphosubstrates (supplemental Fig. S3C), we further supplemented the concentrated hexokinase stock solution with PMSF. The modified extraction buffer completely depleted residual ATP and resulted in substantially enhanced protein phosphatase activity measurements (Fig 3A, columns 3 and 4, supplemental Fig. S6A). Therefore, depletion of endogenous ATP from extracts is critical for widespread measurements of protein phosphatase activity.
Fig. 3.

ATP depletion and salt-detergent dilution are required for reliable subcellular phosphatase activity measurements. A, Adsorbed recombinant phospho-CREB was incubated with (+) or without (–) SE or NE fractions (50,000 cell equivalents) and with (+) or without (−) exogenous hexokinase (Hexo) to deplete intracellular ATP. CREB phosphorylation was measured by phospho-ELISA after incubation for one hour at 30 °C. Interaction between hexokinase and lysate was assessed statistically by two-way ANOVA. Data are shown as the mean of n = 4 independent biological extracts. B, Adsorbed recombinant phospho-p38α was incubated with NE fraction (8,500 cell equivalents) in NE buffer containing the indicated final concentrations of NaCl and NP40. p38α phosphorylation was measured by phospho-ELISA after incubation for one hour at 30 °C. Data are shown as the mean of n = 8 (buffer) or 4 (extract) assay replicates. Difference in means was assessed by two-tailed Student's t test.
We encountered a separate challenge with activity measurements in NE fractions. Efficient extraction of nuclear and transmembrane proteins requires NP40 and NaCl; however, these reagents inhibit protein-protein interactions important for substrate recognition by protein phosphatases. Indeed, extracts in pure NE buffer yielded small and variable decreases in phospho-ELISA signal (Fig. 3B, columns 1 and 3). After converting absorbance to phosphatase activity (see Experimental Procedures), non-diluted extracts yielded an unacceptable technical CV of 20%. We found that 10-fold dilution of NP40 to 0.1% (w/v) and NaCl to 15 mm increased the measured protein phosphatase activity by more than 2-fold and reduced the technical CV to 3% (Fig. 3B, columns 3 and 4). For assays requiring more concentrated NE fractions in a given culture format (supplemental Fig. S4), protein phosphatase activity could be reliably measured with up to 0.3% (w/v) NP40 and 45 mm NaCl, allowing flexibility to adapt measurements to specific experimental settings.
Before quantifying protein phosphatase activity, we altered the temperature and duration of phosphatase reactions to maximize signal (decrease in phospho-ELISA end point) while minimizing technical noise relative to extract-free phosphatase blank wells. We sought conditions where the CV was consistently less than 20% when the extracted phosphatases reduced the phospho-ELISA signal by at least 30%. In vitro dephosphorylation kinetics were linear during the assays (supplemental Fig. S7), ensuring that the extracted phosphatases preserved their activity and did not deplete the available phosphosubstrate on the plate. We calibrated the dynamic range of each assay by measuring protein phosphatase activity in serially diluted SE or NE fractions that maintain the concentration of critical buffer constituents (hexokinase, reducing agents, etc.). SE and NE protein phosphatase activities increased hyperbolically with extract concentration, as expected, and the optimal dynamic range across the panel was well below 50,000 cells (corresponding to ∼20 μg SE fraction and ∼9 μg NE fraction) (Fig. 4A–4L). This sensitivity and overall performance is comparable to quantitative immunoblotting of protein abundance from total cellular extracts (56). NE phosphatase activity assays for MK2 and CREB phosphatases required more extract, which was achieved by diluting to 0.3% (w/v) NP40 and 45 mm NaCl (see above). The reduced sensitivity toward phosphorylated Ser/Thr substrates is consistent with the low relative abundances of PP1 and PP2A holoenzymes observed in the NE fraction (Fig. 2C, supplemental Fig. S2B–S2C). The aggregate sensitivity of the measurement platform readily enables six-plex subcellular activity profiling of extracts from one well of a 12-well plate.
Fig. 4.

Sensitive detection and quantification of substrate-specific subcellular phosphatase activities. A–F, SE phosphatase assays were performed with 2-fold serially diluted AC16-CAR extracts starting at 25,000 (A–C), 50,000 (D–E), or 100,000 (F) cell equivalents. G–L, NE phosphatase assays were performed with 2-fold serially diluted extracts starting at 17,000 (G–I, K) or 51,000 (J, L) cell equivalents. NE fractions for phospho-MK2 (J) and phospho-CREB (K) were collected from cells stimulated with TNF for 30 min. Black arrows denote the optimized extract concentration for each assay. Phosphatase activity data were regressed against cell input with a four-parameter logistic regression curve. Data are shown as the means ± standard error of n = 4 assay replicates.
We evaluated protein phosphatase specificity of the activity assays by using small-molecule inhibitors. With the pan-phosphatase inhibitor sodium pyrophosphate (NaPP) (77, 78), all activity measurements were potently inhibited as expected (Fig. 5A–5F). The tyrosine dephosphorylation of STAT1 was also strongly reduced by sodium orthovanadate (Na3VO4) inhibition of PTPs (79, 80) (Fig. 5A). Reciprocally, the MK2 and CREB phosphatase assays were blocked by microcystin-LR (MCLR), implicating the substrates as targets of PP1, PP2A, or PP4,5,6 phosphatases (81) (Fig. 5B–5C). Moreover, because MCLR does not inhibit acid or alkaline phosphatases (82), these results also exclude contaminating activity from other phosphatases in various cellular subcompartments.
Fig. 5.

Subcellular phosphatase assays are sensitive to specific classes of phosphatase inhibition. A–F, Phosphosubstrates were incubated with AC16-CAR SE (black bars) or NE (gray bars) fractions and the pan phosphatase inhibitor NaPP (10 mm), the Tyr phosphatase inhibitor Na3VO4 (200 μm), the Ser/Thr phosphatase inhibitor microcystin-LR (MCLR, 1 μg/ml), or control (NoTx). NE fractions used for phospho-CREB (B, gray bars) and phospho-MK2 (C, gray bars) were collected from cells stimulated with TNF for 30 min. Cellular equivalents for each assay are listed in supplemental Table S2. Data are shown as the means ± standard error of n = 8 (NoTx) or 4 (NaPP, Na3VO4, MCLR) assay replicates. N. D., not detectable above baseline. Fraction-specific sensitivity differences were assessed by two-tailed Student's t test with Bonferroni correction.
The response of the MAPK phosphatase assays was more complex because of the multiple dephosphorylation reactions involved and the mixed specificity of the antibody detection reagents (20, 83, 84). ERK2 phosphatase measurements in SE and NE fractions were almost entirely MCLR sensitive (Fig. 5D), probably reflecting the phospho-Thr preference of the ELISA antibody (83) or the low abundance of inducible DUSPs for ERK, relative to PP2A, in resting cells (85, 86). Conversely, JNK1 phosphatases were highly vanadate sensitive (Fig. 5E); a phospho-Tyr preference for the phospho-JNK antibody has not been reported, but multiple JNK-selective DUSPs reside in both the cytoplasm and nucleus (5). The observed lack of MCLR sensitivity may stem from the prolyl isomerization of JNK within its pThr-Pro-pTyr activation loop (87), which prevents dephosphorylation by PP2A (88).
Most interesting was the behavior of p38α phosphatases, which were much more vanadate sensitive in the NE fraction (Fig. 5F, gray) but showed similar susceptibility to MCLR as to vanadate in the SE fraction (Fig. 5F, black). These data are consistent with negative p38α regulation by PP2A in the cytoplasm (89) and DUSPs in the nucleus (5). Together, our inhibitor results confirm that the assay platform measures all major classes of protein phosphatases in the SE and NE subcellular fractions.
We benchmarked the assay platform by performing a brief time-course experiment in cells stimulated with EGF to induce phosphorylation of ERK and the up-regulation of DUSPs (90). In parallel with extracts for phosphatase activity, subcellular samples were prepared for phospho-ERK quantification by supplementing SE and NE buffers with phosphatase inhibitors (see Experimental Procedures). In the SE fraction, we observed a rapid increase in ERK phosphorylation within 5 mins that remained elevated for up to two hours (Fig. 6A–6B). ERK2 phosphatase activity also increased in the SE fraction, likely blunting the peak phosphorylation observed, but the increase was transient and could not revert phospho-ERK abundance to pre-stimulus levels (Fig. 6C). Similar transients were observed for p38α and JNK1 phosphatases in the SE fraction (supplemental Fig. S8A–S8B), suggesting activity by a shared set of DUSPs. In contrast, MK2 and CREB phosphatase activities in the SE fraction were slowly reduced after EGF stimulation (supplemental Fig. S8C–S8D), and STAT1 phosphatase activity was rapidly elevated and sustained, perhaps in response to STAT1 phosphorylation induced by EGF (91, 92) (supplemental Fig. S8E). The qualitatively different phosphatase dynamics in the SE fraction provide a further validation of substrate specificity afforded by the assay.
Fig. 6.
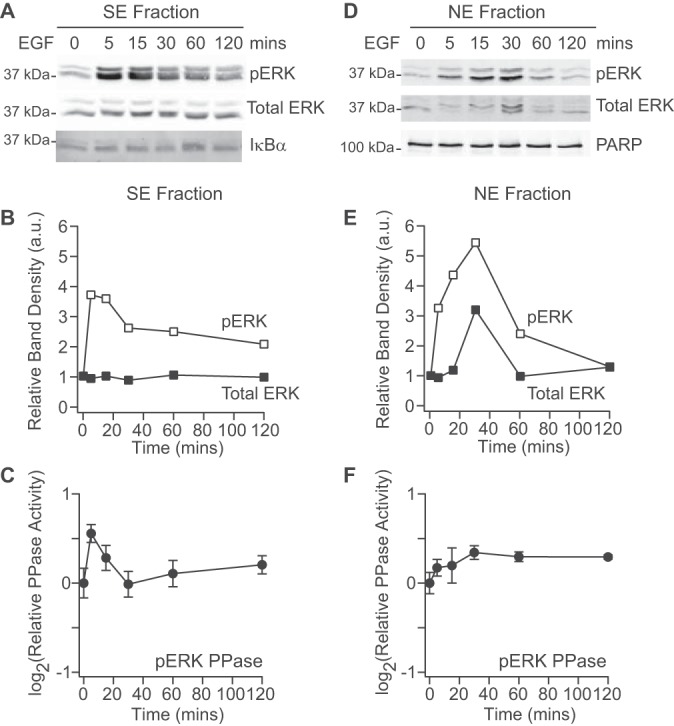
EGF stimulates subcellular ERK phosphorylation and ERK phosphatases. A–B, Immunoblots for phospho-ERK and total ERK in the SE fraction (A) were quantified by densitometry (B) with IκBα used to confirm equal loading. C, ERK2 phosphatase activity quantified in the SE fraction. D–E, Immunoblots for phospho-ERK and total ERK in the NE fraction (D) were quantified by densitometry (E) with PARP used to confirm equal loading. F, ERK2 phosphatase activity quantified in the NE fraction. Phosphatase activities are shown as the means ± standard error of n = 4 biological replicates. Cells were stimulated with 100 ng/ml of EGF for the indicated times.
A very different time course of phospho-ERK abundance was observed in the NE fraction. ERK phosphorylation was slower, peaking at 30 min concomitant with maximum total ERK shuttled into the nucleus (Fig. 6D–6E). Importantly, phospho-ERK in the NE fraction reverted to pre-stimulus levels within two hours, suggesting that negative regulation in this subcompartment was more persistent than in the SE fraction. Corroborating the phospho-ERK dynamics, we found that ERK2 phosphatase activity in the NE fraction was increased and sustained from 30–120 min after EGF stimulation. Moreover, this trajectory was unique among MAPKs, as p38α phosphatase activity in the NE fraction was transiently increased and JNK1 phosphatase activity was slightly decreased (supplemental Fig. S8F–S8G). Among other phosphosubstrates, MK2 and CREB phosphatases were largely unaltered in the NE fraction, whereas STAT1 phosphatases were slowly and transiently activated (supplemental Fig. S8H–S8J). Overall, the measured subcellular ERK2 phosphatase activities reconcile the observed phospho-ERK dynamics, whereas the broader panel of phosphosubstrates in the assay reinforces the more-widespread alterations caused by EGF stimulation.
Viral and Antiviral Regulation of Protein Phosphatases During Acute Infection of Cardiomyocytes with Coxsackievirus B3
To profile subcellular protein phosphatase dynamics in infectious disease, we investigated the interactions between host-cell antiviral signaling and acute infection by CVB3, which perturbs most of the signaling pathways in the assay panel (Fig. 1). Despite its recognized tropism for juvenile cardiomyocytes (93, 94), CVB3 pathogenesis is largely studied in permissive HeLa cells because of the efficiency of viral propagation (40, 95, 96). As an alternative, neonatal mouse HL-1 cells have been used (35, 39), but these cells are atrially derived, and CVB3 infections disproportionately impact the ventricular myocardium (97, 98). A human ventricular cardiomyocyte cell line that is susceptible to CVB3 would be highly desirable for in vitro studies of host-pathogen interactions.
We tackled this challenge by starting with AC16 cells, a clonal human line derived by fusing adult ventricular cardiomyocytes with SV40-transformed fibroblasts (53). Although lacking the typical sarcomeric organization of the myocardium, AC16 cells express several markers of immature cardiomyocytes, including cardiac-specific transcription factors and contractile proteins. SV40 small t antigen alters PP2A function as an unavoidable facet of the line (99), but AC16 cells nonetheless have detectable MCLR-sensitive phosphatase activity at baseline (Fig. 5B–5D, 5F). Given that AC16 cells originate from adult tissue and expression of the CVB3-obligate receptor CAR declines with age (100), we reconstituted CAR by lentiviral transduction and stable selection to generate AC16-CAR cells (see Experimental Procedures). Compared with the parental AC16 line, AC16-CAR cells were much more supportive of intracellular viral protein synthesis upon CVB3 infection, as indicated by expression of the viral capsid protein VP1 (Fig. 7A). We also detected cleavage of eIF4G, a recognized target of the active enteroviral protease 2A (101). Accordingly, CVB3 infection was substantially more toxic to the AC16-CAR line. By fluorescence microscopy, we observed cells with increased caspase-3 cleavage as well as others with compromised membrane integrity (Fig. 7B), indicating concurrent apoptosis and necrosis within the culture (39). Cardiomyocyte apoptosis has been shown to be correlated with CVB3 viral titer (102), and we found that viral titers increased 1000-fold in conditioned media from infected AC16-CAR cells (Fig. 7C–7D). Therefore, with the restoration of CAR, AC16 cells support all stages of the viral life cycle required to amplify an acute infection.
Fig. 7.

Modeling acute CVB3 Infection through engineered AC16-CAR cells. A, Immunoblot of infected and non-infected AC16 cells for V5 epitope-tagged CAR (CAR-V5), viral capsid protein VP1, viral protease target eIF4G, and cleaved caspase-3 (Clv. Casp3), with HSP90 and p38 used as loading controls. B, Immunofluorescence of infected versus non-infected parental and CAR-expressing AC16 cells stained for VP1 (red), Clv. Casp3 (green), LIVE/DEAD stain (white) (39), and nuclei (blue). Green arrowheads indicate apoptotic cells, and white arrowheads indicate necrotic cells. Scale bar is 20 μm. C, Plaque assay of two dilutions of conditioned medium from AC16 cells with or without CAR expression and with or without CVB3 infection. The negative control (unconditioned medium) and positive control (2000 plaque forming units [PFU] of CVB3) are shown on the right. Viral plaques appear white and indicate the presence of single infectious virions, which propagate to surrounding cells. D, Quantification of viral PFUs from n = 3 independent CVB3 infections and plaque assays. Cells were infected with sham or CVB3 at MOI = 10 for 24 h.
We sought to use AC16-CAR cells together with the subcellular phosphatase assay to profile activity changes during CVB3 infection and the associated interferon response. Type I interferons (IFNα, IFNβ) are potently induced by double-stranded RNA (dsRNA) that accompanies viral replication, but CVB3 counteracts the type I response by cleaving sensors and transducers of dsRNA (103). Type II interferon (IFNγ) signaling is initiated by the paracrine action of natural killer cells, which become activated upon CVB3 infection (44). Co-treatment of cells with IFNβ and IFNγ synergistically inhibits CVB3 replication (104), and IFNβ treatment alone has shown some success in clearing persistent CVB3 infections (98). Phosphatases are rare among interferon-stimulated genes (105), but some viruses directly perturb interferon action by upregulating phosphatase activity (106). Whether more-distant crosstalk occurs between phosphatases modulated by interferons and CVB3 has not been examined.
The throughput of the assay enabled a highly replicated (n = 9), fully crossed design of three factors: CVB3 (MOI = 10), IFNβ (30 ng/ml), and IFNγ (50 U/ml). Samples receiving CVB3 were infected for three hours before stimulation with IFNβ, IFNγ, or both, followed by on-plate fractionation and phosphatase-activity profiling of the resulting extracts. Some assays showed skewed or leptokurtic error distributions across biological replicates, requiring the use of nonparametric methods for statistical inference in these instances (see Experimental Procedures). Without antiviral cytokines, we did not detect significant differences in phosphatase activity caused by CVB3 infection alone after correcting for multiple-hypothesis testing (Fig. 8A). Therefore, day-to-day and plate-to-plate variation was accounted for by normalizing data to the median activity of no-cytokine samples in each group (see Experimental Procedures). The full-factorial design of the experiment allowed us to test for main effects of CVB3, IFNβ, and IFNγ, as well as nonlinear interactions between factors that would indicate synergistic or antagonistic regulation.
Fig. 8.

Subcellular phosphatase profiling of CVB3 infection and interferon signaling in AC16-CAR cells. A, Subcellular phosphatase activities are comparable after infection with CVB3 (MOI = 10) for seven hours. No differences were statistically significant after correcting for multiple-hypothesis testing (see Experimental Procedures). B, Heat map of median phosphatase activity changes in AC16-CAR cells treated with or without CVB3 (MOI = 10) for three hours followed by treatment with IFNβ (30 ng/ml), IFNγ (50 U/ml), or both for four hours. Cellular equivalents for each assay are listed in supplemental Table S3. C–F, Box-and-whisker plots of replicated data summarized in (B). Significant main effects at a 10% false-discovery rate (see Experimental Procedures) are highlighted for IFNβ (solid yellow), IFNγ (blue box), and CVB3 (red box). For (A) and (C–F), black bars show log2-transformed median activity levels of n = 9 independent biological replicates. Gray boxes indicate interquartile range. Black whiskers denote values that fall within 1.5 times the interquartile range. Gray dots are outliers beyond 1.5 times the interquartile range.
Overall, we observed various single-factor perturbations consistent with the literature (Fig. 8B). For example, IFNβ independently suppressed ERK2, p38α, and MK2 phosphatase activities in the SE fraction (Fig. 8C–8E, p < 0.01). ERK activity is critical for the type I interferon response (107). Also, the p38-MK2 pathway stabilizes mRNAs with AU-rich elements (108, 109), a characteristic of many interferon-response genes (105) (supplemental Table S4). Reduced phosphatase activity toward these targets may collectively prolong the duration of the type I interferon response. Notably, we did not observe IFNβ-stimulated decreases in all DUSP or Ser/Thr phosphatase targets (Fig. 8B, JNK1 and CREB), reinforcing the specificity of the assay panel.
IFNβ suppression of p38α phosphatase activity was also observed in NE fractions (Fig. 8C, p < 0.001), but there were multiple instances of changes specific to one cellular subcompartment. In contrast to IFNβ, we found that IFNγ up-regulated p38 phosphatase activity only in the NE fraction (p < 0.01). A similar trend was observed for JNK1 phosphatase in the SE fraction (Fig. 8F), although the IFNγ effect did not retain statistical significance after correction for multiple-hypothesis testing at a false-discovery rate of 10%. Long-term (3+ hours) stimulation of macrophages with IFNγ up-regulates MKP5 and MKP7 (110), which dephosphorylate JNK-p38 and endogenously localize to the cytoplasm and nucleus (5). Our findings raise the possibility that these induced DUSPs exhibit different substrate preferences depending on their localization.
Despite median normalization, specific subcellular perturbations were also detected in CVB3-infected cells: ERK2 phosphatase activity in the SE fraction was increased overall (Fig. 8D, p < 0.01) along with the activity of JNK1 phosphatases in the NE fraction (Fig. 8F, p < 0.001). ERK2 phosphatase activity likely reflected the compensatory up-regulation of cytoplasmic DUSPs, such as MKP3, that could be induced by CVB3 activation of the ERK pathway (37). The NE-associated JNK1 phosphatase result was more intriguing given conflicting reports involving the role of JNK activation in CVB3 pathogenesis (38, 111). We therefore pursued follow-on experiments to dissect mechanistically the role of CVB3-associated JNK1 phosphatase activity in the NE fraction.
Nuclear JNK1 Phosphatases Impede CVB3 Pathogenesis
Given the considerable vanadate sensitivity of JNK1 phosphatases in the NE fraction (Fig. 5E), we first sought to identify nuclear DUSPs that contributed to the activity measured by the assay. We cloned inducible shRNAs (supplemental Fig. S9) for four JNK-targeting DUSPs and transduced AC16-CAR cells to assess their individual contributions toward JNK1 phosphatase activity in the NE fraction. Significant changes were not observed with shMKP7 or shDUSP19 perturbations, but knockdown of MKP1 and MKP5 reduced JNK1 phosphatase activity in the NE fraction by ∼50 and ∼30% respectively (supplemental Fig. S10). These results implicate MKP1 and MKP5 as the predominant JNK1 phosphatases in the NE fraction of AC16-CAR cells.
Next, it was important to clarify the role of JNK pathway activity in CVB3 pathogenesis. Prior CVB3 studies involving the JNK pathway relied on a first-generation inhibitor that is now known to inhibit many other kinases (38, 111, 112). We therefore turned to JNK-IN-8 (IN8), a newer covalent JNK inhibitor that is much more selective (113). Rather than block the JNK pathway constitutively, we sought to exploit the covalent nature of IN8 and achieve a slow reactivation of JNK by washout during infection. Three-hour preincubation of cells with IN8 strongly reduced cJun phosphorylation—a surrogate of JNK activity—but phosphorylation was nearly restored to control levels after 20 h of washout (Fig. 9A). By shifting the initial baseline, IN8 washout achieves a fold-change activation of the JNK pathway that can be overlaid on viral and antiviral signaling (Fig. 9B) (114–116).
Fig. 9.

Nuclear JNK1 activity promotes CVB3 protein synthesis in infected AC16-CAR cells. A–B, Immunoblots for p-cJun before and after JNK-IN-8 (IN8) washout (w. o.) (A) were quantified by densitometry (B) with vinculin, HSP90, and tubulin used to confirm equal loading. Fold change in p-cJun relative to the corresponding pre-washout condition is shown in white. C–D, Immunoblots for VP1 with or without IN8 washout, IFNβ treatment, or both in CVB3-infected cells (C) were quantified by densitometry (D) with vinculin, HSP90, and tubulin used to confirm equal loading. Fold change in VP1 relative to the corresponding pre-washout condition is shown in white. E, Immunofluorescence of EGFP-NLS or MKK4/7-EE-NLS and p-cJun in AC16-CAR cells. Scale bar is 20 μm. F, Correlation plots between relative FLAG and VP1 abundance in infected AC16-CAR cells overexpressing EGFP-NLS (top panels) or MKK4/7-EE-NLS (bottom panels). IN8 data are shown as the means ± standard error of n = 4 biological replicates. Differences in p-cJun (B) and VP1 (D) abundance were assessed by Student's t test. FLAG-VP1 Pearson correlations (R) were assessed after Fisher Z transformation and Fisher's method of combined probabilities for each cell line.
As a single factor, CVB3 increased JNK1 phosphatase activity in the NE fraction (Fig. 8F), but there was also a suggestive synergy with IFNβ (interaction p < 0.05), prompting us to perform the IN8 washout experiments in CVB3-infected cells ± IFNβ. The interferon response initiated by IFNβ strongly suppressed translation of VP1 during CVB3 expression, as expected, but we found that VP1 abundance doubled when the JNK pathway was activated by IN8 washout (Fig. 9C–9D). These results suggest that CVB3- and IFNβ-induced activation of JNK phosphatases may contribute to the host antiviral response.
The IN8 washout experiment generally implicates the JNK pathway in CVB3 pathogenesis but cannot assign a specific role for nuclear-localized activity. To do so, we engineered inducible MKK4-EE and MKK7-EE alleles harboring a potent nuclear localization sequence (NLS) that should restrict JNK activation to the nucleus. By immunofluorescence, we confirmed the localization of MKK4-EE-NLS and MKK7-EE-NLS and observed an increased frequency of cells with elevated cJun phosphorylation compared with EGFP-NLS controls (Fig. 9E). Timed induction of JNK activators was reliable under resting conditions but became highly variable during CVB3 infection and IFNβ stimulation because of the shutdown of protein translation during the host-cell interferon response. We exploited this variability to ask whether the amount of induced MKK4/7-EE-NLS corresponded to a proportional increase in VP1 abundance for the same biological replicate. Indeed, VP1 expression was significantly correlated with MKK4/7-EE-NLS but not EGFP-NLS controls, indicating that nuclear JNK activation promotes viral propagation in infected host cells. These follow-on experiments together provide a rationale and molecular basis for the CVB3- and IFNβ-stimulated JNK1 phosphatase activity measured by the assay in the NE fraction.
DISCUSSION
This work considerably extends the premise of substrate-directed phosphatase activity profiling (29) by doubling the number of substrates and subcellular compartments accessible with the method. Phosphoprotein-focused mathematical models of cell signaling often separate rate processes that occur in the cytoplasm and nucleus (117–119). Subcellular measurements of protein phosphatase activity will help to parameterize time- and subcompartment-specific deactivation rates, which determine steady-state signaling (6). Furthermore, it should be easier to hone in on detailed mechanisms of phosphosubstrate regulation (29) by focusing on the specific phosphatases localized to where activity changes were measured (5, 22). In this way, systematic experiments performed with high-throughput methods set the stage for more in-depth mechanistic hypotheses (120, 121).
The expanded phosphatase format revealed cellular ATP as an important confounder in the assay. We had noted increased, rather than decreased, substrate phosphorylation in the assay before (29) but discounted it because it was observed only when JNK phosphatases were measured in the presence of vanadate. After ATP depletion, phospho-JNK dephosphorylation is considerably inhibited by vanadate in both NE and SE fractions (Fig. 5E). Our results corroborate observations in clinical isolates, which illustrated that kinases are as problematic as phosphatases when seeking to accurately capture the cellular phosphoproteome accurately (61). For enzyme-catalyzed depletion of ATP, hexokinase is a useful alternative to apyrase (122) when extracts must be kept at low temperature (123).
Isolation of NE-fractionated protein phosphatases was a tradeoff between extraction efficiency and retention of catalytic activity. Proteins tightly associated with chromatin, including some protein phosphatases (22), likely remain in the nucleus after extraction. However, the conditions required to displace such proteins would undoubtedly interfere with activity in the entire extract. Nuclear MK2 and CREB phosphatase activity measurements were somewhat less sensitive than others in the panel, possibly because of unextracted PP1 isoforms (124). More important than total extraction is the ability to capture stimulus-dependent changes in phosphatase activity, as observed in response to growth factors, interferons, and viruses.
We found that reconstitution of CAR expression was sufficient to render AC16 cells permissive to CVB3. CAR is not universally rate-limiting for CVB3 infection—A549 lung adenocarcinoma cells express as much CAR as permissive HeLa cells, but their ability to propagate the virus is restricted by deficient expression of the DAF coreceptor (125). CVB3 permissiveness is also dictated by more than cell-surface receptors, with many intracellular factors contributing positively or negatively to infectivity (41). For example, HeLa cells become non-permissive upon overexpression of the mitochondrial antiviral signaling protein MAVS (126). AC16 cells were originally isolated from human ventricular cardiomyocytes of an adult (53), and their initial CVB3 restriction likely stems from the natural decline in CAR expression that occurs with age (100). CAR-supplemented AC16 cells provide an improved CVB3 host, which reflects the natural tropism of the virus.
Our crossed experimental design with CVB3 and antiviral factors uncovered multiple changes in protein phosphatase activity that could contribute to systems analyses of virus-host interactions. By coupling protein phosphatase profiles with matched observations of kinase activity and substrate phosphorylation, related data types could be assessed for concordance (127–129). The substrate-focused phosphatase assays are ideal for such a comparison, with the tradeoff that it is more complicated to identify the specific phosphatase(s) involved. Nevertheless, it is possible to dissect mechanisms as we showed for the elevated JNK1 phosphatase activity observed in the NE fraction of AC16-CAR cells. For interferons and other extracellular ligands, transcriptomic profiles of protein phosphatase subunits could provide clues about induced changes in abundance. The challenge is greater for CVB3-induced perturbations, which are largely mediated by the proteases 2A and 3C or by innate antiviral mechanisms. Many protein phosphatases contain high scoring consensus sequences for enteroviral proteases (130), suggesting that perturbations could be highly multifaceted.
The demonstrated generality of the assay platform opens up additional opportunities to interrogate protein phosphatase activities related to CVB3 infection. In epithelia, viral docking rapidly activates Abl and Fyn, and CVB3 crosslinking of DAF in lymphocytes triggers phosphorylation of Lck (32–34). These nonreceptor tyrosine kinases readily autophosphorylate in bacteria and in vitro (131, 132), suggesting that such substrates could be prepared in high yield for Abl-, Fyn-, and Lck-focused phosphatase assays. CVB3 infection also mobilizes Ca2+ stores (133), and the Ca2+-activated phosphatase calcineurin disinhibits NFAT, a transcription factor whose activation in T cells promotes myocarditis (134). A calcineurin-focused NFAT phosphatase assay would enable a further elaboration of the host-cell signaling networks perturbed by CVB3.
The CVB3 genome engages many intracellular signaling pathways to disrupt host-cell functions (135). Decoding viral mechanisms of action on specific host phosphoproteins will be critical for devising strategies to block or offset these mechanisms during CVB3 infection. Reciprocally, one can envision future virus-inspired interventions for diseases of signaling misregulation such as lung cancer, where high CAR expression is important for tumorigenesis (136) and CVB3 has been shown to be oncolytic (137).
Supplementary Material
Acknowledgments
We thank Cheryl Borgman for copyediting this manuscript and for performing the ARED analysis of interferon-stimulated genes as well as Jun Lin and Lixin Wang for assistance with cloning.
Footnotes
Author contributions: M.S., C.M.S., Z.D.C., D.L.B., and K.A.J. designed research; M.S., C.M.S., S.K., and Z.D.C. performed research; M.S., C.M.S., Z.D.C., and D.L.B. contributed new reagents or analytic tools; M.S., C.M.S., and K.A.J. analyzed data; M.S. and K.A.J. wrote the paper.
* This work was supported by the National Institutes of Health (R21-AI105970 to K. A. J., R21-AI114960 to D. L. B.), the American Heart Association (15PRE24480039-SHAH to M. S.), the Pew Charitable Trusts (#2008-000410-006 to K. A. J.), and the David and Lucile Packard Foundation (#2009-34710 to K. A. J.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- DUSP
- dual specificity phosphatase
- ANOVA
- analysis of variance
- ARE
- AU-rich element
- CAR
- coxsackievirus and adenovirus receptor
- Clv
- Casp3 cleaved caspase 3
- CREB
- CAMP responsive element binding protein
- CV
- coefficient of variation
- CVB3
- coxsackievirus B3
- DAF
- decay accelerating factor
- EGF
- epidermal growth factor
- eIF4G
- eukaryotic translation initiation factor 4 gamma
- ERK2
- extracellular signal-regulated kinase 2
- G6P
- glucose-6-phosphate
- Gluc
- glucose
- HK
- or Hexo hexokinase
- HSP90
- heat shock protein 90 kDa
- HRP
- horseradish peroxidase
- IFN
- interferon
- JAK
- Janus kinase
- JNK1
- JUN N-Terminal kinase
- MAPK
- mitogen-activated protein kinase
- MAVS
- mitochondrial antiviral signaling protein
- MCLR
- microcystin-LR
- MK2
- mitogen-activated protein kinase-activated protein kinase 2
- MOI
- multiplicity of infection
- NaPP
- sodium pyrophosphate
- Na3VO4
- sodium orthovanadate
- NE
- NP40 extract
- NP40
- Nonidet P-40
- PFA
- paraformaldehyde
- PFU
- plaque forming unit
- PP1
- protein phosphatase 1
- PP2A
- protein phosphatase 2A
- PTP
- protein tyrosine phosphatase
- RIPA
- radioimmunoprecipitation assay buffer
- SDS
- sodium dodecyl sulfate
- SE
- saponin extract
- STAT1
- signal transducer and activator of transcription 1
- SV40
- Simian virus 40
- VP1
- viral protein 1
- WB
- western blot.
REFERENCES
- 1. Downward J. (2001) The ins and outs of signalling. Nature 411, 759–762 [DOI] [PubMed] [Google Scholar]
- 2. Hunter T. (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80, 225–236 [DOI] [PubMed] [Google Scholar]
- 3. Tonks N. K. (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 [DOI] [PubMed] [Google Scholar]
- 4. Cohen P. T. W. (2002) Protein phosphatase 1 – targeted in many directions. J. Cell Sci. 115, 241–256 [DOI] [PubMed] [Google Scholar]
- 5. Owens D. M., and Keyse S. M. (2007) Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 26, 3203–3213 [DOI] [PubMed] [Google Scholar]
- 6. Heinrich R., Neel B. G., and Rapoport T. A. (2002) Mathematical models of protein kinase signal transduction. Mol. Cell 9, 957–970 [DOI] [PubMed] [Google Scholar]
- 7. Bhalla U. S., Ram P. T., and Iyengar R. (2002) MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 297, 1018–1023 [DOI] [PubMed] [Google Scholar]
- 8. Auger-Messier M., Accornero F., Goonasekera S. A., Bueno O. F., Lorenz J. N., van Berlo J. H., Willette R. N., and Molkentin J. D. (2013) Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ. Res. 112, 48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keyse S. M. (2008) Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 27, 253–261 [DOI] [PubMed] [Google Scholar]
- 10. Zhao Q., Wang X., Nelin L. D., Yao Y., Matta R., Manson M. E., Baliga R. S., Meng X., Smith C. V., Bauer J. A., Chang C.-H., and Liu Y. (2006) MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med. 203, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brewis N., Ohst K., Fields K., Rapacciuolo A., Chou D., Bloor C., Dillmann W., Rockman H., and Walter G. (2000) Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am. J. Physiol. 279, SH1307–SH1318 [DOI] [PubMed] [Google Scholar]
- 12. Arena S., Benvenuti S., and Bardelli A. (2005) Genetic analysis of the kinome and phosphatome in cancer. Cell. Mol. Life Sci. 62, 2092–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi Y. (2009) Serine/threonine phosphatases: Mechanism through structure. Cell 139, 468–484 [DOI] [PubMed] [Google Scholar]
- 14. Zhang Y. Y., Wu J. W., and Wang Z. X. (2011) A distinct interaction mode revealed by the crystal structure of the kinase p38alpha with the MAPK binding domain of the phosphatase MKP5. Sci. Signal. 4, era88. [DOI] [PubMed] [Google Scholar]
- 15. Wood C. D., Thornton T. M., Sabio G., Davis R. A., and Rincon M. (2009) Nuclear localization of p38 MAPK in response to DNA damage. Int. J. Biol. Sci. 5, 428–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gong X., Ming X., Deng P., and Jiang Y. (2010) Mechanisms regulating the nuclear translocation of p38 MAP kinase. J. Cell. Biochem. 110, 1420–1429 [DOI] [PubMed] [Google Scholar]
- 17. Ben-Levy R., Hooper S., Wilson R., Paterson H. F., and Marshall C. J. (1998) Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol. 8, 1049–1057 [DOI] [PubMed] [Google Scholar]
- 18. Lenormand P., Sardet C., Pages G., L'Allemain G., Brunet A., and Pouyssegur J. (1993) Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell Biol. 122, 1079–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J., Tang R., Lv M., Wang Q., Zhang X., Guo Y., Chang H., Qiao C., Xiao H., Li X., Li Y., Shen B., and Zhang J. (2011) Defective anchoring of JNK1 in the cytoplasm by MKK7 in Jurkat cells is associated with resistance to Fas-mediated apoptosis. Mol. Biol. Cell 22, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alessi D. R., Gomez N., Moorhead G., Lewis T., Keyse S. M., and Cohen P. (1995) Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr. Biol. 5, 283–295 [DOI] [PubMed] [Google Scholar]
- 21. Scott J. D., and Pawson T. (2009) Cell signaling in space and time: Where proteins come together and when they're apart. Science 326, 1220–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moorhead G. B., Trinkle-Mulcahy L., and Ulke-Lemee A. (2007) Emerging roles of nuclear protein phosphatases. Nat. Rev. Mol. Cell Biol. 8, 234–244 [DOI] [PubMed] [Google Scholar]
- 23. Sacco F., Perfetto L., Castagnoli L., and Cesareni G. (2012) The human phosphatase interactome: An intricate family portrait. Febs Letters 586, 2732–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kameshita I., Baba H., Umeda Y., and Sueyoshi N. (2010) In-gel protein phosphatase assay using fluorogenic substrates. Anal. Biochem. 400, 118–122 [DOI] [PubMed] [Google Scholar]
- 25. Burridge K., and Nelson A. (1995) An in-gel assay for protein tyrosine phosphatase activity: detection of widespread distribution in cells and tissues. Anal. Biochem. 232, 56–64 [DOI] [PubMed] [Google Scholar]
- 26. McAvoy T., and Nairn A. C. (2010) Serine/threonine protein phosphatase assays. Curr. Protoc. Mol. Biol. CHAPTER, Unit 18.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Montalibet J., Skorey K. I., and Kennedy B. P. (2005) Protein tyrosine phosphatase: enzymatic assays. Methods 35, 2–8 [DOI] [PubMed] [Google Scholar]
- 28. Geladopoulos T. P., Sotiroudis T. G., and Evangelopoulos A. E. (1991) A malachite green colorimetric assay for protein phosphatase activity. Anal. Biochem. 192, 112–116 [DOI] [PubMed] [Google Scholar]
- 29. Bose A. K., and Janes K. A. (2013) A high-throughput assay for phosphoprotein-specific phosphatase activity in cellular extracts. Mol. Cell. Proteomics 12, 797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burch G. E., Sun S. C., Chu K. C., Sohal R. S., and Colcolough H. L. (1968) Interstitial and coxsackievirus B myocarditis in infants and children. A comparative histologic and immunofluorescent study of 50 autopsied hearts. JAMA 203, 1–8 [PubMed] [Google Scholar]
- 31. Cooper L. T., Jr. (2009) Myocarditis. N. Engl. J. Med. 360, 1526–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu P., Aitken K., Kong Y. Y., Opavsky M. A., Martino T., Dawood F., Wen W. H., Kozieradzki I., Bachmaier K., Straus D., Mak T. W., and Penninger J. M. (2000) The tyrosine kinase p56lck is essential in coxsackievirus B3-mediated heart disease. Nat. Med. 6, 429–434 [DOI] [PubMed] [Google Scholar]
- 33. Shenoy-Scaria A. M., Kwong J., Fujita T., Olszowy M. W., Shaw A. S., and Lublin D. M. (1992) Signal transduction through decay-accelerating factor. Interaction of glycosyl-phosphatidylinositol anchor and protein tyrosine kinases p56lck and p59fyn 1. J. Immunol. 149, 3535–3541 [PubMed] [Google Scholar]
- 34. Coyne C. B., and Bergelson J. M. (2006) Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124, 119–131 [DOI] [PubMed] [Google Scholar]
- 35. Garmaroudi F. S., Marchant D., Si X., Khalili A., Bashashati A., Wong B. W., Tabet A., Ng R. T., Murphy K., Luo H., Janes K. A., and McManus B. M. (2010) Pairwise network mechanisms in the host signaling response to coxsackievirus B3 infection. Proc. Natl. Acad. Sci. U.S.A. 107, 17053–17058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jensen E. C. (2013) Quantitative analysis of histological staining and fluorescence using ImageJ. Anat. Rec. 296, 378–381 [DOI] [PubMed] [Google Scholar]
- 37. Huber M., Watson K. A., Selinka H. C., Carthy C. M., Klingel K., McManus B. M., and Kandolf R. (1999) Cleavage of RasGAP and phosphorylation of mitogen-activated protein kinase in the course of coxsackievirus B3 replication. J. Virol. 73, 3587–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Si X., Luo H., Morgan A., Zhang J., Wong J., Yuan J., Esfandiarei M., Gao G., Cheung C., and McManus B. M. (2005) Stress-activated protein kinases are involved in coxsackievirus B3 viral progeny release. J. Virol. 79, 13875–13881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jensen Karin J., Garmaroudi Farshid S., Zhang J., Lin J., Boroomand S., Zhang M., Luo Z., Yang D., Luo H., McManus Bruce M., and Janes Kevin A. (2013) An ERK-p38 subnetwork coordinates host cell apoptosis and necrosis during Coxsackievirus B3 infection. Cell Host Microbe 13, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luo H., Yanagawa B., Zhang J., Luo Z., Zhang M., Esfandiarei M., Carthy C., Wilson J. E., Yang D., and McManus B. M. (2002) Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J. Virol. 76, 3365–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coyne C. B., Bozym R., Morosky S. A., Hanna S. L., Mukherjee A., Tudor M., Kim K. S., and Cherry S. (2011) Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host Microbe 9, 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Q., Zheng Z., Liu Y., Zhang Z., Liu Q., Meng J., Ke X., Hu Q., and Wang H. (2016) 2C Proteins of enteroviruses suppress IKKbeta phosphorylation by recruiting protein phosphatase 1. J. Virol. 90, 5141–5151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seko Y., Takahashi N., Yagita H., Okumura K., and Yazaki Y. (1997) Expression of cytokine mRNAs in murine hearts with acute myocarditis caused by coxsackievirus b3. J. Pathol. 183, 105–108 [DOI] [PubMed] [Google Scholar]
- 44. Huhn M. H., Hultcrantz M., Lind K., Ljunggren H. G., Malmberg K. J., and Flodstrom-Tullberg M. (2008) IFN-gamma production dominates the early human natural killer cell response to Coxsackievirus infection. Cell Microbiol. 10, 426–436 [DOI] [PubMed] [Google Scholar]
- 45. Platanias L. C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 [DOI] [PubMed] [Google Scholar]
- 46. Weil J., Epstein C. J., Epstein L. B., Sedmak J. J., Sabran J. L., and Grossberg S. E. (1983) A unique set of polypeptides is induced by gamma-interferon in addition to those induced in common with alpha-interferon and beta-interferon. Nature 301, 437–439 [DOI] [PubMed] [Google Scholar]
- 47. Raingeaud J., Whitmarsh A. J., Barrett T., Derijard B., and Davis R. J. (1996) MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 16, 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Enslen H., Raingeaud J., and Davis R. J. (1998) Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 273, 1741–1748 [DOI] [PubMed] [Google Scholar]
- 49. Yang X., Boehm J. S., Yang X., Salehi-Ashtiani K., Hao T., Shen Y., Lubonja R., Thomas S. R., Alkan O., Bhimdi T., Green T. M., Johannessen C. M., Silver S. J., Nguyen C., Murray R. R., Hieronymus H., Balcha D., Fan C., Lin C., Ghamsari L., Vidal M., Hahn W. C., Hill D. E., and Root D. E. (2011) A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shin K. J., Wall E. A., Zavzavadjian J. R., Santat L. A., Liu J., Hwang J. I., Rebres R., Roach T., Seaman W., Simon M. I., and Fraser I. D. (2006) A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 13759–13764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Debnath J., Muthuswamy S. K., and Brugge J. S. (2003) Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268 [DOI] [PubMed] [Google Scholar]
- 52. Janes K. A., Wang C. C., Holmberg K. J., Cabral K., and Brugge J. S. (2010) Identifying single-cell molecular programs by stochastic profiling. Nat. Methods 7, 311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davidson M. M., Nesti C., Palenzuela L., Walker W. F., Hernandez E., Protas L., Hirano M., and Isaac N. D. (2005) Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 39, 133–147 [DOI] [PubMed] [Google Scholar]
- 54. Wang L., Brugge J. S., and Janes K. A. (2011) Intersection of FOXO- and RUNX1-mediated gene expression programs in single breast epithelial cells during morphogenesis and tumor progression. Proc. Natl. Acad. Sci. U.S.A. 108, E803-E812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hill M. M., and Hemmings B. A. (2002) Analysis of protein kinase B/Akt. Methods Enzymol. 345, 448–463 [DOI] [PubMed] [Google Scholar]
- 56. Janes K. A. (2015) An analysis of critical factors for quantitative immunoblotting. Sci. Signal. 8, ers2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wiederschain D., Wee S., Chen L., Loo A., Yang G., Huang A., Chen Y., Caponigro G., Yao Y. M., Lengauer C., Sellers W. R., and Benson J. D. (2009) Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504 [DOI] [PubMed] [Google Scholar]
- 58. Bajikar S. S., Fuchs C., Roller A., Theis F. J., and Janes K. A. (2014) Parameterizing cell-to-cell regulatory heterogeneities via stochastic transcriptional profiles. Proc. Natl. Acad. Sci. U.S.A. 111, E626-E635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goldenthal K. L., Hedman K., Chen J. W., August J. T., and Willingham M. C. (1985) Postfixation detergent treatment for immunofluorescence suppresses localization of some integral membrane proteins. J. Histochem. Cytochem. 33, 813–820 [DOI] [PubMed] [Google Scholar]
- 60. Meng T.-C., Fukada T., and Tonks N. K. (2002) Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 9, 387–399 [DOI] [PubMed] [Google Scholar]
- 61. Espina V., Edmiston K. H., Heiby M., Pierobon M., Sciro M., Merritt B., Banks S., Deng J., VanMeter A. J., Geho D. H., Pastore L., Sennesh J., Petricoin E. F., and Liotta L. A. (2008) A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Mol. Cell. Proteomics 7, 1998–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bretscher M. S., and Munro S. (1993) Cholesterol and the Golgi apparatus. Science 261, 1280–1281 [DOI] [PubMed] [Google Scholar]
- 63. Ribbeck K., and Gorlich D. (2001) Kinetic analysis of translocation through nuclear pore complexes. EMBO J. 20, 1320–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zheng C. F., and Guan K. L. (1994) Cytoplasmic localization of the mitogen-activated protein kinase activator MEK. J. Biol. Chem. 269, 19947–19952 [PubMed] [Google Scholar]
- 65. Baeuerle P. A., and Baltimore D. (1988) I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science 242, 540–546 [DOI] [PubMed] [Google Scholar]
- 66. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 67. Paine P. L., Moore L. C., and Horowitz S. B. (1975) Nuclear envelope permeability. Nature 254, 109–114 [DOI] [PubMed] [Google Scholar]
- 68. Masuda K., Shima H., Watanabe M., and Kikuchi K. (2001) MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J. Biol. Chem. 276, 39002–39011 [DOI] [PubMed] [Google Scholar]
- 69. Jagiello I., Van Eynde A., Vulsteke V., Beullens M., Boudrez A., Keppens S., Stalmans W., and Bollen M. (2000) Nuclear and subnuclear targeting sequences of the protein phosphatase-1 regulator NIPP1. J. Cell Sci. 113 Pt 21:3761–3768 [DOI] [PubMed] [Google Scholar]
- 70. Schwertassek U., Buckley D. A., Xu C. F., Lindsay A. J., McCaffrey M. W., Neubert T. A., and Tonks N. K. (2010) Myristoylation of the dual-specificity phosphatase c-JUN N-terminal kinase (JNK) stimulatory phosphatase 1 is necessary for its activation of JNK signaling and apoptosis. FEBS J. 277, 2463–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Van Den Maagdenberg A. M., Bachner D., Schepens J. T., Peters W., Fransen J. A., Wieringa B., and Hendriks W. J. (1999) The mouse Ptprr gene encodes two protein tyrosine phosphatases, PTP-SL and PTPBR7, that display distinct patterns of expression during neural development. Eur. J. Neurosci. 11, 3832–3844 [DOI] [PubMed] [Google Scholar]
- 72. Buschbeck M., Eickhoff J., Sommer M. N., and Ullrich A. (2002) Phosphotyrosine-specific phosphatase PTP-SL regulates the ERK5 signaling pathway. J. Biol. Chem. 277, 29503–29509 [DOI] [PubMed] [Google Scholar]
- 73. Hendriks W. J., Dilaver G., Noordman Y. E., Kremer B., and Fransen J. A. (2009) PTPRR protein tyrosine phosphatase isoforms and locomotion of vesicles and mice. Cerebellum 8, 80–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chirivi R. G., Dilaver G., van de Vorstenbosch R., Wanschers B., Schepens J., Croes H., Fransen J., and Hendriks W. (2004) Characterization of multiple transcripts and isoforms derived from the mouse protein tyrosine phosphatase gene Ptprr. Genes Cells 9, 919–933 [DOI] [PubMed] [Google Scholar]
- 75. Benson J. R., and Hare P. E. (1975) O-phthalaldehyde: fluorogenic detection of primary amines in the picomole range. Comparison with fluorescamine and ninhydrin. Proc. Natl. Acad.Sci. U. S. A. 72, 619–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schulze I. T., and Colowick S. P. (1969) The modification of yeast hexokinases by proteases and its relationship to the dissociation of hexokinase into subunits. J. Biol. Chem. 244, 2306–2316 [PubMed] [Google Scholar]
- 77. Vambutas V., Kaplan D. R., Sells M. A., and Chernoff J. (1995) Nerve growth factor stimulates tyrosine phosphorylation and activation of Src homology-containing protein-tyrosine phosphatase 1 in PC12 cells. J. Biol. Chem. 270, 25629–25633 [DOI] [PubMed] [Google Scholar]
- 78. Resink T. J., Hemmings B. A., Tung H. Y. L., and Cohen P. (1983) Characterisation of a Reconstituted Mg-ATP-Dependent Protein Phosphatase. Eur. J. Biochem. 133, 455–461 [DOI] [PubMed] [Google Scholar]
- 79. Gordon J. A. (1991) Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 201, 477–482 [DOI] [PubMed] [Google Scholar]
- 80. Crans D. C., Smee J. J., Gaidamauskas E., and Yang L. (2004) The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds. Chem. Rev. 104, 849–902 [DOI] [PubMed] [Google Scholar]
- 81. Swingle M., Ni L., and Honkanen R. E. (2007) Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol. Biol. 365, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. MacKintosh C., Beattie K. A., Klumpp S., Cohen P., and Codd G. A. (1990) Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 264, 187–192 [DOI] [PubMed] [Google Scholar]
- 83. Zhou B., and Zhang Z.-Y. (2002) The activity of the extracellular signal-regulated kinase 2 is regulated by differential phosphorylation in the activation loop. J. Biol. Chem. 277, 13889–13899 [DOI] [PubMed] [Google Scholar]
- 84. Askari N., Beenstock J., Livnah O., and Engelberg D. (2009) p38alpha is active in vitro and in vivo when monophosphorylated at threonine 180. Biochemistry 48, 2497–2504 [DOI] [PubMed] [Google Scholar]
- 85. Sun H., Charles C. H., Lau L. F., and Tonks N. K. (1993) MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 75, 487–493 [DOI] [PubMed] [Google Scholar]
- 86. Kucharska A., Rushworth L. K., Staples C., Morrice N. A., and Keyse S. M. (2009) Regulation of the inducible nuclear dual-specificity phosphatase DUSP5 by ERK MAPK. Cell. Signal. 21, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 87. Park J. E., Lee J. A., Park S. G., Lee D. H., Kim S. J., Kim H. J., Uchida C., Uchida T., Park B. C., and Cho S. (2012) A critical step for JNK activation: isomerization by the prolyl isomerase Pin1. Cell Death Differ. 19, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhou X. Z., Kops O., Werner A., Lu P. J., Shen M., Stoller G., Kullertz G., Stark M., Fischer G., and Lu K. P. (2000) Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell 6, 873–883 [DOI] [PubMed] [Google Scholar]
- 89. Lee T., Kim S. J., and Sumpio B. E. (2003) Role of PP2A in the regulation of p38 MAPK activation in bovine aortic endothelial cells exposed to cyclic strain. J. Cell. Physiol. 194, 349–355 [DOI] [PubMed] [Google Scholar]
- 90. Amit I., Citri A., Shay T., Lu Y., Katz M., Zhang F., Tarcic G., Siwak D., Lahad J., Jacob-Hirsch J., Amariglio N., Vaisman N., Segal E., Rechavi G., Alon U., Mills G. B., Domany E., and Yarden Y. (2007) A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 39, 503–512 [DOI] [PubMed] [Google Scholar]
- 91. Fu X. Y., and Zhang J. J. (1993) Transcription factor p91 interacts with the epidermal growth factor receptor and mediates activation of the c-fos gene promoter. Cell 74, 1135–1145 [DOI] [PubMed] [Google Scholar]
- 92. Leaman D. W., Pisharody S., Flickinger T. W., Commane M. A., Schlessinger J., Kerr I. M., Levy D. E., and Stark G. R. (1996) Roles of JAKs in activation of STATs and stimulation of c-fos gene expression by epidermal growth factor. Mol. Cell. Biol. 16, 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dorner A. A., Wegmann F., Butz S., Wolburg-Buchholz K., Wolburg H., Mack A., Nasdala I., August B., Westermann J., Rathjen F. G., and Vestweber D. (2005) Coxsackievirus-adenovirus receptor (CAR) is essential for early embryonic cardiac development. J. Cell Sci. 118, 3509–3521 [DOI] [PubMed] [Google Scholar]
- 94. Noutsias M., Fechner H., de Jonge H., Wang X., Dekkers D., Houtsmuller A. B., Pauschinger M., Bergelson J., Warraich R., Yacoub M., Hetzer R., Lamers J., Schultheiss H. P., and Poller W. (2001) Human coxsackie-adenovirus receptor is colocalized with integrins alpha(v)beta(3) and alpha(v)beta(5) on the cardiomyocyte sarcolemma and upregulated in dilated cardiomyopathy: implications for cardiotropic viral infections. Circulation 104, 275–280 [DOI] [PubMed] [Google Scholar]
- 95. Crowell R. L., and Syverton J. T. (1961) The mammalian cell-virus relationship. VI. Sustained infection of HeLa cells by Coxsackie B3 virus and effect on superinfection. J. Exp. Med. 113, 419–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Carthy C. M., Granville D. J., Watson K. A., Anderson D. R., Wilson J. E., Yang D., Hunt D. W. C., and McManus B. M. (1998) Caspase Activation and Specific Cleavage of Substrates after Coxsackievirus B3-Induced Cytopathic Effect in HeLa Cells. J. Virol. 72, 7669–7675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chow L. H., Gauntt C. J., and McManus B. M. (1991) Differential effects of myocarditic variants of Coxsackievirus B3 in inbred mice. A pathologic characterization of heart tissue damage. Lab. Invest. 64, 55–64 [PubMed] [Google Scholar]
- 98. Kühl U., Pauschinger M., Schwimmbeck P. L., Seeberg B., Lober C., Noutsias M., Poller W., and Schultheiss H.-P. (2003) Interferon-β treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 107, 2793–2798 [DOI] [PubMed] [Google Scholar]
- 99. Hahn W. C., Dessain S. K., Brooks M. W., King J. E., Elenbaas B., Sabatini D. M., DeCaprio J. A., and Weinberg R. A. (2002) Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol. 22, 2111–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kashimura T., Kodama M., Hotta Y., Hosoya J., Yoshida K., Ozawa T., Watanabe R., Okura Y., Kato K., Hanawa H., Kuwano R., and Aizawa Y. (2003) Spatiotemporal changes of coxsackievirus and adenovirus receptor in rat hearts during postnatal development and in cultured cardiomyocytes of neonatal rat. Virchows Arch. 444, 283–292 [DOI] [PubMed] [Google Scholar]
- 101. Lamphear B. J., Yan R., Yang F., Waters D., Liebig H. D., Klump H., Kuechler E., Skern T., and Rhoads R. E. (1993) Mapping the cleavage site in protein synthesis initiation factor eIF-4 gamma of the 2A proteases from human Coxsackievirus and rhinovirus. J. Biol. Chem. 268, 19200–19203 [PubMed] [Google Scholar]
- 102. Saraste A., Arola A., Vuorinen T., Kytö V., Kallajoki M., Pulkki K., Voipio-Pulkki L.-M., and Hyypiä T. (2003) Cardiomyocyte apoptosis in experimental coxsackievirus B3 myocarditis. Cardiovasc. Pathol. 12, 255–262 [DOI] [PubMed] [Google Scholar]
- 103. Mukherjee A., Morosky S. A., Delorme-Axford E., Dybdahl-Sissoko N., Oberste M. S., Wang T., and Coyne C. B. (2011) The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 7, e1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Heim A., Canu A., Kirschner P., Simon T., Mall G., Hofschneider P. H., and Kandolf R. (1992) Synergistic interaction of interferon-beta and interferon-gamma in coxsackievirus B3-infected carrier cultures of human myocardial fibroblasts. J. Infect. Dis. 166, 958–965 [DOI] [PubMed] [Google Scholar]
- 105. Schoggins J. W., Wilson S. J., Panis M., Murphy M. Y., Jones C. T., Bieniasz P., and Rice C. M. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Najarro P., Traktman P., and Lewis J. A. (2001) Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J. Virol. 75, 3185–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. David M., Petricoin E. 3rd, Benjamin C., Pine R., Weber M. J., and Larner A. C. (1995) Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science 269, 1721–1723 [DOI] [PubMed] [Google Scholar]
- 108. Kotlyarov A., Neininger A., Schubert C., Eckert R., Birchmeier C., Volk H. D., and Gaestel M. (1999) MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat. Cell Biol. 1, 94–97 [DOI] [PubMed] [Google Scholar]
- 109. Winzen R., Kracht M., Ritter B., Wilhelm A., Chen C. Y., Shyu A. B., Muller M., Gaestel M., Resch K., and Holtmann H. (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18, 4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Valledor A. F., Arpa L., Sanchez-Tillo E., Comalada M., Casals C., Xaus J., Caelles C., Lloberas J., and Celada A. (2008) IFN-{gamma}-mediated inhibition of MAPK phosphatase expression results in prolonged MAPK activity in response to M-CSF and inhibition of proliferation. Blood 112, 3274–3282 [DOI] [PubMed] [Google Scholar]
- 111. Kim S. M., Park J. H., Chung S. K., Kim J. Y., Hwang H. Y., Chung K. C., Jo I., Park S. I., and Nam J. H. (2004) Coxsackievirus B3 infection induces cyr61 activation via JNK to mediate cell death. J. Virol. 78, 13479–13488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bain J., McLauchlan H., Elliott M., and Cohen P. (2003) The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhang T., Inesta-Vaquera F., Niepel M., Zhang J., Ficarro S. B., Machleidt T., Xie T., Marto J. A., Kim N., Sim T., Laughlin J. D., Park H., LoGrasso P. V., Patricelli M., Nomanbhoy T. K., Sorger P. K., Alessi D. R., and Gray N. S. (2012) Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 19, 140–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Goentoro L., and Kirschner M. W. (2009) Evidence that fold-change, and not absolute level, of beta-catenin dictates Wnt signaling. Mol. Cell 36, 872–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Janes K. A., Reinhardt H. C., and Yaffe M. B. (2008) Cytokine-induced signaling networks prioritize dynamic range over signal strength. Cell 135, 343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lee R. E., Walker S. R., Savery K., Frank D. A., and Gaudet S. (2014) Fold change of nuclear NF-kappaB determines TNF-induced transcription in single cells. Mol. Cell 53, 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nakakuki T., Birtwistle M. R., Saeki Y., Yumoto N., Ide K., Nagashima T., Brusch L., Ogunnaike B. A., Okada-Hatakeyama M., and Kholodenko B. N. (2010) Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell 141, 884–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Swameye I., Muller T. G., Timmer J., Sandra O., and Klingmuller U. (2003) Identification of nucleocytoplasmic cycling as a remote sensor in cellular signaling by databased modeling. Proc. Natl. Acad. Sci. U.S.A. 100, 1028–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Schmierer B., Tournier A. L., Bates P. A., and Hill C. S. (2008) Mathematical modeling identifies Smad nucleocytoplasmic shuttling as a dynamic signal-interpreting system. Proc. Natl. Acad. Sci. U.S.A. 105, 6608–6613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chitforoushzadeh Z., Ye Z., Sheng Z., LaRue S., Fry R. C., Lauffenburger D. A., and Janes K. A. (2016) TNF-insulin crosstalk at the transcription factor GATA6 is revealed by a model that links signaling and transcriptomic data tensors. Sci Signal 9, era59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Albeck J. G., MacBeath G., White F. M., Sorger P. K., Lauffenburger D. A., and Gaudet S. (2006) Collecting and organizing systematic sets of protein data. Nat. Rev. Mol. Cell Biol. 7, 803–812 [DOI] [PubMed] [Google Scholar]
- 122. Liu C.-W., Li X., Thompson D., Wooding K., Chang T-l Tang Z, Yu H., Thomas P. J., and DeMartino G. N. (2006) ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol. Cell 24, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cruz L. A. B., Hebly M., Duong G.-H., Wahl S. A., Pronk J. T., Heijnen J. J., Daran-Lapujade P., and van Gulik W. M. (2012) Similar temperature dependencies of glycolytic enzymes: an evolutionary adaptation to temperature dynamics? BMC Syst. Biol. 6, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Andreassen P. R., Lacroix F. B., Villa-Moruzzi E., and Margolis R. L. (1998) Differential subcellular localization of protein phosphatase-1 alpha, gamma1, and delta isoforms during both interphase and mitosis in mammalian cells. J. Cell Biol. 141, 1207–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bordería A. V., Isakov O., Moratorio G., Henningsson R., Agüera-González S., Organtini L., Gnädig N. F., Blanc H., Alcover A., Hafenstein S., Fontes M., Shomron N., and Vignuzzi M. (2015) Group Selection and Contribution of Minority Variants during Virus Adaptation Determines Virus Fitness and Phenotype. PLoS Pathog. 11, e1004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhang Q.-M., Song W.-Q., Li Y.-J., Qian J., Zhai A.-X., Wu J., Li A.-M., He J.-M., Zhao J.-Y., Yu X., Wei L.-L., and Zhang F.-M. (2012) Over-expression of mitochondrial antiviral signaling protein inhibits coxsackievirus B3 infection by enhancing type-I interferons production. Virol. J. 9, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Janes K. A., Albeck J. G., Peng L. X., Sorger P. K., Lauffenburger D. A., and Yaffe M. B. (2003) A high-throughput quantitative multiplex kinase assay for monitoring information flow in signaling networks: application to sepsis-apoptosis. Mol. Cell. Proteomics 2, 463–473 [DOI] [PubMed] [Google Scholar]
- 128. Gaudet S., Janes K. A., Albeck J. G., Pace E. A., Lauffenburger D. A., and Sorger P. K. (2005) A Compendium of Signals and Responses Triggered by Prodeath and Prosurvival Cytokines. Mol. Cell. Proteomics 4, 1569–1590 [DOI] [PubMed] [Google Scholar]
- 129. Jadwin J. A., Oh D., Curran T. G., Ogiue-Ikeda M., Jia L., White F. M., Machida K., Yu J., and Mayer B. J. (2016) Time-resolved multimodal analysis of Src Homology 2 (SH2) domain binding in signaling by receptor tyrosine kinases. Elife 5, e11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Blom N., Hansen J., Blaas D., and Brunak S. (1996) Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci. 5, 2203–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Jullien P., Bougeret C., Camoin L., Bodeus M., Durand H., Disanto J. P., Fischer S., and Benarous R. (1994) Tyr394 and Tyr505 are autophosphorylated in recombinant Lck protein-tyrosine kinase expressed in Escherichia coli. Eur. J. Biochem. 224, 589–596 [DOI] [PubMed] [Google Scholar]
- 132. Seeliger M. A., Young M., Henderson M. N., Pellicena P., King D. S., Falick A. M., and Kuriyan J. (2005) High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 14, 3135–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bozym R. A., Morosky S. A., Kim K. S., Cherry S., and Coyne C. B. (2010) Release of intracellular calcium stores facilitates coxsackievirus entry into polarized endothelial cells. PLoS Pathog. 6, e1001135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Huber S. A., and Rincon M. (2008) Coxsackievirus B3 induction of NFAT: requirement for myocarditis susceptibility. Virology 381, 155–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Esfandiarei M., and McManus B. M. (2008) Molecular biology and pathogenesis of viral myocarditis. Annu. Rev. Pathol. 3, 127–155 [DOI] [PubMed] [Google Scholar]
- 136. Qin M., Escuadro B., Dohadwala M., Sharma S., and Batra R. K. (2004) A novel role for the coxsackie adenovirus receptor in mediating tumor formation by lung cancer cells. Cancer Res. 64, 6377–6380 [DOI] [PubMed] [Google Scholar]
- 137. Miyamoto S., Inoue H., Nakamura T., Yamada M., Sakamoto C., Urata Y., Okazaki T., Marumoto T., Takahashi A., Takayama K., Nakanishi Y., Shimizu H., and Tani K. (2012) Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 72, 2609–2621 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.