

Abstract
Human cytomegalovirus (HCMV) is a significant cause of disease in immune-compromised adults and immune naïve newborns. No vaccine exists to prevent HCMV infection, and current antiviral therapies have toxic side effects that limit the duration and intensity of their use. There is thus an urgent need for new strategies to treat HCMV infection. Repurposing existing drugs as antivirals is an attractive approach to limit the time and cost of new antiviral drug development. Virus-induced changes in infected cells are often driven by changes in cellular kinase activity, which led us to hypothesize that defining the complement of kinases (the kinome), whose abundance or expression is altered during infection would identify existing kinase inhibitors that could be repurposed as new antivirals. To this end, we applied a kinase capture technique, multiplexed kinase inhibitor bead-mass spectrometry (MIB-MS) kinome, to quantitatively measure perturbations in >240 cellular kinases simultaneously in cells infected with a laboratory-adapted (AD169) or clinical (TB40E) HCMV strain. MIB-MS profiling identified time-dependent increases and decreases in MIB binding of multiple kinases including cell cycle kinases, receptor tyrosine kinases, and mitotic kinases. Based on the kinome data, we tested the antiviral effects of kinase inhibitors and other compounds, several of which are in clinical use or development. Using a novel flow cytometry-based assay and a fluorescent reporter virus we identified three compounds that inhibited HCMV replication with IC50 values of <1 μm, and at doses that were not toxic to uninfected cells. The most potent inhibitor of HCMV replication was OTSSP167 (IC50 <1.2 nm), a MELK inhibitor, blocked HCMV early gene expression and viral DNA accumulation, resulting in a >3 log decrease in virus replication. These results show the utility of MIB-MS kinome profiling for identifying existing kinase inhibitors that can potentially be repurposed as novel antiviral drugs.
Developing a new antiviral drug can take over a decade and cost more than a billion dollars before approval for use in patients (1). Even then, the majority of drugs in development will not meet the criteria for Food and Drug Administration (FDA) approval. This arduous development process delays new treatments from reaching the clinic and greatly increases healthcare costs. Repurposing of existing drugs for use as antivirals provides an alternative to the traditional drug development process and leverages the fact that viruses manipulate many of the same cellular pathways dysregulated in other diseases states. Drugs targeting these mutual signaling events may have unintended uses as novel antiviral therapies. There are currently thousands of FDA-approved drugs whose effect on virus replication has not been examined. As these drugs have already been tested for safety and bioavailability in humans, they could be rapidly repurposed for clinical use (2). Thus repurposing of FDA-approved drugs as antivirals is a rapid, cost-effective means to identify new treatments for viral infections.
Human cytomegalovirus (HCMV)1 is a pervasive public health issue (3). Primary infection during pregnancy is the leading cause of congenital birth defects, and reactivation of latent infection during immunosuppression can lead to significant morbidity and mortality (4). The few drugs available to treat HCMV infection are associated with severe side effects, and no vaccines for HCMV currently exist (5). In addition, the emergence of drug-resistant HCMV strains has become increasingly common (6). Thus new antiviral drugs are greatly needed to curtail HCMV disease.
The extensive manipulation of cellular signaling pathways by HCMV suggests that drug repurposing may be an especially useful approach to identify new antiviral drugs. HCMV has a prolonged replication cycle that is tightly integrated into the state of the infected cell. Thus, HCMV actively manipulates a multitude of cellular signaling pathways to facilitate virus replication, including inhibition of cellular intrinsic defenses and activation of pathways that control protein synthesis and metabolism (7–14). These changes generate a cellular environment conducive for virus replication and serve as potential targets to limit HCMV disease. Recent studies have taken advantage of high throughput screening opportunities to search for protein kinase inhibitors (15, 16) or other compounds that may block HCMV replication (17).
Manipulation of host kinase activity accounts for many of the changes in cellular signaling observed during HCMV infection. HCMV inhibits several cellular kinases critical for the innate immune response such as protein kinase R, which potently suppresses HCMV replication when activated (13). The HCMV TRS1 and IRS1 proteins antagonize protein kinase R activation, ensuring continued synthesis of viral proteins and limited expression of interferon-dependent genes (18). Conversely, infection also activates cellular kinases that drive signaling pathways that promote virus replication. Specifically, HCMV activates the AMPK and mTOR kinases to increase glycolysis and fatty acid synthesis to generate anabolic metabolites required for the production of infectious virus (19–22). Inhibiting AMPK or mTOR activity dramatically decreases virus replication (21, 23). Thus, the coordinated activation and inhibition of cellular kinases is a critical aspect of HCMV replication.
Dysregulated kinase activity underlies multiple disease states such as cancer, autoimmunity and diabetes. As a result, kinase inhibitors are a promising class of drugs for treatment of a wide range of diseases, and could potentially be repurposed for use as HCMV antivirals (24, 25). For instance, multiple cancer cell types show increased mTOR activity and an ATP-competitive mTOR kinase inhibitor Torin1 was developed as an effective cancer treatment. Based on the increase in mTOR activity during HCMV infection, we previously found that a specific mTOR inhibitor, Torin1, inhibited lytic replication of representative alpha-, beta-, and gammaherpesvirus (23). Further, transplant recipients treated with the mTOR inhibitor rapamycin showed decreased incidence and severity of HCMV disease (26). These examples highlight the clinical utility of repurposing existing kinase inhibitors targeting HCMV-induced kinases as novel antiviral therapeutics.
Although the above examples demonstrate the importance of a small subset of cellular kinases in HCMV replication, the full complement of virus-induced changes to the host kinome are unknown. The sheer number of human kinases (∼500) presents a significant hurdle to measuring each kinase during infection using standard approaches, particularly those where antibodies or other reagents are unavailable. Kinome profiling was developed to facilitate the quantitative measurement of changes in hundreds of kinases from a single sample (27). Multiplexed kinase inhibitor beads coupled with mass spectrometry (MIB-MS), is an adaption of the original KinoBead method described by Daub, Kuster and others (28–33). Using immobilized pan kinase inhibitors for kinase capture, MIB-MS can be used to quantify differences in kinase expression/abundance and in some instances, kinase activity between samples (34–36). Similar examples of immobilized kinase inhibitor capture have proven the utility of this methodology to study the kinome (For review see (29)). Coupled with quantitative mass spectrometry (MS), the difference in the amount of captured kinase from each sample provides a global and unbiased view of kinome perturbations between samples. Additionally, methods such as KinActiv (25), and targeted MRM-based approaches (38) can be applied to profile changes in the activation state of the kinome.
Our research group and others have applied MIB-MS kinome profiling to define kinome adaptations to select kinase inhibitors, to acquired drug resistance and to epigenetic inhibitors (27, 35, 39, 40). MIB-MS proteomics has also been used to characterize the kinome of model organisms (41). In infectious disease, kinome profiling provided an in-depth picture of how Ebola virus infection altered liver cell signaling networks, and identified new host targets for novel antiviral drugs (42, 43). These examples demonstrate the utility of kinome profiling for identifying changes in kinase activity associated with multiple disease states.
The objective of this study was to use MIB-MS kinome profiling to identify temporal alterations in the host kinome induced by HCMV infection. Our ultimate goal was to elucidate virus-induced changes to the host kinome that could be targeted with existing kinase inhibitors to limit HCMV replication. Using two different HCMV strains, we compared the abundance/activity of >240 kinases in uninfected and infected cells over a time course of infection. Both lab-adapted AD169 strain and the TB40/E clinical strain induced remarkably consistent kinome changes, which were predicted to affect multiple signaling pathways and a wide variety of cellular processes. Our MIB-MS results directed our efforts to test a number of kinase inhibitors for their antiviral effects. Using a simple assay to quantify the effects of these inhibitors on HCMV replication, we identified three kinase inhibitors currently in clinical development or use that significantly decreased HCMV replication. One such compound, OTSSP167, potently inhibited HCMV replication at nanomolar concentrations not toxic to uninfected cells. These data demonstrate the utility of MIB-MS kinome profiling for defining novel virus-induced kinome changes and for identifying host pathways that can be targeted by existing or emerging drugs to limit virus replication.
MATERIALS AND METHODS
Cells and Viruses
Primary human MRC-5 fibroblasts were obtained from the ATCC and cultured in DMEM containing 10% fetal bovine serum and penicillin/streptomycin. In all experiments, cells were used between passage 7 and 21. The HCMV AD169inGFP strain, which contains a GFP expression cassette inserted in the UL21.5 locus (44), was used as the wild type HCMV strain. The pp28-GFP virus has been described previously (45). This virus expresses the HCMV pp28 protein as a GFP fusion protein from its native location in the HCMV genome under the control of its endogenous promoter, and replicates with similar kinetics and efficiency as wild type HCMV. Unless otherwise noted. Infections were performed at a multiplicity of infection (MOI) of one. Cell free HCMV was quantified using the TCID50 method on primary human fibroblasts as previously described (46).
For TGFβ treatment, MRC-5 fibroblasts were serum-starved for 24 h. Cells were infected with HCMV AD169inGFP at an MOI of 3. Forty-eight hours after infection cells were treated with 4 μg of human TGF-β1 (Cell Signaling Technology, Danver, MA; #5154) for 4 h prior to harvest.
MIB-MS Kinome Profiling
Cell pellets were lysed in MIB-MS lysis buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 1 mm EDTA, 1 mm EGTA, 10 mm NaF, 2.5 mm NaV04 plus Protease Inhibitor Mixture (Roche, Basel, Switzerland) and Phosphatase Inhibitor Mixture 2 (Sigma, St. Louis, MO; # P5726) and 3 (Sigma P0044). Samples were sonicated and clarified by centrifugation at 14,000 × g and supernatants filtered through a 0.2 μm filter (Corning, Roche, Basel, Switzerland; # 431219). Protein amounts were quantified by Bradford protein assay and the concentration of protein was normalized across samples prior to adjusting salt concentration to 1 m NaCl. MIB-MS columns composed of 4 immobilized kinase inhibitors (PP58, Purvalanol B, VI16832, and UNC21474), were prepared by mixing equal proportions of immobilized kinase inhibitors in a final volume of 200 μl settled beads. Columns were equilibrated with MIB-MS high salt buffer (50 mm HEPES, pH 7.5, 1 m NaCl, 0.5% Triton X-100, 1 mm EDTA, and 1 mm EGTA) prior to passing samples over MIBs columns by gravity flow. MIB columns were sequentially washed with MIB-MS high salt buffer, MIB-MS low salt buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 1 mm EDTA, and 1 mm EGTA), and SDS wash buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 0.1% SDS, 1 mm EDTA, and 1 mm EGTA). Proteins were eluted from MIB columns by boiling for 15 min in elution buffer (100 mm Tris-HCl, pH 6.8, 0.5% SDS, 1% β-mercaptoethanol) twice. dl-DTT was added to a final concentration of 5 mm and samples were incubated at 60 °C for 25 min. Samples were cooled to room temperature on ice and iodoacetamide was added to a final concentration of 20 mm. Samples were incubated for 30 min at room temperature in the dark. dl-DTT was added to bring the final concentration to 10 mm and samples were incubated at room temperature for 5 mins in the dark. Samples were then concentrated to a final volume of ∼100 μl in 10K Amicon Ultra centrifugal concentrators and proteins purified by methanol chloroform extraction. Samples were re-suspended in 50 mm HEPES, pH 8.0, and digested with sequencing grade porcine trypsin (Promega, Madison, WI) overnight at 37 °C. Samples were extracted with ethyl acetate 3 times to remove residual detergent and desalted using Pierce C-18 spin columns according to the manufacturer's protocol.
LC/MS/MS Analysis
The peptide samples were analyzed by LC/MS/MS using an Easy nLC 1000 coupled to a QExactive HF mass spectrometer (Thermo Scientific, Waltham, MA). Samples were injected onto an Easy Spray PepMap C18 column (75 μm id × 25 cm, 2 μm particle size) (Thermo Scientific) and separated over a 2 h method. The gradient for separation consisted of 5–32% mobile phase B at a 250 nl/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in ACN. The QExactive HF was operated in data-dependent mode where the 15 most intense precursors were selected for subsequent fragmentation. Resolution for the precursor scan (m/z 400–1600) was set to 120,000 with a target value of 3 × 106 ions. MS/MS scans resolution was set to 15,000 with a target value of 2 × 104 ions. The normalized collision energy was set to 27% for HCD. Peptide match was set to preferred, and precursors with unknown charge or a charge state of 1 and ≥ 8 were excluded.
Data Analysis
Raw data files were processed using MaxQuant version 1.5.3.17 and searched against the reviewed Uniprot human database (containing 20,203 entries, downloaded Aug 2015), appended with a Uniprot HCMV AD169 or TB40/E database and contaminants database, using Andromeda within MaxQuant. Enzyme specificity was set to trypsin, up to two missed cleavage sites were allowed, carbamidomethylation of C was set as a fixed modification and deamidation of N, Q, oxidation of M, phospho of S, T, Y, and acetyl of N terminus were set as variable modifications. The mass tolerance for precursor (main search) and product ions was set to 6 ppm and 20 ppm, respectively. A 1% FDR was used to filter all data. Match between runs was enabled, and a minimum of two unique peptides was required for label-free quantitation (LFQ) using the LFQ intensities. Data are available via ProteomeXchange (www.proteomexchange.org, project ID PXD005276) with identifier PXD005276.
Experimental Design and Statistical Rationale
Two biological replicates for each time point after infection with HCMV AD169 and two biological replicates for each time point after HCMV TB40/E infection (0, 24, 48, 72 h) were used for MIB-MS experiments, in sum providing four independent replicates for each time point. Assessment of individual MaxQuant analyses conducted for both strain and replicate showed consistent results between strains and replicates; therefore, the data sets from both strains and replicates were merged to provide a more robust analysis. We selected a set of 25 proteins in an unbiased manner by ranking them in terms of their variability across all samples from largest to smallest based upon median absolute deviation, a robust measure of variability. We then performed hierarchical clustering of the log transformed protein intensities from each of these 25 most variable proteins using 1 - the Spearman's correlation as the distance metric. The clustering profile indicates that samples cluster strongly together by time point rather than virus type, indicating similar temporal patterns of change.
Statistical testing based upon these samples was performed to identify those kinases whose patterns showed strong temporal changes in expression that were consistent across all replicates. Statistical analyses were performed in the R statistical programming language version 3.3.1. Biological duplicates from each strain were searched separately in MaxQuant, and LFQ values from all four MaxQuant runs were merged into a single data set consisting of n = 16 samples (four time points per run). To reduce our data set to a subset of consistently observed kinases, we removed those kinases that were found to have less than or equal to two peptides in all four MaxQuant runs, in addition to kinases that had greater than 35% missing values (LFQ value of 0) across samples in the merged data set.
To determine which kinases showed consistent temporal trends in expression, we utilized linear mixed models (47,48) to model the log LFQ for each particular kinase with respect to time, accounting for correlation between samples within each run as well potential inter-run heterogeneity in kinase trends. Specifically, we utilized a linear mixed model with a run-level random intercept and a run-level random slope with respect to time, treating time as a continuous variable. Similar to previous studies, Log LFQ values were assumed to be approximately normally distributed (49). Mock samples were treated as a zero hour time point. p values for the significance of the estimated time trend coefficient were computed using the summary function from the lmerTest (50) package on the fitted model, testing the null hypothesis that the slope of the time trend is equal to zero. p values from each kinase were corrected for multiple testing using the Benjamini-Hochberg procedure (51) kinases with adjusted p values less than 0.05 were considered to have a significant trend in expression over time. Hierarchical clustering was utilized to display changes in observed log LFQ intensity with respect to the set of kinases showing such significant time trend (1 - Spearman Correlation distance metric). Pairwise changes between a single time point and the mock sample were similarly computed using linear mixed models, testing the null hypothesis that the difference in log LFQ between a given time point and the mock sample is 0, where within-run correlation is accounted for with a run-level random intercept. Raw and adjusted p values were reported for each kinase, as well as model estimates for time (log LFQ scale) for the trend analysis or difference in log LFQ for the pairwise analyses. Prior to statistical analysis, missing kinases were imputed by first calculating the covariance matrix between samples using only pairwise complete observations. Then, for each kinase with missing values, a single sample was randomly drawn from a multivariate normal distribution with the calculated covariance matrix and mean predicted from the trend between time and the non-missing LFQ values for that kinase (linear mixed model). Missing values in the given kinase were replaced with the corresponding elements from this random sample.
Analysis of Protein Expression
Analysis of proteins levels by Western blot was performed essentially as described previously (52). Briefly, infected cells were pelleted and stored at −80 °C until use. Cell lysates were prepared by resuspending cell pellets in RIPA buffer (50 mm Tris-HCl pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, and complete protease inhibitor (Roche)) and incubation on ice for 15 min. Cellular debris was removed by centrifugation at 21,000 × g for 10 min. Total protein concentration was determined by Bradford assay. Equal amounts of protein were separated on SDS-PAGE gels and transferred to nitrocellulose or PVDF membranes. Membranes were blocked in Tris-buffered saline containing 0.1% Tween (TBS-T) and 5% nonfat milk or 5% bovine serum albumin (BSA) for phosphoblots for 1 h at room temperature. Blots were then incubated overnight with the indicated antibodies. Rabbit polyclonal antibodies were diluted in TBS-T containing 5% BSA and incubated with membranes at 4 °C overnight. Mouse monoclonal antibodies were diluted in TBS-T containing 1% BSA and incubated with membranes for at least one hour at room temperature. After washing with TBS-T, membranes were incubated with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies and visualized by enhanced chemiluminesence (ECL) using the BioRad (Hercules, CA) ChemiDoc system. The following commercial antibodies were used in this study: MELK (#2274, Cell Signaling Technologies); eIF4B (#3592, Cell Signaling Technologies) phospho-eIF4B Ser406; (#5399, Cell Signaling Technologies); β-actin (sc-47778, Santa Cruz Biotechnology); ICP36 (UL44; 10D8, Virusys, Taneytown, MD), tubulin (DM1A, Sigma), TGFBR2 (#A304–414A, Bethyl, Laboratories, Montgomery, TX), Ephrin A2 (#6997, Cell Signaling Technology), Ephrin B4 (#14960, Cell Signaling Technology), phosphorylated Smad 2/3 (#8828, Cell Signaling Technology), total Smad 2/3 (#3122, Cell Signaling Technology), IE1 (84), TRS1 (60) and pp28 (65) were kindly provided by Dr. Tom Shenk (Princeton University).
Quantification of Viral DNA
Viral DNA was quantified by real time PCR (qPCR) previously described (56). Briefly, MRC5 fibroblasts were infected with HCMV at an MOI of 0.05 and harvested by scraping. The cell pellet was resuspended in lysis buffer (400 nm NaCl, 10 mm Tris-HCl pH 8.0 and 10 mm EDTA). Proteinase K (40 μg/ml) and SDS (final concentration 0.16%) was then added and the cells were incubated at 37 °C overnight. The lysates were extracted with phenol followed by incubation with 40 μg/ml RNase A at 37 °C for 1 h. The lysate was again extracted with phenol followed by a chloroform extraction. The DNA was precipitated with ethanol and resuspended in 10 mm Tris-HCl, pH 8.0. Quantitative PCR was performed using primers specific for the HCMV UL99 gene (UL99F 5′-GTGTCCCATTCCCGACTCG-3′, UL99R 5′-TTCACAACGTCCACCCACC-3′) and GAPDH (GAPDHF 5′-CTGTTGCTGTAGCCAAATTCGT, GAPDHR 5′-ACCCACTCCTCCACCTTTGAC-3′). Viral DNA abundance was determined using the absolute abundance method by comparison to a standard curve of AD169 BAC DNA ranging from 101 to 109 copies. GAPDH copy number was quantified using the same approach with a standard curve consisting of the same dilution series of the GAPDH amplicon. Viral DNA abundance in each sample was normalized to the abundance of GAPDH to control for variation in loading.
Quantification of mRNA
Viral or cellular mRNA levels were quantified by reverse transcriptase real time PCR (qRT-PCR) as described before (57). Briefly, cell pellets were resuspended in 1 ml of Trizol (Thermo Fisher Scientific). Total RNA was purified according to the manufacturer's specifications (with the exception of a >1 h incubation of isopropanol at −20 °C). Contaminating DNA was removed using the Turbo DNase kit according to the manufacturer directions (Thermo Fisher Scientific). One microgram of DNase-treated RNA was reverse transcribed using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative reverse transcriptase real time PCR (qRT-PCR) was performed using SYBR green master mix (Thermo Fisher Scientific) and the following primers: IE1F 5′-CAAGTGACCGAGGATTGCAA-3′, IE1R 5′-CACCATGTCCACTCGAACCTT-3′; UL44F 5′-GCGTGCAAGTCTCGACTAAGGAGC-3′, UL44R 5′-AAGTACTGTGCCTCTTAGTCGGGGG-3′; UL99F 5′-GTGTCCCATTCCCGACTCG-3′, UL99R 5′-TTCACAACGTCCACCCACC-3′; HO1F 5′-CGTGCAGAGAATTCTGAGTTC-3′, HO1R 5′-AGACGCTTTACGTAGTGCTG-3′). The abundance of each transcript was determined using the absolute abundance method and a standard curve consisting of 10-fold serial dilutions of the specific amplicon cloned into the pCRBlunt vector (Invitrogen, Carlsbad, CA). Serial dilutions from 108 to 101 were used for each gene, and the R2 value of each standard curve was > 0.98. The relative increase in HO1 mRNA levels after TGFβ treatment was determined using the ΔΔCt method, with uninfected, untreated cells serving as the reference sample.
Antiviral Drug Screen
MRC-5 fibroblasts were seeded into 96-well plates. At confluency, the media was replaced with serum-free DMEM for 24h. Cells were infected with HCMV-pp28GFP at an MOI of 1 for one hour, and drugs diluted in serum-free media were added to the cells at the time the inoculum was removed. Compounds were purchased from Selleckchem (Munich, DE), and included: Alisertib (S1133), PF 03814735 (S2725), LY2603618 (S2626), OTSSP167 (S7159), SBI-0206965 (S7885), TG003 (S7320), Tivatinib (S2753), MRT67307 (S7948), Saracatinib (S1006), Sotrastaurin (S2791), BMS-777607 (S1561), OTX015 (S7360), Dinaciclib (S2768). At 96 h after infection the cells were washed with PBS, trypsinized, and transferred to a round bottom plate. Cells were fixed in 4% paraformaldehyde for 10 min at room temperature, washed twice with PBS, and then resuspended in 200 μl of FACS buffer. A Guava EasyCyte HT flow cytometer (EMD Millipore) was used to determine the number of GFP positive cells. The GFP-positive gate was set so that less than 1% of uninfected cells crossed the threshold. Drug treated wells were normalized to vehicle treated wells for each appropriate dilution. IC50 values were calculated using GraphPad Prism software.
Cellular Toxicity Assay
Lactate dehydrogenase release was used to assess drug toxicity to uninfected cells as per the manufacturer's directions (Sigma). For the initial screen of kinase inhibitors, MRC-5 cells were serum starved overnight before addition of the drugs. Ninety-six hours after addition of the drugs, LDH activity in the supernatant was measured by absorbance at 450 nm and normalized to the activity in untreated media.
RESULTS
MIB-MS Kinome Profiling Identifies Host Signaling Pathways Altered by HCMV Infection
We utilized MIB/MS profiling to identify time-dependent changes in the human kinome throughout HCMV infection using the laboratory strain, AD169, and the clinical strain TB40/E (58). Our MIB-MS columns were composed of a four inhibitor (PP58, Purvalanol B, VI16832, and UNC21474) bead mix that was optimized for kinome capture as previously reported (27) (East, unpublished observations). A schematic of the kinome profiling workflow is shown in Fig. 1. Primary human fibroblasts were serum-starved prior to infection to decrease basal signaling and allow for better quantification of virus-induced changes in the kinome. Infected cell lysates were harvested at 24, 48, or 72 h after infection, and kinases were enriched from cell lysates with MIB columns as described above. The captured kinases were digested and identified by LC-MS/MS and the expression/abundance of each kinase relative to the uninfected control at each time point after infection was determined using label-free quantification (MaxQuant-see Methods).
Fig. 1.
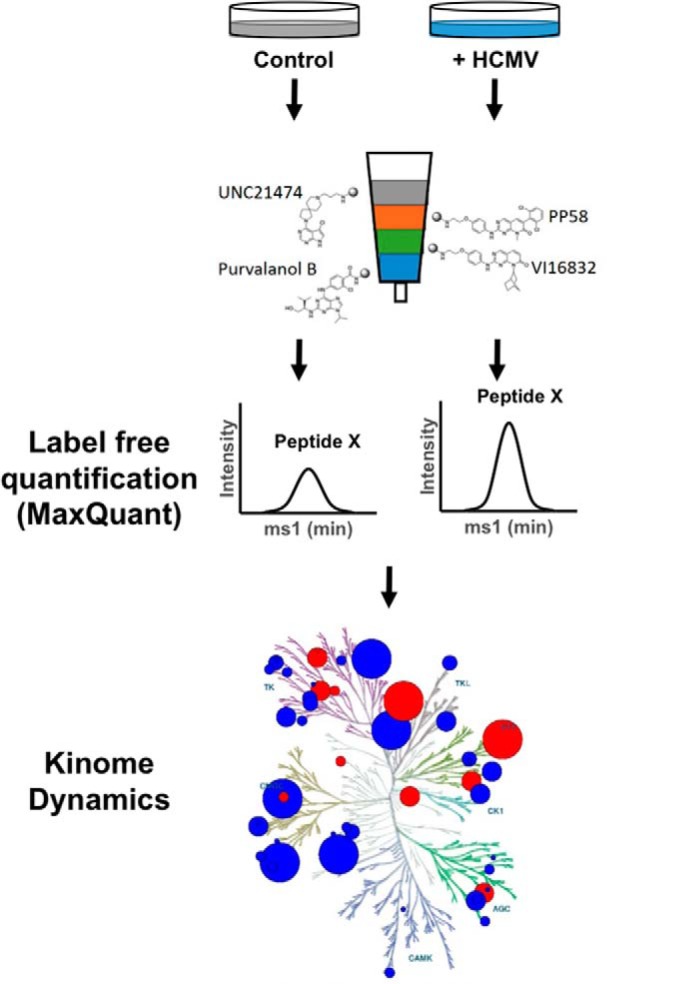
Cartoon showing workflow of kinome profiling of HCMV infected cells. Primary human fibroblasts were infected with HCMV at a multiplicity of 3, or left uninfected for 24, 48, or 72 h. Lysates were passed over multiplexed inhibitor beads to enrich for kinases. The recovered kinases were digested with trypsin, and the tryptic peptides were analyzed by LC/MS/MS. Label-free quantification was used to determine the abundance of each recovered kinase. Kinome dynamics resulting from HCMV were determined by comparing the abundance of each kinase in infected samples relative to uninfected control. *Kinome tree illustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
In addition to identifying changes in the kinome in response to HCMV infection, this approach allowed us to directly compare HCMV strains associated with clinical infections to their lab-adapted counterparts. Infection with either HCMV strain significantly altered the abundance/activity of multiple cellular kinases as identified by MIB-MS, and these changes were observed to be highly consistent between both strains. These strains were first analyzed individually, and the data sets from both strains combined to provide a more robust analysis. The full list kinases quantified during infection is shown in supplemental Tables S1 and S2. Two hundred forty-four kinases were present in all samples, and only these kinases were included in the subsequent analyses. To generate a high confidence list of kinases whose recovery with MIB-MS was changed by infection, we used linear mixed models (See Statistical Analysis in Methods) to identify kinases consistently altered at multiple times after infection. The graphs showing the linear mixed model analysis for each quantified kinase are shown in supplemental Fig. S1. Using this conservative analysis, we found 53 kinases that were more abundantly recovered after MIB-MS from infected lysates at all times after infection (Fig. 2A). Significant among these included the cyclin-dependent kinases (CDK 1, 7, 9, 10), receptor and non-receptor tyrosine kinases (INS, MET, MERTK, HCK, LYN), and lipid kinases (PIP4K2C, PIP4K2B, PIK3C3, PI4K2B, PIK3R4). We also identified 51 kinases that were consistently recovered at reduced levels from infected cell lysates (Fig. 2A, supplemental Table S3). This group included PDGFR (A/B), FGF (1/2), TGF-β (1/2), and other receptor tyrosine kinases (e.g. Ephrin RTKs).
Fig. 2.

Host kinome changes quantified over a time course of HCMV infection. A, Heat map showing the quantified kinases significantly altered during HCMV infection. Fibroblasts were infected with AD169 or TB40E at a multiplicity of one, and cellular kinases were identified and quantified by MIB-MS kinome profiling. The heat map shows the change in kinase quantification for kinases significantly altered at multiple times after infection. B, Graph showing the number of cellular kinases significantly changed at each time after HCMV infection as quantified by MIBS-MS as compared with the uninfected sample. C, Percentage of kinases (of 244 total) at each time point with a significant increase, decrease, or no change in recovery with MIB-MS after HCMV infection.
The graphs showing the fold change for each kinase during infection compared with uninfected cells are shown in supplemental Figs. S2 and S3. Some kinases were consistently altered at all times after infection (supplemental Table S3), whereas others were significantly changed only at discreet times after infection (supplemental Tables S4). Several of these kinase changes, such as increased AMPK and p70 S6 kinase (p70S6K), have been previously observed during HCMV infection (21, 22). At 24 h after infection, 64 kinases were significantly altered compared with uninfected cells (Fig. 2B). The percentage of kinases changed during infection at each time point is shown in Fig. 2C. Overall, more kinases were affected at 48 and 72 h, with 138 and 139 kinases showing quantitative differences in capture by MIB-MS, respectively. Together these data show that HCMV infection significantly changes the abundance/activity of multiple cellular kinases in a temporal manner.
To identify signaling pathways altered by HCMV infection, we performed bioinformatics analysis to identify signaling networks significantly altered in our kinome data. We found that infection increased or decreased signaling through multiple biological pathways and processes. Kinases that were detected in greater abundance by MIB-MS after infection were associated with multiple signaling pathways, including the AMPK, mTOR and ERK/MAPK signaling networks (supplemental Table S5). This data was consistent with previous studies showing that HCMV infection induces the AMPK, mTOR, and ERK kinases. Similarly, multiple pathways were negatively impacted by infection (supplemental Table S6), such as the NFkB and PDGFR pathways, in line with the previously reported inhibition of NFkB and PDGFR signaling pathways by HCMV. Thus MIB-MS kinome profiling accurately identified known HCMV-induced changes in cellular signaling pathways important for HCMV replication.
Novel Cellular Pathways Affected by HCMV Infection
Our kinome profiling data identified multiple signaling pathways affected by HCMV infection. For instance, the ephrin (Eph) receptor kinases are a large family of tyrosine kinases that play important roles in cell-to-cell communication, gene expression, and cell growth (59). HCMV infection significantly decreased the amount of EphA4, EphB2, EphB3, and EphB4 quantified by MIB-MS at 24 h after infection (Fig. 3A). In contrast, we found that EphA2 was slightly elevated at 24 h after infection and slowly decreased at 48 and 72 h. To determine if the changes in Eph receptor capture by MIBs were because of changes in Eph kinase activity or Eph receptor abundance, we measured the expression of EphA2 and EphB4 by Western blot analysis. Infected cells expressed less EphB4 protein (Fig. 3B) compared with uninfected controls, whereas EphA2 levels initially increased, before declining to levels seen in uninfected cells by 72 h after infection. The change in EphA2 and EphB4 protein levels detected by Western blots after infection closely matched the changes in EphA2 and EphB4 quantified by MIB-MS kinome profiling. Interestingly, the decrease in EphB4 protein abundance was not reflected in EphB4 transcript abundance (Fig. 3C), suggesting that post-transcriptional regulatory events limit EphB4 abundance in HCMV infected cells.
Fig. 3.

HCMV infection alters multiple novel signaling pathways. A, Fold change in the amount of ephrin (Eph) kinases recovered with MIBs over a time course of HCMV infection relative to uninfected cells. Primary human fibroblasts were infected with either AD169 or TB40E as in Fig. 1. The results are the average from all infections combined. B, MRC-5s were serum-starved for 24 h before infection with AD169 at a multiplicity of three. The levels of the Ephrin A2 (EPHA2), Ephrin B4 (EPHB4), and IE1 proteins were measured by Western blot over a time course of infection. The levels of tubulin were measured as a loading control. C, As in (B), levels of EphB4 mRNA levels were measured by qRT-PCR. D, Same as in (A). E, MRC-5s were infected with AD169 at a MOI of 3 for 48 h. Cells were then treated with 4 μg of TGF-β1 for 4 h. Levels of Smad2/3-P, Smad2, TGF-β receptor II, and pTRS1 were determined by Western blot. F, MRC-5s were infected as in (D). Cells were then treated with 4 μg of TGF-β1 for 24 h. Levels of HO1 mRNA (a Smad2/3-responsive transcript) were measured by qRT-PCR. G, Fold change in MELK kinase as in A. H, H, Western blot for MELK expression, eIF4B Ser406, total eIF4B and actin expression as in B. I, Western blot results for phosphorylation of the MELK substrate eIF4B (Ser406) and total eIF4B.
Interestingly, we observed a decrease in the amount of TGFβ receptor subunits TGFβRI and TGFβRII quantified by MIB-MS from infected cell lysates (Fig. 3D). Binding of TGFβ to the TGFβ receptor results in phosphorylation of SMAD family transcription factors (60). To determine if HCMV infection inhibited TGFβ receptor signaling, we measured SMAD phosphorylation in response to TGFβ treatment in mock and HCMV infected cells (Fig. 3E). As expected, TGFβ treatment induced SMAD phosphorylation in uninfected cells. However, no SMAD phosphorylation was observed in response to TGFβ treatment of HCMV infected cells, despite the maintained or slightly elevated levels of TGFβR2. In addition we found that HCMV infection prevented the TGFβ-dependent increase in the HO1 mRNA observed in uninfected cells (Fig. 3F). Taken together, these results suggest that HCMV infection inhibits TGFβ signaling.
We also observed an increase in the serine/threonine kinase MELK after infection. MELK is an AMPK family member involved in the regulation of replication stress and cell proliferative responses (61, 62). MELK activity increases in proportion to MELK expression (52), and MELK has been reported to phosphorylate multiple proteins involved in cell cycle, transcriptional and translational regulation (63–65). MIB-MS analysis identified increased MELK at 24–48 h, which decreased after 72 h of infection (Fig. 3G). To confirm the changes in MELK expression, we performed Western blots on lysates after infection (Fig. 3H). As determined by Western blotting, MELK protein levels followed the MIB-MS pattern, with the highest levels observed at 24–48 h compared with uninfected cells. Recently MELK was shown to phosphorylate eIF4B on Ser406 (64). Using a phosphospecific antibody to this site (Ser406), we found increased eIF4B Ser406 phosphorylation after infection that mirrored the changes in MELK and eIF4B protein expression (Fig. 3H). These data identify two previously unreported effects of HCMV infection on host signaling pathways, demonstrating the utility of MIB-MS kinome profiling for identifying cellular signaling pathways altered by HCMV infection.
Development of a Novel Assay to test HCMV Antiviral Efficacy
Based on our MIB-MS kinome profile data, we identified several drugs currently in clinical use or development that could be used to target HCMV-induced kinases. We therefore developed a rapid assay to assess the ability of these drugs as inhibitors of HCMV replication (Fig. 4A). Our assay utilizes a recombinant virus (pp28-GFP) expressing the HCMV pp28 protein fused to the green fluorescent protein (GFP) on its carboxyl terminus (45). The pp28-GFP fusion protein is expressed from its native location in the HCMV genome under the control of its endogenous promoter. Although pp28 expression is essential for HCMV replication, we previously showed that the pp28-GFP virus replicates with similar kinetics and to equivalent levels as wild type HCMV (45). As pp28 transcription requires prior viral DNA replication, inhibiting the HCMV lytic cycle at any step prior to viral DNA replication (e.g. entry, uncoating, immediate early and early gene expression, etc.), will result in decreased pp28-GFP expression.
Fig. 4.

Analysis of potential HCMV antivirals. A, Schematic of method. Fibroblasts were treated with infected with pp28-GFP at an MOI of 1 in a 96-well plate format. Cells were treated with seven concentrations of each drug ranging from 10 μm to 2.4 nm at the time of infection. Cells were harvested at 96 h after infection, and the GFP fluorescence of each well was determined using a Guava HTS flow cytometer. The change in GFP fluorescence at each dose of drug was used to calculate the 50% inhibitory concentration (IC50). B, Fibroblasts were infected as in A, and treated with a range of concentrations of PAA, a well-characterized inhibitor of viral DNA replication. The IC50 was determined as in A. The observed IC50 value and previously reported IC50 are shown. C, Graph showing the IC50 data for drugs that significantly inhibited pp28-GFP fluorescence. D, The dose of each drug that resulted in 50% cell death (LD50) was determined using the LDH release assay. Toxicity data is shown for drugs that inhibit pp28-GFP fluorescence. TG003 is included in graphs C and D as an example of a drug with no effect on pp28-GFP fluorescence or cell viability. The graphs show the mean and standard deviation of at least two experiments.
To test the applicability of this assay, we examined the effect of a known inhibitor of HCMV DNA replication, phosphonoacetic acid (PAA) (66, 67), on pp28-GFP expression. Primary human fibroblasts were infected with the pp28-GFP virus and treated with a range of concentrations of PAA at the time of infection. At 96 h after infection, pp28-GFP fluorescence was measured using a 96-well flow cytometer. The percent inhibition of pp28-GFP fluorescence at each concentration was used to calculate the 50% inhibitory concentration (IC50). PAA decreased the fluorescence of pp28-GFP infected cells, and the observed IC50 value of 26.8 μg/ml closely matched the previously reported IC50 of 20 μg/ml (Fig. 4B and (66)). These data confirm that the pp28-GFP expression assay closely recapitulates inhibition of HCMV replication as determined by traditional molecular techniques.
Inhibitors of Kinases Identified by MIB-MS Have Antiviral Activity
We next used the pp28-GFP assay to measure the potential antiviral activity of specific kinase inhibitors. The inhibitors were selected based on three criteria. First, we selected kinases increased in at least one time point after HCMV infection, as detected by MIB-MS. From this list, we chose kinase inhibitors that had been tested in humans or were currently in clinical trials. A few additional kinase inhibitors were selected based on preclinical data suggesting their potential entry into clinical trials. The list of the 13 kinase inhibitors tested are shown in Table I. These compounds were examined in the pp28-GFP assay for their ability to inhibit HCMV replication.
Table I. Kinase inhibitors tested for HCMV inhibition. List of kinase inhibitors and their target(s). The stage of clinical testing is listed for those drugs currently in clinical trials. Inhibition of HCMV replication (IC50 values) was determined using the pp28-GFP assay (N.D. = drug does not reach 50% inhibition at concentrations tested). Cellular toxicity (LD50 values) was determined by LDH assay in uninfected primary human fibroblasts.
| Drug name | Target | Clinical stage | IC50 | LD50 |
|---|---|---|---|---|
| OTSSP167 | MELK | Phase I/II | 5.4 nm | 98 nm |
| Dinaciclib | CDK1, 2, 5, 9 | Phase 3 | 20 nm | >10 μm |
| OTX015 | BRD2,3,4 | Phase 1 | 8.4 μm | >10 μm |
| LY2603618 | Chk1 | 10 μm | >10 μm | |
| Tivantinib | c-Met | Phase 3 | >10 μm | >10 μm |
| Alisertib | AURKA | Phase 3 | >10 μm | >10 μm |
| Saracatinib | c-Src, v-Abl, lyn | Phase 1/2 | >10 μm | >10 μm |
| BMS-777607 | c-Met, Axl, Ron, TYRO3 | Phase 1/2 | >10 μm | >10 μm |
| Sotrastaurin | PKC (pan) | Phase 1/2 | >10 μm | >10 μm |
| TG003 | CLK1,2,4 | >10 μm | >10 μm | |
| SBI-0206965 | ULK1/2 | >10 μm | >10 μm | |
| PF-03814735 | AURKA, AURKB Flt1, FAK, TrkA | Phase 1 | N.D. | 1.7 μm |
| UNC2025 | MERTK | N.D. | 1.2 μm | |
| MRT67307 | IKK(epsilon), TBK1 | N.D. | 1.1 μm |
Nine of these inhibitors had little or no effect on pp28-GFP expression, suggesting that their kinase targets are not critical for virus replication. However, four compounds significantly decreased pp28-GFP expression (Fig. 4C). The bromodomain and extraterminal (BET) proteins BRD2, BRD3, and BRD4 are known to bind kinase inhibitors (68) and were detected by MIB-MS after HCMV infection. Although not a kinase inhibitor, OTX015 binds and inhibits the BRD2, BRD3, and BRD4 proteins (69–71). Consistent with a role for BRD2 in HCMV replication, OTX015 inhibited HCMV replication with an IC50 of 8.5 μm, although some toxicity was observed in uninfected cells.
The Chk1 kinase plays a central role in the DNA damage response, which is induced by HCMV infection and required for efficient virus replication (72). Incubation with the Chk1 inhibitor LY2603618 inhibited HCMV replication with an IC50 of ∼10 μm. Dinaciclib, a CDK1,2,5,9 inhibitor in clinical testing for the treatment of solid tumors (73), also inhibited pp28-GFP expression with an IC50 of 20 nm with limited toxicity to uninfected cells (Fig. 4C, 4D, Table I). We confirmed the specificity of dinaciclib by showing that dinaciclib specifically prevented the binding of CDKs to MIB beads (supplemental Fig. S4, supplemental Tables S7 and S8).
MELK is believed to play a critical role in cell cycle regulation, and HCMV specifically modulates the cell cycle to support replication. OTSSP167 was discovered as a potent MELK inhibitor and was the first commercially available MELK inhibitor. Although the specificity of OTSSP167 is uncertain, it is currently in phase I/II clinical trials for the treatment of solid tumors and leukemias, is well tolerated and bioavailable in vivo (74). OTSSP167 decreased pp28-GFP fluorescence with an IC50 of < 5.4 nm which was >18 times lower than the toxic dose observed for fibroblasts (Fig. 4C, 4D, Table I). These results suggest that compounds that inhibit MELK, select CDKs, Chk1, and the BRD family of proteins may have potential as antiviral drugs. In addition, these data demonstrate the utility of MIB-MS kinome profiling to identify specific kinase inhibitors currently used in humans that could be repurposed to treat HCMV disease.
The OTSSP167 is a Potent Inhibitor of HCMV Replication
Dinaciclib and OTSSP167 exhibited IC50 concentrations significantly lower than the IC50 of the current frontline HCMV antiviral gancyclovir (approx. 1.7 μm (75). Based on the low IC50 value of OTSSP167, we further characterized the effect of OTSSP167 on the HCMV lytic cycle. We began by confirming the antiviral efficacy of OTSSP167 on the production of infectious wild type HCMV. Infected cells were treated with a range of OTSSP167 concentrations at the time of infection, and the abundance of cell-free HCMV in culture supernatants at 96 h after infection was measured by the TCID50 assay. OTSSP167 inhibited the production of infectious virus in a dose-dependent manner (Fig. 5A), further confirming that decreased pp28-GFP expression correlates with decreased HCMV replication. We next measured the effect of OTSSP167 at its IC90 (20 nm, as determined by the pp28-GFP assay) on virus replication over a single cycle of virus growth. OTSSP167 was added at the time of infection, and replenished every 48 h. Importantly this concentration of drug was not toxic to uninfected cells at 48 h after treatment (data not shown). Infectious cell-free virus was first detected in untreated cell supernatants at 72 h after infection, however no virus was observed in OTSSP167-treated cells at this time point (Fig. 5B). The amount of cell free virus produced by untreated cells increased at 96 and 120 h after infection, however virus yield in OTSSP167-treated cells was reduced by > 2 orders of magnitude at these later times. To better define the step in replication blocked by OTSSP167, we measured the expression of representative HCMV immediate early (IE1), early (pUL44) and late (pp28) proteins in OTSSP167-treated infected cells (Fig. 5C). IE1 protein levels were slightly reduced at 6 h after infection in OTSSP167-treated cells, but recovered to similar levels observed in untreated cells by 24 h after infection and remained constant thereafter. In contrast, the expression of the early protein UL44 and late protein pp28 was reduced and delayed in OTSSP167-treated cells compared with untreated cells. As expression of pUL44 is required for viral DNA accumulation, we next examined the levels of viral DNA in OTSSP167-treated cells (Fig. 5D). We found that viral DNA accumulated to lower levels in OTSSP167-treated cells compared with controls. The levels of HCMV proteins correlated with the abundance of viral transcripts; IE1 mRNA levels were similar in treated and untreated cells, whereas OTSSP167 reduced the accumulation of UL44 and pp28 (UL99) mRNAs (Fig. 5E). Together these data confirm that OTSSP167 inhibits the HCMV lytic cycle at low nanomolar concentrations that are not toxic to uninfected cells. In addition, these results confirm the accuracy of the pp28-GFP assay, and demonstrate the utility of kinome profiling for identifying drugs that may be repurposed as novel antivirals.
Fig. 5.

OTSSP167 inhibits HCMV infection. A, Primary fibroblasts were infected with HCMV and treated with varying concentrations of OTSSP167 at the time the inoculum was removed. The amount of virus in cell free supernatants was quantified by the TCID50 assay at 96 h after infection. B, Cells were treated with 20 nm OTSSP167 at the time of infection with HCMV (MOI = 1), and cell free virus was quantified as in A at the indicated times after infection. C, Cells were infected as B, and harvested at the indicated times. Viral protein expression was measured by Western blot. Tubulin protein expression was used as a loading control. The results are representative of three independent experiments. D, Cells were infected with HCMV at an MOI of 0.05, and treated either OTSSP167 (20 nm) or PAA (200 μg/ml) when the inoculum was removed. Intracellular viral DNA abundance was measured by qPCR at 96 h after infection. E, Cells were infected as in C. The abundance of the indicated viral transcripts was measured by qRT-PCR (black boxes = untreated; open boxes = OTSSP167). All experiments were performed at least three times. The graphs in A, B, D an E show the mean and standard error of three independent experiments.
DISCUSSION
In this study we sought to apply MIB-MS kinome profiling to define changes in the host kinome during HCMV infection. Our ultimate goal was to use this methodology to identify kinase inhibitors that could potentially be repurposed as novel HCMV antivirals. Although other studies have approached the impact of HCMV infection on the proteome more broadly (76, 77), our studies were specifically focused on characterizing the kinome. The results of this study identified global changes in the kinome after infection with either the lab-adapted AD169 strain or the clinical strain TB40E. Both strains induced surprisingly similar changes to the kinome, suggesting that both lab-adapted and clinical strains modify host signaling pathways in a similar manner.
Changes in cellular kinase activity induced by HCMV infection have previously been identified on a case-by-case basis using traditional molecular virology approaches. Although useful, the vast number of cellular kinases prohibits a comprehensive analysis of the kinome in response to infection using standard techniques. Using MIB-MS kinome profiling, we were able to measure the effect of infection on >250 cellular kinases in a single sample (28–30). This provided a global view of kinome reprogramming in HCMV infected cells, and identified cellular signaling pathways that were significantly affected by HCMV infection. Many of the kinome changes observed matched previous reports, confirming that MIB-MS profiling accurately identifies changes in kinase expression/abundance during HCMV infection (9, 21, 78). Importantly, we identified perturbations in several signaling pathways that have not previously been examined during infection, highlighting the utility of kinome profiling for identifying novel kinases/signaling pathways that may play critical roles in HCMV replication.
Our results also confirmed previously reported changes to cellular signaling pathways during infection (21, 22, 78–82), and identified several new pathways manipulated by HCMV. This included multiple receptor kinases, some that were up-regulated and many that were downregulated after infection. We validated several of the changes by Western blotting in particular the decreased expression of the Eph receptor (EphA2, B4) tyrosine kinases. Membrane bound ephrins bind their cognate Eph receptors on neighboring cells to induce bidirectional signaling events that affect cell adhesion and growth (83). The decreased expression of multiple Eph receptors in infected cells suggests that HCMV might limit ephrin mediated cell-to-cell communication. Interesting, the study of Weekes et al. also detected significant down-regulation of Eph proteins including EphB2 after HCMV infection (77). Downregulation of this and/or other Eph proteins may be involved in immune evasion as suggested. Conversely, we found that a specific subset of ephrins continue to be expressed during infection. Given the variety of functions ephrins play in cell biology (59), our results warrant additional studies to understand the role of ephrin signaling during HCMV infection.
Similarly, infection decreased the amount of TGFβ receptor subunits recovered with MIBs beads. This likely reflects a decrease in TGFβ receptor kinase activity, as the expression of the TGFβ receptor remains constant during infection, however infected cells fail to induce SMAD phosphorylation in response to TGFβ treatment. The mechanism of TGFβ receptor inhibition is currently unclear, as is the underlying biological significance of its inhibition in primary human fibroblasts. However, inhibition of TGFβ signaling might play an important role in HCMV replication in other cell types. HCMV induces an M1 macrophage-like phenotype in infected monocytes, which promotes virus replication (84). Because TGFβ signaling promotes M2 macrophage differentiation (85), blocking TGFβ receptor signaling could drive infected monocytes to differentiate into M1 macrophages, potentially promoting HCMV replication and spread in infected hosts.
We also identified multiple kinases whose quantification after MIB-MS was increased after infection. These kinases may be crucial for increased replication and/or host cell survival. For example, MELK is believed to be important for increased cell proliferation and survival in cancer (53). Previous studies found that MELK regulates cell cycle progression, and HCMV specifically modifies the cell cycle to support virus replication. This suggests that MELK activation promotes HCMV replication and early gene expression by modulating the cell cycle. The temporal increase in MELK detected by MIB-MS was confirmed by Western blotting. Moreover, this change was consistent with the temporal increase and decrease in the MELK substrate eIF4B (86). Whether MELK phosphorylation of this protein affects its stability remains to be determined.
We also detected an increase in a number of CDKs, including CDK9 after HCMV infection. Consistent with previous studies that CDK inhibitors inhibit viral early gene expression, DNA accumulation and late gene expression (81, 82), we found that dinaciclib, a broad CDK inhibitor, potently inhibits HCMV replication. These results warrant further investigation of cell cycle inhibitors as HCMV antivirals.
Using our newly developed assay to quantify virus replication, we tested inhibitors against the induced kinases as potential antivirals. Although applied on a small scale, the potential for this assay to be used in high-throughput screens is evident from our studies. Based on our kinome profiling results, and focusing on drugs in clinical use or development, we identified three kinase inhibitors that significantly decreased HCMV replication. The fact that 3 of only 13 tested drugs inhibited HCMV replication (Table I) suggests that a more extensive analysis would likely identify additional kinase inhibitors that limit virus replication.
The potential for HCMV-induced kinases to serve as targets for antiviral drugs was highlighted by our finding that the reported MELK inhibitor OTSSP167, potently decreased HCMV replication by limiting early gene transcription and viral DNA accumulation. Additional studies revealed that OTSSP167 inhibited the production of infectious progeny virus at nontoxic concentrations in a dose-dependent manner by inhibiting HCMV early protein expression and viral DNA accumulation. Importantly OTSSP167 inhibited HCMV replication at a nontoxic dose that is clinically achievable in humans. Because OTSSP167 has already been evaluated for safety and efficacy in humans, these data suggest a potential novel use as an HCMV antiviral. Currently the mechanism by which OTSSP167 inhibits early gene transcription is unknown. Although a recent study questioned the specificity of OTSSP167 and showed that it could inhibit additional kinases including the Aurora kinase (87), our experiments with the Aurora inhibitor Alisertib failed to recapitulate the effects of OTSSP167, suggesting that inhibition of Aurora alone was insufficient to block viral replication. Additional studies are necessary to determine if the antiviral effects of OTSSP167 is through inhibition of MELK or other kinases. Despite concerns about its specificity, OTSSP167 demonstrated the greatest potency and lowest toxicity of the compounds tested herein.
Together these data demonstrate the utility of kinome profiling for identifying changes in cellular kinase activity and/or expression during HCMV infection. These data also show that defining HCMV-induced kinome reprogramming is a powerful approach for identifying drugs in clinical use or development that may be repurposed as novel HCMV antivirals. Future studies to identify cell-type specific changes in kinase activity in response to HCMV infection may reveal additional cellular kinases that could also be targeted to limit HCMV disease. Moreover, this approach may be useful for identifying common changes to cellular signaling pathways in response to infection with additional viruses, potentially allowing for the development of broad spectrum antiviral drugs. Ultimately this approach could save millions of dollars and many years of testing compared with the traditional antiviral drug development process.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifier PXD005276.
Supplementary Material
Acknowledgments
We thank the members of the Moorman, Graves, Heise, Baric, de Silva and Lemon laboratories for helpful suggestions and comments. We also wish to thank the University of North Carolina virology community for their continued support. We wish to thank Dr. Stephen Frye of the gift of UNC2025.
Footnotes
Author contributions: K.C.A., H.A.V., L.G., and N.J.M. designed research; K.C.A., E.M.L., H.A.V., E.L., I.M.M., T.S.G., M.P.E., and L.E.H. performed research; N.R. and G.L.J. contributed new reagents or analytic tools; K.C.A., E.M.L., H.A.V., N.R., E.L., T.S.G., M.P.E., L.E.H., L.G., and N.J.M. analyzed data; K.C.A., H.A.V., L.G., and N.J.M. wrote the paper.
* This work was funded by NIH grants to N.J.M. (R01AI103311 and R21AI123811) and startup funds from the North Carolina University Cancer Research Fund. K.C.A was funded by awards from the National Science Foundation Graduate Research Fellowship grant (DGE-1144081) and the UNC Virology Training Grant (T32 AI07419). The UNC Proteomics Core Facility is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- HCMV
- human cytomegalovirus
- FDA
- Food and Drug Administration
- MIB-MS
- multiplexed kinase inhibitor bead - mass spectrometry
- MRM
- multiple reaction monitoring
- FDR
- false discovery rate
- LFQ
- label free quantification
- qRT-PCR
- quantitative reverse transcriptase real time polymerase chain reaction
- qPCR
- quantitative real time polymerase chain reaction
- TCID50
- tissue culture infectious dose 50%
- LDH
- lactate dehydrogenase assay
- IC50
- 50% inhibitory concentration.
REFERENCES
- 1. DiMasi J. A., Grabowski H. G., and Hansen R. W. (2016) Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 47, 20–33 [DOI] [PubMed] [Google Scholar]
- 2. Marschall M., and Stamminger T. (2009) Molecular targets for antiviral therapy of cytomegalovirus infections. Future Microbiol. 4, 731–742 [DOI] [PubMed] [Google Scholar]
- 3. van der Bij W., and Speich R. (2001) Management of cytomegalovirus infection and disease after solid-organ transplantation. Clin. Infect. Dis. 33, S32–S37 [DOI] [PubMed] [Google Scholar]
- 4. Griffiths P., Baraniak I., and Reeves M. (2015) The pathogenesis of human cytomegalovirus. J. Pathol. 235, 288–297 [DOI] [PubMed] [Google Scholar]
- 5. Jacobsen T., and Sifontis N. (2010) Drug interactions and toxicities associated with the antiviral management of cytomegalovirus infection. Am. J. Health Syst. Pharm. 67, 1417–1425 [DOI] [PubMed] [Google Scholar]
- 6. Eid A. J., and Razonable R. R. (2010) New developments in the management of cytomegalovirus infection after solid organ transplantation. Drugs 70, 965–981 [DOI] [PubMed] [Google Scholar]
- 7. Munger J., Bajad S. U., Coller H. A., Shenk T., and Rabinowitz J. D. (2006) Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Munger J., Bennett B. D., Parikh A., Feng X. J., McArdle J., Rabitz H. A., Shenk T., and Rabinowitz J. D. (2008) Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26, 1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shenk T., and Alwine J. C. (2014) Human Cytomegalovirus: Coordinating Cellular Stress, Signaling, and Metabolic Pathways. Annu. Rev. Virol. 1, 355–374 [DOI] [PubMed] [Google Scholar]
- 10. Bain M., and Sinclair J. (2007) The S phase of the cell cycle and its perturbation by human cytomegalovirus. Rev. Med. Virol. 17, 423–434 [DOI] [PubMed] [Google Scholar]
- 11. Sanchez V., and Spector D. H. (2008) Subversion of cell cycle regulatory pathways. Curr. Top. Microbiol. Immunol. 325, 243–262 [DOI] [PubMed] [Google Scholar]
- 12. Yurochko A. D. (2008) Human cytomegalovirus modulation of signal transduction. Curr. Top. Microbiol. Immunol. 325, 205–220 [DOI] [PubMed] [Google Scholar]
- 13. Vincent H. A., Ziehr B., and Moorman N. J. (2016) Human cytomegalovirus strategies to maintain and promote mRNA translation. Viruses 8, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walsh D., Mathews M. B., and Mohr I. (2013) Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 5, ea012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beelontally R., Wilkie G. S., Lau B., Goodmaker C. J., Ho C. M., Swanson C. M., Deng X., Wang J., Gray N. S., Davison A. J., and Strang B. L. (2017) Identification of compounds with anti-human cytomegalovirus activity that inhibit production of IE2 proteins. Antiviral Res. 138, 61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khan A. S., Murray M. J., Ho C. M., Zuercher W. J., Reeves M. B., and Strang B. L. (2017) High throughput screening of a GlaxoSmithKline protein kinase inhibitor set identifies an inhibitor of human cytomegalovirus replication that prevents CREB and histone H3 post-translational modification. J. Gen. Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mercorelli B., Luganini A., Nannetti G., Tabarrini O., Palu G., Gribaudo G., and Loregian A. (2016) Drug repurposing approach identifies inhibitors of the prototypic viral transcription factor IE2 that block human cytomegalovirus replication. Cell Chem. Biol. 23, 340–351 [DOI] [PubMed] [Google Scholar]
- 18. Ziehr B., Vincent H. A., and Moorman N. J. (2016) Human cytomegalovirus pTRS1 and pIRS1 antagonize protein kinase R to facilitate virus replication. J. Virol. 90, 3839–3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson R. A., Wang X., Ma X. L., Huong S. M., and Huang E. S. (2001) Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J. Virol. 75, 6022–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moorman N. J., Cristea I. M., Terhune S. S., Rout M. P., Chait B. T., and Shenk T. (2008) Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3, 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McArdle J., Moorman N. J., and Munger J. (2012) HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog. 8, e1002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terry L. J., Vastag L., Rabinowitz J. D., and Shenk T. (2012) Human kinome profiling identifies a requirement for AMP-activated protein kinase during human cytomegalovirus infection. Proc. Natl. Acad. Sci. U.S.A. 109, 3071–3076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moorman N. J., and Shenk T. (2010) Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J. Virol. 84, 5260–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perwitasari O., Yan X., O'Donnell J., Johnson S., and Tripp R. A. (2015) Repurposing kinase inhibitors as antiviral agents to control influenza a virus replication assay. Drug Dev. Technol. 13, 638–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mohr E. L., McMullan L. K., Lo M. K., Spengler J. R., Bergeron E., Albarino C. G., Shrivastava-Ranjan P., Chiang C. F., Nichol S. T., Spiropoulou C. F., and Flint M. (2015) Inhibitors of cellular kinases with broad-spectrum antiviral activity for hemorrhagic fever viruses. Antiviral Res. 120, 40–47 [DOI] [PubMed] [Google Scholar]
- 26. Marty F. M., Bryar J., Browne S. K., Schwarzberg T., Ho V. T., Bassett I. V., Koreth J., Alyea E. P., Soiffer R. J., Cutler C. S., Antin J. H., and Baden L. R. (2007) Sirolimus-based graft-versus-host disease prophylaxis protects against cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation: a cohort analysis. Blood 110, 490–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duncan J. S., Whittle M. C., Nakamura K., Abell A. N., Midland A. A., Zawistowski J. S., Johnson N. L., Granger D. A., Jordan N. V., Darr D. B., Usary J., Kuan P. F., Smalley D. M., Major B., He X., Hoadley K. A., Zhou B., Sharpless N. E., Perou C. M., Kim W. Y., Gomez S. M., Chen X., Jin J., Frye S. V., Earp H. S., Graves L. M., and Johnson G. L. (2012) Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oppermann F. S., Gnad F., Olsen J. V., Hornberger R., Greff Z., Keri G., Mann M., and Daub H. (2009) Large-scale proteomics analysis of the human kinome. Mol. Cell. Proteomics 8, 1751–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daub H. (2015) Quantitative proteomics of kinase inhibitor targets and mechanisms. ACS Chem. Biol. 10, 201–212 [DOI] [PubMed] [Google Scholar]
- 30. Graves L. M., Duncan J. S., Whittle M. C., and Johnson G. L. (2013) The dynamic nature of the kinome. Biochem. J. 450, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruprecht B., Zecha J., Heinzlmeir S., Medard G., Lemeer S., and Kuster B. (2015) Evaluation of Kinase Activity Profiling Using Chemical Proteomics. ACS Chem. Biol. 10, 2743–2752 [DOI] [PubMed] [Google Scholar]
- 32. Bantscheff M., Eberhard D., Abraham Y., Bastuck S., Boesche M., Hobson S., Mathieson T., Perrin J., Raida M., Rau C., Reader V., Sweetman G., Bauer A., Bouwmeester T., Hopf C., Kruse U., Neubauer G., Ramsden N., Rick J., Kuster B., and Drewes G. (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044 [DOI] [PubMed] [Google Scholar]
- 33. Wissing J., Jansch L., Nimtz M., Dieterich G., Hornberger R., Keri G., Wehland J., and Daub H. (2007) Proteomics analysis of protein kinases by target class-selective prefractionation and tandem mass spectrometry. Mol. Cell. Proteomics 6, 537–547 [DOI] [PubMed] [Google Scholar]
- 34. Zawistowski J. S., Graves L. M., and Johnson G. L. (2014) Assessing adaptation of the cancer kinome in response to targeted therapies. Biochem. Soc. Trans. 42, 765–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stuhlmiller T. J., Miller S. M., Zawistowski J. S., Nakamura K., Beltran A. S., Duncan J. S., Angus S. P., Collins K. A., Granger D. A., Reuther R. A., Graves L. M., Gomez S. M., Kuan P. F., Parker J. S., Chen X., Sciaky N., Carey L. A., Earp H. S., Jin J., and Johnson G. L. (2015) Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 11, 390–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johnson G. L., Stuhlmiller T. J., Angus S. P., Zawistowski J. S., and Graves L. M. (2014) Molecular pathways: adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin. Cancer Res. 20, 2516–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Graves L. M., and Litchfield D. W. (2011) “Going KiNativ”: probing the native kinome. Chem. Biol. 18, 683–684 [DOI] [PubMed] [Google Scholar]
- 38. Miao W., Xiao Y., Guo L., Jiang X., Huang M., and Wang Y. (2016) A high-throughput targeted proteomic approach for comprehensive profiling of methylglyoxal-induced perturbations of the human kinome. Anal. Chem. 88, 9773–9779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cooper M. J., Cox N. J., Zimmerman E. I., Dewar B. J., Duncan J. S., Whittle M. C., Nguyen T. A., Jones L. S., Ghose Roy S., Smalley D. M., Kuan P. F., Richards K. L., Christopherson R. I., Jin J., Frye S. V., Johnson G. L., Baldwin A. S., and Graves L. M. (2013) Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS ONE 8, e66755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kurimchak A. M., Shelton C., Duncan K. E., Johnson K. J., Brown J., O'Brien S., Gabbasov R., Fink L. S., Li Y., Lounsbury N., Abou-Gharbia M., Childers W. E., Connolly D. C., Chernoff J., Peterson J. R., and Duncan J. S. (2016) Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 16, 1273–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Werth E. G., McConnell E. W., Gilbert T. S., Couso Lianez I., Perez C. A., Manley C. K., Graves L. M., Umen J. G., and Hicks L. M. (2016) Probing the global kinome and phosphoproteome in Chlamydomonas reinhardtii via sequential enrichment and quantitative proteomics. Plant J. [DOI] [PubMed] [Google Scholar]
- 42. Kouznetsova J., Sun W., Martinez-Romero C., Tawa G., Shinn P., Chen C. Z., Schimmer A., Sanderson P., McKew J. C., Zheng W., and Garcia-Sastre A. (2014) Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg. Microbes Infect. 3, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johansen L. M., DeWald L. E., Shoemaker C. J., Hoffstrom B. G., Lear-Rooney C. M., Stossel A., Nelson E., Delos S. E., Simmons J. A., Grenier J. M., Pierce L. T., Pajouhesh H., Lehar J., Hensley L. E., Glass P. J., White J. M., and Olinger G. G. (2015) A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci. Transl. Med. 7, e290ra289. [DOI] [PubMed] [Google Scholar]
- 44. Wang D., Bresnahan W., and Shenk T. (2004) Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci. U.S.A. 101, 16642–16647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moorman N. J., Sharon-Friling R., Shenk T., and Cristea I. M. (2010) A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics 9, 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arend K. C., Ziehr B., Vincent H. A., and Moorman N. J. (2016) Multiple transcripts encode full-length human cytomegalovirus IE1 and IE2 proteins during lytic infection. J. Virol. 90, 8855–8865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Charles E. McCulloch S. R. S., John M. Neuhaus (2008) Generalized linear mixed models., Wiley [Google Scholar]
- 48. Bates D., et al. (2014) lme4: Linear mixed-effects models using Eigen and S4, R package version 1.7 [Google Scholar]
- 49. Clough T., Thaminy S., Ragg S., Aebersold R., and Vitek O. (2012) Statistical protein quantification and significance analysis in label-free LC-MS experiments with complex designs. BMC Bioinformatics 13, eS6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuznetsova A., Per Bruun Brockhoff and Rune Haubo Bojesen Christensen (2015) Package 'lmerTest'. R package version [Google Scholar]
- 51. Benjamini Y., and Yosef Hochberg. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statistical Soc. 289–300 [Google Scholar]
- 52. Lenarcic E. M., Ziehr B. J., and Moorman N. J. (2015) An unbiased proteomics approach to identify human cytomegalovirus RNA-associated proteins. Virology 481, 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu H., Shen Y., and Shenk T. (1995) Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69, 7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Romanowski M. J., and Shenk T. (1997) Characterization of the human cytomegalovirus irs1 and trs1 genes: a second immediate-early transcription unit within irs1 whose product antagonizes transcriptional activation. J. Virol. 71, 1485–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Silva M. C., Yu Q. C., Enquist L., and Shenk T. (2003) Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77, 10594–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ziehr B., Lenarcic E., Cecil C., and Moorman N. J. (2016) The eIF4AIII RNA helicase is a critical determinant of human cytomegalovirus replication. Virology 489, 194–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lenarcic E. M., Ziehr B., De Leon G., Mitchell D., and Moorman N. J. (2014) Differential role for host translation factors in host and viral protein synthesis during human cytomegalovirus infection. J. Virol. 88, 1473–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sinzger C., Hahn G., Digel M., Katona R., Sampaio K. L., Messerle M., Hengel H., Koszinowski U., Brune W., and Adler B. (2008) Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89, 359–368 [DOI] [PubMed] [Google Scholar]
- 59. Kania A., and Klein R. (2016) Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 17, 240–256 [DOI] [PubMed] [Google Scholar]
- 60. Massague J. (2012) TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kig C., Beullens M., Beke L., Van Eynde A., Linders J. T., Brehmer D., and Bollen M. (2013) Maternal embryonic leucine zipper kinase (MELK) reduces replication stress in glioblastoma cells. J. Biol. Chem. 288, 24200–24212 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62. Ganguly R., Hong C. S., Smith L. G., Kornblum H. I., and Nakano I. (2014) Maternal embryonic leucine zipper kinase: key kinase for stem cell phenotype in glioma and other cancers. Mol. Cancer Therap. 13, 1393–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Beullens M., Vancauwenbergh S., Morrice N., Derua R., Ceulemans H., Waelkens E., and Bollen M. (2005) Substrate specificity and activity regulation of protein kinase MELK. J. Biol. Chem. 280, 40003–40011 [DOI] [PubMed] [Google Scholar]
- 64. Yang Y., Begley M., Huang H. T., Lako A., Eck M. J., Gray N. S., Mitchison T. J., Cantley L. C., and Zhao J. J. (2016) Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proc. Natl. Acad. Sci. U.S.A. 113, 9810–9815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Y., Lee Y. M., Baitsch L., Huang A., Xiang Y., Tong H., Lako A., Von T., Choi C., Lim E., Min J., Li L., Stegmeier F., Schlegel R., Eck M. J., Gray N. S., Mitchison T. J., and Zhao J. J. (2014) MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. eLife 3, e01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Huang E. S. (1975) Human cytomegalovirus. IV. Specific inhibition of virus-induced DNA polymerase activity and viral DNA replication by phosphonoacetic acid. J. Virol. 16, 1560–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Overall J. C. Jr, Kern E. R., and Glasgow L. A. (1976) Effective antiviral chemotherapy in cytomegalovirus infection of mice. J. Infect. Dis. 133, SA237–SA244 [DOI] [PubMed] [Google Scholar]
- 68. Ember S. W., Zhu J. Y., Olesen S. H., Martin M. P., Becker A., Berndt N., Georg G. I., and Schonbrunn E. (2014) Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 9, 1160–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Coude M. M., Braun T., Berrou J., Dupont M., Bertrand S., Masse A., Raffoux E., Itzykson R., Delord M., Riveiro M. E., Herait P., Baruchel A., Dombret H., and Gardin C. (2015) BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 6, 17698–17712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Asangani I. A., Wilder-Romans K., Dommeti V. L., Krishnamurthy P. M., Apel I. J., Escara-Wilke J., Plymate S. R., Navone N. M., Wang S., Feng F. Y., and Chinnaiyan A. M. (2016) BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol. Cancer Res. 14, 324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Boi M., Gaudio E., Bonetti P., Kwee I., Bernasconi E., Tarantelli C., Rinaldi A., Testoni M., Cascione L., Ponzoni M., Mensah A. A., Stathis A., Stussi G., Riveiro M. E., Herait P., Inghirami G., Cvitkovic E., Zucca E., and Bertoni F. (2015) The BET Bromodomain Inhibitor OTX015 Affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 21, 1628–1638 [DOI] [PubMed] [Google Scholar]
- 72. Xiaofei E., and Kowalik T. F. (2014) The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses 6, 2155–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen P., Lee N. V., Hu W., Xu M., Ferre R. A., Lam H., Bergqvist S., Solowiej J., Diehl W., He Y. A., Yu X., Nagata A., VanArsdale T., and Murray B. W. (2016) Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Therap. 15, 2273–2281 [DOI] [PubMed] [Google Scholar]
- 74. Chung S., Suzuki H., Miyamoto T., Takamatsu N., Tatsuguchi A., Ueda K., Kijima K., Nakamura Y., and Matsuo Y. (2012) Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget 3, 1629–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mar E. C., Cheng Y. C., and Huang E. S. (1983) Effect of 9-(1,3-dihydroxy-2-propoxymethyl)guanine on human cytomegalovirus replication in vitro. Antimicrob. Agents Chemother. 24, 518–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stern-Ginossar N., Weisburd B., Michalski A., Le V. T., Hein M. Y., Huang S. X., Ma M., Shen B., Qian S. B., Hengel H., Mann M., Ingolia N. T., and Weissman J. S. (2012) Decoding human cytomegalovirus. Science 338, 1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weekes M. P., Tomasec P., Huttlin E. L., Fielding C. A., Nusinow D., Stanton R. J., Wang E. C., Aicheler R., Murrell I., Wilkinson G. W., Lehner P. J., and Gygi S. P. (2014) Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157, 1460–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Johnson R. A., Ma X. L., Yurochko A. D., and Huang E. S. (2001) The role of MKK1/2 kinase activity in human cytomegalovirus infection. J. Gen. Virol. 82, 493–497 [DOI] [PubMed] [Google Scholar]
- 79. Cojohari O., Peppenelli M. A., and Chan G. C. (2016) Human cytomegalovirus induces an atypical activation of Akt to stimulate the survival of short-lived monocytes. J. Virol. 90, 6443–6452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Peppenelli M. A., Arend K. C., Cojohari O., Moorman N. J., and Chan G. C. (2016) Human cytomegalovirus stimulates the synthesis of select Akt-dependent antiapoptotic proteins during viral entry to promote survival of infected monocytes. J. Virol. 90, 3138–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sanchez V., and Spector D. H. (2006) Cyclin-dependent kinase activity is required for efficient expression and posttranslational modification of human cytomegalovirus proteins and for production of extracellular particles. J. Virol. 80, 5886–5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zydek M., Hagemeier C., and Wiebusch L. (2010) Cyclin-dependent kinase activity controls the onset of the HCMV lytic cycle. PLoS Pathog. 6, e1001096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pasquale E. B. (2010) Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 10, 165–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chan G., Bivins-Smith E. R., Smith M. S., and Yurochko A. D. (2008) Transcriptome analysis of NF-kappaB- and phosphatidylinositol 3-kinase-regulated genes in human cytomegalovirus-infected monocytes. J. Virol. 82, 1040–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhang F., Wang H., Wang X., Jiang G., Liu H., Zhang G., Wang H., Fang R., Bu X., Cai S., and Du J. (2016) TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang Y., Begley M., Li Q., Huang H. T., Lako A., Eck M. J., Gray N. S., Mitchison T. J., Cantley L. C., and Zhao J. J. (2016) Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proc. Natl. Acad. Sci. U.S.A. 113, 9810–9815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ji W., Arnst C., Tipton A. R., Bekier M. E. 2nd, Taylor W. R., Yen T. J., and Liu S. T. (2016) OTSSP167 Abrogates mitotic checkpoint through inhibiting multiple mitotic kinases. PLoS ONE 11, e0153518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
{kind=link}
{kind=link}
{kind=link}
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifier PXD005276.