

Abstract
Context: Mutations in ANKH cause the highly divergent conditions familial chondrocalcinosis and craniometaphyseal dysplasia. The gene product ANK is supposed to regulate tissue mineralization by transporting pyrophosphate to the extracellular space.
Objective: We evaluated several family members of a large consanguineous family with mental retardation, deafness, and ankylosis. We compared their skeletal, metabolic, and serological parameters to that of the autosomal recessive progressive ankylosis (ank) mouse mutant, caused by a loss-of-function mutation in the murine ortholog Ank.
Participants: The studied patients had painful small joint soft-tissue calcifications, progressive spondylarthropathy, osteopenia, mild hypophosphatemia, mixed hearing loss, and mental retardation.
Results: After mapping the disease gene to 5p15, we identified the novel homozygous ANK missense mutation L244S in all patients. Although L244 is a highly conserved amino acid, the mutated ANK protein was detected at normal levels at the plasma membrane in primary patient fibroblasts. The phenotype was highly congruent with the autosomal recessive progressive ankylosis (ank) mouse mutant. This indicates a loss-of-function effect of the L244S mutation despite normal ANK protein expression. Interestingly, our analyses revealed that the primary step of joint degeneration is fibrosis and mineralization of articular soft tissues. Moreover, heterozygous carriers of the L244S mutation showed mild osteoarthritis without metabolic alterations, pathological calcifications, or central nervous system involvement.
Conclusion: Beyond the description of the first human progressive ankylosis phenotype, our results indicate that ANK influences articular soft tissues commonly involved in degenerative joint disorders. Furthermore, this human disorder provides the first direct evidence for a role of ANK in the central nervous system.
ANKH is essential in the calcium-phosphor homeostasis. Autosomal recessive defects lead to hypophosphatemia, mixed hearing loss, and mental retardation, emphasizing the importance of ANKH in the brain.
Degenerative joint disorders like ankylosing spondylitis frequently lead to the formation of osteophytes, which finally can result in osseous ankylosis, the bridging of joints by mineralized tissue (1). This process can lead to conductive hearing loss if it affects the articulations of the hearing ossicles (2). Although the most common trigger is chronic middle ear inflammation, hearing ossicle ankylosis is also observed in otosclerosis (MIM 166800) and in several Mendelian disorders, e.g. multiple synostoses syndrome (MIM 186500), proximal symphalangism syndrome (MIM 185800), and Keutel syndrome (MIM 245150) (3, 4, 5).
Mixed hearing loss has also been observed in the dominantly inherited craniometaphyseal dysplasia [CMD (MIM 123000)]. Although conductive hearing loss in this disease is due to stapes ankylosis and fixation of other middle ear ossicles, the sensorineural component is secondary to os petrosum sclerosis (6, 7). CMD is due to mutations in exons 8–10 of the gene ANKH, encoding the putative pyrophosphate (PPi) transporter ANK (8).
Mutations in ANKH exons 1, 2, and 12, however, cause a clinically different, but also dominantly inherited disease, familial chondrocalcinosis type 2 [CCAL2 (MIM 118600)] (9, 10). These patients suffer from gout-like joint pain or chronic arthropathy without hearing problems. The joint inflammation is caused by calcium pyrophosphate dihydrate (CPPD) crystal deposits (11).
In contrast, the recessive murine progressive ankylosis (ank) phenotype, characterized by stiffening of several joints including the articulations of the hearing ossicles, is elicited by a nonsense mutation in exon 11 of the murine ortholog Ank (p.E440X) (12, 13). Deposition of calcium phosphate in joints of ank mice together with the finding of increased intracellular and diminished extracellular PPi levels in ank fibroblasts led to the conclusion that the Ank transmembrane protein transports PPi via the plasma membrane (13). PPi has been established as a negative regulator of calcium phosphate deposition (14, 15). Accordingly, the deposition of CPPD crystals in CCAL2 was assumed to be due to enhanced ANK activity. In vitro investigations revealed that mutations in CMD cause a loss of PPi transport function, whereas the CCAL2 mutant behaved like the wild-type protein (12).
Ank is broadly expressed, and highest levels are found in brain, muscle, and cartilage (13, 16). In neurons, Ank was detected in the cell body and in dendrites (17). The relevance of this neuronal expression pattern, however, remained unclear because neither human ANKH mutations nor the loss of the murine Ank lead to a consistent neurological phenotype.
Here we report the first human autosomal recessive disorder associated with a novel ANKH missense mutation. This novel syndrome is characterized by sensorineural and conductive hearing loss in association with mental retardation, spinal ankylosis, and periarticular calcification of small joints in homozygous patients and a mild arthropathy in heterozygous individuals. A detailed phenotypic comparison showed remarkable similarities with the murine ank phenotype. Our results imply a crucial role of ligaments in the pathogenesis of ankylosis, an important function of ANK in the central nervous system (CNS), and a loss of function of the mutated ANK protein despite normal plasma membrane expression.
Patients and Methods
Patients
We evaluated six patients from two generations from a large sibship of Turkish ethnicity (V:1, V:2, V:8, V:10/index, VI:8, and VI:10). There is double consanguinity in the family (Fig. 1). The patients suffered from mental retardation, mixed hearing loss (sensorineural hearing loss >40 dB and variable degree of stapes fixation, stapes ankylosis, or incus/malleus fixation in the epitympanium), ankylosis, periarticular ligament ossification, enthesopathy, and dentinogenesis imperfecta. Additional family members (V:5, V:7, V:11, V:12, VI:3, VI:4, VI:5, VI:6, VI:7, VI:9, and VI:11) were included in the study. Patients and controls (home-biobank) consented to laboratory, skeletal, DNA, and fibroblast analysis.
Fig. 1.

Pedigree of the family with mental retardation, deafness, ankylosis, and altered bone metabolism. Individuals represented as filled symbols exhibit the syndromic phenotype with infantile onset; individuals represented by a half-filled symbol developed arthrosis during late adulthood. Patient VI:5 (quarter-filled symbol) demonstrated mixed hearing loss and speech delay. Family members V:8, V:10, V:11, V:12, VI:8, and VI:10 were used for homozygosity mapping.
Genotyping and linkage analysis
Genomic DNA was extracted from peripheral blood lymphocytes using standard salting out procedures (18, 19). Genotyping was performed using the Affymetrix 10K single-nucleotide polymorphism (SNP) array according to the manufacturer’s protocols. Subsequently, linkage analysis was performed by the Genehunter software (20).
An autosomal recessive mode of inheritance was assumed with full penetrance and a disease allele frequency of 0.001. The Marshfield SNP map (sex averaged) was used for SNP localization and allele frequencies from the AFFY Ref Caucasian database.
Mutation analysis
Primers for amplification and sequencing of all exons and intron-exon boundaries of ANKH were designed with primer3plus and amplified under standard PCR conditions. Primer sequences and conditions for amplification are provided in Supplemental Table 1 (published on The Endocrine Society’s Journals Online web site at http://jcem.endojournals.org). Sequence analysis was performed with the ABI PRISM Big Dye Terminator Cycle Sequencing V2.0 Ready Reaction kit and the ABI PRISM 3730 DNA analyzer (Applied Biosystems, Foster City, CA). To determine the presence of the c.731C→T mutation in control individuals, an amplification refractory mutation system approach was performed. Primer sequences are provided in Supplemental Table 2. ANKH mRNA splicing was evaluated by using lymphocyte RNA. Peripheral blood was sampled and RNA isolated using the Paxgene blood RNA kit (PreAnalytiX, Hombrechtikon, Switzerland). cDNA was synthesized with random hexamers according to standard protocols, and PCR was performed with primers provided in Supplemental Table 1.
Analysis of blood samples
In all patients, routine laboratory analysis was performed, including serum electrolytes, calcium, ionized calcium, magnesium, phosphate, alkaline phosphatase (ALP), PTH, calcitonin, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol. Calcium and phosphorus excretion was measured in urine samples collected over 12 h.
Serum samples of ank mice were analyzed for calcium, phosphorus, ALP, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol. Pyrophosphate was measured enzymatically in heparinized human plasma samples using uridine diphosphoglucose (UDPG) pyrophosphorylase and [14C]UDPG as previously described (21). Each sample was diluted 5-fold in pyrophosphatase-free water and assayed with and without UDPG pyrophosphorylase with the difference representing the signal due to pyrophosphate.
For the methods for the expression construct, cell culture, plasmid transfection and immunostaining (22), protein analysis (23, 24), and histological and microcomputed tomography analysis (13, 25), see Supplemental Data.
Results
Mental retardation, deafness, and ankylosis in a large Turkish family
Two generations of a large consanguineous Turkish sibship were extensively examined (V:1, V:2, V:5, V:7, V:8, V:10/index, V:11, V:12, and VI:3–VI:11), including clinical, otological, audiological, biochemical, and genetic studies (Fig. 1). Six individuals (V:1, V:2, V:8, V:10, VI:8, and VI:10) had a disease onset in early childhood. Two of these were children (VI:8, 17 yr, and VI:10, 10 yr), whereas the other four were adults (V:1, V:2, V:8, and V:10, age 26–47 yr). Affected individuals suffered from mental retardation, mixed hearing loss with variable degree of sensorineural hearing loss (>40–65 dB), in association with stapes fixation, stapes ankylosis, and incus/malleus fixation in the epytympanium. Computed tomography (CT) and magnetic resonance imaging (MRI) scanning showed normal middle ear formation and no os petrosum hyperostosis (Fig. 2C, Supplemental Fig. 1G, and Table 1). In addition, these individuals had joint pain and movement restrictions first affecting the small finger joints at the age of approximately 17 yr followed by involvement of the small joints of the feet. All these individuals demonstrated dentinogenesis imperfecta and subtle dysmorphic features including a long, narrow face and a long prominent nose (Fig. 2, A and B, and Table 1). Three of the six individuals had short stature (between −2.5 and −3.0 sd). Based on a nonverbal evaluation of intelligence (nonverbal intelligence test), the individuals V:1, V:2, and V:8 had a mean IQ level of 64, 56, and 59, respectively (none of these individuals were surgically treated for stapes fixation before adulthood). IQ evaluation in individuals after stapes fixation surgery showed an IQ of 61 in V:10, an IQ of 66 in VI:8, and an IQ of 65 in VI:10. The intelligence level was average and above average in V:7, VI:6, and VI:9. Cranial MRI showed appropriate myelinization, no atrophy, normal cortical gyration, and normal basal ganglia and cerebellar structures (Supplemental Fig. 1, A–G).
Fig. 2.

Stapes ankylosis, teeth abnormalities, and periarticular calcification are observed in association with mental retardation, deafness, ankylosis, and altered bone metabolism. A, Affected individuals showed mild facial dysmorphism with long face, prominent nose, and dentinogenesis imperfecta. B, Enlarged presentation of teeth abnormalities revealed, including yellow stippeling and dentinogenesis imperfecta. C, MRI image of the skull (T2-weighted transverse section) demonstrated a well aerated middle ear cleft, the head of the malleus and the body and long process of the incus. The cochlea was well surrounded by bone. No sign of otosclerosis or skull hyperostosis were found. D, Radiography of the lumbar spine displayed periarticular calcification of the vertebral joints with ankylosis and beaking. E, Radiography of the hand revealed periarticular calcification beginning at the entheses at the age of 18 yr as a typical sign of the syndrome. No mineralized structures within the articular cartilage were detectable. F, No calcification of articular cartilage and ligaments were identified by radiography of the knee from a homozygous individual at the age of 28 yr.
TABLE 1.
Phenotype of investigated family members
| Patient | Age (yr) | Sex | Genetic status | Mental retardation | Sensineural hearing loss | Conductive hearing loss | Stapes fixation | Arthrosis/joint pain | Teeth anomalies | Short stature | Hypophosphatemia |
|---|---|---|---|---|---|---|---|---|---|---|---|
| V:1 | 47 | F | Hom | + (IQ 64) | ++4 | + | NA | + | NA | + | + |
| V:2 | 45 | F | Hom | ++ (IQ 56) | − | + | NA | + | NA | − | + |
| V:5 | 31 | F | Het | − | − | − | − | −/+ | − | − | − |
| V:7 | 40 | F | Het | − | − | − | − | + | − | − | − |
| V:8 | 34 | M | Hom | + (IQ 59) | ++4 | + | + | + | + | + | + |
| V:10 | 26 | F | Hom | + (IQ 61) | + | ++4 | + | + | + | + | + |
| V:11 | 36 | M | Het | − | − | − | − | − | − | − | NA |
| V:12 | 38 | F | Het | − | − | − | − | + | − | − | − |
| VI:3 | 2 | F | Het | − | − | − | − | − | − | − | − |
| VI:4 | 3 | M | WT | − | − | − | − | − | − | − | NA |
| VI:5 | 5 | M | Het | −2 | +1 | − | − | − | − | − | − |
| VI:6 | 20 | M | Het | − | − | − | − | − | − | − | − |
| VI:7 | 6 | F | Het | − | − | − | − | − | − | − | − |
| VI:8 | 10 | F | Hom | + (IQ 66) | +4 | ++4 | + | − | + | − | + |
| VI:9 | 8 | M | WT | − | − | − | − | − | − | − | − |
| VI:10 | 17 | F | Hom | + (IQ 65) | +4 | + | + | −/+ | + | −3 | + |
| VI:11 | 6 | F | WT | − | − | − | − | − | − | − | − |
F, Female; Het, heterozygous; Hom, homozygous; M, male; NA, not accessible; WT, wild type; +, present; −, absent; ++, severe; +/−, present in some cases.
Congenital sensorineural hearing loss.
Delayed speech development.
Growth curve follows 2 sd.
Hearing loss of more than 50 dB.
Some radiological features of affected family members have been described previously (26). At the axial skeleton, progressive spondylarthropathy and severe ligamental calcifications were similar to the bamboo spine found in ankylosing spondylitis (Fig. 2D). Periarticular calcifications became first evident in the interphalangeal joints of hands and feet in combination with enthesopathy during the second decade (Fig. 2E). Only later in adulthood did ligament calcification at the tuber ischiadicum and the trochanter major become prominent. Although these calcifications resulted in pain and movement restriction of the spine, the interphalangeal joints, and the elbow joint, the larger hip and knee joints remained functional (Fig. 2F). No severely affected individuals showed soft tissue calcifications (no periarterial, renal, or CNS calcifications) or symphalangism.
Several other individuals in the family had milder arthropathy with a similar pattern of joint involvement but onset after the third decade of life. These individuals did not develop joint stiffening, ankylosis, tooth anomalies, hearing loss, and mental retardation (IV:1, IV:2, IV:5, V:5, V:7, V:11, and V:12) (Table 1). Patient VI:5 had congenital, isolated sensineural deafness without associated ankylosis (and without metabolic abnormalities).
The homozygous ANKH missense mutation c.731T→C cosegregates with mental retardation, deafness, and ankylosis
To identify the genetic defect underlying the features of mental retardation, deafness, and ankylosis, the branch consisting of individuals V:11–V:12, their offspring VI:6–VI:11, V:8, and V:10 were selected for combined linkage-homozygosity mapping (Fig. 1). Affected individuals V:10, VI:8, and VI:10 were chosen for initial analysis because their clinical signs were highly similar. Genotyping revealed only one significant homozygous region that was confined to the affected individuals. This region was delimited by the SNPs rs1512508 and rs1898162 on the short arm of chromosome 5 (5p15.2-15.1) (Supplemental Table 2). A maximum multipoint LOD score of 4.01 was calculated for the region between rs986986 and rs966430. Within the critical 10.1-Mb region, 23 RefSeq genes are present (UCSC Human Genome Database). Among these genes, ANKH was an excellent candidate gene because it is associated with bone and cartilage pathologies (8, 9, 13). Sequence analysis of all 12 exons and exon-intron boundaries of the ANKH gene revealed the transition c.731T→C in exon 6 (Fig. 3A). This nucleotide change causes an amino acid substitution p.L244S, affecting a conserved amino acid (Fig. 3B). Although an effect on binding of splice enhancers was predicted, no aberrant splicing was detected in lymphoblastoid cells. Using amplification refractory mutation system analysis, we confirmed that the sequence alteration was not present in 218 Turkish and 200 Caucasian control alleles.
Fig. 3.

Mental retardation, deafness, ankylosis, and altered bone metabolism cosegregates with homozygous ANKH mutation c.731T→C. A, Electropherogram showing the ANKH mutation c.731T→C in the heterozygous (het) and homozygous (hom) state. On the protein level, the mutation caused the amino acid substitution p.L244S. B, Alignment of ANK protein sequences of various species demonstrated that the p.L244S substitution affects an evolutionarily conserved amino acid. wt, Wild type.
Importantly, the sequence variant cosegregated with the disease in the family presented here (Table 1). All patients homozygous for the L244S mutation showed the full clinical picture with, ankylosis, hearing loss, mental retardation, and onset during childhood (Table 2). Most of the heterozygous individuals demonstrated late-onset arthrosis from the third decade of life without ankylosis and mental retardation.
TABLE 2.
Comparison of ANK-associated phenotypes
| Involved organ | Tissue | Phenotype | Mental retardation, deafness, and ankylosis ANK L244S human, recessive | CMD (6 8 29 37 ) ANK F377Del human, dominant | CC (9 ) ANK M48T human, dominant | Ank KI/KI (32 ) Ank F377Del mouse, recessive | ank (13 27 36 38 ) Ank E440X mouse, recessive |
|---|---|---|---|---|---|---|---|
| CNS | Nervous system | Mental retardation | ++ | − | − | ND | ND |
| Auditory pathways | Nervous system | Sensorineural hearing loss | ++ | +/− (deafness secondary to compression) | − | ND | ND |
| Middle ear | Conductive hearing loss (stapes fixation) | +++ (early onset) | ++ | − | ND | +++ (early onset) | |
| Small joints | Articular cartilage | Calcification | +/− (periarticular) | − | +++ | +++ (early onset) | +++ (early onset) |
| Type of mineral | ND | − | CPPD | ND | HA | ||
| Capsule | Inflammation | ++ | − | ND | ND | ++ | |
| Knee joint | Articular cartilage | Calcification | − | − | +++ (late onset) | ND | − |
| Ligament | Calcification | +1 | − | ND | ND | +++ | |
| Synovium/ligament | Fibrosis | + | − | ND | ND | +++ | |
| Synovial fluid | Crystal | ND | ND | +++ (CPPD) | ND | ND | |
| Vertebra | Intervertebral disc | Osteophytes | +++ | − | ++ | ND | +++ (late onset) |
| Trabecular bone | Osteopenia | − | + | − | ND | − | |
| Skull | Vault | Hyperostosis | − | +++ | − | + | + |
| Hypermineralization | − | ND | − | + | + (local) | ||
| Mandible | Hyperostosis | − | + | − | + | ND | |
| Long bones | Trabecular bone | Osteopenia | + | + | + | +++ | +++ |
| Cortical bone | Osteopenia | + | − | + | +++ | +++ | |
| Growth plate | Abnormality | ND | ND | ND | ND | − | |
| Teeth | Dentin density | Reduction | + | ND | ND | + | − |
| Cementum | Expanded | ND | ND | ND | ND | + | |
| Stature | Small | + | − | − | − | − |
CC, Chondrocalcinosis; HA, hydroxyapatite; ND, not determined; −, no effect; +, weak effect; +++, strong effect.
Ligament calcification is present of the tuber ischiadicum and trochanter major.
Although patient VI:5 carried the ANKH mutation heterozygously, he presented with neonatal hearing loss (sensorineural hearing loss >65 dB on the right and deafness on the left). Another ANKH mutation, aberrant mRNA splicing, or a lower transcript level of the nonmutated ANKH allele was not found in patient VI:5. Because this patient does not exhibit any skeletal signs and does not have biochemical abnormalities suggesting altered bone metabolism, his severe hearing loss is either acquired or due to defects in another gene. Chromosomal copy number variations and mutations in GJB2 were excluded.
The mutation L244S does not affect ANK cell surface expression
By cell surface biotinylation of patient and control fibroblasts, we found no difference in cell surface expression of endogenous ANK-L244S (Supplemental Fig. 3B). Normal plasma membrane distribution of Ank-L244S was further confirmed by biotinylation of transfected HeLa cells (Supplemental Fig. 4D).
Altered bone metabolism in patients with mental retardation, deafness, and ankylosis
In patients presenting with mental retardation, deafness, and ankylosis, alterations of bone mass and metabolism were observed. Serum analysis revealed increased serum ALP, slightly decreased 25-hydroxycholecalciferol, increased 1,25-dihydroxycholecalciferol, and low serum phosphate levels in homozygous individuals (Table 3). These abnormalities were more pronounced in the pediatric age than in adults. Heterozygous individuals showed normal values of ALP, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol. Bone densitometry values were available for three homozygous mutation carriers showing an average Z-score of −0.8 ± 0.9 at the spine and −1.8 ± 1.2 at the femoral neck (controls, 0–1; osteoporosis, <−2). Plasma PPi was normal in homozygous patients (three homozygous patients, 1.8–2.6; control range, 1.9–5.5; n = 30), illustrating no substantial effect of the L244S mutation on circulating PPi levels.
TABLE 3.
Serum measurements in patients with mental retardation, deafness, and ankylosis and in ank mice
| Serum parameter | Control mice | Heterozygous ank mice | Homozygous ank mice | ANKH L244S homozygous | ANKH L244S heterozygous | Human reference values |
|---|---|---|---|---|---|---|
| Ca (mmol/liter) | 2.43 ± 0.29 (n = 35) | 2.58 ± 0.26 (n = 26) | 2.56 ± 0.24 (n = 20) | 2.3 ± 0.15 (n = 10) | 2.3 ± 0.11 (n = 6) | 2.20–2.65 |
| P (mmol/liter) | 4.44 ± 1.24 (n = 35) | 4.49 ± 1.16 (n = 26) | 4.70 ± 1.10 (n = 20) | 0.8 ± 0.09 (n = 14) | 1.03 ± 0.15 (n = 7) | 0.85–1.4 |
| ALP (U/liter), 8 wk | 42.9 ± 8 (n = 14; P ≤ 0.001) | ND | 59.7 ± 7.4 (n = 14; P ≤ 0.001) | 80 ± 17 (n = 4 adults) | 61 ± 13 (n = 4 adults) | 40–120 |
| ALP (U/liter), 4 wk | ND | ND | ND | 166 ± 30 (n = 6 children) | ND | 40–120 |
| PTH (pg/ml) | ND | ND | ND | 6.1 ± 1.7 (n = 10) | 5.4 ± 1.6 (n = 4) | 1–6.5 |
| 25OH-VitD (nmol/liter) | 155.5 ± 14.5 (4 pools) | 155.5 ± 14.5 (4 pools) | 138.3 ± 6.5 (3 pools) | 31.5 ± 6.1 (n = 8) | 45 ± 18.2 (n = 5) | 35–100 |
| 1,25-OHD3 (pmol/liter), 8 wk | 244 (pool) | 244 (pool) | 260 (pool) | 177 ± 21 (n = 4 children) | ND | 50–150 |
| 1,25-OHD3 (pmol/liter), 12 wk | 109 (pool) | 123 (pool) | 243 (pool) | 162 ± 6.2 (n = 4 adults) | 136 ± 26 (n = 5) | 50–150 |
Values are given as mean ± sd. Bold values indicate statistically significant differences. Statistical significance for mouse experiments was calculated with t test. 1,25-OHD3, 1,25-dihydroxycholecalciferol; 25OH-VitD, 25-hydroxycholecalciferol; n, number of measurements.
The phenotype of ank mice closely corresponds to the human disorder caused by the L244S mutation
Because the L244S mutation did not alter stability or cellular localization of the ANK protein, we wanted to learn more about the functional consequences by comparing the human disorder with the phenotype of ank mutant mice that harbor a clear loss-of-function mutation. Like the L244S homozygous patients, ank mutants showed no significant morphological changes in brain (Supplemental Fig. 2). Although ankylosis and hearing ossicle fixation in patients were reminiscent, the lack of articular cartilage calcification was not in line with the reported findings in ank mouse mutants. Therefore, proximal interphalangeal toe and knee joints of ank mice were investigated histologically at different time points. The lateral ligament in interphalangeal joints at the age of 28 d revealed infiltration by mononuclear cells, formation of cartilaginous tissue, and initiation of articular cartilage calcification (Fig. 4A). In contrast, even in 16-wk-old ank mutants, the articular cartilage of the knee did not show any indications of calcification or chondrocyte cell death (Fig. 4B). However, progressive fibrotic changes around the cruciate ligaments were observed from 6 wk onward (Supplemental Fig. 5).
Fig. 4.
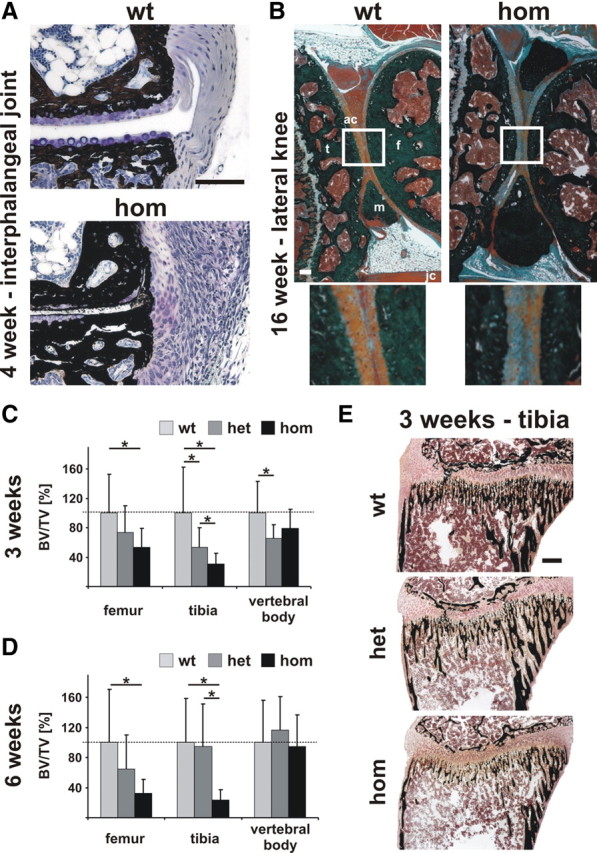
The ank mouse mutants develop ligament transformations and early-onset osteopenia in long bones but not in vertebrae. A, Interphalangeal joints of 4-wk-old control mice were composed of mineralized bone (black), adjacent articular cartilage with chondrocytes (purple), and lateral ligaments (light blue). In ank mice, articular cartilage of interphalangeal joints appeared partially calcified starting from the articular surface. Moreover, periarticular ligaments were transformed into fibrocartilaginous tissue and showed infiltration with mononuclear cells. Images depict von Kossa/toluidine-stained serial sections in transverse plane. Scale bar, 100 μm. B, In longitudinal sections of lateral knee from 16-wk-old homozygous ank mutant articular cartilage (ac) remained unmineralized. Images depict von Kossa/Masson/Goldner staining. Individual parts of the knee joint are indicated: tibia (t), femur (f), meniscus (m), and joint capsule (jc). Scale bar, 500 μm. C, Quantification of trabecular bone by micro-CT of 3-wk-old ank mice showed a decrease in BV/TV of about 40 and 60% in femur and tibia, respectively. BV/TV in vertebral bodies was reduced by about 30%. D, Trabecular bone measurement by micro-CT of 6-wk-old ank mice revealed BV/TV reduction in long bones of hetero- and homozygous animals. However, normal BV/TV levels were measured in vertebral bodies. E, Calcified tissue histology of tibia at 3 wk of age illustrated osteopenia in ank mice. Histology was performed according to von Kossa/Kernechtrot, which stains mineralized tissue in black and soft tissue in shades of red. Scale bar, 100 μm. het, Heterozygous; hom, homozygous, wt, wild type.
Previous reports showed that ank mice exhibit osteopenia after onset of the ankylosis phenotype (12), in the stage of reduced locomotive activity (27). To delineate a primary role of Ank in bone mass regulation, we assessed trabecular bone in long bones and vertebral bodies before and after the onset of phalangeal ankylosis at 3 and 6 wk of age, respectively (Supplemental Table 3). Already at 3 wk of age, homozygous ank mice showed a 40 and 60% reduction in bone volume (BV) fraction BV/[trabecular volume (TV)] in femur and tibia, respectively (Fig. 4, C–E). In vertebral bodies, the reduction of BV/TV was approximately 30% (Fig. 4, C–E, and Supplemental Fig. 6A). At 6 wk of age, homozygous ank mice demonstrated a much more severe reduction in BV/TV of approximately 60–70% in femur and tibia (Fig. 4, D and E, and Supplemental Fig. 6B). However, the BV/TV reduction in the vertebral bodies of hetero- and homozygous ank was no longer significant. Although the bone mineral density changes were more severe in ank mice, the pattern was highly similar to the here-described clinical syndrome, especially the strong involvement of the long bones.
In ank mice, a significant increase in ALP activity was measured (Table 3). Moreover, 3-month-old animals showed increased levels of 1,25-dihydroxycholecalciferol and slightly reduced levels of 25-hydroxycholecalciferol, mirroring the findings in affected homozygous individuals.
Discussion
Here we report on a novel autosomal recessive disorder caused by the mutation L244S in the ANK protein in a single large family. In homozygous mutation carriers, the phenotype is characterized by mixed hearing loss associated with mental retardation and progressive ankylosis of the spine and the small joints of hands and feet. Patients with this particular mutation have a variable degree of sensorineural deafness, middle ear ossicle fixation, dental anomalies, ligament calcifications resulting in enthesopathy, osteopenia, increased 1,25-dihydroxycholecalciferol, and mild hypophosphatemia. In heterozygous individuals, mild arthropathy was detectable in adulthood without any other clinical or metabolic features. Only some of the skeleto-articular features of this novel recessive disease entity are reminiscent of the two other ANKH-related pathologies: autosomal dominant familial CCAL2 and autosomal dominant CMD (Table 2).
In CCAL2, deposition of CPPD crystals in joint cartilage, menisci, and bursae leads to joint problems in the third to fourth decade (28). Not only the disease onset but also the pattern of the clinical signs and symptoms differ from our patients with homozygous L244S mutation. Although CCAL2 primarily affects knee, wrist, and symphysis pubis, our patients first have pain and periarticular calcifications at the interphalangeal joints and the spine without symptoms in large joints. ANKH-heterozygous individuals, however, develop an arthropathy with a later onset in the third to fourth decade similar to CCAL2 but without calcifications. In contrast, CMD does not involve the joints. The joint phenotype of ank mutant mice, albeit being more severe, corresponds well with its human counterpart. It also initially shows periarticular soft tissue calcifications. Although articular cartilage in small joints subsequently mineralizes and degenerates, the knee joint does not show cartilage mineralization even at late stages.
Combined hearing loss has been observed in CMD and is attributed to stapes ankylosis and os petrosum hyperostosis, which not only facilitates middle-ear ossicle fixation due to changes in middle ear morphology but also impairs the function of the vestibulocochlear nerve (6, 7, 29). No hearing impairment has been reported in CCAL2.
It is currently unclear why homozygous individuals had mild hypophosphatemia and mildly elevated PTH, 1,25-dihydroxycholecalciferol, and ALP levels, which was also partially found in ank mutants. Because extracellular phosphate concentrations are known to promote pathological and physiological mineralization, this can be viewed as a countermeasure to dampen pathological calcification. Alternatively, loss of Ank could also lead to altered PTH signaling, 25(OH) vitamin D3 hydroxylation, and phosphate reabsorption in the kidney. Interestingly, mutations in the pyrophosphate-generating enzyme ENPP1 (ectonucleotide pyrophosphatase/phosphodiesterase 1) have been reported to cause hypophosphatemic rickets (30). However, Ank expression in the kidney seems to be restricted to the collecting duct, whereas PTH-mediated phosphate reabsorption rather takes place in earlier parts of the nephron (31).
ANK loss of function leads to long-bone osteopenia in our patients, in ank mutants, and in CMD patients, which at the same time present with a striking skull hyperostosis. This finding is currently explained by accelerated bone resorption in long bones rather than the calvaria (32, 33). This is supported by increased osteoclast numbers per trabecular bone surface in ank long bones (32). In patients affected by ankylosing spondylitis and corresponding animal models, osteopenia as a consequence of joint destruction has been well established (1, 34). Normal trabecular bone values in ank vertebrae at 6 wk of age suggest that osteopenia in long bones is secondary to joint stiffness and that the joint pathology does not induce systemic bone loss at this stage. Therefore, we speculate that the bone phenotype upon loss of Ank is caused by either mechanical unloading or a local pro-osteoclastic stimulus released from transforming joints.
An intriguing finding is the presence of mental retardation in all patients harboring the homozygous mutation. Even after surgical intervention and a significant improvement in hearing, the patients scored below lowest normal (subnormal) IQ level. Detailed CT and MRI imaging of the CNS showed no structural alterations or calcifications in humans, and no macroscopic and histological brain abnormalities were seen ank mutants either. Cosegregation of a second mutation causing mental retardation in our consanguineous family is theoretically possible. However, the homozygous region in our family does not overlap with a candidate region for mental retardation on chromosome 5 (MIM 611091). Other known disorders related to mental retardation were also ruled out.
Only few reports indicate a CNS involvement in ANKH-related disorders. In one family, chondrocalcinosis cosegregated with seizures, and one CMD patient was judged to be mentally retarded (8, 35). This could indicate that mental retardation develops only if homozygous ANKH loss-of-function mutations are present. Because for many behavioral tests in mice, an intact locomotion is prerequisite it is difficult to assess brain function in ank mutants. Studies on primary neuronal cultures from mice cortex showed expression of Ank with a predominant presence of Ank in the cell body and some localization in dendrites, suggesting that Ank is involved in synaptic transmission and/or in modulation of electrical activity (17). Areas of the central nervous system with high metabolic activity showed high Ank/ANK levels, and pharmacologically induced seizures enhanced Ank expression (17). This is in line with the high ANK expression in heart and skeletal muscle, which also have high metabolic rates (13, 16). Therefore, Guo et al. (16) proposed that Ank/ANKH expression may be necessary to maintain proper intracellular PPi levels at conditions of high cellular metabolic activity.
The close similarity of the here-described human phenotype and the phenotype of the ank mouse mutant caused by a truncating mutation implies a recessive loss-of-function effect of the L244S mutation. Most likely, the milder phenotype in heterozygous carriers can be explained by haploinsufficiency. This hypothesis is supported by the fact that heterozygous ank mutants also show mild bone anomalies. Because we excluded degradation of ANK-L244S, the loss of function must be due to a more complex alteration. According to the generally accepted model, it is assumed that ANK transports PPi at the plasma membrane to control tissue mineralization and that an impairment of this transport induces pathological articular cartilage calcification (36). Previous studies identified ANK at the plasma membrane of different cell types after transient overexpression (12, 13). Although PPi concentrations in patient sera were in the normal range, this does not exclude altered PPi levels in the microenvironments of the affected tissues due to abnormal ANK function at the plasma membrane.
Depending on the topology model, the residue L244 resides either within transmembrane helix 7 of ANK or in a loop of unknown function. All CMD mutations, which were shown to entail dominant-negative loss-of-function effect, induce strong amino acid changes either by altering the charge or size of the side chain and they cluster in a protein region close to transmembrane helices 9 and 10 of the model proposed by Nürnberg et al. (8). The absence of a clear dominant-negative effect could imply that ANK-L244S, in contrast to CMD mutations, is no longer incorporated into a multimeric protein complex.
In summary, we report on the association of mental retardation, deafness, and ankylosis with abnormal bone metabolism in a large consanguineous family. This is the first autosomal recessive human ankylosis disease due to ANKH mutations. We demonstrate striking similarities between the human syndrome and the recessive ank mouse mutant phenotype, which implies a loss-of-function effect of the L244S mutation. Our data offer the first clinical example for a functional relevance of ANK in the brain. Furthermore, the human as well as the ank mutant phenotype provides clear evidence for the significance of Ank in bone metabolism and the primary role of articular soft tissues in the development of the joint degeneration warranting further investigation.
Acknowledgments
We thank Denise Pankalla and Claire Schlack for technical assistance in ank mouse histology and genotyping. We acknowledge Fokje Zijlstra for cell isolation and technical laboratory support. We acknowledge Dr. I. Braakman, University Medical Center Utrecht for providing PDI antibodies. In memoriam Prof. Endre Morava.
Footnotes
This project was supported by the Sonderforschungsbereich 577 A4 of the Deutsche Forschungsgemeinschaft. P.M.T.D. is the recipient of VICI Grant 865.07.002 of The Netherlands Organization for Scientific research (NWO), which is acknowledged.
Disclosure Summary: The authors report no conflict of interest.
First Published Online October 13, 2010
E.M. and J.K. made equal contributions to this work.
H.K. and R.A.W. made equal contributions to this work.
Author affiliations are shown at the bottom of the next page.
For editorial see page 72
Abbreviations: ALP, Alkaline phosphatase; CCAL2, chondrocalcinosis type 2; CMD craniometaphyseal dysplasia; CNS, central nervous system; CPPD calcium pyrophosphate dihydrate; CT, computed tomography; MRI, magnetic resonance imaging; PPi pyrophosphate; SNP, single-nucleotide polymorphism; UDPG, uridine diphosphoglucose.
Department of Pediatrics (E.V., J.M.D., H.L.C.-v.d.G.) and Laboratory of Metabolic, Endocrine, and Genetic Diseases (E.M., D.L., R.A.W.), Department of Laboratory Medicine, Institute for Genetic and Metabolic Disease; Department of Neurology (C.C., M.A.W.) and Nijmegen Center for Molecular Life Sciences (H.K.), Donders Institute of Brain, Cognition, and Behavior, Radboud University Nijmegen, 6500 HB Nijmegen, The Netherlands; Institute for Medical Genetics (J.K., S.S., U.K.), Charité-Universitätsmedizin Berlin, 10117 Berlin, Germany; Max Planck Institute for Molecular Genetics (J.K., U.K.), FG Development and Disease, and Faculty of Biology, Chemistry, and Pharmacy (J.K.), Free University of Berlin, 14195 Berlin, Germany; Departments of Physiology (J.H.R., P.M.T.D.), Otorhinolaryngology and Head and Neck Surgery (C.C., H.K.), and Human Genetics (K.V., S.V., A.H.), Radboud University Nijmegen Medical Center, 6500 HB Nijmegen, The Netherlands; Departments of Pediatrics (P.v.S.) and Rheumatology (A.B., A.d.J.), Rijnstate Hospital, 6815 AD Arnhem, The Netherlands; Department of Rheumatology (A.B.), Jeroen Bosch Hospital, 5211 NL ’s-Hertogenbosch, The Netherlands; and Renal Division (C.W.O.), Emory University School of Medicine, Atlanta, Georgia 30303
References
- 1.Thomas GP, Brown MA 2010. Genetics and genomics of ankylosing spondylitis. Immunol Rev 233:162–180 [DOI] [PubMed] [Google Scholar]
- 2.Raveh E, Hu W, Papsin BC, Forte V 2002. Congenital conductive hearing loss. J Laryngol Otol 116:92–96 [DOI] [PubMed] [Google Scholar]
- 3.Declau F, van Spaendonck M, Timmermans JP, Michaels L, Liang J, Qiu JP, van de Heyning P 2007. Prevalence of histologic otosclerosis: an unbiased temporal bone study in Caucasians. Adv Otorhinolaryngol 65:6–16 [DOI] [PubMed] [Google Scholar]
- 4.Brown DJ, Kim TB, Petty EM, Downs CA, Martin DM, Strouse PJ, Moroi SE, Milunsky JM, Lesperance MM 2002. Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin. Am J Hum Genet 71:618–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parmar H, Blaser S, Unger S, Yoo SJ, Papsin B 2006. Petrified ears in a patient with Keutel syndrome: temporal bone CT findings. Pediatr Radiol 36:241–243 [DOI] [PubMed] [Google Scholar]
- 6.Beighton P, Hamersma H, Horan F 1979. Craniometaphyseal dysplasia: variability of expression within a large family. Clin Genet 15:252–258 [DOI] [PubMed] [Google Scholar]
- 7.Kornak U, Brancati F, Le Merrer M, Lichtenbelt K, Höhne W, Tinschert S, Garaci FG, Dallapiccola B, Nürnberg P 2009. Three novel mutations in the ANK membrane protein cause craniometaphyseal dysplasia with variable conductive hearing loss. Am J Med Genet 152A:870–874 [DOI] [PubMed]
- 8.Nürnberg P, Thiele H, Chandler D, Höhne W, Cunningham ML, Ritter H, Leschik G, Uhlmann K, Mischung C, Harrop K, Goldblatt J, Borochowitz ZU, Kotzot D, Westermann F, Mundlos S, Braun HS, Laing N, Tinschert S 2001. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet 28:37–41 [DOI] [PubMed] [Google Scholar]
- 9.Pendleton A, Johnson MD, Hughes A, Gurley KA, Ho AM, Doherty M, Dixey J, Gillet P, Loeuille D, McGrath R, Reginato A, Shiang R, Wright G, Netter P, Williams C, Kingsley DM 2002. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet 71:933–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams CJ, Zhang Y, Timms A, Bonavita G, Caeiro F, Broxholme J, Cuthbertson J, Jones Y, Marchegiani R, Reginato A, Russell RG, Wordsworth BP, Carr AJ, Brown MA 2002. Autosomal dominant familial calcium pyrophosphate dihydrate deposition disease is caused by mutation in the transmembrane protein ANKH. Am J Hum Genet 71:985–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halverson PB, Derfus BA 2001. Calcium crystal-induced inflammation. Curr Opin Rheumatol 13:221–224 [DOI] [PubMed] [Google Scholar]
- 12.Gurley KA, Reimer RJ, Kingsley DM 2006. Biochemical and genetic analysis of ANK in arthritis and bone disease. Am J Hum Genet 79:1017–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho AM, Johnson MD, Kingsley DM 2000. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 289:265–270 [DOI] [PubMed] [Google Scholar]
- 14.Fleisch H, Bisaz S 1962. Mechanism of calcification: inhibitory role of pyrophosphate. Nature 195:911 [DOI] [PubMed]
- 15.Terkeltaub RA 2001. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol 281:C1–C11 [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, Hsu DK, Feng SL, Richards CM, Winkles JA 2001. Polypeptide growth factors and phorbol ester induce progressive ankylosis (ank) gene expression in murine and human fibroblasts. J Cell Biochem 84:27–38 [DOI] [PubMed] [Google Scholar]
- 17.Yepes M, Moore E, Brown SA, Hanscom HN, Smith EP, Lawrence DA, Winkles JA 2003. Progressive ankylosis (Ank) protein is expressed by neurons and Ank immunohistochemical reactivity is increased by limbic seizures. Lab Invest 83:1025–1032 [DOI] [PubMed] [Google Scholar]
- 18.Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A 1989. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res 17:8390 [DOI] [PMC free article] [PubMed]
- 19.Miller SA, Dykes DD, Polesky HF 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215 [DOI] [PMC free article] [PubMed]
- 20.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES 1996. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- 21.O'Neill WC, Sigrist MK, McIntyre CW 2010. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol Dial Transplant 25:187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deen PM, Van Balkom BW, Savelkoul PJ, Kamsteeg EJ, Van Raak M, Jennings ML, Muth TR, Rajendran V, Caplan MJ 2002. Aquaporin-2: COOH terminus is necessary but not sufficient for routing to the apical membrane. Am J Physiol Renal Physiol 282:F330–F340 [DOI] [PubMed] [Google Scholar]
- 23.Robben JH, Knoers NV, Deen PM 2005. Characterization of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus in a polarized cell model. Am J Physiol Renal Physiol 289:F265–F272 [DOI] [PubMed] [Google Scholar]
- 24.Deen PM, Croes H, van Aubel RA, Ginsel LA, van Os CH 1995. Water channels encoded by mutant aquaporin-2 genes in nephrogenic diabetes insipidus are impaired in their cellular routing. J Clin Invest 95:2291–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolanczyk M, Kühnisch J, Kossler N, Osswald M, Stumpp S, Thurisch B, Kornak U, Mundlos S 2008. Modelling neurofibromatosis type 1 tibial dysplasia and its treatment with lovastatin. BMC Med 6:21 [DOI] [PMC free article] [PubMed]
- 26.Branten AJ, de Jong AJ, Janssen M 2009. Familial presentation of bamboo spine, peripheral calcifications, and normal sacroiliac joints. Scand J Rheumatol 38:156–157 [DOI] [PubMed] [Google Scholar]
- 27.Sweet HO, Green MC 1981. Progressive ankylosis, a new skeletal mutation in the mouse. J Hered 72:87–93 [DOI] [PubMed] [Google Scholar]
- 28.Richette P, Bardin T, Doherty M 2009. An update on the epidemiology of calcium pyrophosphate dihydrate crystal deposition disease. Rheumatology (Oxford) 48:711–715 [DOI] [PubMed] [Google Scholar]
- 29.Taylor DB, Sprague P 1989. Dominant craniometaphyseal dysplasia: a family study over five generations. Australas Radiol 33:84–89 [DOI] [PubMed] [Google Scholar]
- 30.Lorenz-Depiereux B, Schnabel D, Tiosano D, Häusler G, Strom TM 2010. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 86:267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carr G, Sayer JA, Simmons NL 2007. Expression and localisation of the pyrophosphate transporter, ANK, in murine kidney cells. Cell Physiol Biochem 20:507–516 [DOI] [PubMed] [Google Scholar]
- 32.Chen IP, Wang CJ, Strecker S, Koczon-Jaremko B, Boskey A, Reichenberger EJ 2009. Introduction of a Phe377del mutation in ANK creates a mouse model for craniometaphyseal dysplasia. J Bone Miner Res 24:1206–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HJ, Minashima T, McCarthy EF, Winkles JA, Kirsch T 2010. Progressive ankylosis protein (ANK) in osteoblasts and osteoclasts controls bone formation and bone remodeling. J Bone Miner Res 25:1771–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rauner M, Stupphann D, Haas M, Fert I, Glatigny S, Sipos W, Breban M, Pietschmann P 2009. The HLA-B27 transgenic rat, a model of spondyloarthritis, has decreased bone mineral density and increased RANKL to osteoprotegerin mRNA ratio. J Rheumatol 36:120–126 [DOI] [PubMed] [Google Scholar]
- 35.McKee S, Pendleton A, Dixey J, Doherty M, Hughes A 2004. Autosomal dominant early childhood seizures associated with chondrocalcinosis and a mutation in the ANKH Gene. Epilepsia 45:1258–1260 [DOI] [PubMed] [Google Scholar]
- 36.Gurley KA, Chen H, Guenther C, Nguyen ET, Rountree RB, Schoor M, Kingsley DM 2006. Mineral formation in joints caused by complete or joint-specific loss of ANK function. J Bone Miner Res 21:1238–1247 [DOI] [PubMed] [Google Scholar]
- 37.Tinschert S, Braun HS 1998. Craniometaphyseal dysplasia in six generations of a German kindred. Am J Med Genet 77:175–181 [DOI] [PubMed] [Google Scholar]
- 38.Fong H, Foster BL, Sarikaya M, Somerman MJ 2009. Structure and mechanical properties of Ank/Ank mutant mouse dental tissue—an animal model for studying periodontal regeneration. Arch Oral Biol 54:570–576 [DOI] [PMC free article] [PubMed] [Google Scholar]