

Abstract
Context:
Thyroid cancer is usually cured by timely thyroidectomy; however, the treatment of patients with advanced disease is challenging because their tumors are mostly unresponsive to conventional therapies. Recently, the malignancy has attracted much interest for two reasons: the dramatic increase in its incidence over the last three decades, and the discovery of the genetic mutations or chromosomal rearrangements causing most histological types of thyroid cancer.
Objective:
This update reviews the molecular genetics of thyroid cancer and the clinical trials evaluating kinase inhibitors (KIs) in patients with locally advanced or metastatic disease. The update also reviews studies in other malignancies, which have identified mechanisms of efficacy, and also resistance, to specific KIs. This information has been critical both to the development of effective second-generation drugs and to the design of combinatorial therapeutic regimens. Finally, the update addresses the major challenges facing clinicians who seek to develop more effective therapy for patients with thyroid cancer.
Results:
PubMed was searched from January 2000 to November 2013 using the following terms: thyroid cancer, treatment of thyroid cancer, clinical trials in thyroid cancer, small molecule therapeutics, kinase inhibitors, and next generation sequencing.
Conclusions:
A new era in cancer therapy has emerged based on the introduction of KIs for the treatment of patients with liquid and solid organ malignancies. Patients with thyroid cancer have benefited from this advance and will continue to do so with the development of drugs having greater specificity and with the implementation of clinical trials of combined therapeutics to overcome drug resistance.
The incidence of thyroid cancer has risen sharply over the last three decades, especially in developed countries. In the United States, the incidence is increasing at a more rapid rate than that of any other malignancy. It is estimated that there will be 60 220 new cases of thyroid cancer in the United States in 2013, with a 3-fold higher frequency in women (1). The increase, almost all due to papillary thyroid carcinoma (PTC), was initially thought the result of more frequent use of diagnostic medical imaging such as ultrasound, computed tomographic scans, and associated fine-needle aspiration biopsy. Other investigators, however, have questioned this hypothesis, noting that the increased incidence involves PTCs of all sizes and clinical stages, raising the question of whether environmental or other as yet undefined factors account for the increase (2–4). Intriguingly, there has been a substantial change in the prevalence of genetic mutations in PTCs over the last few decades, supporting the possibility of a variation over time in the underlying etiological factors (5). This review addresses the treatment of thyroid cancer patients with a class of drugs referred to as kinase inhibitors (KIs), which include small molecule KIs directed against tyrosine kinases and less often serine/threonine kinases. These drugs target specific oncogenic proteins bearing structural alterations present in tumor cells but not normal cells. This is in contrast to cytotoxic chemotherapeutic agents, which attack specific molecular targets (such as RNA and DNA, topoisomerase II [anthracyclines], or microtubules [taxanes]) in normal cells as well as tumor cells.
Classification of Thyroid Cancer
Thyroid cancers derived from the follicular cells represent a biological and clinical spectrum ranging from the differentiated thyroid carcinomas (DTCs), PTC, and follicular thyroid carcinoma (FTC) to poorly differentiated thyroid carcinoma (PDTC) and anaplastic thyroid carcinoma (ATC) (6, 7). There is a great range of biological aggressiveness among these cancers, perhaps more so than with any solid organ malignancy. The 25-year cause-specific survival is 95% for patients with PTC and 66% for patients with FTC (8). The 10-year survival in patients with PDTC is approximately 40%, and ATC is virtually incurable, with a median survival of 4–12 months from the time of diagnosis (9).
Medullary thyroid carcinoma (MTC) arises from the neural crest-derived parafollicular C cells of the thyroid and accounts for 3–5% of thyroid cancers. Most often, MTC occurs sporadically, but in 25% of cases it is hereditary, presenting as multiple endocrine neoplasia (MEN) type 2A (∼80%), MEN2B (∼5%), or familial MTC (∼15%). Treatment of MTC at an early stage is associated with a 25-year cause-specific survival of 80%; however, 10-year survival is less than 50% in patients with distant metastases (8, 10). The prognosis is especially favorable in family members with hereditary MTC who are diagnosed by direct DNA analysis and treated by early thyroidectomy before MTC develops or while it is still confined to the thyroid gland (11).
The Molecular Genetics of Thyroid Cancer
Over the last three decades, advances in molecular biology and biotechnology have markedly changed the management of many types of cancer. The emergence of next-generation DNA sequencing enabled the identification in liquid and solid tumors of somatic or germline mutations or chromosomal rearrangements driving oncogenesis. Figures 1 and 2 highlight the major signaling pathways and the most common mutations in thyroid cancers originating from follicular or parafollicular cells (12). These mutations are the basis for the design of clinical trials of KIs in thyroid cancer.
Figure 1.
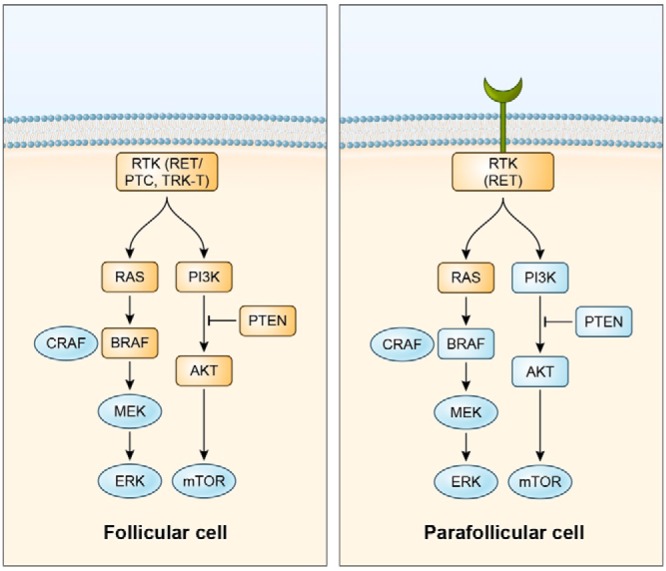
Oncogenic lesions in the growth factor signaling cascade are depicted (orange boxes) in follicular (left) and parafollicular (right) cell-derived thyroid neoplasms. PTCs are characterized by the presence, in a fraction of cases, of gene rearrangements (RET/PTC or TRK-T) leading to the oncogenic conversion of receptor tyrosine kinases (RTKs) RET or NTRK1. In follicular-cell derived thyroid cancers overall, most of the other mutations target either the “MAPK” (gain-of-function mutations in RAS or BRAF) or the “AKT” (gain-of-function mutations of PI3K or AKT or loss-of-function mutations of PTEN) arm of the RTK signaling cascade. Gene rearrangements of the PPARγ nuclear receptor (to PAX8 or less commonly to CREB3L2) represent another genetic lesion commonly (∼35%) found in FTC; this is not represented here because it appears to be functionally distinct (targeting transcriptional regulation rather than cytoplasmic signal transduction) from the other reported lesions. In most cases, parafollicular cell-derived thyroid cancers (MTC) feature, in a mutually alternative fashion, activating point mutations of RET or RAS. Familial MTC is virtually always caused by germline RET mutations.
Figure 2.

Subtypes of thyroid neoplasms, arising from either follicular cells or parafollicular cells, are depicted together with genes bearing the most prevalent oncogenic driving lesions (see text for details). RTK, receptor tyrosine kinase. Arrow indicates area of parafollicular thyroid C cells.
Mutations or chromosomal rearrangements in the MAPK and phosphatidylinositol 3-kinase (PI3K)/AKT pathways cause the most common forms of thyroid cancer. For example, 70% of PTCs have mutually exclusive activating point mutations of HRAS, KRAS, NRAS, or BRAF, or chromosomal rearrangements of genes encoding the receptor tyrosine kinases RET (REarranged during Transfection) or NTRK1, named RET/PTCs or TRK-Ts, respectively (13). The most common mutation, BRAFV600E, is present in 40–50% of PTCs, including 10–15% of follicular variant PTCs, 60% of classic PTCs, and greater than 90% of tall cell variant PTCs. The mutation does not occur in benign thyroid nodules or other histological types of thyroid cancer, except for PDTCs and ATCs derived from PTCs. Mutations in HRAS, KRAS, or most commonly NRAS occur in 25% of FTCs, 15% of PTCs (almost always the follicular variant of PTC), and 20–40% of follicular adenomas (FAs). BRAFV600E mutations are present in approximately 15% of PDTCs. ATCs may share RAS and BRAF mutations with PTC and FTC. Most ATCs have mutations in p53 and CTNNB1 (B-catenin) (14). Very recently, mutations in the telomerase (TERT) promoter were identified in thyroid cancer, mainly in ATC (15).
Chromosomal rearrangements are a characteristic feature of PTC. In the most common rearrangements, RET/PTCs, there is a fusion of the 3′-terminal portion of RET, coding for the tyrosine kinase domain, and the 5′-terminal sequence of unrelated genes, leading to constitutive activation of RET kinase. There have been several RET/PTC rearrangements reported, the most common being RET/PTC1 and RET/PTC3 (16). The prevalence of RET/PTC in PTC ranges from 20–40%, partly depending on geographic variability and the known higher incidence of these rearrangements in children and older individuals subjected to radiation exposure, particularly at an early age (17, 18). Other less common rearrangements in PTC associated with radiation exposure include the AKAP9/BRAF fusion, the PAX8/PPARγ fusion, and others (19–21). The PAX8/PPARγ fusion most commonly (10–65%) occurs in FTCs and in 0–50% of FAs, depending on the methodology and the number of tumors studied (22, 23). The FTC developing in patients with Cowden's syndrome features loss of the tumor suppressor gene, phosphatase and tensin homolog (PTEN) (24).
The mutational profile of metastatic DTCs is characterized by enrichment of mutations in PI3KCA and AKT1, which has important implications related not only to prognosis but also to the design of therapeutic regimens for patients with advanced disease (25). The progression of DTC to PDTC and ATC is most likely the result of additional genetic mutations developing after the primary oncogenic event, which provide the tumor with a more aggressive growth initiative. Alternatively, the cancer phenotype may be dictated by the differential nature of the stem cells capable of initiating PTCs, FTCs, and ATCs (26).
In a recent molecular study of 27 Hurthle cell tumors, classified by the World Health Organization as an oncocytic variant of FTC, there were no mutations detected in BRAF, PI3KCA, or PTEN and no rearrangements in RET/PTC or PAX8/PPARγ, although three patients had a RAS mutation (27, 28). The global copy number alterations in Hurthle cell tumors, primarily large regions of gain on chromosomes 5, 7, 12, and 17, were quite different from those found in PTC and FTC. Also, differential gene expression profiles showed no correlation between Hurthle cell carcinomas and PTC or FTC. These findings suggest that Hurthle cell tumors are a unique class of thyroid malignancies, quite different from DTCs at the genetic level. Further molecular studies are needed to identify possible therapeutic targets (28).
Virtually all patients with MEN2A, MEN2B, and familial MTC have germline mutations in the RET proto-oncogene, and approximately 50% of sporadic MTCs harbor somatic RET mutations. Somatic mutations in HRAS, KRAS, and rarely NRAS occur in 10–40% of sporadic MTCs and are almost always mutually exclusive with RET mutations (29–31).
Conventional Treatment of Patients With Thyroid Cancer
Total thyroidectomy, with or without resection of lymph nodes in the central neck or the lateral neck, is the primary treatment for patients with DTC, PDTC, MTC, and ATC (if technically feasible). After thyroidectomy, patients receive maintenance levothyroxine. Patients with DTC and PDTC who have persistent disease, or are at high risk for recurrent disease, are treated with radioactive iodine. Only 25% of patients with advanced DTC or MTC respond to single agent or combinatorial cytotoxic chemotherapy, and responses are characterized by partial remissions of short duration and significant toxicity (32, 33). However, regimens of chemotherapy combined with molecularly targeted therapeutics have shown efficacy in other tumors, examples being the combination of the epidermal growth factor receptor (EGFR) antibody, cetuximab, with irinotecan in patients with metastatic colon cancer and the combination of the human epidermal growth factor receptor 2 (HER2) KI lapatinib with capecitabine in patients with advanced breast cancer (34–36). Similar approaches are being initiated in patients with thyroid cancer. Cytotoxic chemotherapy consisting of taxanes, anthracyclines, or platin, combined with external beam radiation therapy, preferably intensity-modulated radiation therapy, may offer benefit in selected patients with ATC and in some patients with advanced PDTC (37). No prospective randomized trials of systemic chemotherapy, molecularly targeted agents, or vascular stabilizing agents have shown prolonged survival or improvement in quality of life in patients with ATC (37). The primary use of external beam radiation therapy in patients with advanced DTC or MTC is to treat isolated metastases, especially of bone or brain.
The Introduction of Molecular Targeted Therapeutics for Patients With Advanced Stage Malignancies
The discoveries of the genetic mutations and chromosomal rearrangements causing various cancers stimulated the pharmaceutical industry to develop small molecule inhibitors, rationally designed to block specific targets, most commonly protein kinases. The rationale was that KIs would exploit the state of “oncogene addiction,” whereby cancer cells depend upon the constitutive activation of an oncogene for maintenance of their malignant phenotype (38). This new class of drugs was thought to have distinct advantages over cytotoxic chemotherapy due to the selective rather than general effect on the body's cells. Generally, this proved to be true, although the selective effect was relative because most KIs target more than one protein and their associated toxicity can be appreciable, especially if two KIs with overlapping target profiles are administered simultaneously. Generally, the KIs have been well tolerated, and adverse events have been managed, either by treatment of symptoms or by dose alterations. The long-term effects of individual KIs are unknown, and significant late toxicity, if evident, may limit clinical utility.
The first successful treatment of a malignancy based on targeting a mutant oncoprotein kinase was in BCR-ABL, Philadelphia chromosome positive, chronic myelogenous leukemia (CML), where the tyrosine KI, imatinib, induced complete cytogenetic responses at 60 months in 87% of patients (39).
The Efficacy of KIs in Patients With Advanced Thyroid Cancer
The studies with CML gave hope and even expectation that effective KIs could be developed for other liquid and solid tumors where the initiating oncogenic event was known and targetable. Armed with an arsenal of promising small molecule therapeutics, clinical oncologists initiated clinical trials to evaluate specific drugs in patients with a number of advanced malignancies. The results, however, have been less impressive than those seen with CML, the characteristic responses being partial remission of growth or stable disease for variable time periods followed by tumor progression.
Based on observations in preclinical studies, clinical trials of KIs were initiated in patients with advanced PTC, FTC, PDTC, and ATC. Almost all trials evaluated a single drug, and typical responses were partial remissions, ranging from 0 to 63%, or stable disease (as determined by response evaluation criteria for solid tumors [RECIST] (40–65) (Tables 1–4). Until this year, the U.S. Food and Drug Administration (FDA) had approved no KI, alone or in combination with another therapeutic, for treatment of patients with advanced PTC, FTC, PDTC, or ATC. Recently completed, however, was a double-blind, randomized, phase III trial evaluating the efficacy and safety of the KI, sorafenib, in patients with progressive, locally advanced or metastatic 131I refractory DTC. There was a statistically significant improvement in progression-free survival (PFS) of patients treated with sorafenib, compared to placebo, leading to FDA approval of sorafenib for patients with advanced DTC (Table 4) (66, 67).
Table 1.
Phase II Clinical Trials of KIs in Patients with DTC
| Drug | Study | No. of Patients | PR, % | PFS, mo | Ref. |
|---|---|---|---|---|---|
| Axitinib | Phase II | 45 | 31 | NA | 40 |
| Gefitinib | Phase II | 18 | 0 | 3.7 | 54 |
| Motesanib | Phase II | 93 | 14 | 10 | 41 |
| Pazopanib | Phase II | 37 | 49 | 11.7 | 42 |
| Selumetinib | Phase II | 32 | 3 | 8 | 57 |
| Phase II | 8 | 63 | 6a | 65 | |
| Sorafenib | Phase II | 30 | 23 | 19.7 | 43 |
| Phase II | 41 | 15 | 15 | 58 | |
| Phase II | 31 | 25 | 14 | 61 | |
| Sorafenib plus tipifarnib | Phase II | 22 | 4.5 | 20 | 55 |
| Sunitinib | Phase II | 29 | 28 | NA | 44 |
| Vandetanib | |||||
| Randomized phase II trial | Phase II | 72 | 8 | 11.1 | 59 |
| Control | 73 | 5 | 5.9 | 59 |
Abbreviations: PR, partial response (RECIST); NA, not available.
Patients were only followed for 6 months after treatment.
Table 2.
Phase II Clinical Trials of KIs in Patients with MTC
| Drug | Study | No. of Patients | PR, % | PFS, mo | Ref. |
|---|---|---|---|---|---|
| Cabozantinib | Phase II | 37 | 29 | NA | 50 |
| Imatinib | Phase II | 9 | 0 | NA | 53 |
| Imatinib | Phase II | 15 | 0 | NA | 52 |
| Motesanib | Phase II | 91 | 2 | 12 | 45 |
| Sorafenib | Phase II | 16 | 6 | 17.9 | 46 |
| Sorafenib plus tipifarnib | Phase II | 13 | 38 | 15 | 55 |
| Sunitinib | Phase II | 6 | 50 | NA | 44 |
| Vandetanib (300 mg/d) | Phase II | 30 | 20 | 27.9 | 48 |
| Vandetanib (100 mg/d) | Phase II | 19 | 16 | NA | 49 |
Abbreviations: PR, partial response (RECIST); NA, not available.
Table 3.
Phase II Clinical Trials of KIs in Patients with ATC
| Drug | Study | No. of Patients | PR, % | PFS, mo | Ref. |
|---|---|---|---|---|---|
| Imatinib | Phase II | 8 | 25 | NA | 60 |
| Pazopanib | Phase II | 15 | 0 | NA | 62 |
| Sorafenib | Phase II | 20 | 10 | 1.9 | 63 |
| Phase II | 4 | 0 | NA | 58 | |
| Axitinib | Phase II | 2 | 50 | NA | 40 |
| Gefitinib | Phase II | 5 | 0 | NA | 54 |
Abbreviations: PR, partial response (RECIST); NA, not available.
Table 4.
Phase III Clinical Trials of KIs in Patients with DTC and MTC
| Cancer | Study | E/C | HR | PFS (E/C) | Ref. |
|---|---|---|---|---|---|
| MTC | |||||
| Vandetanib (300 mg/d) | Phase III | (231/100) | 0.46a | 30.5/19.3 | 51 |
| Cabozantinib (140 mg/d) | Phase III | (219/111) | 0.28b | 11.2/4.0 | 64 |
| DTC | |||||
| Sorafnib (400 mg/d) | Phase III | (207/210) | 0.58c | 10.8/5.8 | 66 |
Abbreviations: E/C, experimental group/placebo group; HR, hazard ratio.
95% confidence interval (CI), 0.31–0.69; P = .0001.
95% CI, 0.19 to 0.40; P < .001.
95% CI, 0.45–0.75; P < .0001.
There are significant preclinical data supporting the efficacy of combinatorial therapy in patients with DTC and MTC. The simultaneous administration of PI3K and MAPK kinase (MEK) inhibitors has shown efficacy in the treatment of PI3K- and RAS-mutant cancer models (68). Concurrent genetic alterations in the RAS/BRAF/MAPK and PI3K/AKT/mammalian target of rapamycin (mTOR) signaling pathways are common in aggressive thyroid carcinomas, and mutated RAS and PTEN strongly cooperated in vivo, driving formation of aggressive thyroid carcinomas in a conditional transgenic mouse model (25, 69–72). Consistently, several reports demonstrate efficacy of combined inhibition of RAS/BRAF/MAPK and PI3K/AKT/mTOR pathways in preclinical thyroid cancer models. Dual targeting of MEK (AZD6244) or BRAF (PLX4032) with AKT (MK2206), or MEK (RDEA119, AZD6244) with mTOR (temsirolimus, rapamycin) showed potent synergism in thyroid cancer cells, including ATC cells (73–75). Similarly, combination of a PI3K/mTOR KI (NVP-BEZ235) with a multitargeted KI (RAF265), inhibiting vascular endothelial growth factor receptor 2, RET, and RAF kinases (RAF265), was effective against DTC and MTC cell lines both in vitro and in mouse xenografts (76). In RET mutant MTC cells, a MEK KI, AZD6244, synergized with a multikinase KI with activity against RET (sorafenib) and was able to prevent rebound ERK phosphorylation upon RET inhibition (77). Finally, mTOR pathway activation is common in human MTC, and combined targeting of RET (AST487) and mTORC1/mTORC2 kinases (INK128) not only suppressed growth but also killed RET mutant MTC cells (78, 79).
A recent novel approach in clinical trial design demonstrated the ability of the KI selumetinib to reverse refractoriness to radioactive iodine therapy in patients with DTC (57, 65). Treatment was based on 124I positron-emission tomography, performed before and after 4 weeks of selumetinib, to quantify that iodine uptake in tumor cells was sufficient to administer the subsequent desired dose of 131I. Redifferentiation in response to selumetinib was successful in 12 of 20 patients, eight of whom while receiving selumetinib were treated with 2000 cGy or more of 131I. There were confirmed and prolonged partial remissions in five patients and stable disease in three patients associated with an 89% mean reduction in thyroglobulin levels. The brief exposure to selumetinib before 131I treatment is a particular appeal of this regimen (65). The results of this study are the basis for a larger clinical trial evaluating this therapeutic regimen in patients with DTC.
Clinical trials of KIs in patients with ATC have been discouraging and were characterized by either a lack of responses or infrequent responses, few of which met RECIST criteria for partial or complete remissions (Table 3). The low response rates are almost certainly due to the lack of a targetable mutation in ATC cells and the markedly increased growth rate of most of the cells. In a recent case report, a patient with a rapidly progressive ATC, which contained a BRAFV600E mutation, experienced a near-complete response after 4 weeks of vemurafenib, a selective mutant BRAF KI (80).
Clinical trials of vascular destabilizing agents, either alone or in combination with chemotherapy, have shown little efficacy in patients with ATC; however, it is of interest that there have been rare cases of complete remissions for prolonged periods (81–84). With single agent KIs showing minimal efficacy in patients with ATC, investigators explored in preclinical models the therapeutic possibilities of combinatorial therapy with one or more KIs or a KI combined with a chemotherapeutic agent. For example, the dual PI3K/mTOR inhibitor NVP-BEZ235 (BEZ235) showed efficacy as a single agent in thyroid cancer cells, and a combination of BEZ235 and paclitaxel demonstrated synergistic effects against ATC in vitro (85). Similarly, the combination of imatinib and docetaxel significantly enhanced apoptosis of ATC cells compared to treatment with either single agent (86). Also, preclinical studies in ATC cellular models demonstrated efficacy, either alone or in combination, of targeted agents of classes other than KIs such as all-trans-retinoic acid, the proteasome inhibitor bortezomib, histone deacetylase inhibitors, and peroxisomal proliferator-activated receptor-γ (PPAR-γ) agonists (87–91).
Although pazopanib, a multikinase inhibitor, induced no RECIST responses in patients with ATC, recent preclinical studies exploited combinations of this drug with agents of different classes in ATC models (62, 92). A series of studies demonstrated efficacy of KIs against aurora family kinases in ATC cells and pazopanib, with activity against vascular endothelial growth factor receptor and aurora A kinases, and produced synergistic antitumor effects in ATC models when combined with the microtubule inhibitor, paclitaxel, an effect that was attributed to its aurora A inhibitory activity (93–95). A proof-of-concept of the validity of this approach was obtained in an ATC patient (92). These data served as the basis for an ongoing clinical trial [Radiation Therapy Oncology group 0912, “A Randomized Phase II Study of Concurrent Intensity Modulated Radiation Therapy (IMRT), Paclitaxel and Pazopanib (NSC 737754)/Placebo, for The Treatment of Anaplastic Thyroid Cancer”] (96). Another promising therapeutic regimen combined paclitaxel with efatutazone (RS5444), a highly potent agonist of PPAR-γ. In preclinical studies, efatutazone inhibited ATC cell proliferation by up-regulation of RhoB and p21 and potentiated apoptosis when combined with paclitaxel (87, 97). In a recent phase I trial of patients with advanced ATC, this combinatorial regimen was found to be safe and feasible. Furthermore, one of 15 patients achieved a durable response, and five patients had stable disease (98).
Phase II clinical trials of several KIs in patients with advanced MTC have also shown partial remissions ranging from 0 to 50% (Table 2). Recently, a phase III prospective, randomized, placebo-controlled, double-blind clinical trial of vandetanib, a multitargeted KI, in patients with locally advanced or metastatic disease demonstrated significant improvement in PFS, compared to placebo (51). The FDA approved vandetanib for the treatment of patients with advanced MTC and has since approved a second KI, cabozantinib, after a phase III study also demonstrating significant improvement in PFS compared to placebo (64).
Rather than review the experience with individual clinical trials of various KIs in thyroid cancer, the reader is referred to recent excellent articles on the subject (99–101).
A major focus of the pharmaceutical industry has been and will continue to be the development of KIs that bind to a specific target with great avidity and minimal associated toxicity. With the “one drug one target” approach, however, it is unlikely that any single KI will be curative because multiple interconnected pathways are active in cancer cells, and blockade of a single node in a network will result in compensatory activation of other pathways, thereby overcoming drug effects (102). Understanding the activation of alternate pathways is key to developing effective combinatorial therapy for patients with thyroid cancer.
Challenges Facing Clinical Investigators Seeking to Develop More Effective Therapeutics for Thyroid Cancer
We have reached a plateau in the treatment of patients with advanced thyroid cancer, and most of the “low hanging fruit” has been plucked (103). The stark reality is that regardless of the initial response to a single KI, acquired drug resistance ultimately develops, leading to disease progression and death. Molecular studies of tumor cell lines, tumor xenografts, and tumor and matched normal tissues collected from patients participating in clinical trials of KIs will be fundamental to understanding the mechanisms of resistance and to the development of more effective combinatorial therapy. Unfortunately, to date serial tumor and normal tissue samples have been collected infrequently, if at all, from patients participating in clinical trials of KIs.
Overcoming mechanisms of resistance to KIs
We have little understanding of the mechanism(s) of resistance developing in thyroid cancers treated with a KI; however, it is instructive to review what has been learned from studies of other malignancies that feature driver oncogenic mutations targeting protein kinases similar to those found commonly in thyroid cancer. Overall, there are two major types of resistance to KIs, depending on whether it is present before treatment (“primary” or “innate” resistance) or it develops after an initial response to treatment (“secondary” resistance). In both cases, underlying molecular mechanisms can be mutational or nonmutational but are based on a rewiring of intracellular signaling or gene transcription mechanisms (102). A prototypical example of resistance formation to a KI is represented by CML, where resistance is due most often to secondary point mutations in BCR-ABL (particularly in the so-called “gate-keeper” T315 residue) that prevent imatinib binding to the ABL kinase. Less often, resistance is due to genomic amplification of BCR-ABL, the development of secondary clonal abnormalities, constitutive activation of downstream signaling molecules, or insufficient plasma levels of imatinib, either due to increased binding proteins or to pharmacokinetic variability (104). It is worth mentioning that RET mutations targeting the V804 residue corresponding to ABL T315 confer resistance to the RET KI, vandetanib, in vitro and thus may mediate resistance to this KI in patients with MTC (105).
Alternatively, secondary resistance to KIs can be driven by second-site mutations. These mutations target signaling components that typically function either parallel to or downstream from the drug target, thereby bypassing drug effects. For instance, secondary resistance of colorectal cancer patients to cetuximab (a monoclonal antibody directed against the EGFR tyrosine kinase) involves secondary mutations in KRAS (106, 107). Similarly, secondary resistance of lung adenocarcinoma cells to EGFR KIs includes not only second-site EGFR mutations (primarily in the T790 residue corresponding to T315 in ABL), but also amplification of the MET kinase and mutations in the downstream RAS, BRAF, or PIK3CA genes (108). It is noteworthy, that these same mechanisms of resistance may exist in both lung and colorectal cancer cells even prior to treatment (109, 110).
Based on the high prevalence of the BRAFV600E mutation in PTCs and in a fraction of ATCs, the results of treatment with the BRAF KI, vemurafenib, in patients with BRAF mutant melanoma and colon cancer are particularly instructive. In a clinical trial of vemurafenib in patients with BRAFV600E-mutated melanomas, the overall response rate exceeded 70% and lasted for more than 7 months before resistance developed (111). Thus, BRAFV600E mutant melanomas are primarily sensitive to a BRAF KI. Conversely, melanomas bearing only wild-type BRAF alleles are primarily resistant to a BRAF KI because they paradoxically activate RAF proteins and MAPK signaling, thereby inducing vigorous cellular proliferation (112). However, even if initially responsive to vemurafenib, BRAFV600E mutant melanomas develop secondary resistance through mechanisms including: overexpression of PDGFRα and IGF-1R receptors, secondary NRAS and MEK1 mutations, overexpression of the serine/threonine kinase COT, expression of a drug-resistant splicing variant of the BRAFV600E kinase, as well as non-cell autonomous mechanisms, such as exposure to tumor stromal-derived hepatocyte growth factor (113–117). Differently from melanoma, BRAFV600E-mutated colorectal cancer features primary resistance to a BRAF KI, and treatment with vemurafenib is relatively ineffective. Mechanistically, the inhibition of BRAF relaxes a negative feedback loop that normally suppresses the activity of EGFR, a receptor that is abundantly expressed in colon cells but not in melanocytes. The released EGFR activates RAS, which induces the formation of RAF protein dimers, against which vemurafenib is ineffective (118). These findings suggest that advanced BRAFV600E-mutated colorectal carcinomas would respond to a BRAF inhibitor, such as vemurafenib, combined with an EGFR inhibitor, such as the kinase-targeted antibody cetuximab or the KI gefitinib (119). Importantly, a similar mechanism was demonstrated in vitro in BRAFV600E-mutant thyroid carcinoma cells that rapidly adapted to treatment with BRAF KIs, secondary to induction of EGFR-family members HER3 and HER2. Importantly, the HER2/HER3 inhibitor lapatinib restored sensitivity to BRAF blockade, highlighting the importance of these studies in the design of logical combinatorial regimens (120).
The foregoing examples emphasize the marked variability in response to the same KI in three different epithelial tumors with the same BRAFV600E mutation and demonstrate the marked variability and tissue specificity in mechanisms of resistance (104, 121). These studies also demonstrate that assays to identify phosphorylation of receptor tyrosine kinases or other signaling proteins may be deceptive because the design of effective combinatorial therapies will depend on a thorough understanding of the lineage-specific determination of response to specific KIs (120).
Finally, this spectrum of studies serves to elucidate how molecular analysis of tumor cells can clarify the causes of treatment failure, information of great value in the development of subsequent generation KIs and the design of effective combinatorial therapy. For instance, in the case of CML, understanding mechanisms of resistance to imatinib was critical to the development of dasatinib, nilotinib, and bosutinib, second- and third-generation KIs with activity against all known imatinib-resistant mutations except T315I (104, 123). More recently ponatinib, a potent inhibitor of BCR-ABL and known resistant mutants, including T315I, was approved by the FDA for the treatment of CML (124). The current question in patients with CML is whether any single agent should be used as first-line therapy or, alternatively, whether a combination of drugs should be administered (and in what sequence) in an attempt to prevent outgrowth of resistant cells. There are early examples of the effectiveness of combined imatinib and nilotinib in the treatment of patients with imatinib-refractory blast phase CML who were initially unresponsive to, or could not tolerate, either agent alone (125, 126).
Dealing with tumor heterogeneity
Cancers are regarded as “ecosystems” of evolving clones, according to a Darwinian-like process of somatic evolution, and may therefore feature spatial mutational variation within different regions of the same tumor (127, 128). Intratumor genetic heterogeneity is a common feature of solid malignancies, with some genetic alterations presenting as subclonal events displayed only by a fraction of cells within the tumor. Such heterogeneity leads to a sampling bias and errors in interpretation of mutational data from a single tumor biopsy. Moreover, intratumor heterogeneity is particularly relevant to KIs. On one hand, it facilitates the development of resistance to a KI, where treatment induces selective pressure leading to the emergence of drug-resistant malignant cell clones. On the other hand, the presence of a specific oncoprotein in a minority of cancer cells, rather than the majority, would argue against using KI-based therapies directed against that particular oncoprotein (129). It follows that it is important to analyze at high resolution multiple biopsies from the primary tumor and metastases to ascertain the homogeneity of a given mutation in the tumor mass. This not only confirms the existence of heterogeneity but also may provide useful information regarding disease progression, prognosis, and response to therapy. For example, the molecular analysis of serial biopsies of Barrett's esophagus, a premalignant condition, defines a pattern of heterogeneity predictive of progression from a benign state to esophageal carcinoma (130).
The homogeneous or heterogeneous presence of some oncogenic mutations in thyroid cancer is still a matter of debate. However, it is important to consider that varying results of studies may be due to differences in the sensitivity or specificity of analytical techniques. Some studies have questioned the clonal nature of the BRAFV600E mutations in PTC, showing that a BRAF mutation is frequently an oligoclonal event (131, 132). Instead, a recent study using both immunohistochemistry and mass spectrometry genotyping demonstrated homogeneous staining for mutated BRAF protein in the vast majority of thyroid cancer cases, pointing to BRAFV600E as a clonal event in thyroid cancer (133). Similarly, another study applying next-generation sequencing found that in PTC, the BRAF mutant allele frequency was consistent with a 36–88% fraction of cells with a heterozygous mutation, therefore supporting its clonal nature. The frequency of BRAF mutant alleles was lower in individual ATC samples, with the frequency of a mutant allele varying from 5 to 38% (134). The clonal nature of the BRAF mutation fits with preclinical evidence showing that, in both constitutive and conditional transgenic mice, BRAFV600E expression in thyroid follicular cells is able to initiate thyroid carcinogenesis (135, 136).
A number of studies reported a heterogeneous distribution of RET/PTC rearrangements within individual PTCs. In particular, by applying several methodologies with different sensitivity, Zhu and colleagues (137) proposed to subclassify PTCs in samples bearing clonal, subclonal, or nonclonal RET/PTC, thereby underscoring the importance of calibrating RET/PTC detection sensitivity to justify application of RET/PTC-targeted therapy. In the first group of tumors (“clonal RET/PTC”), most cells were positive for the rearrangement, these tumors being expected to respond to RET/PTC-targeted therapies. The second group of tumors had “subclonal RET/PTC,” present only in a minor portion of tumor cells. Finally, the third group of tumors had “nonclonal RET/PTC,” present only in a minute fraction of cells within the tumor, making it unlikely that these tumors would respond to RET/PTC inhibitors (137).
As far as RET mutations in MTC, a recent next-generation sequencing study found that allelic frequency of RET mutations varied from 39 to 46% (78–92% of cells with heterozygous mutations) and allelic frequency of RAS mutations varied from 34–47% (68–94% of cells with heterozygous mutations) (134).
The Goal of Personalized Medicine in Thyroid Cancer
The concept of personalized medicine can be applied to patients with cancer, and thyroid cancer in particular, considering the causal mutations described in the various types of tumors and the specific KIs that already have demonstrated varying degrees of efficacy in clinical trials. The treatment of patients with advanced disease presents a complex problem as new mutations are acquired as the tumor spreads from the primary to regional and distant sites. These mutations will not necessarily be the same in different patients with tumors of the same histology, and it may be challenging to detect them by standard sequencing techniques. However, characterization of the mutational profile is critical in the design of treatment with either single agent or combinatorial therapy. Questions arise regarding the practicality of such a personalized approach, pertaining both to the time required for the molecular analysis and the cost. Two pilot studies have addressed this issue.
In the first study, 86 patients with refractory metastatic cancer from nine centers had tissue samples submitted centrally for molecular profiling (MP) in two formats: formalin-fixed tissue blocks for immunohistochemistry and fluorescent in situ hybridization assays, and fast-frozen tissue for oligonucleotide microarray gene expression assays. All studies were performed in a Clinical Laboratory Improvement Amendment-approved laboratory. The MP approach was deemed beneficial if the individual patient had a PFS greater than 1.3 times the length of the PFS on prior therapy. In 84 of 86 patients, a molecular target was identified, and 66 patients were treated according to MP results. Eighteen (27%) of the 66 patients had a longer PFS compared to the immediately prior regimen on which they had experienced progression (138).
In a second pilot of two patients, fresh tumor tissues were thoroughly analyzed by: paired-end whole-genome sequencing, targeted exome sequencing, and paired-end transcriptome sequencing (139). Results of molecular studies were available within 24 days of biopsy at a cost of $3,600. A mock tumor board of specialty oncologists evaluated the results and considered therapeutic regimens that might benefit the patient. Analysis of molecular data from a patient with colorectal cancer revealed findings of biological interest but clinical insignificance, whereas analysis of a second patient with melanoma showed a mutational profile that would qualify her for an upcoming clinical trial (139). Thus, for patients with advanced stages of cancer, it is possible to obtain informative tumor genotyping at a reasonable cost and in a time frame that allows decision-making. There are issues with such studies related to informed consent, patient attrition, computational analysis of data, and interpretation of results. The most significant problem seems to be whether or not a clinical trial of a specific small molecule therapeutic would be available for a patient with a specific mutation (139). This matter is highly important considering the inefficiency of current cancer therapies, which benefit a minority of patients at an enormous cost.
Future Directions in the Treatment of Thyroid Cancer
Clinical investigators have also considered therapies directed at cancer-associated mechanisms other than oncogene addiction, such as: the identification of synthetic lethal drug combinatorial therapy, primarily through technology such as RNA interference screening using functional cell models, high throughput screens with chemical libraries, and RNA interference libraries in combination with chemical inhibitors (140). There have also been significant advances with cancer vaccines, as exemplified by studies in patients with melanoma and prostate cancer (122, 141). Altogether, these approaches hold great promise for the treatment of many cancers, especially thyroid cancer where most of the driving oncogenic events are known and druggable. Investigators involved in this emerging era of genomic medicine will face great challenges; however, the possibility of markedly improving the treatment of patients, and perhaps curing some of them, is real and is worth a commitment to the effort.
Acknowledgments
No funding was received for this study.
Disclosure Summary: S.A.W. has nothing to declare. M.S. has received research support from AstraZeneca PLC, and Roche. He has also participated in a collaborative study with Ariad Pharmaceuticals, Inc.
Footnotes
- ATC
- anaplastic thyroid carcinoma
- CML
- chronic myelogenous leukemia
- DTC
- differentiated thyroid carcinoma
- EGFR
- epidermal growth factor receptor
- FA
- follicular adenoma
- FTC
- follicular thyroid carcinoma
- HER2
- human epidermal growth factor receptor 2
- KI
- kinase inhibitor
- MEK
- MAPK kinase
- MEN
- multiple endocrine neoplasia
- MP
- molecular profiling
- MTC
- medullary thyroid carcinoma
- mTOR
- mammalian target of rapamycin
- PDTC
- poorly differentiated thyroid carcinoma
- PFS
- progression-free survival
- PI3K
- phosphatidylinositol 3-kinase
- PPAR-γ
- peroxisomal proliferator-activated receptor-γ
- PTC
- papillary thyroid carcinoma
- PTEN
- phosphatase and tensin homolog
- RECIST
- response evaluation criteria for solid tumors
- RET
- rearranged during transfection.
References
- 1. American Cancer Society. Cancer Facts and Figures 2012. Atlanta, GA: American Cancer Society; 2012. [Google Scholar]
- 2. Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA. 2006;295:2164–2167. [DOI] [PubMed] [Google Scholar]
- 3. Chen AY, Jemal A, Ward EM. Increasing incidence of differentiated thyroid cancer in the United States, 1988–2005. Cancer. 2009;115:3801–3807. [DOI] [PubMed] [Google Scholar]
- 4. Enewold L, Zhu K, Ron E, et al. Rising thyroid cancer incidence in the United States by demographic and tumor characteristics, 1980–2005. Cancer Epidemiol Biomarkers Prev. 2009;18:784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jung CK, Little MP, Lubin JH, et al. The increase in thyroid cancer incidence during the last four decades is accompanied by a high frequency of BRAF mutations and a sharp increase in RAS mutations [published online December 20, 2013]. J Clin Endocrinol Metab. doi:10.1210/jc.2013–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liska J, Altanerova V, Galbavy S, Stvrtina S, Brtko J. Thyroid tumors: histological classification and genetic factors involved in the development of thyroid cancer. Endocr Regul. 2005;39:73–83. [PubMed] [Google Scholar]
- 7. Robbins J, Merino MJ, Boice JD Jr, et al. Thyroid cancer: a lethal endocrine neoplasm. Ann Intern Med. 1991;115:133–147. [DOI] [PubMed] [Google Scholar]
- 8. Schlumberger MJ, Filetti S, Hay I. Nontoxic Diffuse and Nodular Goiter and Thyroid Neoplasia. 12th ed Philadelphia, PA: Elsevier; 2011. [Google Scholar]
- 9. Are C, Shaha AR. Anaplastic thyroid carcinoma: biology, pathogenesis, prognostic factors, and treatment approaches. Ann Surg Oncol. 2006;13:453–464. [DOI] [PubMed] [Google Scholar]
- 10. Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006;107:2134–2142. [DOI] [PubMed] [Google Scholar]
- 11. Skinner MA, Moley JA, Dilley WG, Owzar K, Debenedetti MK, Wells SA Jr. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med. 2005;353:1105–1113. [DOI] [PubMed] [Google Scholar]
- 12. Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol. 2011;7:569–580. [DOI] [PubMed] [Google Scholar]
- 13. Fagin JA. The Jeremiah Metzger Lecture: intelligent design of cancer therapy: trials and tribulations. Trans Am Clin Climatol Assoc. 2007;118:253–261. [PMC free article] [PubMed] [Google Scholar]
- 14. Lee J, Hwang JA, Lee EK. Recent progress of genome study for anaplastic thyroid cancer. Genomics Inform. 2013;11:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu X, Bishop J, Shan Y, et al. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer. 2013;20:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155:645–653. [DOI] [PubMed] [Google Scholar]
- 17. Santoro M, Thomas GA, Vecchio G, et al. Gene rearrangement and Chernobyl related thyroid cancers. Br J Cancer. 2000;82:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148:936–941. [DOI] [PubMed] [Google Scholar]
- 19. Ciampi R, Knauf JA, Kerler R, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest. 2005;115:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leeman-Neill RJ, Brenner AV, Little MP, et al. RET/PTC and PAX8/PPARγ chromosomal rearrangements in post-Chernobyl thyroid cancer and their association with iodine-131 radiation dose and other characteristics. Cancer. 2013;119:1792–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ricarte-Filho JC, Li S, Garcia-Rendueles ME, et al. Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. J Clin Invest. 2013;123:4935–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kroll TG, Sarraf P, Pecciarini L, et al. PAX8-PPARγ1 fusion oncogene in human thyroid carcinoma [published correction appears in Science. 2000;289(5484):1474]. Science. 2000;289:1357–1360. [DOI] [PubMed] [Google Scholar]
- 23. Nikiforova MN, Lynch RA, Biddinger PW, et al. RAS point mutations and PAX8-PPAR γ rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metab. 2003;88:2318–2326. [DOI] [PubMed] [Google Scholar]
- 24. Nelen MR, Kremer H, Konings IB, et al. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet. 1999;7:267–273. [DOI] [PubMed] [Google Scholar]
- 25. Ricarte-Filho JC, Ryder M, Chitale DA, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Todaro M, Iovino F, Eterno V, et al. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 2010;70:8874–8885. [DOI] [PubMed] [Google Scholar]
- 27. DeLellis R, Lloyd R, Heitz P, Eng C. Pathology and Genetics of Tumors of Endocrine Organs (IARC WHO Classification of Tumors). Lyon, France: World Health Organization, IARC Press; 2004. [Google Scholar]
- 28. Ganly I, Ricarte Filho J, Eng S, et al. Genomic dissection of Hurthle cell carcinoma reveals a unique class of thyroid malignancy. J Clin Endocrinol Metab. 2013;98:E962—E972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lakhani VT, You YN, Wells SA. The multiple endocrine neoplasia syndromes. Annu Rev Med. 2007;58:253–265. [DOI] [PubMed] [Google Scholar]
- 30. Moura MM, Cavaco BM, Pinto AE, Leite V. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab. 2011;96:E863—E868. [DOI] [PubMed] [Google Scholar]
- 31. Ciampi R, Mian C, Fugazzola L, et al. Evidence of a low prevalence of RAS mutations in a large medullary thyroid cancer series. Thyroid. 2013;23:50–57. [DOI] [PubMed] [Google Scholar]
- 32. Sherman SI. Cytotoxic chemotherapy for differentiated thyroid carcinoma. Clin Oncol (R Coll Radiol). 2010;22:464–468. [DOI] [PubMed] [Google Scholar]
- 33. Matuszczyk A, Petersenn S, Bockisch A, et al. Chemotherapy with doxorubicin in progressive medullary and thyroid carcinoma of the follicular epithelium. Horm Metab Res. 2008;40:210–213. [DOI] [PubMed] [Google Scholar]
- 34. Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. [DOI] [PubMed] [Google Scholar]
- 35. Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743. [DOI] [PubMed] [Google Scholar]
- 36. Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679–692. [DOI] [PubMed] [Google Scholar]
- 37. Smallridge RC, Ain KB, Asa SL, et al. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104–1139. [DOI] [PubMed] [Google Scholar]
- 38. Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. [DOI] [PubMed] [Google Scholar]
- 39. Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. [DOI] [PubMed] [Google Scholar]
- 40. Cohen EE, Rosen LS, Vokes EE, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol. 2008;26:4708–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sherman SI, Wirth LJ, Droz JP, et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med. 2008;359:31–42. [DOI] [PubMed] [Google Scholar]
- 42. Bible KC, Suman VJ, Molina JR, et al. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 2010;11:962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gupta-Abramson V, Troxel AB, Nellore A, et al. Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol. 2008;26:4714–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carr LL, Mankoff DA, Goulart BH, et al. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res. 2010;16:5260–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schlumberger MJ, Elisei R, Bastholt L, et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J Clin Oncol. 2009;27:3794–3801. [DOI] [PubMed] [Google Scholar]
- 46. Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28:2323–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De Souza JA, Busaidy N, Zimrin A, et al. Phase II trial of sunitinib in medullary thyroid cancer (MTC). J Clin Oncol. 2010;28(suppl):5504. [Google Scholar]
- 48. Wells SA Jr, Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robinson BG, Paz-Ares L, Krebs A, Vasselli J, Haddad R. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Endocrinol Metab. 2010;95:2664–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kurzrock R, Sherman SI, Ball DW, et al. Activity of XL184 (cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29:2660–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Groot JW, Zonnenberg BA, van Ufford-Mannesse PQ, et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3466–3469. [DOI] [PubMed] [Google Scholar]
- 53. Frank-Raue K, Fabel M, Delorme S, Haberkorn U, Raue F. Efficacy of imatinib mesylate in advanced medullary thyroid carcinoma. Eur J Endocrinol. 2007;157:215–220. [DOI] [PubMed] [Google Scholar]
- 54. Pennell NA, Daniels GH, Haddad RI, et al. A phase II study of gefitinib in patients with advanced thyroid cancer. Thyroid. 2008;18:317–323. [DOI] [PubMed] [Google Scholar]
- 55. Hong DS, Cabanillas ME, Wheler J, et al. Inhibition of the Ras/Raf/MEK/ERK and RET kinase pathways with the combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in medullary and differentiated thyroid malignancies. J Clin Endocrinol Metab. 2011;96:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. [DOI] [PubMed] [Google Scholar]
- 57. Hayes DN, Lucas AS, Tanvetyanon T, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18:2056–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kloos RT, Ringel MD, Knopp MV, et al. Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol. 2009;27:1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leboulleux S, Bastholt L, Krause T, et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol. 2012;13:897–905. [DOI] [PubMed] [Google Scholar]
- 60. Ha HT, Lee JS, Urba S, et al. A phase II study of imatinib in patients with advanced anaplastic thyroid cancer. Thyroid. 2010;20:975–980. [DOI] [PubMed] [Google Scholar]
- 61. Schneider TC, Abdulrahman RM, Corssmit EP, Morreau H, Smit JW, Kapiteijn E. Long-term analysis of the efficacy and tolerability of sorafenib in advanced radio-iodine refractory differentiated thyroid carcinoma: final results of a phase II trial. Eur J Endocrinol. 2012;167:643–650. [DOI] [PubMed] [Google Scholar]
- 62. Bible KC, Suman VJ, Menefee ME, et al. A multiinstitutional phase 2 trial of pazopanib monotherapy in advanced anaplastic thyroid cancer. J Clin Endocrinol Metab. 2012;97:3179–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Savvides P, Nagaiah G, Lavertu P, et al. Phase II trial of sorafenib in patients with advanced anaplastic carcinoma of the thyroid. Thyroid. 2013;23:600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Elisei R, Schlumberger MJ, Müller SP, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ho AL, Grewal RK, Leboeuf R, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368:623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brose MS, Nutting C, Jarzarb B, et al. Sorafenib in locally advanced or metastatic patients with radioactive iodine-refractory differentiated thyroid cancer: the phase III DECISION trial. J Clin Oncol. 2013;31(suppl):4. [DOI] [PubMed] [Google Scholar]
- 67. FDA Approves Nexavar to Treat Type of Thyroid Cancer. Silver Spring, MD: US Dept of Health and Human Services; November 22, 2013. [Google Scholar]
- 68. Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. García-Rostán G, Costa AM, Pereira-Castro I, et al. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005;65:10199–10207. [DOI] [PubMed] [Google Scholar]
- 70. Liu Z, Hou P, Ji M, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab. 2008;93:3106–3116. [DOI] [PubMed] [Google Scholar]
- 71. Miller KA, Yeager N, Baker K, Liao XH, Refetoff S, Di Cristofano A. Oncogenic Kras requires simultaneous PI3K signaling to induce ERK activation and transform thyroid epithelial cells in vivo. Cancer Res. 2009;69:3689–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2008;93:278–284. [DOI] [PubMed] [Google Scholar]
- 73. Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW. Dual inhibition of mitogen-activated protein kinase kinase and mammalian target of rapamycin in differentiated and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009;94:4107–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu D, Xing J, Trink B, Xing M. BRAF mutation-selective inhibition of thyroid cancer cells by the novel MEK inhibitor RDEA119 and genetic-potentiated synergism with the mTOR inhibitor temsirolimus. Int J Cancer. 2010;127:2965–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu R, Liu D, Xing M. The Akt inhibitor MK2206 synergizes, but perifosine antagonizes, the BRAF(V600E) inhibitor PLX4032 and the MEK1/2 inhibitor AZD6244 in the inhibition of thyroid cancer cells. J Clin Endocrinol Metab. 2012;97:E173—E182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clin Cancer Res. 2011;17:6482–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Koh YW, Shah MH, Agarwal K, et al. Sorafenib and Mek inhibition is synergistic in medullary thyroid carcinoma in vitro. Endocr Relat Cancer. 2012;19:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gild ML, Landa I, Ryder M, Ghossein RA, Knauf JA, Fagin JA. Targeting mTOR in RET mutant medullary and differentiated thyroid cancer cells. Endocr Relat Cancer. 2013;20:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tamburrino A, Molinolo AA, Salerno P, et al. Activation of the mTOR pathway in primary medullary thyroid carcinoma and lymph node metastases. Clin Cancer Res. 2012;18:3532–3540. [DOI] [PubMed] [Google Scholar]
- 80. Rosove MH, Peddi PF, Glaspy JA. BRAF V600E inhibition in anaplastic thyroid cancer. N Engl J Med. 2013;368:684–685. [DOI] [PubMed] [Google Scholar]
- 81. Dowlati A, Robertson K, Cooney M, et al. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res. 2002;62:3408–3416. [PubMed] [Google Scholar]
- 82. Gramza AW, Balasubramaniam S, Fojo AT, Ward J, Wells SA. Phase I/II trial of crolibulin and cisplatin in solid tumors with a focus on anaplastic thyroid cancer: phase I results. J Clin Oncol. 2013;31:6074. [Google Scholar]
- 83. Mooney CJ, Nagaiah G, Fu P, et al. A phase II trial of fosbretabulin in advanced anaplastic thyroid carcinoma and correlation of baseline serum-soluble intracellular adhesion molecule-1 with outcome. Thyroid. 2009;19:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sosa JA, Elisei R, Jarzab B, et al. Randomized safety and efficacy study of fosbretabulin with paclitaxel/carboplatin against anaplastic thyroid carcinoma [published online September 10, 2013]. Thyroid. doi:10.1089/thy.2013.0078. [DOI] [PubMed] [Google Scholar]
- 85. Lin SF, Huang YY, Lin JD, Chou TC, Hsueh C, Wong RJ. Utility of a PI3K/mTOR inhibitor (NVP-BEZ235) for thyroid cancer therapy. PLoS One. 2012;7:e46726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim E, Matsuse M, Saenko V, et al. Imatinib enhances docetaxel-induced apoptosis through inhibition of nuclear factor-κB activation in anaplastic thyroid carcinoma cells. Thyroid. 2012;22:717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Copland JA, Marlow LA, Kurakata S, et al. Novel high-affinity PPARγ agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene. 2006;25:2304–2317. [DOI] [PubMed] [Google Scholar]
- 88. Greenberg VL, Williams JM, Cogswell JP, Mendenhall M, Zimmer SG. Histone deacetylase inhibitors promote apoptosis and differential cell cycle arrest in anaplastic thyroid cancer cells. Thyroid. 2001;11:315–325. [DOI] [PubMed] [Google Scholar]
- 89. Koh CS, Ku JL, Park SY, et al. Establishment and characterization of cell lines from three human thyroid carcinomas: responses to all-trans-retinoic acid and mutations in the BRAF gene. Mol Cell Endocrinol. 2007;264:118–127. [DOI] [PubMed] [Google Scholar]
- 90. Mitsiades CS, McMillin D, Kotoula V, et al. Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J Clin Endocrinol Metab. 2006;91:4013–4021. [DOI] [PubMed] [Google Scholar]
- 91. Wunderlich A, Roth S, Ramaswamy A, et al. Combined inhibition of cellular pathways as a future therapeutic option in fatal anaplastic thyroid cancer. Endocrine. 2012;42:637–646. [DOI] [PubMed] [Google Scholar]
- 92. Isham CR, Bossou AR, Negron V, et al. Pazopanib enhances paclitaxel-induced mitotic catastrophe in anaplastic thyroid cancer. Sci Transl Med. 2013;5:166ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Arlot-Bonnemains Y, Baldini E, Martin B, et al. Effects of the Aurora kinase inhibitor VX-680 on anaplastic thyroid cancer-derived cell lines. Endocr Relat Cancer. 2008;15:559–568. [DOI] [PubMed] [Google Scholar]
- 94. Sorrentino R, Libertini S, Pallante PL, et al. Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. J Clin Endocrinol Metab. 2005;90:928–935. [DOI] [PubMed] [Google Scholar]
- 95. Wunderlich A, Fischer M, Schlosshauer T, et al. Evaluation of aurora kinase inhibition as a new therapeutic strategy in anaplastic and poorly differentiated follicular thyroid cancer. Cancer Sci. 2011;102:762–768. [DOI] [PubMed] [Google Scholar]
- 96. A Randomized Phase II Study of Concurrent Intensity Modulated Radiation Therapy (IMRT), Paclitaxel and Pazopanib (NSC 737754)/Placebo, for The Treatment of Anaplastic Thyroid Cancer. ClinicalTrialsgov 2013:NCT01236547. [Google Scholar]
- 97. Marlow LA, Reynolds LA, Cleland AS, et al. Reactivation of suppressed RhoB is a critical step for the inhibition of anaplastic thyroid cancer growth. Cancer Res. 2009;69:1536–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Smallridge RC, Copland JA, Brose MS, et al. Efatutazone, an oral PPAR-γ agonist, in combination with paclitaxel in anaplastic thyroid cancer: results of a multicenter phase 1 trial. J Clin Endocrinol Metab. 2013;98:2392–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Harris PJ, Bible KC. Emerging therapeutics for advanced thyroid malignancies: rationale and targeted approaches. Expert Opin Investig Drugs. 2011;20:1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lerch C, Richter B. Pharmacotherapy options for advanced thyroid cancer: a systematic review. Drugs. 2012;72:67–85. [DOI] [PubMed] [Google Scholar]
- 101. Haugen BR, Sherman SI. Evolving approaches to patients with advanced differentiated thyroid cancer. Endocr Rev. 2013;34:439–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bernards R. A missing link in genotype-directed cancer therapy. Cell. 2012;151:465–468. [DOI] [PubMed] [Google Scholar]
- 103. Fagin JA, Tuttle RM, Pfister DG. Harvesting the low-hanging fruit: kinase inhibitors for therapy of advanced medullary and nonmedullary thyroid cancer. J Clin Endocrinol Metab. 2010;95:2621–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Stein B, Smith BD. Treatment options for patients with chronic myeloid leukemia who are resistant to or unable to tolerate imatinib. Clin Ther. 2010;32:804–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Carlomagno F, Guida T, Anaganti S, et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene. 2004;23:6056–6063. [DOI] [PubMed] [Google Scholar]
- 106. Diaz LA Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci USA. 2012;109:E2127–E2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. [DOI] [PubMed] [Google Scholar]
- 111. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hey F, Pritchard C. A new mode of RAF autoregulation: a further complication in the inhibitor paradox. Cancer Cell. 2013;23:561–563. [DOI] [PubMed] [Google Scholar]
- 113. Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480:387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Solit DB, Jänne PA. Translational medicine: primed for resistance. Nature. 2012;483:44–45. [DOI] [PubMed] [Google Scholar]
- 119. Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. [DOI] [PubMed] [Google Scholar]
- 120. Montero-Conde C, Ruiz-Llorente S, Dominguez JM, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17:1616–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. [DOI] [PubMed] [Google Scholar]
- 123. Amsberg GK, Koschmieder S. Profile of bosutinib and its clinical potential in the treatment of chronic myeloid leukemia. Onco Targets Ther. 2013;6:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367:2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhu GR, Ji O, Ji JM, et al. Combining nilotinib and imatinib improves the outcome of imatinib-resistant blast phase CML. Acta Haematol. 2012;127:152–155. [DOI] [PubMed] [Google Scholar]
- 126. Gómez-Almaguer D, Tarín-Arzaga L, Cantú-Rodríguez O, Ceballos-López A. More about imatinib and nilotinib combination therapy in chronic myeloid leukemia. Acta Haematol. 2013;129:18–19. [DOI] [PubMed] [Google Scholar]
- 127. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368:842–851. [DOI] [PubMed] [Google Scholar]
- 129. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. [DOI] [PubMed] [Google Scholar]
- 130. Maley CC, Galipeau PC, Finley JC, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–473. [DOI] [PubMed] [Google Scholar]
- 131. Gandolfi G, Sancisi V, Torricelli F, et al. Allele percentage of the BRAF V600E mutation in papillary thyroid carcinomas and corresponding lymph node metastases: no evidence for a role in tumor progression. J Clin Endocrinol Metab. 2013;98:E934–E042. [DOI] [PubMed] [Google Scholar]
- 132. Guerra A, Sapio MR, Marotta V, et al. The primary occurrence of BRAF(V600E) is a rare clonal event in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2012;97:517–524. [DOI] [PubMed] [Google Scholar]
- 133. Ghossein RA, Katabi N, Fagin JA. Immunohistochemical detection of mutated BRAF V600E supports the clonal origin of BRAF-induced thyroid cancers along the spectrum of disease progression. J Clin Endocrinol Metab. 2013;98:E1414–E1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nikiforova MN, Wald AI, Roy S, Durso MB, Nikiforov YE. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J Clin Endocrinol Metab. 2013;98:E1852–E1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Knauf JA, Ma X, Smith EP, et al. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res. 2005;65:4238–4245. [DOI] [PubMed] [Google Scholar]
- 136. Chakravarty D, Santos E, Ryder M, et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest. 2011;121:4700–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhu Z, Ciampi R, Nikiforova MN, Gandhi M, Nikiforov YE. Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: effects of the detection methods and genetic heterogeneity. J Clin Endocrinol Metab. 2006;91:3603–3610. [DOI] [PubMed] [Google Scholar]
- 138. Von Hoff DD, Stephenson JJ Jr, Rosen P, et al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28:4877–4883. [DOI] [PubMed] [Google Scholar]
- 139. Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Brough R, Frankum JR, Costa-Cabral S, Lord CJ, Ashworth A. Searching for synthetic lethality in cancer. Curr Opin Genet Dev. 2011;21:34–41. [DOI] [PubMed] [Google Scholar]
- 141. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]