

Abstract
Context:
Gordon Holmes syndrome (GHS) is characterized by cerebellar ataxia/atrophy and normosmic hypogonadotropic hypogonadism (nHH). The underlying pathophysiology of this combined neurodegeneration and nHH remains unknown.
Objective:
We aimed to provide insight into the disease mechanism in GHS.
Methods:
We studied a cohort of 6 multiplex families with GHS through autozygosity mapping and whole-exome sequencing.
Results:
We identified 6 patients from 3 independent families carrying loss-of-function mutations in PNPLA6, which encodes neuropathy target esterase (NTE), a lysophospholipase that maintains intracellular phospholipid homeostasis by converting lysophosphatidylcholine to glycerophosphocholine. Wild-type PNPLA6, but not PNPLA6 bearing these mutations, rescued a well-established Drosophila neurodegenerative phenotype caused by the absence of sws, the fly ortholog of mammalian PNPLA6. Inhibition of NTE activity in the LβT2 gonadotrope cell line diminished LH response to GnRH by reducing GnRH-stimulated LH exocytosis, without affecting GnRH receptor signaling or LHβ synthesis.
Conclusion:
These results suggest that NTE-dependent alteration of phospholipid homeostasis in GHS causes both neurodegeneration and impaired LH release from pituitary gonadotropes, leading to nHH.
Gordon Holmes syndrome (GHS) (MIM 212840) is characterized by cerebellar ataxia/atrophy and normosmic hypogonadotropic hypogonadism (nHH), which was first described in 1908 (1). The closely associated Boucher-Neuhäuser syndrome (BNS) (MIM 215470) has chorioretinal dystrophy as an additional feature (2). BNS is believed to be a continuum of GHS. Neurological characteristics of these syndromes become gradually obvious during mostly the second or third decade and appear to be a progressive neurodegeneration. nHH, on the other hand, manifests itself as pubertal failure. Inactivating mutations in 3 ubiquitination-related genes, RNF216 (3), OTUD4 (3), and STUB1 (4), were reported in some patients with GHS, indicating genetic heterogeneity of this syndrome. Very recently, when we were preparing this manuscript for publication, a report appeared demonstrating that that BNS and GHS are caused by mutations in PNPLA6 (5). However, none of these studies provide an explanation of how these gene deficiencies relate to neurodegeneration or nHH. To shed light into the disease mechanism in these syndromes, we studied a group of multiplex families with GHS or BNS through autozygosity mapping and whole-exome sequencing and identified loss-of-function mutations in PNPLA6. Complementary studies provided significant insight into how the clinical features of these syndromes are brought about.
Subjects and Methods
Case presentations
We studied a cohort of 6 multiplex families with GHS or BNS. The ethics committee of the Cukurova University Faculty of Medicine approved this study, and full informed consent was obtained from each participant.
Family 1
The proband (II-1) is a 51-year-old Turkish man who was noted to have micropenis and undescended testicles as a child and poor sexual development as a teenager. He has 2-mL testes in the scrotum and a 4.5-cm prepubertal penis. His unsteady gait started at age 37; speech difficulty followed 1 year later.
His affected sister (II-3) is a 46-year-old woman with ataxia and primary amenorrhea. Only after starting estrogen replacement at age 19 did she begin to have breast development. She had an unsteady gait from childhood and developed speech difficulties and tremor in her 20s.
An electromyography showed sensory and motor neuropathy in both siblings. They had normal ophthalmological features.
Family 2
The clinical features of the Dutch siblings have been previously reported (2). Briefly, a 25-year-old man was seen for his poor sexual development. He had a very small scrotum and penis. A standard GnRH stimulation test did not elicit any LH response. Furthermore, after a daily sc injection of GnRH at 0.1 mg for 1 week, plasma LH levels remained below the detection limit of 0.1 mIU/mL.
His 2-years-younger sister was first seen at age 21 for primary amenorrhea and poor sexual development. On examination, a disturbed balance and gait was evident in both patients. Both siblings were diagnosed with atypical chorioretinal dystrophy at 23 years of age.
Family 3
The proband (II-1) is an 18-year-old-male Turkish patient who presented with absence of pubertal development. He had testicles of 1 mL with a penis length of 2 cm. He developed nystagmus, ataxic gait, and dysarthria at age 12. His 15-year-old brother also suffers from absence of pubertal development. He had a testicular volume of 1.5 mL and a penis length of 3 cm. He developed nystagmus without other cerebellar signs at age 14. These siblings had normal ophthalmological features.
The pedigrees of the 3 families are shown in Figure 1A. All 6 patients had a normal sense of smell by conventional testing. All patients but the younger brother in family 3 have cerebellar atrophy on brain magnetic resonance imaging (MRI) (Figure 1B). The patients had otherwise normal anterior pituitary functions with no evidence of structural lesions on MRI of olfactory bulbs and sulci or hypothalamus and pituitary regions. Their off-treatment hormonal profiles showed low or normal serum gonadotropins in the face of severely decreased serum estradiol or testosterone levels, clearly indicating HH. Additionally, GnRH stimulation test results, when available, were consistent with HH (Supplemental Table 1).
Figure 1. Pedigrees of the families with genotypes and cerebellar atrophy evident on MRI.

A, In each family, genotype status for each of 2 alleles of PNPLA6 is given below individuals in the nuclear family available to genetic study. B, Severe cerebellar atrophy evident on MRI of patient (II-3) in family 1 (left) and patient (II-1) in family 3 (right). For brain images of patients in family 2 see Ref. 2. Abbreviation: WT, wild-type.
Laboratory Methods
Genetic studies
For genome-wide single-nucleotide polymorphism (SNP) analysis we used 250K NspI SNP microarrays (Affymetrix) and analyzed data using AutoSNPa software (AutoSNPa.org). For exome sequencing, samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using Illumina HiSeq 2000 Sequencer (Macrogen). The reads were mapped against UCSC hg18. For Sanger sequencing, PCR-amplified exons and splice junctions of PNPLA6 were sequenced on an ABI PRISM 3130 autosequencer (Applied Biosystems).
To study functional effects of human mutations, we used a pCMV6_XL6 expression vector (SC115919; Origene) containing a human cDNA encoding PNPLA6 transcript variant 2 (NM_006702.2). Each of the 3 mutations found in patients were introduced into this cDNA by site-directed mutagenesis using the Quikchange Lightning site-directed mutagenesis kit according to the instructions of the manufacturer (Stratagene).
Neuropathy target esterase activity
LβT2 cells were plated at a density of 2 million cells per well in poly-lysine–treated 12-well plates and incubated overnight in complete culture medium with or without 3μM chlorpyrifos oxon (CPO) (Accustandard), an inhibitor of neuropathy target esterase (NTE) activity (6, 7). Next day, cells were washed twice with cold PBS and snap-frozen on dry ice. NTE activity was detected according to Ref. 8 with some modifications. Briefly, the cells were resuspended in 200 μL assay buffer (20mM Tris base, 0.2mM EDTA [pH 8]) and sonicated 10 seconds using an ultrasonic cell disruptor (Thermo Fisher Scientific) (amplitude = 1). Supernatants were collected after a 5-minute centrifugation at 12 000g. The assay detects the conversion of phenyl valerate into phenol, which is read spectrophotometrically. Because the NTE as well as cellular acetylcholinesterases convert phenyl valerate into phenol, NTE activity is calculated as the difference between the absorbance in a sample treated with paraoxon (PA), an inhibitor of acetylcholinesterases, from the absorbance of a sample treated with PA plus CPO (which inhibits both types of enzymes). In short, 50 μL lysate was used to determine NTE activity in the presence of 0.07 mg/mL PA (AccuStandard) or PA plus 0.170mM CPO in a microplate incubated 30 minutes at 37°C in the presence of the substrate (0.7 g/mL phenyl valerate, kindly provided by Marion Ehrich, Virginia Polytechnic Institute). The reaction was terminated by adding 50 μL 5% sodium dodecyl sulfate, 0.02% 4-aminoantipyrine, 0.5M Tris base (pH 9.6), followed by 50 μL 0.4% potassium ferricyanide. After 15 minutes of color development, absorbance was measured at 510 nm using a Spectramax M5 plate reader (Molecular Devices). The absorbance was plotted against a phenol standard curve. Protein content was determined using the 660-nm protein assay (Thermo Fisher Scientific). NTE activity was expressed as nanomoles phenyl valerate converted per minute per milligram protein.
GnRH-stimulated exocytosis
To assess the effect of NTE deficiency on GnRH-stimulated exocytosis, we used the fluorescent dye FM1–43, an amphipathic molecule that allows the visualization of membranes during the exocytosis/endocytosis cycle by virtue of its ability to reversibly partition into lipid membranes. LβT2 cells were plated at a density of 400 000 cells per well in poly-lysine–treated glass coverslips inside a 6-well plate and incubated in DMEM without phenol red containing 1% serum in the absence or presence of 3μM CPO to inhibit NTE activity. Twenty-four hours later, the cells were washed once in Hanks' balanced salt solution (HBSS) and loaded with 5μM FM1–43FX in HBSS for 10 minutes at room temperature followed by 10 minutes incubation with the dye in the presence or absence of 50nM GnRH. Cells were rinsed 6 times with cold HBSS for 8 minutes each time and fixed with 4% paraformaldehyde/PBS for 20 minutes at 4°C followed by 10 minutes incubation with 0.1 μg/mL Hoechst 33258 (to label cell nuclei) and 0.5 μg/mL wheat germ agglutinin (WGA) (Invitrogen) (to label cell membrane glycoproteins) in PBS. Coverslips were mounted on glass slides for later microscopy (9).
Cells were imaged using a Leica SP5 AOBS laser scanning confocal with a HCX Pl APO lambda blue 63 × 1.4 objective with pixels of 120 × 120 nm2 and 1 μm distance between planes. Three fluorescence channels (Hoechst 33258 excitation 405 nm/emission 410–450 nm, FM1–43FX excitation 488/emission 500–500 nm, and WGA-Alexa 633 excitation 633 nm/emission 650–700 nm) were acquired in sequential mode. To avoid bias, fields to be imaged were selected based on the 4′,6-diamidino-2-phenylindole channel with no prior information regarding the FM1–43FX distribution. Images were processed in ImageJ (10) by applying conditions similar to the ones used for pituitary lactotrophs (9): background subtraction of an image processed by smoothing the images (Gaussian blur, σ = 10), intensity threshold and analyzed particles filtered by size (100–1000 nm) and by circularity factor 0.7 to 1. Cellular contours were identified using the WGA signal and applied as a mask on top of the images to identify particles on a per-cell basis.
The procedures used for Drosophila transgenic line studies, GnRH receptor (GnRHR) signaling in Hela cells, culture of LβT2 cells, transfection of small interfering RNAs (siRNAs), RT-PCR, proliferation assays, LH immunoassay, measurement of intracellular calcium levels, and LH immunofluorescence are described in the Supplemental Materials and Methods.
Results
PNPLA6 mutations are detected in individuals with pubertal failure and cerebellar ataxia
All living members of family 1 were subjected to genome-wide SNP analysis, and we identified 3 regions of homozygosity. These regions were a 2.4-Mb region on chromosome 5 from 104.4 to 106.8 Mb, a 2.1-Mb region on chromosome 8 from 99.3 to 101.4 Mb, and a 2.0-Mb region on chromosome 19 from 5.7 to 7.7 Mb. A subsequent whole-exome sequencing identified a mutation in PNPLA6 (HUGO Gene Nomenclature Committee: 16268, NM_006702) on the homozygous region on chromosome 19. This mutation (PNPLA6-F1M) was a homozygous c.C3380G leading to p.Ser1127Cys in the catalytic domain of the NTE and was confirmed by Sanger sequencing. This mutation segregated perfectly within the nuclear family according to the rules of autosomal recessive inheritance. Upon screening for mutations in PNPLA6 in the 5 additional families with GHS/BNS in our cohort, we identified compound heterozygous mutations in 2 families. In family 2, mutation 1 (PNPLA6-F2M1) was a c.1127insG leading to a frameshift and a stop codon (p.Asp376GlyfsX18), which was inherited from the mother. The resultant protein is predicted to lack the entire catalytic domain and the C-terminal portion of the regulatory domain. The other heterozygous mutation, the family 2 mutation 2 (PNPLA6-F2M2), was a c.C3295T leading to a p.Arg1099Cys, which was inherited from the father.
In affected brothers from family 3, we identified 2 mutations, each inherited from one parent: a C-to-T change at cDNA 3931 creating the mutation p.Arg1311Trp from the mother and a Gly832fsX13 mutation due to insertion of TGTGGGCCTGGGG at cDNA 2494 from the father.
SIFT and Polypene2 predict that these substitutions affect protein function. They were not found in 100 ethnically matched healthy adult controls and were not seen in public databases including dbSNP, 1000 genomes, and Exome Variant Server. Mutations and their inheritance can be seen in Figure 1A. Mutation details including mutation pictures can be found in Supplemental Figure 1. Most notably, no mutations in the recently reported GHS genes, RNF216, OTUD4, and STUB1, were detected in the 3 families studied (3, 4). Neither these genes nor PNPLA6 exhibited mutations in 3 other multiplex families of our cohort affected by nHH and ataxia.
PNPL6 mutants fail to rescue neurodegeneration defects caused by loss of sws in Drosophila
Due to NTE being evolutionarily conserved, we used a fly model to determine whether the identified mutations interfere with the ability of PNPLA6 to rescue the neurodegenerative defect caused by loss of the Drosophila ortholog SWS. Transgenic lines were created using the PhiC31 system to achieve equal expression levels (11). The UAS-PNPLA6 constructs were then induced with the pan-neuronal Appl-GAL4 driver and the flies analyzed at 14 days of age. As shown in Figure 2, a paraffin head section from a control fly expressing only the Appl-GAL4 demonstrated the wild-type phenotype (Figure 2A). An sws fly mutant1 showed the characteristic vacuoles described in this mutant (Figure 2B). Expressing wild-type PNPLA6 in the sws1 mutant significantly reduced the lesions (Figure 2C), although it did not completely revert the phenotype to wild-type. In contrast, expression of PNPLA6-F1M did not rescue the degeneration but seemed to enhance the vacuolization (Figure 2D). Because this indicated a dominant-negative effect, we expressed this construct in a wild-type background but could not detect a phenotype (data not shown). Expressing PNPLA6-F2M1 (Figure 2E) or PNPLA6-F2M2 (Figure 2F) in sws1 also failed to rescue the vacuolization when compared with the sws mutant alone. To quantify the effects on the vacuolization, we measured the area of vacuoles (in a double-blind analysis) in a distinct and easily identifiable brain region, the deutocerebral neuropil (dn in Figure 2A). As shown in Figure 2G, this revealed a significant reduction of the vacuolization when the wild-type PNPLA6 construct was expressed in sws1 but not with PNPLA6-F2M1 or PNPLA6-F2M2. This study also confirmed that expression of PNPLA6-F1M significantly enhanced the vacuolization. Expression of the constructs was confirmed by Western blots (Figure 2H).
Figure 2. Expression of mutant PNPLA6 does not rescue the neuronal degeneration in sws1.

A, Paraffin head section from a control fly expressing only the Appl-GAL4 driver construct. B, A head section from an sws1 control fly reveals the vacuoles characteristic for this mutant. C–F, Expressing wild-type PNPLA6 in sws1 (C) reduced vacuole formation, whereas expressing PNPLA6-F1M (D), PNPLA6-F2M1 (E), or PNPLA6-F2M2 (F) in sws1 did not result in fewer vacuoles. The arrows point to some of the vacuoles found in these flies. Abbreviations: cb, central brain; dn, deutocerebral neuropil; ol, optic lobes; re, retina. Scale bar, 50 μm. G, Mean area of vacuoles in square micrometers in the different genotypes tested. Whereas expression of the wild-type PNPLA6 reduced the vacuolization significantly (P < .05), PNPLA6-F1M significantly enhanced it (P < .01). The other 2 constructs had no significant effect on the vacuolization in sws1. SEMs and the number of brain hemispheres analyzed are indicated. All flies were 14-day-old males. H, Western blot showing similar expression levels of the different constructs with the exception of PNPLA6-F2M1, which cannot be detected by the antibody. Homogenates from 5 fly heads were loaded in each lane. Flies were 1- to 3-day-old males. A loading control using antitubulin is shown below.
Human PNPLA6 does not change GnRHR signaling
We first ascertained whether mutated PNPLA6 influenced GnRHR signaling. Results are shown in Supplemental Results along with related Supplemental Figure 2.
Inhibition of NTE activity reduces the LH response to GnRH
To generate a model of pituitary dysfunction similar to that seen in our patients with GHS, we used the immortalized mouse pituitary gonadotrope cell line LβT2 (12). We could transfect siRNAs into LβT2 cells with only moderate (50%) efficiency, resulting in about 40% decrease in PNPLA6 mRNA abundance (Figure 3A) and no change in NTE activity (Figure 3B). We determined the maximal inhibitory concentration of the NTE inhibitor CPO (Figure 3C). We chose 3μM as a dose that does not affect cell proliferation (Figure 3D) and found that this dose not only suppresses NTE activity in LβT2 cells by nearly 70% (Figure 3C) but also inhibits significantly the LH response to GnRH (Figure 3E). This effect is not due to inhibition of acetylcholinesterase activity because no Ache mRNA or enzymatic activity was detected in LβT2 cells (Supplemental Figure 3, A and B, respectively). Also, CPO-dependent inhibition of GnRH-induced LH release was due neither to inhibition of GnRH-dependent intracellular calcium mobilization (Figure 3F) nor suppression of LβT2 gene expression (Figure 3G).
Figure 3. Inhibition of NTE activity in LβT2 cells decreases GnRH-induced LH release.

A, Treatment of LβT2 cells with a pool of siRNAs against Pnpla6 significantly, but moderately, reduced Pnpla6 mRNA levels. B, The treatment failed to decrease NTE activity. C, Identification of the maximal inhibitory concentration of CPO on NTE activity. D, Treating LβT2 cells overnight with 3μM CPO does not change LβT2 cell proliferation (proliferation was calculated as a percentage of proliferation seen in the untreated group). E–G, Treating LβT2 cells with 3μM CPO reduces GnRH-induced LH release (E), without altering either the effect of GnRH on the intracellular Ca2+ concentration (F) or the effect of GnRH on LHβ mRNA content (G) (n = 4–6 independent observations per group). Columns are means, and vertical bars are SEMs. *, P < .05; ***, P < .001 vs control groups (Student's t test).
Inhibition of NTE activity decreases GnRH-stimulated LH exocytosis in pituitary gonadotropes
To determine whether NTE is involved in vesicle recycling in mammalian cells, and more specifically in pituitary gonadotropes, we evaluated the effect of NTE deficiency in LβT2 cells after a single exposure to GnRH. Individual exocytic events can be optically monitored using the fluorescent dye FM1–43 in both neurons and neuroendocrine cells (9). Under basal conditions LβT2 cells show very little FM1–43FX dye uptake, indicating that, as shown earlier in pituitary lactotropes (9), membrane recycling is very slow under resting conditions in LβT2 cells. Exposing the cells to GnRH resulted in a striking increase in the number of FM43-labeled vesicular membranes (Figure 4, A and B), indicating that GnRH activates the exocytosis/endocytosis cycle in pituitary gonadotropes. CPO dramatically prevented this activation. Immunohistofluorescence detection of LH in cells treated with GnRH demonstrated that most FM1–43–labeled granules also contained LH (Figure 4C). The reason for the increase in basal vesicular content after CPO-only treatment is unclear. A potential explanation is that CPO diminishes only vesicular exocytosis without affecting endocytosis. This would result in a net increase in vesicular content under resting conditions.
Figure 4. Inhibition of NTE activity represses GnRH-induced membrane recycling in LβT2 cells.
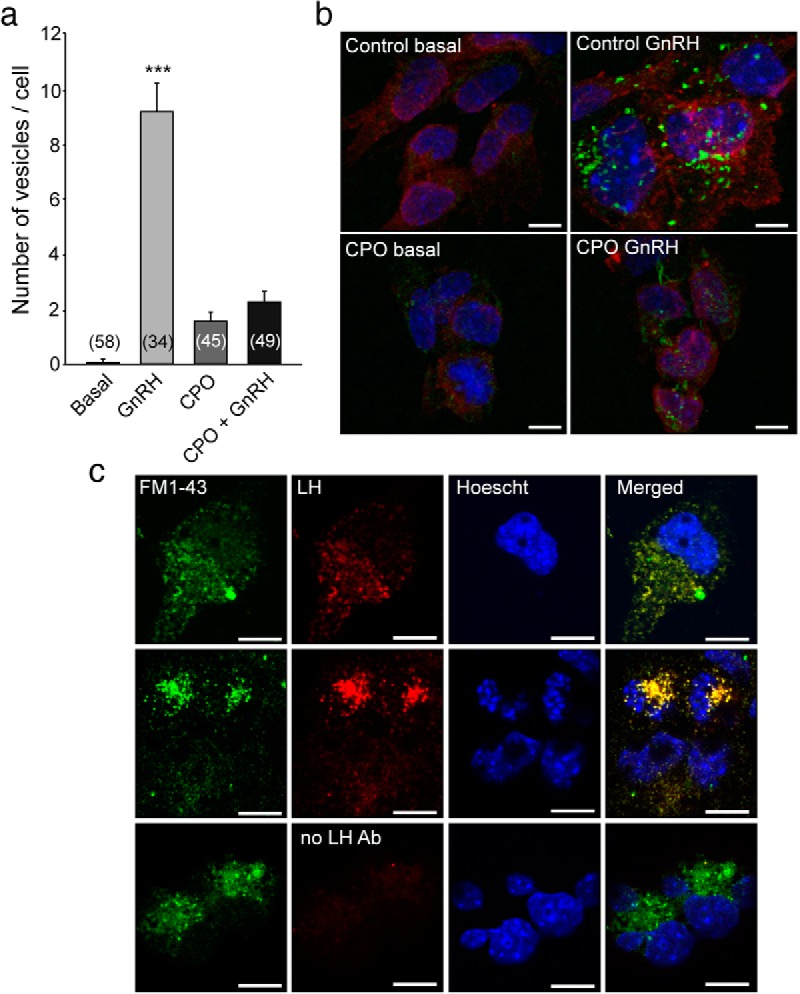
Membrane recycling was studied in LβT2 cells using the membrane dye FM1–43FX. A and B, Under basal conditions, the FM dye shows very little incorporation into the cell. When cells are stimulated with GnRH, the FM dye gets rapidly incorporated into vesicular-like structures (GnRH group in A, and control GnRH in B), indicating an increase in membrane dynamics. When NTE activity is blocked with CPO, the stimulatory effect of GnRH on vesicular recycling is inhibited. C, Vesicular structures labeled with FM1–43 contain LHβ as detected by immunofluorescence. In A, columns are means, and vertical bars are SEMs. ***, P < .001 vs all other groups (ANOVA followed by the Student-Newman-Keuls multiple-comparison test for unequal replications). Numbers in bars indicate the number of cells analyzed per treatment. B, High magnification of a representative group of cells for each treatment showing the FM1–43FX dye (labeling recently formed vesicles, in green), WGA (labeling glycoproteins of the cell membrane, in red), and Hoechst 33258 (labeling cell nuclei, in blue). Scale bars, 10 μm. C, Examples of cells with colocalization of the FM1–43FX dye (green) and LHβ immunoreactivity (red). Nuclei were identified by staining with Hoechst 33258 (blue). Scale bars, 10 μm.
Discussion
In this study, we identified loss-of-function mutations in the PNPLA6 gene in patients with GHS or BNS from 3 unrelated families. Because mutations in the same gene, ie, PNPLA6, account for patients from all 3 families, the conclusion that GHS and BNS are indeed the same entity (2) is now confirmed. Henceforth, we refer to the phenotype as GHS because Gordon Holmes was the first to describe this clinical constellation (1).
PNPLA6 encodes NTE, a lysophospholipase that converts lysophosphatidylcholine (LPC) to glycerophosphocholine. PNPLA6 is expressed in neurons throughout the brain, particularly in the cortex, Purkinje cells of the cerebellum, and hippocampus (13).
The first human mutations in PNPLA6 were reported in 2008 (14). The patients in that report had a specific neurodegenerative phenotype without cerebellar involvement, which was named hereditary spastic paraplegia (SPG39 HSP) (14, 15). The authors did not specify the status of puberty/reproductive function in their patients. However, in their family 1, male and female patients parented children, which suggests, unlike in our patients, absence of an overt reproductive dysfunction (14). Therefore, it appears that these 2 different phenotypes are due to a deficit of the same gene; ie, SPG39 HSP and GHS are allelic disorders. Very recently, an article describing PNPLA6 mutations as the cause of GHS/BNS as well as HSP was published (5). However, like previous articles (3, 4) reporting GHS caused by gene mutations, this report also does not address the underlying pathophysiology of this syndrome.
NTE is an evolutionarily conserved protein that can also be found in invertebrates (16) including yeast (17). In Drosophila, it is encoded by the Swiss-cheese (sws) gene, and the SWS protein shows 61% overall identity to human and mouse NTE in its catalytic domain. The Drosophila sws mutant is characterized by progressive degeneration of the adult nervous system, associated with apoptotic cell death and the formation of spongiform lesions (Swiss cheese appearance) (18). Therefore, we used the fly model to determine whether the identified mutations in our patients interfere with the function of PNPLA6 in the nervous system. Whereas wild-type human PNPLA6 significantly reduced the degeneration in sws, the mutant constructs did not, which clearly demonstrates the deleterious character of these mutations and points to the underlying cause of neurodegeneration in GHS.
However, establishing a causal link between PNPLA6 and the reproductive component of the syndrome is less straightforward because there is no existing literature describing an involvement of PNPLA6 in hypothalamo-pituitary-gonadal function.
When we stimulated one of our probands with GnRH in an extended fashion, the patient did not show a noticeable LH response, suggesting that the response of the pituitary gland to GnRH is compromised. Several others also made similar observations in multiple patients with GHS (19–23).
Given the fact that the LH response to extended exogenous GnRH stimulation in human patients is poor and that overt hypogonadism is absent in brain-specific knockout mice (24, 25), we focused on the pituitary as the site of dysfunction leading to nHH.
We first ascertained whether mutated PNPLA6 influenced GnRHR signaling. Because GnRHR is primarily a Gq/11-coupled receptor, we expressed wild-type and mutated PNPLA6 plus human GnRHR in HeLa cells also transfected with an nuclear factor of activated T-cells (NFAT)1c-Emerald fluorescent protein reporter gene. Translocation of NFAT-EFP from the cytoplasm to the nucleus was used as a readout for GnRHR-mediated activation of the Ca2+/calmodulin/calcineurin/NFAT pathway. As a second approach, fluorescence immunohistochemistry was used to quantify phosphorylated PKR like ERK, as a readout for GnRHR-mediated activation of the Raf/MAPK kinase/ERK pathway. We observed the expected stimulatory and inhibitory effects with GnRH and the GnRHR antagonist cetrorelix, but no significant effects of either wild-type or mutated PNPLA6, indicating that NTE actions do not involve GnRH signaling through its receptor (Supplemental Figure 2).
Then we created a state of NTE deficiency in LβT2 pituitary gonadotropes using a pharmacological approach to model GHS. Although CPO is more potent in inhibiting acetylcholinesterase than NTE (6, 7), acetylcholinesterases have been associated with increased LH secretion instead of a reduction (26). In addition, in this study, no Ache mRNA or enzymatic activity was detected in LβT2 cells (Supplemental Figure 3, A and B, respectively). In the present study, we observed that GnRH stimulation of NTE-deficient LβT2 cells resulted in diminished LH release without altering LHβ mRNA levels. These observations indicate that the reduction in LH response was not due to impaired LH synthesis. Furthermore, the lack of alterations in intracellular calcium in response to GnRH in CPO-treated cells supports the contention that NTE does not directly participate in GnRHR signaling.
Having largely excluded potential causes in afferent receptor signaling and hormone synthesis, we next investigated the regulated secretory pathway, which involves packing of peptide hormones by neuroendocrine cells into secretory vesicles for release upon calcium stimulation. In this process, fusion of the cargo vesicles to the cell membrane and release of the contents (exocytosis) is followed by endocytosis as part of a recycling mechanism (27). There is ample evidence that intracellular lipids, particularly phospholipids, are involved in this mechanism (27, 28). In yeast, LPC homeostasis and vesicular transport are linked via the phospholipid-binding protein, sec14p (17, 28–30). An inactivating mutation of the Sec14p gene disrupts vesicular transport, resulting in cell death. These effects are overcome by the overexpression of exogenous NTE (17). In addition, excess LPC has been shown to be toxic to the yeast secretory pathway (31). Based on these observations, we hypothesized that the inhibition of LH release seen in CPO-treated LβT2 cells could be due to impaired vesicular transport. When we inhibited NTE activity using CPO in cultured LβT2 gonadotropes, we detected a significant reduction in GnRH-stimulated LH exocytosis, which suggests that, consistent with its evolutionary conservation, NTE contributes to maintaining membrane trafficking in mammalian cells. It would not be surprising that altered membrane trafficking shares a common biological pathway with ubiquitination (3, 4) so that alteration of either component would result in the same phenotype. Collectively, our findings suggest that NTE deficiency caused by deleterious PNPLA6 mutations interferes with gonadotropin release from pituitary gonadotropes, leading to nHH and pubertal failure. This site of action notwithstanding, the present study does not rule out the concomitant compromise of hypothalamic systems regulating GnRH output and/or GnRH neurons themselves.
In conclusion, deficiency of PNPLA6-encoded NTE may cause a delayed neurodegeneration with altered intracellular phospholipid homeostasis particularly affecting cerebellum and a reproductive failure likely to be due to impaired gonadotropin release, thus collectively accounting for GHS.
Acknowledgments
We thank Dr Marion Ehrich for kindly providing the phenyl valerate used to detect NTE activity as well as Dr R. J. Richardson for helping us develop the NTE activity protocol in our lab. We thank Pamela Mellon, University of California San Diego, for the LβT2 cell line and Dr. Anda Cornea, director of the Oregon National Primate Research Center Imaging and Morphology Core.
This work was supported by the Scientific and Technological Research Council of Turkey (109S455) and the Turkish Society for Pediatric Endocrinology and Diabetes (to A.K.T.) and by the European Society for Pediatric Endocrinology, Sabbatical Leave Programme (to A.K.T.) and National Science Foundation (IOS1121691 to S.R.O). The Oregon National Primate Research Center Imaging and Morphology Core is supported by Grant NS061800.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by the Scientific and Technological Research Council of Turkey (109S455) and the Turkish Society for Pediatric Endocrinology and Diabetes (to A.K.T.) and by the European Society for Pediatric Endocrinology, Sabbatical Leave Programme (to A.K.T.) and National Science Foundation (IOS1121691 to S.R.O). The Oregon National Primate Research Center Imaging and Morphology Core is supported by Grant NS061800.
Footnotes
- BNS
- Boucher-Neuhäuser syndrome
- CPO
- chlorpyrifos oxon
- GHS
- Gordon Holmes syndrome
- GnRHR
- GnRH receptor
- HBSS
- Hanks' balanced salt solution
- HSP
- hereditary spastic paraplegia
- LPC
- lysophosphatidylcholine
- MRI
- magnetic resonance imaging
- NFAT
- nuclear factor of activated T-cells
- nHH
- normosmic hypogonadotropic hypogonadism
- NTE
- neuropathy target esterase
- PA
- paraoxon
- siRNA
- small interfering RNA
- SNP
- single-nucleotide polymorphism
- WGA
- wheat germ agglutinin.
References
- 1. Holmes G. A form of familial degeneration of the cerebellum. Brain. 1907;30:466–489. [Google Scholar]
- 2. Rump P, Hamel BC, Pinckers AJ, van Dop PA. Two sibs with chorioretinal dystrophy, hypogonadotrophic hypogonadism, and cerebellar ataxia: Boucher-Neuhäuser syndrome. J Med Genet. 1997;34:767–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Margolin DH, Kousi M, Chan YM, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368:1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi CH, Schisler JC, Rubel CE, et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23:1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Synofzik M, Gonzalez MA, Lourenco CM, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. 2014;137:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehrich M, Correll L, Veronesi B. Acetylcholinesterase and neuropathy target esterase inhibitions in neuroblastoma cells to distinguish organophosphorus compounds causing acute and delayed neurotoxicity. Fundam Appl Toxicol. 1997;38:55–63. [DOI] [PubMed] [Google Scholar]
- 7. Kropp TJ, Richardson RJ. Relative inhibitory potencies of chlorpyrifos oxon, chlorpyrifos methyl oxon, and mipafox for acetylcholinesterase versus neuropathy target esterase. J Toxicol Environ Health A. 2003;66:1145–1157. [DOI] [PubMed] [Google Scholar]
- 8. Johnson MK, Glynn P. Neuropathy target esterase (NTE) and organophosphorus-induced delayed polyneuropathy (OPIDP): recent advances. Toxicol Lett. 1995;82–83:459–463. [DOI] [PubMed] [Google Scholar]
- 9. Johnson JM, Betz WJ. The color of lactotroph secretory granules stained with FM1–43 depends on dye concentration. Biophys J. 2008;94:3167–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bateman JR, Lee AM, Wu CT. Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics. 2006;173:769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turgeon JL, Kimura Y, Waring DW, Mellon PL. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol Endocrinol. 1996;10:439–450. [DOI] [PubMed] [Google Scholar]
- 13. Moser M, Stempfl T, Li Y, Glynn P, Büttner R, Kretzschmar D. Cloning and expression of the murine sws/NTE gene. Mech Dev. 2000;90:279–282. [DOI] [PubMed] [Google Scholar]
- 14. Rainier S, Bui M, Mark E, et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. 2008;82:780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rainier S, Albers JW, Dyck PJ, et al. Motor neuron disease due to neuropathy target esterase gene mutation: clinical features of the index families. Muscle Nerve. 2011;43:19–25. [DOI] [PubMed] [Google Scholar]
- 16. Lush MJ, Li Y, Read DJ, Willis AC, Glynn P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem J. 1998;332:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernández-Murray JP, McMaster CR. Phosphatidylcholine synthesis and its catabolism by yeast neuropathy target esterase 1. Biochim Biophys Acta. 2007;1771:331–336. [DOI] [PubMed] [Google Scholar]
- 18. Mühlig-Versen M, da Cruz AB, Tschäpe JA, et al. Loss of Swiss cheese/neuropathy target esterase activity causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila. J Neurosci. 2005;25:2865–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fok AC, Wong MC, Cheah JS. Syndrome of cerebellar ataxia and hypogonadotrophic hypogonadism: evidence for pituitary gonadotrophin deficiency. J Neurol Neurosurg Psychiatry. 1989;52:407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baroncini A, Franco N, Forabosco A. A new family with chorioretinal dystrophy, spinocerebellar ataxia and hypogonadotropic hypogonadism (Boucher-Neuhäuser syndrome). Clin Genet. 1991;39:274–277. [DOI] [PubMed] [Google Scholar]
- 21. Tojo K, Ichinose M, Nakayama M, et al. A new family of Boucher-Neuhäuser syndrome: coexistence of Holmes type cerebellar atrophy, hypogonadotropic hypogonadism and retinochoroidal degeneration: case reports and review of literature. Endocr J. 1995;42:367–376. [DOI] [PubMed] [Google Scholar]
- 22. Quinton R, Barnett P, Coskeran P, Bouloux PM. Gordon Holmes spinocerebellar ataxia: a gonadotrophin deficiency syndrome resistant to treatment with pulsatile gonadotrophin-releasing hormone. Clin Endocrinol (Oxf). 1999;51:525–529. [DOI] [PubMed] [Google Scholar]
- 23. Seminara SB, Acierno JS Jr, Abdulwahid NA, Crowley WF Jr, Margolin DH. Hypogonadotropic hypogonadism and cerebellar ataxia: detailed phenotypic characterization of a large, extended kindred. J Clin Endocrinol Metab. 2002;87:1607–1612. [DOI] [PubMed] [Google Scholar]
- 24. Akassoglou K, Malester B, Xu J, Tessarollo L, Rosenbluth J, Chao MV. Brain-specific deletion of neuropathy target esterase/swisscheese results in neurodegeneration. Proc Natl Acad Sci U S A. 2004;101:5075–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moser M, Li Y, Vaupel K, et al. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol Cell Biol. 2004;24:1667–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herman AP, Krawczyska A, Bochenek J, et al. The effect of rivastigmine on the LPS-induced suppression of GnRH/LH secretion during the follicular phase of the estrous cycle in ewes. Anim Reprod Sci. 2013;138:203–212. [DOI] [PubMed] [Google Scholar]
- 27. Park JJ, Loh YP. How peptide hormone vesicles are transported to the secretion site for exocytosis. Mol Endocrinol. 2008;22:2583–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bankaitis VA, Mousley CJ, Schaaf G. The Sec14 superfamily and mechanisms for crosstalk between lipid metabolism and lipid signaling. Trends Biochem Sci. 2010;35:150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cleves AE, McGee TP, Whitters EA, et al. Mutations in the CDP-choline pathway for phospholipid biosynthesis bypass the requirement for an essential phospholipid transfer protein. Cell. 1991;64:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mousley CJ, Tyeryar KR, Vincent-Pope P, Bankaitis VA. The Sec14-superfamily and the regulatory interface between phospholipid metabolism and membrane trafficking. Biochim Biophys Acta. 2007;1771:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xie Z, Fang M, Bankaitis VA. Evidence for an intrinsic toxicity of phosphatidylcholine to Sec14p-dependent protein transport from the yeast Golgi complex. Mol Biol Cell. 2001;12:1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]