

Abstract
Context:
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is a rare cause of primary adrenal Cushing's syndrome (CS). ARMC5 germline mutations have been identified recently in PBMAH.
Objective:
To determine the prevalence of ARMC5 mutations and analyze genotype-phenotype correlation in a large cohort of unrelated PBMAH patients with subclinical or clinical CS.
Patients and Methods:
ARMC5 was sequenced in 98 unrelated PBMAH index cases. PBMAH was identified by bilateral adrenal nodular enlargement on computed tomography scan. The effect on apoptosis of ARMC5 missense mutants was tested in H295R and HeLa cells. Clinical and hormonal data were collected including midnight and urinary free cortisol levels, ACTH, androgens, renin/aldosterone ratio, cortisol after overnight dexamethasone suppression test, cortisol and 17-hydroxyprogesterone after ACTH 1-24 stimulation and illegitimate receptor responses. Computed tomography and histological reports were analyzed.
Results:
ARMC5-damaging mutations were identified in 24 patients (26%). The missense mutants and the p.F700del deletion were unable to induce apoptosis in both H295R and HeLa cell lines, unlike the wild-type gene. ARMC5-mutated patients showed an overt CS more frequently, compared to wild-type patients: lower ACTH, higher midnight plasma cortisol, urinary free cortisol, and cortisol after dexamethasone suppression test (P = .003, .019, .006, and <.001, respectively). Adrenals of patients with mutations were bigger and had a higher number of nodules (P = .001 and <.001, respectively).
Conclusions:
ARMC5 germline mutations are common in PBMAH. Index cases of mutation carriers show a more severe hypercortisolism and larger adrenals. ARMC5 genotyping may help to identify clinical forms of PBMAH better and may also allow earlier diagnosis of this disease.
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is an adrenal disorder characterized by bilateral benign adrenocortical nodules associated with variable levels of cortisol excess. In contrast to micronodular adrenal hyperplasia, adrenal nodules observed in patients with PBMAH are usually detectable by abdominal imaging; by definition, nodules in PBMAH are larger than 1 cm (1). PBMAH can lead to overt Cushing's syndrome (CS), most frequently diagnosed in patients between 40 and 70 years of age (2). Mild forms of CS can also be observed, characterized by subclinical CS and frequently associated with smaller enlargement of the adrenal glands. Frequently, these patients are identified in the course of an investigation for an incidentally discovered adrenal lesion. Because of the variability of the disease, the prevalence of PBMAH is not well established, especially in patients with mild clinical presentation (1, 3).
The pathophysiology of PBMAH is not completely understood. In the past 20 years, several studies have shown that cortisol secretion is regulated in part by abnormal expression of various membrane receptors (4). Cortisol stimulation after food ingestion (due to abnormal expression of the gastric inhibitory polypeptide receptor) or upright posture (due to vasopressin or β-adrenergic receptors) has been reported (5–8). Recently, Louiset et al (9) have shown that cortisol secretion was regulated by ACTH produced locally by clusters of steroidogenic cells in hyperplastic tissue. For this reason, the term “primary bilateral macronodular adrenal hyperplasia” seems more appropriate instead of the previously used “ACTH-independent macronodular adrenal hyperplasia” (10).
The bilateral nature of the nodules supports the hypothesis of a germline genetic predisposition, as frequently demonstrated for micronodular adrenal hyperplasia (11–13). Moreover, even if most cases appear to be sporadic, familial cases have been well documented, suggesting the involvement of germline hereditary factors in PBMAH (3, 8, 14, 15). Until recently, no genetic events were known except within rare families presenting with multiple tumor syndromes (3, 16–18).
We recently identified frequent inactivating mutations of the armadillo repeat containing 5 (ARMC5) gene in PBMAH (19). The investigation of another cohort of PBMAH patients recently reported ARMC5 gene variants in 15 of 34 patients (20). Subsequently, an ARMC5 mutation was reported in eight of 10 kindreds presenting with clear familial presentation of PBMAH (21–23). ARMC5 is likely to be a tumor suppressor gene. Little is known about the function of ARMC5. In vitro studies have suggested that wild-type ARMC5 is able to stimulate apoptosis (19).
In the first series of patients reporting ARMC5 mutations in PBMAH, only patients who had undergone surgery had been included (19). Operated patients are probably more prone to present with severe hypercortisolism and/or significant adrenal enlargement, compared with PBMAH patients who are not operated. This series was not large enough to allow a genotype-phenotype correlation. In particular, patients with ARMC5 mutations that have not yet been operated on were not studied in the first cohort. Thus, the aim of the present study was to expand our original cohort and to also include index cases of patients that have not undergone adrenalectomy to allow for a comprehensive genotype-phenotype correlation in a total of 98 patients with PBMAH.
Patients and Methods
Patients
A total of 98 consecutive unrelated patients with PBMAH were investigated. PBMAH was defined morphologically as a bilateral adrenal enlargement with nodules larger than 1 cm or extensive thickening on computed tomography (CT) scans. The clinical signs or consequences of CS were recorded from the patients' files. For those operated, pathological examination was also recorded.
Hormonal investigations
Hormonal investigations were performed as previously described (24), including early morning plasma ACTH, 24-hour urinary free cortisol (UFC), total plasma T, dehydroepiandrosterone sulfate (DHEA-S), early morning and midnight plasma cortisol, aldosterone and renin levels in the supine position, and early morning plasma cortisol after overnight dexamethasone suppression test (oral dexamethasone 1 mg at midnight). In addition, aberrant membrane receptor expression was investigated by evaluating the cortisol response during upright test, after food intake, after LHRH injection, and after metoclopramide administration in 51, 51, 27, and 26 patients, respectively (4). The response was considered as illegitimate for an increase of 25% in plasma cortisol at the peak (25). Clinical or overt CS was defined by the presence of physical signs of CS and abnormal cortisol results (ie, increased midnight cortisol and/or increased cortisol after dexamethasone suppression test and/or increased UFC and/or decreased ACTH <10 pg/mL). Subclinical CS was defined by an abnormal cortisol test in the absence of physical signs. The plasma cortisol and 17-hydroxyprogesterone (17OHP) response after iv injection of 250 mg ACTH 1-24 was investigated in 74 and 68 patients, respectively, as previously described (25) (Table 1).
Table 1.
Clinical and Hormonal Characteristics of PBMAH Patients
| Characteristics | Total Cohorta,b | Mutateda,b | Wild-Typea,b | P Valuec |
|---|---|---|---|---|
| Number of patients | 98 | 24 | 68 | |
| Age, y | 53 [30–75] | 49 [30–73] | 55 [32–75] | .046 |
| Sex (F/M), % | 64.3/35.7 | 50/50 | 69.1/30.9 | .137 |
| CS, % | .007 | |||
| Clinical | 43 | 71 | 35 | |
| Subclinical | 47 | 29 | 53 | |
| Absent | 10 | 0 | 11.8 | |
| Hypertension, %d | 69.3 | 94.7 | 63.5 | .009 |
| Diabetes, %d | 33.7 | 52.9 | 30.2 | .093 |
| Hypokalemia, %d | 12.9 | 17.6 | 11.3 | .441 |
| ACTH, %e | .003 | |||
| <10 pg/mL | 60 | 92 | 53 | |
| 10 to 15 pg/mL | 30 | 8 | 34 | |
| >15 pg/mL | 9 | 0 | 12 | |
| UFC (% of increase above upper limit of normal)e | 204 [11–2433] | 355 [11–1626] | 167 [16–2433] | .006 |
| Plasma cortisol after 1 mg dexamethasone overnight test, μg/dLe | 9.9 [0.3–36.7] | 18.6 [3.6–36.7] | 6.3 [0.3–2.7] | <.001 |
| Plasma cortisol at midnight, μg/dLe | 12.1 [1.4–71.7] | 20 [4.6–71.7] | 9.4 [1.4–42.8] | .019 |
| Morning plasma cortisol, μg/dLe | 14.1 [4.7–57.1] | 16.9 [5.5–34] | 13.8 [4.7–57.1] | .245 |
| Plasma cortisol after ACTH 1–24 250 μg, μg/dLe | 39.2 [11.1–123] | 48.9 [19.8–81.3] | 37 [11.1–123] | 0.058 |
| Basal 17OHP, ng/dLe | 99 [0–726] | 132 [0–594] | 99 [0–726] | .28 |
| 17OHP after ACTH 1–24 250 μg, ng/dLe | 990 [132–7128] | 1089 [264–5578] | 990 [132–7128] | .79 |
| Aldosterone/direct renin ratio, pmol/L/μIU/mLe | 22 [0–331] | 33 [0–331] | 19 [0–133] | .45 |
| Food response, %d | 17.6 | 0 | 28.1 | <.001 |
| Upright response, %d | 52.9 | 68.8 | 46.9 | .221 |
Abbreviations: F, female; M, male.
Quantitative variables are presented as means [minimum-maximum]; qualitative variables are presented as relative counts percentage.
Of the 98 patients, 24 were classified as ARMC5-mutated, 68 as ARMC5-wild type, and six were excluded for the comparison, presenting variants of uncertain significance.
Comparison of mutated patients vs wild-type patients.
Data about hypertension were available for 88 patients (19 mutated, 63 wild-type), diabetes for 86 (17 mutated, 63 wild-type), hypokalemia for 85 (17 mutated, 62 wild-type), and abnormal response for 51 (16 mutated, 32 wild-type).
ACTH was available for 97 patients, UFC for 94, morning plasma cortisol for 74, cortisol after suppression test for 77, cortisol at midnight for 67, aldosterone/renin ratio for 85, basal 17OHP for 80, cortisol after ACTH for 74, and 17OHP after ACTH for 68.
Genetic testing
DNA was collected from peripheral blood leukocytes or tumor tissue. Written informed consent was obtained for genetic analysis. The study was approved by the Institutional Review Board of Cochin Hospital and the University Hospital of Munich.
Morphological evaluation
An adrenal CT scan study was performed in all patients and read by a senior radiologist blinded from the mutational status. The maximum diameter and the number of nodules were measured on the CT scans for each adrenal gland, and the sums of these parameters for the two glands were calculated.
Pathological evaluation
Forty-six patients underwent adrenalectomy. Histological reports confirmed the diagnosis of macronodular adrenal hyperplasia in all operated patients. The adrenal weight measured after fat removal was obtained from the pathology reports.
ARMC5 sequencing analysis
DNA was extracted from leukocytes or tumor tissue as previously reported (26). The ARMC5 coding and the flanking intronic sequences were amplified by PCR both in leukocytes and tumor DNA from 30 patients, only in leukocytes DNA from 62 patients, and only in tumoral DNA in eight patients. Both strands of the amplified products were directly sequenced with forward and reverse primers as previously described (19). All mutations were confirmed twice in two independent experiments. The in silico software Polyphen-2 http://genetics.bwh.harvard.edu/pph2/) (27) and SIFT version 2 http://sift.jcvi.org/www/SIFT_enst_submit.html) was (28) was utilized to predict the pathogenic potential of the missense variants. The software Mutalyser (Mutalyser 2.0.3; https://mutalyzer.nl/name-checker/) was utilized to check the sequence variant nomenclature according to Human Genome Variation Society version 2.0 (29).
Cell culture
Human HeLa cells and adrenocortical cancer cells (H295R) were cultured and transfected as previously described (19). Mutagenesis of F700del, Y736S, R362L, R315W, L331P, C657R, C139R, I664S, L754P, L778P, and R454W was performed using QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) from FLAG-tagged wild-type ARMC5 expression vector (Origen RC226267).
Immunofluorescence staining
Cultured cells were fixed in chilled methanol/ethanol solution (vol/vol) for 15 minutes, followed by 10-minute permeabilization with 0.1% Triton X-100 (Sigma). Blocking was performed at room temperature for 45 minutes with serum goat 10% (Invitrogen). Samples were incubated with primary antibodies (Abs) diluted in the blocking solution, cleaved caspase 3 Ab (1:250, D9661; Cell Signaling Technology) and Flag Ab (1:150, F-1804; Sigma-Aldrich), overnight at 4°C. Then, cells were washed 3 times in PBS and incubated with secondary Abs, Alexia fluor 647 goat antimouse (1:750, A; Life Technologies) and Alexia fluor 488 goat antirabbit (1/750, A11008; Life Technologies) for 1 hour at room temperature. Samples were washed three times with PBS. The nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole), and after mounting with Fluoromount G (DAKO), images were acquired using a fluorescence microscope (Zeiss Axiovert 200M; Carl Zeiss).
Statistical analysis
Quantitative variables were summarized by their mean [range], and ARMC5-mutated and ARMC5-wild-type patients were compared using the Student's t test. The UFC values were compared using Wilcoxon test. Qualitative variables were summarized by their absolute counts (and relative counts in percentages); ARMC5-mutated and ARMC5-wild-type patients were compared using the Fisher exact test. The prevalence of described ARMC5 variants in the PBMAH patients and in the general population (Exome Variant Server, NHLBI GO Exome Sequencing Project, version 0.0.29; http://evs.gs.washington.edu/EVS/) was compared using the χ2 test. All tests were two-sided. A P value of .05 or less was considered statistically significant. The data were analyzed using R version 3.0.0 (www.R-project.org).
Results
Patient characteristics
The characteristics (age, sex, clinical Cushing, hormonal investigations) of the 98 index case PBMAH patients are summarized in Tables 1 and 2. Mean size and number of nodes on CT scan as well as adrenal weight for operated patients are described in Table 2.
Table 2.
Adrenal Morphology of PBMAH Patients
| Characteristics | Total Cohort | Mutated | Wild-Type | P Valuea |
|---|---|---|---|---|
| Number of patients | 98 | 24 | 68 | |
| Surgery, %b | 46.9 | 83.3 | 33.8 | <.001 |
| Right adrenal weight, gc | 36 [6–86] | 42 [21–65] | 29 [6–86] | .099 |
| Left adrenal weight, gc | 50 [9–121] | 65 [26–121] | 37 [9–107] | .007 |
| Total weight, gc | 81 [0–331] | 107 [53–187] | 50 [23–166] | .001 |
| Right adrenal size, mmd | 32 [9–100] | 45 [10–100] | 29 [9–77] | .019 |
| Left adrenal size, mmd | 34 [9–130] | 53 [17–100] | 30 [9–130] | .005 |
| Total size, mmd | 66 [28–200] | 98 [33–200] | 59 [28–180] | .003 |
| No. of nodules, %d | <.001 | |||
| 2 or less | 57.0 | 15.8 | 68.9 | |
| 3 | 12.8 | 5.3 | 14.8 | |
| 4 | 5.8 | 5.3 | 4.9 | |
| 5 or more | 24.4 | 73.7 | 11.5 |
Quantitative variables are presented as means [minimum-maximum].
Comparison of mutated patients vs wild-type patients.
Three patients excluded for the comparison, presenting variants of uncertain significance, underwent surgery.
For right, left, total adrenal weight, respectively, data available for 34, 37 and 30 of the 46 operated patients.
For right, left, total adrenal size and number of nodules, respectively, data available for 83, 81, 81, and 86 patients of the total cohort.
ARMC5 sequencing
Fifty-three patients from the 98 index cases (54%) had an ARMC5 coding sequence variation in comparison with the reference genome (GRCh37).
Twenty-three patients (23.5%) harbored only missense variants previously reported in variation databases with a prevalence superior to 1% (Exome Variant Server, http://evs.gs.washington.edu/EVS/; 1000 genomes project [30], http://www.1000genomes.org/) and predicted to be benign by in silico models (rs151069902/p.F14Y, rs35923277/p.I170V, rs142376949/p.P507L) (data not shown). These patients and the 45 patients without polymorphisms were considered as wild type.
Five patients carried four germline missense mutations reported in the databases that were predicted to probably be damaging. The prevalence of these variations was significantly higher in the 98 patients than was reported in the Exome Variant Server database for the variants p.P731R (1 vs 0.4%; P = .04) and p.R454W (rs372473237) (0.5 vs 0.02%; P = .046) but did not differ for the variants p.A692S (rs201162311) (0.5 vs 0.09%; P = .17) and p.Q408R (rs141923065) (0.5 vs 0.4%; P = .56). One patient carried a missense substitution, p.S669T, not reported in the databases and predicted as benign by in silico models. Due to these uncertainties and to avoid misclassification, these six patients were not included in the comparison.
Twenty-four patients (24.5%) had an ARMC5 variant leading to a probably damaging protein or a lack of protein integrity (Table 3 and Supplemental Table 1). One patient (H15) carried a germline deletion of the region, previously identified by single nucleotide polymorphism array (19). Of these 24 patients, 19 had been operated, and tumor tissue was available. In a total of 31 nodules examined, two ARMC5 alterations were found on the tumor DNA; the second hit was a mutation in 21 cases and a loss of heterozygosity in 10 cases (Supplemental Table 2). For three patients (nos. 11, 13, and 15), germline DNA was not available, but the germline mutation was determined as the common mutation found in two nodules from the same patient. For one patient (no. 16) the germline mutation was not determined because the DNA from only one nodule was available.
Table 3.
Characteristics of Mutated Patients
| Patient ID | Germline ARMC5 Mutation | Clinical Features |
Biological Evaluation |
Biological Evaluation |
CT |
Surgery, Histology |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age, y | Sex | CS | HTA | Midnight Cortisol, μg/dLa | ACTH, pg/mLb | Value of UFC Above Upper Limit of Normal, % | Cortisol After Overnight DXM Test, μg/dLc | Food Response | Upright Response | Total Adrenal Nodules, n | Left Adrenal Size, mm | Right Adrenal Size, mm | Y/N | Total Adrenal Weight, g | ||
| 1 | p.Q86X | 34 | F | Clinical | Y | 15.3 | <5 | 414 | 23.6 | N | Y | >5 | 30 | 45 | Y | 66 |
| 2 | p.R267X | 41 | F | Clinical | NA | NA | <5 | NA | NA | NA | NA | NA | NA | NA | Y | 124 |
| 3 | p.L548P | 64 | M | Clinical | Y | 42.7 | 6 | 962 | 27.2 | N | Y | >5 | 85 | 55 | Y | 157 |
| 5 | p.R267X | 30 | F | Clinical | Y | 24.8 | <5 | 509 | 26.1 | N | N | >5 | 80 | 80 | Y | 119 |
| 6 | p.G57Efs*80 | 48 | F | Clinical | Y | 23.4 | <5 | 131 | 27.4 | N | Y | >5 | 100 | 100 | Y | 84 |
| 7 | p.R267X | 51 | F | Clinical | Y | 17.9 | <10 | 276 | 22.4 | NA | NA | >5 | NA | NA | Y | 109 |
| 8 | p.(A702_S706del) | 45 | M | Clinical | Y | 23.2 | <10 | 1626 | 36.7 | N | Y | >5 | NA | NA | Y | 119 |
| 9 | p.R267X | 70 | F | Clinical | Y | 13.1 | <5 | 136 | 16 | N | Y | >5 | 30 | 50 | Y | 81 |
| 10 | p.R898W | 41 | M | Clinical | Y | NA | <5 | 438 | 29 | NA | NA | >5 | NA | NA | Y | 186 |
| 11 | p.F700del | 46 | M | Clinical | Y | 16.2 | <5 | 227 | 18 | NA | NA | NA | NA | NA | Y | 80 |
| 12 | p.R898W | 55 | M | Subclinical | Y | 5.1 | <5 | 150 | 3.6 | N | N | 3 | 32 | 35 | Y | 72 |
| 13 | p.A296Cfs*34 | 63 | M | Clinical | Y | 14.5 | 8 | 336 | 22.9 | N | Y | >5 | NA | NA | Y | 85 |
| 14 | p.R619X | 53 | M | Clinical | Y | 16.6 | <5 | 326 | 25.1 | N | Y | >5 | 45 | 35 | Y | 142 |
| 15 | p.C657R | 56 | M | Clinical | NA | NA | <5 | 119 | NA | N | Y | >5 | 70 | 50 | Y | 166 |
| 16 | p.E430 × or p.A110Pfs*26 | 51 | M | Clinical | NA | NA | <5 | NA | NA | N | Y | NA | NA | NA | Y | NA |
| 17 | micro DEL | 52 | F | Subclinical | NA | 23.3 | <5 | 71 | 8.9 | NA | NA | NA | NA | NA | Y | 53 |
| 18 | p.A104Gfs*7 | 73 | F | Clinical | NA | NA | <5 | NA | 15 | N | Y | 2 | 29 | 31 | Y | 55 |
| 26 | p.R898W | 46 | F | Subclinical | Y | 4.6 | 5 | 113 | 4.6 | NA | NA | 4 | 17 | 16 | N | |
| 27 | p.I664S | 38 | F | Subclinical | Y | 10.2 | <5 | 72 | 10.9 | N | N | >5 | 90 | 10 | Y (Left) | |
| 28 | p.L754P | 41 | F | Clinical | Y | NA | <5 | 444 | NA | NA | NA | NA | NA | NA | Y | NA |
| 29 | p.R764X | 54 | M | Subclinical | Y | 9.5 | 11 | 34 | 12 | N | Y | >5 | 54 | 61 | N | |
| 30 | p.R764X | 32 | M | Clinical | Y | 71.7 | 9 | 1050 | 27.6 | NA | NA | >5 | 40 | 35 | Y | 117 |
| 31 | p.A106Rfs*31 | 42 | M | Subclinical | Y | 7.6 | 6 | 11 | 5.6 | N | N | 2 | 65 | 30 | N | |
| 32 | p.A104Gfs*7 | 58 | F | Subclinical | N | NA | 10 | 20 | 7.5 | N | N | 2 | 35 | 42 | N | |
Abbreviations: F, female; M, male; DXM, dexamethasone; NA, not available; Y, yes; N, no. Asterisks show locations of the various mutations on the ARMC5 protein described according to the variant NM_001105247 (http://ncbi.nlm.nih.gov/).
Normal midnight cortisol level, <7.5 μg/dL.
Normal ACTH level, 10–60 pg/mL.
Normal cortisol level after overnight dexamethasone test, 1.8 μg/dL (<50 nmol/L).
In total, in these 24 patients with ARMC5 mutations, 36 different mutations were found by the sum of the germline and somatic DNA analysis in addition to one germline microdeletion and somatic loss of heterozygosity (Figure 1). Sixteen of these mutations were frame-shift mutations resulting in one-base or larger insertions or deletions. Eight are substitutions leading to nonsense mutations. Two were short amino acid deletions [p.F700del and p.(A702_S706del)]. Ten missense substitutions that were not previously described in public databases were predicted probably damaging by in silico models (Supplemental Tables 1 and 2), in keeping with the functional studies presented below. Finally, mutations p.R267X, p.R764X, p.R898W, and p.A104Gfs*6 were shared by 4, 2, 3, and 2 patients, respectively, and can therefore be considered as hotspots.
Figure 1. ARMC5 somatic or germline mutation distributions.

The location of the various mutations is shown on the ARMC5 protein described according to the variant NP_001098717 (http://www.ncbi.nlm.nih.gov/). The location of domains is according to UniProtKB (Uniprot Q96C12-1, http://www.uniprot.org/). Above, Germline mutations. Below, Somatic mutations and the two mutations found on somatic DNA for which germline or somatic level was not determinate (italic font with dotted line). °, Mutations described for several patients; each symbol represents one primary bilateral macronodular adrenal hyperplasia patient.
ARMC5 mutation in vitro analysis
We have previously shown that the expression of wild-type ARMC5 causes cell death and that certain missense mutants of ARMC5 (ie, p.R898W and p.L548P) fail to induce apoptosis (19). To demonstrate the pathogenic role of the other eight missense substitutions (three germline and five somatic) and the germline deletion p.F700del, we investigated their function on apoptosis in H295R and HeLa cells. Expression of the FLAG-wild type ARMC5 induces apoptosis 12–14 hours after transfection as shown by co-staining of cleaved caspase 3 and ARMC5-FLAG in H295R (Figure 2) and HeLa cells (Supplemental Figure 1). On the contrary, cells transfected with ARMC5 mutants maintain normal morphology and do not produce the apoptotic marker cleaved caspase 3, 8 and 12 hours after transfection (Supplemental Figures 2 and 3). This is in keeping with the in silico analysis, suggesting a deleterious effect of these mutations.
Figure 2. Study of ARMC5 mutant-induced apoptosis in H295R cells.

The figure shows the immunofluorescence staining of FLAG and the cleaved caspase 3 in H295R cells 14 hours after transfection with the wild-type or mutant ARMC5 constructs. H295R cells were transfected with different ARMC5-FLAG constructs (wild-type). The cells were double-labeled with antibodies against the FLAG tag (red staining) and cleaved caspase 3 (green staining), in addition to nuclei staining by DAPI (blue). Double staining and altered morphology were observed with wild-type ARMC5, whereas transected cells with mutant ARMC5 did not express cleaved caspase 3 (white arrows).
ARMC5-mutated patients present a more severe disease
The characteristics of the 24 patients with a clearly pathogenic ARMC5 mutation are described in Tables 1, 2, and 3 and Supplemental Table 3. The characteristics of the 68 wild-type patients are shown in Supplemental Table 4 (and those of the six patients who were not included in the comparison are shown in Supplemental Table 5). The ARMC5-mutated patients were diagnosed earlier with PBMAH than the wild-type patients (mean age at diagnosis, 49 vs 55 y, respectively; P = .046) (Supplemental Figure 4). The prevalence of clinical CS was higher in the mutated group (71 vs 35%; P = .007). Hypertension was also more frequent in the mutant group (95 vs 63%; P = .009).
ACTH plasma levels were more often suppressed in the mutant group than in the wild-type group (92 vs 53%, respectively; P = .003). Mean midnight plasma cortisol level was higher (20 vs 9.7 μg/dL, respectively; P = .019) as well as the UFC (355 vs 167% above the upper limit of normal; P = .006). Plasma cortisol was less suppressed after dexamethasone overnight in the mutant group (18.6 vs 6.3 μg/dL, respectively; P < .001) (Table 1 and Supplemental Figure 4). No difference in other hormonal parameters, such as adrenal androgens (DHEA-S, androstenedione) or mineralocorticoid hormones, was found between the groups. In addition, 17OHP (1089 vs 990 ng/dL; P = .79) and plasma cortisol (48,9 vs 37 μg/dL; P = .058) did not differ between the two groups upon ACTH 1-24 stimulation.
Interestingly, no mutated patients, but nine patients without mutations presented with food-dependent CS (0 vs 28.1%, respectively; P = .02). No significant differences were seen between the mutant and wild-type groups for responses to the upright position (Table 1), the LHRH injection (11.1 vs 22.2%, respectively; P = .581), or metoclopramide administration (29.4 vs 33.3%, respectively; P = 1) (data not shown).
Twenty of the 46 operated patients carried an ARMC5 alteration. Total adrenal weight (available for 17 mutated patients and 12 wild-type patients) was significantly higher in the mutant group than the wild-type group (mean weight of both adrenal, 107 vs 50 g, respectively; P = .001). Likewise, total adrenal size measured by CT for 75 patients was significantly higher in the mutant group than in the wild-type group (total size, 95 vs 59 mm, respectively; P = .003). The number of nodules was statistically higher in the mutant group than in the wild-type group (P < .001). The number of nodules was greater than five for 73.6% of the ARMC5-mutated patients, whereas this was the case for only 11.5% of the ARMC5 wild-type patients (Table 2). Figure 3 summarizes different examples of bilateral adrenal hyperplasia, depending on ARMC5 status.
Figure 3. CT scan of ARMC5 mutated and wild-type PBMAH patients.
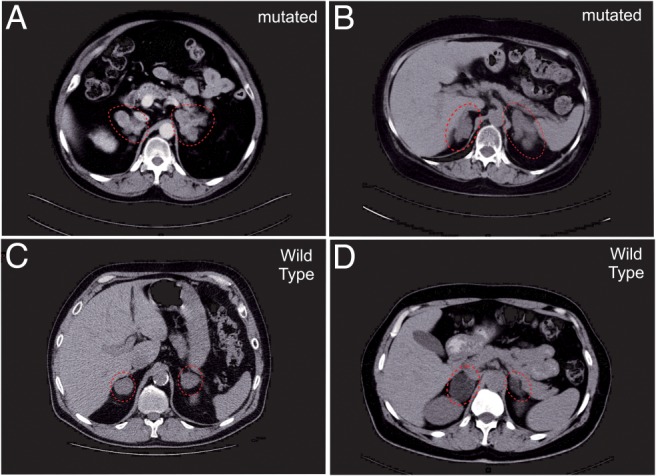
A and B, Patients with mutations of ARMC5 had massive enlargement of their adrenal glands (dotted red circle) and multiple nodules on their CT scans [A, patient 8 with p.(A702_S706del); B, patient 9 with p.R267X]. C and D, Wild-type patients have smaller adrenal hyperplasia (C, patient 97; D, patient 74).
Discussion
This cohort reflects the heterogeneity of PBMAH, a disease with various degrees of cortisol secretion and adrenal enlargement. As demonstrated in our initial study of adult patients with apparently sporadic PBMAH leading to the identification of ARMC5 as a causative gene, the bilateral nature of the disease is likely due to its genetic origin (19). In the first study of PBMAH patients, all selected for surgery, the prevalence of ARMC5 gene defects was 55%. Indeed, after this initial report, the study of large families with PBMAH from Brazil, Australia, and Germany reported the frequent occurrence of ARMC5 mutations in familial forms of the disease (21–23). In these three studies, eight of the 10 families investigated carried an ARMC5 mutation. The analysis by Faucz et al (20) of the NIH series of 33 adult index case patients mainly with an apparently sporadic form of the disease found ARMC5 mutations in 21% of the patients.
The present study represents the largest series of index cases with PBMAH investigated so far. It shows that part of the heterogeneity of the disease could be related to the ARMC5 status of the patient. By extending the initial cohort (66 new cases), we were now able to investigate 98 index cases of PBMAH who were not selected on the basis of the severity of the disease or included because of surgical treatment. The main biological and clinical characteristics were comparable between patients from the different centers. This series is therefore representative of the spectrum of PBMAH patients now seen in an endocrinology department. Indeed, several patients are now found after the investigation of incidentalomas and most patients present with subclinical CS (1, 3) and are not always referred to the surgeon. In this series, we found a frequency of ARMC5 alterations of 25%, in agreement with the NIH cohort (20). This clearly shows that ARMC5 is frequently involved in PBMAH, regardless of the possible classification of the disease in subcategories.
In the current study, we described four new germline ARMC5 variants in five index cases among the newly investigated patients, in addition to five new somatic mutations. Taken together with our initial study, we identified 14 different germline pathogenic mutations and 19 somatic mutations; one mutation was found in two different patients in germline and in somatic DNA. For two mutations in the same patient, because only somatic DNA was available, we were unable to identify which were the germline and the somatic events. Additionally, we described a germline deletion and loss of heterozygosity in tumors (Ref. 19 and the present study). We confirmed that the 10 missense mutations and the one amino acid deletion are pathogenic, as shown in vitro by the loss of ability to induce apoptosis with the mutants ARMC5 in comparison to wild-type (Ref. 19 and the present study).
Interestingly, patients with ARMC5 mutations presented more frequently with overt CS, as shown by the significant increase of midnight plasma cortisol level and UFC, the suppression of ACTH level, and the absence of cortisol suppression after overnight dexamethasone suppression test. Considering the operated patients, we confirmed that the adrenal weight of ARMC5-mutated patients is higher than in patients without ARMC5 gene defects, as suggested by the initial study (19). This observation was in line with the CT scan data of nonoperated patients demonstrating a larger adrenal size and a higher number of nodules in ARMC5-mutated index cases. Notably, the plasma cortisol after the dexamethasone suppression test and the number of adrenal nodules remained independently associated with ARMC5 mutation in multivariate analysis (data not shown).
ARMC5 mutation carriers were also younger than the wild-type PBMAH patients. It is possible that this is because patients with PBMAH caused by ARMC5 mutations are the only ones in whom the disease is genetic and that in all other cases the disease is acquired. However, the bilateral nature of the disease and previous studies (3) suggest that this is not the case; in fact, almost certainly there are other genes yet to be identified in other forms of PBMAH, such as food-dependent CS. Thus, we favor the hypothesis that ARMC5-mutated patients present at a younger age because they experience more rapid growth of their tumors, as suggested by this and all other studies so far that show increased weight and number of nodules in the adrenal glands of affected patients (19, 20). This remains to be proven in prospective follow-up studies of these patients.
In transient transfection experiments, contrary to ARMC5 wild-type, all ARMC5 mutants tested have lost the ability to lead to cell death (19), and ARMC5 inactivation by small interfering RNA leads to decreased steroidogenesis (19). It is likely that excess cortisol is due to adrenal enlargement. Steroidogenesis has been shown to be relatively inefficient in PBMAH tissue, explaining the discrepancy between the enlargement of adrenal glands and the mild hypercortisolism or even its absence (31, 32). High cortisol and 17OHP increases after ACTH 1-24 stimulation were observed, in agreement with previous studies (25, 33), but this was not associated with ARMC5 status. Concurrent secretion of mineralocorticoids, such as aldosterone, or androgens such as T, DHEA-S, or androstenedione, has been described in PBMAH (34–36). In the present series, levels of these steroids were not associated with ARMC5 status. Therefore, the higher frequency of hypertension in ARMC5-mutated patients may not be explained by mineralocorticoid excess at least in these series.
CS and adrenal tumors are more frequent in female patients (37). A female predominance is indeed observed in the present series of PBMAH patients. However, the same is not seen in the subgroup of ARMC5-mutated patients. This is in agreement with the study of Faucz et al (20). Larger studies will be needed to study gender effects of ARMC5 mutations.
Illegitimate membrane receptor expression is observed in most PBMAH patients (25). In this study, abnormal cortisol response during the upright posture test was found for 54% of the patients, with no difference with regard to the ARMC5 defect. Abnormal response after LHRH or metoclopramide administration is less frequent and is not associated with ARMC5 status. In contrast, no food response in cortisol secretion was observed in patients with ARMC5 mutation, whereas this was seen in 28.1% of the wild-type ARMC5 patients. This supports our initial hypothesis (15) that ARMC5 is associated with a particular pattern of illegitimate receptors that does not cause food-dependent CS and is consistent with the variable molecular expression profile of adrenal lesions from patients with this from of PBMAH (38).
The functional role of ARMC5 protein and its associated signaling pathway is still poorly known. The protein contains repeated armadillo domains, as β-catenin which plays a main role in adrenal tumorigenesis (39, 40). The mechanism of inactivation of the gene (one germline event and one other occurring at the somatic level) suggests that ARMC5 is a tumor suppressor gene. Overexpression of the wild-type ARMC5 quickly stimulates apoptosis in vitro in H295R and HeLa cells. On the contrary, the germline missense mutations p.R898W and p.L548P did not stimulate apoptosis 12 to 14 hours after transfection (19). In the present study, we confirmed in these two different cell lines a loss of apoptosis in the transfected cells with the new missense mutations (and p.F700del) in comparison with the wild-type ARMC5. This in vitro study is important (along with the in silico analysis) to demonstrate the deleterious nature of the new mutants, especially the missense mutations causing the change of a single amino acid.
In conclusion, PBMAH is an heterogeneous disease because it can be associated with different degrees of hypercortisolism (from subclinical to overt CS), with different radiological aspects (from massive nodular hyperplasia to macronodular adrenal hyperplasia), and with different illegitimate receptors (33). The present study shows that PBMAH due to ARMC5 defects defines a specific subtype with larger and multinodular adrenal tumors, more frequent clinically overt CS, and may not be associated with food-dependent CS. ARMC5 genetic analysis appears to also help in the clinical management of PBMAH patients, with ARMC5-mutated patients needing surgery more frequently. Many questions remain, including what one does with the identification of ARMC5 mutation carriers, and whether these patients should be treated medically or surgically. Larger and long-term prospective studies are needed to answer these questions.
Acknowledgments
We thank the Cochin Hospital cell bank (Profs J. Chelly and M. Delpech), tumor bank (Prof B. Terris), and oncogenetic unit (Prof E. Clauser) for their help in sample collection; the Cochin Imaging Facility; and all the members of the Genomics and Signaling of Endocrine Tumors team for helpful discussions. We also thank the patients, their families, and the physicians and staff involved in patient management for their active participation.
This study was supported in part by the Agence Nationale de la Recherche (ANR-10-Blan-1136), the COMETE Network, Programme Hospitalier de Recherche Clinique Grant AOM95201, the Assistance Publique-Hôpitaux de Paris (Clinical Research Center Grant Genhyper P061006), ENSAT-CANCER Health-F2-2010-259735 (FP7 program), Inserm (S.E. is receiving a Poste Accueil, and G.A. is receiving a Contrat d'Interface), the E-RARE program (Genomics of cAMP signaling alterations in adrenal Cushing project), the Conny-Maeva Charitable Foundation, the Brou de Laurière Fondation, and the Intramural Program of the Eunice Kennedy Shriver, National Institute of Child Health & Human Development (Z01 HD008920-04).
Disclosure Summary: The authors have nothing to declare.
Funding Statement
This study was supported in part by the Agence Nationale de la Recherche (ANR-10-Blan-1136), the COMETE Network, Programme Hospitalier de Recherche Clinique Grant AOM95201, the Assistance Publique-Hôpitaux de Paris (Clinical Research Center Grant Genhyper P061006), ENSAT-CANCER Health-F2-2010-259735 (FP7 program), Inserm (S.E. is receiving a Poste Accueil, and G.A. is receiving a Contrat d'Interface), the E-RARE program (Genomics of cAMP signaling alterations in adrenal Cushing project), the Conny-Maeva Charitable Foundation, the Brou de Laurière Fondation, and the Intramural Program of the Eunice Kennedy Shriver, National Institute of Child Health & Human Development (Z01 HD008920-04).
Footnotes
- Ab
- antibody
- ARMC5
- armadillo repeat containing 5
- CS
- Cushing's syndrome
- CT
- computed tomography
- DAPI
- 4′,6-diamidino-2-phenylindole
- DHEA-S
- dehydroepiandrosterone sulfate
- 17OHP
- 17-hydroxyprogesterone
- PBMAH
- primary bilateral macronodular adrenal hyperplasia
- UFC
- urinary free cortisol.
References
- 1. Swain JM, Grant CS, Schlinkert RT, et al. . Corticotropin-independent macronodular adrenal hyperplasia: a clinicopathologic correlation. Arch Surg. 1998;133:541–545; discussion 545–546. [DOI] [PubMed] [Google Scholar]
- 2. Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:245–259. [DOI] [PubMed] [Google Scholar]
- 3. Hsiao HP, Kirschner LS, Bourdeau I, et al. . Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab. 2009;94:2930–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lacroix A, Bourdeau I, Lampron A, Mazzuco TL, Tremblay J, Hamet P. Aberrant G-protein coupled receptor expression in relation to adrenocortical overfunction. Clin Endocrinol (Oxf). 2010;73:1–15. [DOI] [PubMed] [Google Scholar]
- 5. Lacroix A, Bolté E, Tremblay J, et al. . Gastric inhibitory polypeptide-dependent cortisol hypersecretion–a new cause of Cushing's syndrome. N Engl J Med. 1992;327:974–980. [DOI] [PubMed] [Google Scholar]
- 6. Groussin L, Perlemoine K, Contesse V, et al. . The ectopic expression of the gastric inhibitory polypeptide receptor is frequent in adrenocorticotropin-independent bilateral macronodular adrenal hyperplasia, but rare in unilateral tumors. J Clin Endocrinol Metab. 2002;87:1980–1985. [DOI] [PubMed] [Google Scholar]
- 7. Horiba N, Suda T, Aiba M, et al. . Lysine vasopressin stimulation of cortisol secretion in patients with adrenocorticotropin-independent macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 1995;80:2336–2341. [DOI] [PubMed] [Google Scholar]
- 8. Vezzosi D, Cartier D, Régnier C, et al. . Familial adrenocorticotropin-independent macronodular adrenal hyperplasia with aberrant serotonin and vasopressin adrenal receptors. Eur J Endocrinol. 2007;156:21–31. [DOI] [PubMed] [Google Scholar]
- 9. Louiset E, Duparc C, Young J, et al. . Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med. 2013;369:2115–2125. [DOI] [PubMed] [Google Scholar]
- 10. Lacroix A. Heredity and cortisol regulation in bilateral macronodular adrenal hyperplasia. N Engl J Med. 2013;369:2147–2149. [DOI] [PubMed] [Google Scholar]
- 11. Groussin L, Jullian E, Perlemoine K, et al. . Mutations of the PRKAR1A gene in Cushing's syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab. 2002;87:4324–4329. [DOI] [PubMed] [Google Scholar]
- 12. Groussin L, Horvath A, Jullian E, et al. . A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab. 2006;91:1943–1949. [DOI] [PubMed] [Google Scholar]
- 13. Horvath A, Boikos S, Giatzakis C, et al. . A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet. 2006;38:794–800. [DOI] [PubMed] [Google Scholar]
- 14. Findlay JC, Sheeler LR, Engeland WC, Aron DC. Familial adrenocorticotropin-independent Cushing's syndrome with bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 1993;76:189–191. [DOI] [PubMed] [Google Scholar]
- 15. Miyamura N, Taguchi T, Murata Y, et al. . Inherited adrenocorticotropin-independent macronodular adrenal hyperplasia with abnormal cortisol secretion by vasopressin and catecholamines: detection of the aberrant hormone receptors on adrenal gland. Endocrine. 2002;19:319–326. [DOI] [PubMed] [Google Scholar]
- 16. Burgess JR, Harle RA, Tucker P, et al. . Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg. 1996;131:699–702. [DOI] [PubMed] [Google Scholar]
- 17. Yamakita N, Murai T, Ito Y, et al. . Adrenocorticotropin-independent macronodular adrenocortical hyperplasia associated with multiple colon adenomas/carcinomas which showed a point mutation in the APC gene. Intern Med. 1997;36:536–542. [DOI] [PubMed] [Google Scholar]
- 18. Matyakhina L, Freedman RJ, Bourdeau I, et al. . Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical Cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab. 2005;90:3773–3779. [DOI] [PubMed] [Google Scholar]
- 19. Assié G, Libé R, Espiard S, et al. . ARMC5 mutations in macronodular adrenal hyperplasia with Cushing's syndrome. N Engl J Med. 2013;369:2105–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Faucz FR, Zilbermint M, Lodish MB, et al. . Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab. 2014;99:E1113–E1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alencar GA, Lerario AM, Nishi MY, et al. . ARMC5 mutations are a frequent cause of primary macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2014;99:E1501–E1509. [DOI] [PubMed] [Google Scholar]
- 22. Gagliardi L, Schreiber AW, Hahn CN, et al. . ARMC5 mutations are common in familial bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2014;99:E1784–E1792. [DOI] [PubMed] [Google Scholar]
- 23. Elbelt U, Trovato A, Kloth M, et al. . Molecular and clinical evidence for an ARMC5 tumor syndrome: concurrent inactivating germline and somatic mutations are associated with both primary macronodular adrenal hyperplasia and meningioma. J Clin Endocrinol Metab. 2015;100:E119–E128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bertherat J, Contesse V, Louiset E, et al. . In vivo and in vitro screening for illegitimate receptors in adrenocorticotropin-independent macronodular adrenal hyperplasia causing Cushing's syndrome: identification of two cases of gonadotropin/gastric inhibitory polypeptide-dependent hypercortisolism. J Clin Endocrinol Metab. 2005;90:1302–1310. [DOI] [PubMed] [Google Scholar]
- 25. Libé R, Coste J, Guignat L, et al. . Aberrant cortisol regulations in bilateral macronodular adrenal hyperplasia: a frequent finding in a prospective study of 32 patients with overt or subclinical Cushing's syndrome. Eur J Endocrinol. 2010;163:129–138. [DOI] [PubMed] [Google Scholar]
- 26. Assié G, Letouzé E, Fassnacht M, et al. . Integrated genomic characterization of adrenocortical carcinoma. Nat Genet. 2014;46:607–612. [DOI] [PubMed] [Google Scholar]
- 27. Adzhubei IA, Schmidt S, Peshkin L, et al. . A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13. [DOI] [PubMed] [Google Scholar]
- 30. 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, et al. . An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sasano H, Suzuki T, Nagura H. ACTH-independent macronodular adrenocortical hyperplasia: immunohistochemical and in situ hybridization studies of steroidogenic enzymes. Mod Pathol. 1994;7:215–219. [PubMed] [Google Scholar]
- 32. Aiba M, Hirayama A, Iri H, et al. . Adrenocorticotropic hormone-independent bilateral adrenocortical macronodular hyperplasia as a distinct subtype of Cushing's syndrome. Enzyme histochemical and ultrastructural study of four cases with a review of the literature. Am J Clin Pathol. 1991;96:334–340. [DOI] [PubMed] [Google Scholar]
- 33. Bourdeau I, D'Amour P, Hamet P, Boutin JM, Lacroix A. Aberrant membrane hormone receptors in incidentally discovered bilateral macronodular adrenal hyperplasia with subclinical Cushing's syndrome. J Clin Endocrinol Metab. 2001;86:5534–5540. [DOI] [PubMed] [Google Scholar]
- 34. Yamada Y, Sakaguchi K, Inoue T, et al. . Preclinical Cushing's syndrome due to adrenocorticotropin-independent bilateral adrenocortical macronodular hyperplasia with concurrent excess of gluco- and mineralocorticoids. Intern Med. 1997;36:628–632. [DOI] [PubMed] [Google Scholar]
- 35. Hayashi Y, Takeda Y, Kaneko K, et al. . A case of Cushing's syndrome due to ACTH-independent bilateral macronodular hyperplasia associated with excessive secretion of mineralocorticoids. Endocr J. 1998;45:485–491. [DOI] [PubMed] [Google Scholar]
- 36. Goodarzi MO, Dawson DW, Li X, et al. . Virilization in bilateral macronodular adrenal hyperplasia controlled by luteinizing hormone. J Clin Endocrinol Metab. 2003;88:73–77. [DOI] [PubMed] [Google Scholar]
- 37. Grumbach MM, Biller BM, Braunstein GD, et al. . Management of the clinically inapparent adrenal mass (“incidentaloma”). Ann Intern Med. 2003;138:424–429. [DOI] [PubMed] [Google Scholar]
- 38. Bourdeau I, Antonini SR, Lacroix A, et al. . Gene array analysis of macronodular adrenal hyperplasia confirms clinical heterogeneity and identifies several candidate genes as molecular mediators. Oncogene. 2004;23:1575–1585. [DOI] [PubMed] [Google Scholar]
- 39. Berthon A, Stratakis CA. From β-catenin to ARM-repeat proteins in adrenocortical disorders. Horm Metab Res. 2014;46:889–896. [DOI] [PubMed] [Google Scholar]
- 40. Berthon A, Martinez A, Bertherat J, Val P. Wnt/β-catenin signalling in adrenal physiology and tumour development. Mol Cell Endocrinol. 2012;351:87–95. [DOI] [PubMed] [Google Scholar]