

Abstract
Context and Objective:
Intraportal islet transplantation can restore insulin production in type 1 diabetes patients, but its effect is subject to several interfering processes. To assess the influence of β-cell loss before and during engraftment, we searched for a real-time marker of β-cell destruction. Previous studies showed that 65-kDa isoform of glutamate decarboxylase (GAD65) is discharged by chemically damaged rat β-cells. We therefore examined the utility of the GAD65 assay to detect and quantify destruction of human β-cells in vitro and in vivo.
Design and Participants:
A time-resolved fluorescence immunoassay was used to measure GAD65 discharge from β-cells after administration of toxins or after intraportal transplantation. The study in patients involved type 1 diabetes recipients of 56 implants.
Results:
GAD65 was discharged from cultured human β-cells between 4 and 24 hours after acute insult and proportional to the number of dying cells. It was also detected in plasma during the first 24 hours after intraportal transplantation of human islet cell grafts. Diabetic nude rat recipients without hyperglycemic correction exhibited higher plasma GAD65 levels than those with normalization. In type 1 diabetes recipients of grafts with 2–5 × 106 β-cells per kilogram of body weight, five of six with plasma GAD65 greater than 1 ng/mL failed to increase plasma C-peptide by greater than 0.5 ng/mL at posttransplant month 2, whereas five of six with undetectable plasma GAD 65 and 15 of 19 with intermediate levels did result in such increase.
Conclusion:
Plasma GAD65 qualifies as a marker for early β-cell loss after intraportal transplantation. Further studies are needed to extend its clinical utility.
The analytical tools for predicting, classifying, and monitoring type 1 diabetes have steadily expanded as an array of genetic, hormonal, metabolic, and immune markers (1–3). However, a real-time clinical marker for cell destruction analogous to aminotransferases for liver damage or cardiac troponins for myocardial infarction is missing for pancreatic islets. Its availability would allow detection of (pre)clinical phases of β-cell losses and thus locate key events in the natural history of type 1 diabetes; this would help determine populations for preventive or therapeutic interventions. One way to identify candidate markers consists of examining discharge of β-cell proteins from damaged β-cells. The first protein reported with such potential was the 65-kDa isoform of glutamate decarboxylase (GAD65), known to be specifically expressed in human and rat β-cells (4): alloxan- or streptozotocin (STZ)-induced destruction of cultured rat β-cells resulted in the discharge of GAD65 in the medium, whereas the protein was not detected in the medium of viable cells (5).
To further investigate the clinical significance and utility of this finding, the initial enzymatic assay was replaced by more sensitive immune assays (6–9). A subsequent study confirmed that β-cell injury in the rat induced by exposure to alloxan or STZ resulted in an increase in plasma GAD65 at 5 and 10 hours after the in vivo administration (10). Measurement of plasma GAD remained, however unsuccessful, for assessing β-cell destruction caused by (auto)immune reactivity such as after transplantation (11). This may be due to the relatively low concentrations of released GAD65 and its short half-time (12). Therefore, we further improved the sensitivity of our GAD65 time-resolved fluorescence immunoassay (TRFIA) (9) and chose as the first clinical test condition one with an expected high β-cell loss in a narrow time window, namely shortly after intraportal injection of islet cell grafts. This loss is assumed to occur as a result of unfavorable conditions in the implant site such as acute hypoxia, thrombus formation, and inflammation (13, 14). It will probably also vary with the quality of the graft. Its extent is unknown but has been estimated up to 50% of the initial mass (15, 16). The present study examined the utility of the GAD65 assay to detect and quantify an acute destruction process in human β-cells, in vitro and in vivo. Its ability to assess early β-cell loss in type 1 diabetic recipients of an intraportal islet cell graft supports further development of GAD65 discharge as marker in β-cell replacement.
Materials and Methods
Animals
Male Wistar rats (200–300 g, Janvier) were used for in vitro and in vivo cytotoxicity studies. Male Rowett nude rats (aged 7–10 wk; Harlan) served as recipients of human islet-grafts. Animals were housed and handled according to Belgian regulation. Protocols were approved by the local ethical committee (CPG-ID 07–274-3).
Preparation of β-cells and β-cell grafts
Rat β-cells were fluorescence-activated cell sorting purified and cultured as aggregates in serum-free Ham's F10 medium (Gibco) (17). Human islet cells were prepared at our β-Cell Bank, cultured for maximally 4 weeks in serum-free Ham's F10 medium and characterized for β-cell number, endocrine purity, and viability before clinical transplantation (18, 19). Qualitatively acceptable preparations that could not be included in a clinical graft were released for approved projects. Protocols were approved by the local ethical committee (CME 2005/118 2010/193).
Measurement of GAD65 discharge after β-cell destruction in vitro
After 16 hours of culture, rat β-cell aggregates were exposed for 30 minutes to 5 mmol/L STZ (Sigma) and further cultured for 24 hours without the toxin. Because human β-cells are not sensitive to STZ (20), H2O2 (500 μmol/L, 2 h exposure) (Merck) was used to damage human islet cells. Culture medium was collected at 4, 8, and 24 hours and analyzed for its GAD65 content; after 24 hours cells were analyzed for viability (21) and GAD65 content.
Measurement of GAD65 discharge after β-cell destruction in rodents
The first model consisted of male Wistar rats that received a diabetogenic dose of STZ (60 mg/kg). Animals were killed 4, 6, 8, 10, 12, and 24 hours later to collect plasma for glucose, insulin, and GAD65 assays and pancreatic tissue for assessing changes in insulin and GAD65 staining (Supplemental Materials and Methods).
In the second model, we implanted human β-cell preparations [50–60 × 106 β-cells per kilogram body weight (BW) at 40 to 58% purity] in the liver of nude rats with established diabetes after STZ injection 4–7 days earlier. Intraportal injection of islet cell grafts is considered associated with β-cell destruction. Plasma was collected for GAD65, C-peptide, and glucose assays.
Detection of β-cell destruction in type 1 diabetic graft recipients
Between January 2002 and February 2012, 49 nonuremic C-peptide-negative type 1 diabetes patients received islet cell grafts under ATG, mycophenolate mofetil, and tacrolimus (protocol NCT00623610, ethical approval 98/059D) (18, 22). Methylprednisolone was administered iv at the time of each transplantation. Informed consent was obtained from all candidate recipients. The present study was conducted on 26 patients, with 16 being excluded for presence of GAD65 autoantibodies (GADA) at levels that interfere with the GAD65 assay and seven because no plasma was available at the selected time points.
Graft characterization and transplantation procedure have been previously described (18, 23, 24). Each patient received one to five grafts. Each graft was composed of cultured islet cell preparations from one to six donors and characterized for its total β-cell number and purity, its endocrine cell composition, and its C-peptide and GAD65 content. Plasma GAD65, C-peptide, and glucose levels were determined before and during the first 24 hours after the transplantation. Recipients were further followed up for their plasma C-peptide levels at glycemia 120–220 mg/dL. When a graft contained less than 2 × 106 β-cells per kilogram of BW at the time of injection, an additional infusion was given within 1 month; in such a case, GAD65 levels were measured after each injection and the C-peptide increment was determined as the difference between the level at the time of the last injection and the level 2 months later. These grafts were not included in the present analysis (Figure 1).
Figure 1. Flow diagram of subgroups analyzed for GAD65 levels and implant function.

ND, undetectable.
GAD65 and human C-peptide assay
GAD65 was quantified using a TRFIA (9). The assay's functional sensitivity is 0.174 ng/mL. The assay shows negative interference by GADA, even at relatively low titers (90 World Health Organization units per milliliter; reference range: <23 World Health Organization units per milliliter). Human C-peptide was measured by a commercial TRFIA kit (PerkinElmer) adapted to an AutoDelfia automate (PerkinElmer).
Statistical analysis
Data represent means ± SEM. Statistical analysis was carried out using GraphPad Prism5. Significance of differences was calculated with an unpaired two-tailed t test. Correlations were assessed by a Spearman test. Outliers were identified by fitting curves with nonlinear regression (ROUT method, Q = 1%) (25).
Results
GAD65 discharge during toxin-induced necrosis of rat and human β-cells in vitro
When rat β-cells were exposed for 30 minutes to 5 mM STZ, more than 80% of the cells died in the subsequent 24 hours, mainly by necrosis (21, 26). During this period, GAD65 was discharged in the medium, as was also previously observed (5, 6). STZ-treated preparations discharged 11-fold more GAD65 than control cells, leading to 87% reduction in cellular GAD65 content (Table 1). Although their hourly GAD65 discharge did not significantly exceed the control values during the first 4 hours, they exceeded those of control cells by 17-fold between 4 and 8 hours and remained elevated between 8 and 24 hours (Table 1); in absolute values, the highest discharge was measured between 4 and 8 hours. The sum of total medium GAD65 discharge and residual cellular GAD65 content was comparable in control and STZ cells, which indicates that their respective change during a necrotic process is inversely related, expressing the degree of cell death without being influenced by major intra- or extracellular degradation. Over 24 hours, the medium of STZ-treated and control cells contained, respectively, 80% and 8% of this sum, which correlates with the percentages of dead cells counted at the end of this period.
Table 1.
GAD65 Discharge by Necrotic Rat β-Cells and Human Islet Cells in Culture
| Cells |
Medium |
||||
|---|---|---|---|---|---|
| end 24 h |
0–4 h |
4–8 h |
8–24 h |
Total |
|
| pg/103 Cells | pg/103 Cells per Hour | pg/103 Cells per 24 Hours | |||
| Rat cells | |||||
| Control | 39.3 ± 1.3 | 0.3 ± 0.3 | 0.2 ± 0.1 | 0.1 ± 0.1 | 3.3 ± 1.4 |
| STZ | 5.0 ± 0.9a | 0.6 ± 0.3 | 3.4 ± 0.3a | 1.3 ± 0.3b | 37.2 ± 5.7b |
| Human cells | |||||
| Control | 41.7 ± 0.7 | 0.22 ± 0.09 | 0.01 ± 0.01 | 0.04 ± 0.02 | 1.52 ± 0.56 |
| H2O2 | 19.6 ± 3.0b | 0.52 ± 0.18 | 0.67 ± 0.10c | 0.95 ± 0.05c | 20.02 ± 3.03c |
Rat and human β-cells were incubated with and without toxins (rat: STZ 5 mM, 30 min; human: H2O2 500 μM, 2 h), washed, and cultured for 24 hours without toxin. Culture medium was sampled at 4, 8, and 24 hours. Data represent means ± SEM of four independent experiments. Statistically significant differences between control and toxin conditions were calculated by Student t test.
P < .001 vs control at same time point;
P < .05 vs control at same time point;
P < .01 vs control at same time point.
GAD65 was also discharged by human β-cell preparations after exposure to H2O2 (500 μM for 2 h). This condition resulted in 72% ± 12% dead cells after 24 hours vs 31% ± 15% in control cells (P < .05). As in rat β-cells, the sum of medium GAD65 content and residual cellular content was comparable in toxin-treated and control cells, with a significantly higher fraction in the medium in the cytotoxic condition (50% vs 3%, Table 1). The toxic condition caused a rise in GAD65 discharge between 4 and 24 hours after its exposure. Control preparations showed some GAD65 discharge during the first 4 hours after plating (∼2% cellular content) but not thereafter, probably the result of damaged β-cells at the start.
GAD65 discharge in plasma after STZ injection in rats and preceding onset of diabetes
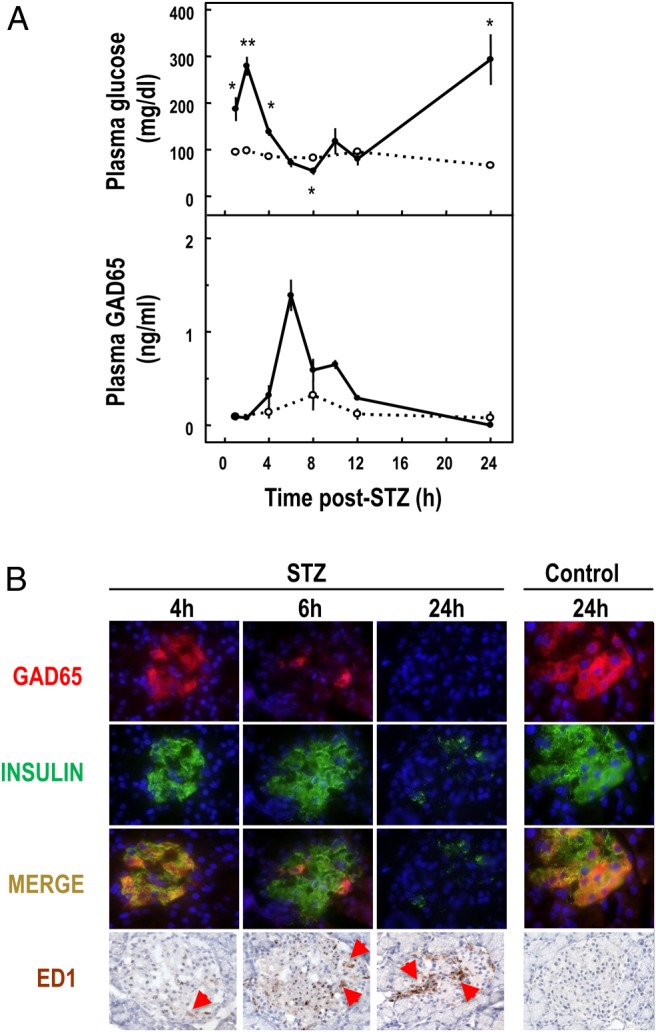
Injection of STZ 60 mg/kg in rats caused a discharge of GAD65 in plasma between 4 and 12 hours later, with peak levels between 4 and 8 hours (Figure 2A). The area under the curve of GAD65 was 2.8 ± 0.9 ng/mL in the control rats and 7.9 ± 0.9 ng/mL in the STZ rats (P < .05). Immunostaining for GAD65 in pancreatic islets showed a marked disappearance of the protein from 6 hours onward, the time at which macrophages became detectable in the islets (Figure 2B). The disappearance of insulin staining occurred later, explaining the installation of a sustained diabetic state between 12 and 24 hours (Figure 2).
Figure 2. A, Plasma glucose and GAD65 concentrations in rats injected at time 0 with STZ (60 mg/kg body weight, full circles) or with solvent (open circles). Data represent mean ± SEM of three (control) or four (STZ) animals. Statistical significance of difference between control and STZ: *, P < .05, **, P < .01. B, Immunofluorescent double staining for GAD65 (red) and insulin (green) or immunostaining for macrophages (ED1, brown) on pancreatic sections of STZ-treated and control rats. Nuclei are stained with 4′,6′-diamino-2-phenylindole (blue) in fluorescence or hematoxylin.
GAD65 discharge after intraportal injection of human islet cell grafts in diabetic nude rats
Intraportal injection of human islet cell grafts corrected within 1 week the basal hyperglycemia in four of seven STZ-diabetic nude rat recipients (Figure 3A). The three other recipients remained hyperglycemic but still presented detectable human C-peptide levels (2.2 ± 0.8 ng/mL), which was, however, significantly lower than in normoglycemic animals (6.7 ± 1.1 ng/mL, P < .05). When normoglycemic and hyperglycemic animals were compared for their graft composition, no differences were noticed in the number of implanted β-cells or hormone content, but hyperglycemic animals received implants with a 2-fold higher number of nongranulated cells (Supplemental Table 1).
Figure 3. Plasma glucose, C-peptide, and GAD65 levels in STZ-diabetic nude rats after intraportal transplantation of human islet cell grafts.

A, Animals were divided into two subgroups according to their 2-hour fasting glycemia at PT week 1: the normoglycemic group (open circles) consisted of four animals receiving grafts from three different preparations, and the hyperglycemic group (filled circles) consisted of three animals receiving grafts from two different preparations. B, Plasma GAD65 levels in the two subgroups in function of time after the transplant. Statistical significance of difference between subgroups: *, P < .05; †, P < .01.
All seven recipients exhibited a rise in plasma GAD65 levels within the first 12 hours after transplantation, reaching peak levels between 0.5 and 1 hour (Figure 3B). The levels were significantly higher for three hyperglycemic animals (Figure 3B). Over the first 3 hours, the area under the curve of GAD65 content was more than 3 times higher (20 ± 3 vs 6 ± 1, P < .01), whereas the GAD65 content of the initial grafts was similar (Supplemental Table 1). During the first 30 minutes, hyperglycemic animals released 14% of its initial graft GAD65 content into the plasma, compared with only 4% for normoglycemic animals. The higher GAD65 discharge observed for hyperglycemic animals indicates a higher β-cell loss and explains the failure of these recipients to normalize hyperglycemia. Plasma GAD65 levels at 1 hour after transplantation did not significantly correlate with total cell or β-cell number in the initial grafts but correlated with their number of duct cells (Supplemental Figure 1).
Discharge of GAD65 after intraportal injection of human islet cell grafts in type 1 diabetes patients
We measured plasma GAD65 in 26 type 1 diabetic recipients in whom a total of 56 intraportal islet cell grafts had been injected (seven patients received a single injection and 19 more than one graft). In none of them was plasma GAD65 detected before transplantation; for 37 of the 56 injections, the protein was measured 1 hour later. GAD65 plasma levels correlated positively with the number of β-cells (expressed per kilogram of BW) in the grafts (P = .001, Figure 4A), which indicates that β-cell loss is inherent to our procedure, being proportional to the initial β-cell mass. Absence of detectable GAD65 appears to be at least in part attributable to a small size of the injected β-cell mass: GAD65 discharge was undetectable for the majority of grafts (13 of 25) less than 2 × 106 β-cells per kilogram, whereas this was the case for only 6 of 31 of larger grafts (Figure 1). Regression analysis identified five outliers with plasma GAD65 levels that were disproportionately high for the β-cell number in the graft (Figure 4A, filled circles), thus indicative for a disproportionally high β-cell loss. Plasma GAD65 levels were also correlated with the graft C-peptide content (Figure 4A, P < .001) and showed the same five outliers.
Figure 4. A, Correlation between plasma GAD65 levels at 60 minutes PT in type 1 diabetic patients with β-cell mass in graft as expressed by β-cell number and C-peptide content. Open circles represent grafts with plasma GAD65 1 ng/mL or less; filled circles represent grafts with plasma GAD65 greater than 1 ng/ mL, which were identified as outliers by ROUT (Robust regression and Outlier removal) test (set Q = 1%); filled square represents a graft with plasma GAD65 greater than 1 ng/ mL, which was not identified as an outlier; the full lines represent regression lines without omission of outliers and the dashed lines their 95% confidence interval (CI). Statistical significance of the correlation: left panel, P = .001; right panel, P < .001. After omission of outliers: left panel, r2 = 0.179; right panel, r2 = 0.401; P < .001 for both. B, Correlation between plasma GAD65 levels at PT minute 60 and plasma C-peptide increment (differences between before the transplant and PT month 2). Open circles represent grafts with low plasma GAD65 levels (≤1 ng/mL or < 0.04 ng/mL when GAD65 levels were normalized to transplanted β-cell mass) and C-peptide increment of 0.5 ng/mL or less; open squares represent grafts with low plasma GAD65 and C-peptide increment greater than 0.5 ng/mL; filled circles represent grafts with disproportionally plasma GAD65 levels (outliers in panel A); filled square represents a graft with plasma GAD65 greater than 1 ng/ mL, which was not identified as an outlier in panel A. Statistical significance of the correlation: left panel, P = .0050; right panel, P = .0035.

For 23 of the 37 conditions with detectable plasma GAD65 after transplantation, multiple blood samples were available over the first 24 hours, showing that the protein was already measured after 5 minutes and reached its maximal level at minutes 15–30, indicating that it not only originated in the injected medium but was also released from cells in the liver. On the basis of the peak plasma GAD65 levels and the corresponding GAD65 content of the grafts, and assuming a circulating volume of 39 mL per kilogram of BW (27), we calculated how much of the cellular protein had been released in the plasma at that time. Values largely varied with median of 10% (interquartile range 8%–14%) and a maximum of 47%. These values were more than 5-fold higher than the levels that are expected in the injection medium (<2% cellular GADA65 discharged within 4 h of in vitro handling, Table 1). In view of the variability in the peak values, all measurements at later time points were expressed as a percentage of the minute 15 value to determine the disappearance rate of the plasma GAD65 (Supplemental Figure 2).
Correlation between plasma GAD65 levels after islet cell transplantation and its metabolic outcome
A graft size with at least 2 × 106 β-cells per kilogram of BW is one condition to bring C-peptide-negative recipients to C-peptide levels greater than 0.5 ng/mL at posttransplant (PT) month 2, with an associated metabolic effect (18). Another variable with functional impact is probably the degree of β-cell loss during implantation, for which GAD65 levels at PT minute 60 might serve as marker. We plotted these levels against the increment in plasma C-peptide for recipients of grafts containing at least 2 × 106 β-cells per kilogram of BW (Figure 4B).
The five outliers with disproportionally high plasma GAD65 did not achieve a C-peptide rise greater than 0.5 ng/mL (filled circles in Figure 4B). Their plasma GAD65 levels (>1 ng/mL) can be used as marker for implants with lower chance for metabolic effect (Figure 1): only one of six implants showed a C-peptide increment greater than 0.5 ng/mL, whereas this was the case in 20 of 25 with lower GAD65 levels. Most notably, this patient received the highest β-cell mass (5.3 × 106 β-cells per kilogram of BW, with the highest associated C-peptide content), which made his high GAD65 level not an outlier (Figure 4A, filled square). This observation confirms that plasma GAD65 levels after transplantation should be interpreted in light of the β-cell mass in the graft (Figure 4B).
A positive functional outcome of the implants was correlated with low GAD65 values: five of six implants with undetectable GAD65 discharge achieved a C-peptide increment greater than 0.5 ng/mL (Figure 1), and 12 of 12 implants with a C-peptide increase greater than 1 ng/mL exhibited a GAD65 level less than 0.75 ng/mL or less than 0.4 ng/mL when GAD65 levels were normalized to transplanted β-cell mass (Figure 4B).
Discussion
Since the first report on GAD65 as an extracellular marker for dying β-cells (5), studies have confirmed this concept in conditions of toxin-induced destruction of rat β-cells (6, 10), but no in vitro or in vivo data have appeared for human β-cells. Lack of a sensitive assay has been an obstacle to examine the potential clinical relevance of the early findings. Development of a time-resolved fluorescence immunoassay (9) allowed us to detect and follow discharge of GAD65 from damaged human β-cells, during culture, and shortly after transplantation.
When human islet cell preparations were exposed to a H2O2 concentration that killed 70% of the cells during the subsequent 24 hours, approximately 50% of the cellular GAD65 content was discharged in the medium, mainly between 4 and 24 hours. These data are similar to those for rat β-cells after a brief exposure to a toxic STZ concentration. The GAD65 in culture medium and plasma thus represents a quantitative marker for β-cell death after acute damage.
Discharge of GAD65 from human islet cell preparations was also detected and quantified over 24 hours after their intraportal transplantation in nude rats. GAD65 levels ranged between 0.5 and 10 ng/mL, with peak values between 0.5 and 1 hour after injection, indicating that presence of the protein in circulation was not the result of its presence in the medium of injected grafts but of damage during lodging in the intrahepatic veins. According to the GAD65 levels, the degree of postinjection damage is variable, with an impact on metabolic outcome. Implants that did not correct hyperglycemia within 1 week were associated with 3.7-fold higher GAD65 discharge than those with correction. Failure to achieve normoglycemia was also associated with a higher number of duct cells in the grafts, whereas the respective β-cell numbers were similar. Because human duct cells were found to exert a potent factor VII-dependent procoagulant activity related to their expression of tissue factor (28), it is conceivable that a higher number causes more extensive thrombotic events that contribute to early damage of neighboring β-cells.
GAD65 was also discharged in plasma after intraportal islet cell transplantation in type 1 diabetes patients. GAD65 levels ranged from 0.17 (detection limit) to 4.6 ng/mL, in general considerably lower than in nude rat recipients of human islet cell grafts. This difference can be attributed to the more than 10-fold lower β-cell dose in patients (up to 5 × 106 β-cells per kilogram of BW vs 50–60 × 106 in rats). Levels in patients were positively correlated with the number of β-cells in the grafts, suggesting an early loss of β-cells that is inherent to the procedure (graft preparation and implantation) and proportional to the initial β-cell mass. This explains, at least in part, why grafts with less than 2 × 106 β-cells per kilogram of BW result in more GAD65-negative cases than those with a higher number. Because this lower dose does not meet the minimum that is set for achieving a functional hormone release (18), we examined the utility of GAD65 measurements for grafts with a higher cell dose (2–5 × 106 β-cells per kilogram of BW). In this range, six implants caused a GAD65 level of 1 ng/mL or greater at PT minute 60; five of six failed to increase C-peptide levels by 0.5 ng/mL, whereas 20 of 25 of implants with lower GAD65 levels exerted this effect. It was also noticed that all implants (n = 12) with the best outcome at PT month 2 (C-peptide > 1 ng/mL) exhibited a GAD65 level less than 0.75 ng/mL. Plasma GAD65 levels during the first 4 hours after the intraportal injection thus represent a clinically relevant marker for the degree of early β-cell loss. The values need to be interpreted against the size of the graft, for which β-cell number and C-peptide content per kilogram of BW can serve as parameter.
We were unable to examine whether GAD65 discharge in patients is lower for grafts with a lower percentage of duct cells, as was the case in rat recipients. The percent duct cells in clinically used islet cell preparations is in the higher range [mean of 47% (18)] because we have to select quantity above purity to reach the total β-cell number that is necessary for achieving a functional hormone release. This high contamination cannot be avoided, at least not with our current methods (18, 29), making it a likely cause for early β-cell loss in patients. When the amount of GAD65 in the plasma is compared with the initial GAD65 content in the grafts, this loss appears to vary from 5% to 47%. This figure might be an underestimation because only part of the GAD65 from damaged or dead β-cells might end up in circulation. Losses up to 50% have been considered to occur shortly after implantation and are attributed to a combination of causes, including hypoxia, inflammation, mechanical stress, and drug toxicity (11, 13, 15, 16). The GAD65 assay may help quantify this loss and thus also be useful in assessing the effects of antiinflammatory treatment as used in other transplant protocols (30, 31). This will, however, require standardized procedures in which the size and viability of the initial β-cell mass is also evaluated.
The clinical utility of the GAD65 assay is at present limited: although it may allow early identification of implants that will fail to reach the PT month 2 readout for functional β-cells, this represents only a small application for a marker intended to have a much larger scope, such as detecting episodes of β-cell destruction at later time points, as can occur under (auto)immune, inflammatory, or drug aggression. This would help select and guide interventions that preserve metabolic end points. The implementation of GAD65 assays is further restricted by the presence of circulating GAD65 autoantibodies due to direct interference with the measurements and altering the marker's half-life (32). Although we have not yet undertaken longitudinal studies in transplanted patients, the current data indicate that further development is needed to increase the analytical sensitivity of the GAD65 assay. Additional candidate markers should also be evaluated. We recently reported two other β-cell-selective proteins that are discharged by damaged rat and human β-cells, eg, doublecortin and phosphatase 1-inhibitor (33, 34). Promising in terms of analytical sensitivity and ease of assay development, is the use of nucleic acid-type biomarkers eg, β-cell-selective microRNA miR-375 (35), and the ratio of unmethylated/methylated (prepro)insulin promotor (insulin demethylation index) (36–39). The latter set of biomarkers will now require clinical validation. Current clinical immunoassays at best achieve detection limits in the low femtomolar range, perhaps even lower for immuno-PCR-based assays (40), but can be operated at high throughput, high precision, and relatively low cost, rendering protein-type biomarkers like GAD65 still a relevant choice for further development.
Acknowledgments
We thank Evilien Vekens, Lei Jiang, Katrijn Verhaeghen, and Olivier Costa for their contribution to the improvement of the GAD65 assay.
The clinical trial registration number was NCT00623610.
This study was supported by Juvenile Diabetes Research Foundation Grants 4-2001-434, 2005-132-1327, and 1-PNF-2014-181-A-V, by European Union Grant FP7-Health-2009-1.4-1 (to the Center for β Cell Therapy in Diabetes; www.betacelltherapy.org), by the Research Foundation Flanders Grants G.0374.08N and G.0492.12N, and by the Willy Gepts Fund, University Hospital Brussels (2011). B.K. and G.A.M. are recipients of a senior clinical research fellowship (Fundamenteel Klinisch Onderzoeksmandaat) from the Research Foundation Flanders.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This study was supported by Juvenile Diabetes Research Foundation Grants 4-2001-434, 2005-132-1327, and 1-PNF-2014-181-A-V, by European Union Grant FP7-Health-2009-1.4-1 (to the Center for β Cell Therapy in Diabetes; www.betacelltherapy.org), by the Research Foundation Flanders Grants G.0374.08N and G.0492.12N, and by the Willy Gepts Fund, University Hospital Brussels (2011). B.K. and G.A.M. are recipients of a senior clinical research fellowship (Fundamenteel Klinisch Onderzoeksmandaat) from the Research Foundation Flanders.
Footnotes
- BW
- body weight
- GAD65
- 65-kDa isoform of glutamate decarboxylase
- PT
- posttransplant
- STZ
- streptozotocin
- TRFIA
- time-resolved fluorescence immunoassay.
References
- 1. Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clin Chem. 2011;57:176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bingley PJ, Bonifacio E, Williams AJ, Genovese S, Bottazzo GF, Gale EA. Prediction of IDDM in the general population: strategies based on combinations of autoantibody markers. Diabetes. 1997;46:1701–1710. [DOI] [PubMed] [Google Scholar]
- 3. Roep BO, Peakman M. Diabetogenic T lymphocytes in human type 1 diabetes. Curr Opin Immunol. 2011;23:746–753. [DOI] [PubMed] [Google Scholar]
- 4. Karlsen AE, Hagopian WA, Petersen JS, et al. . Recombinant glutamic acid decarboxylase (representing the single isoform expressed in human islets) detects IDDM-associated 64,000-M(r) autoantibodies. Diabetes. 1992;41:1355–1359. [DOI] [PubMed] [Google Scholar]
- 5. Smismans A, Ling Z, Pipeleers D. Damaged rat β-cells discharge glutamate decarboxylase in the extracellular medium. Biochem Biophys Res Commun. 1996;228:293–297. [DOI] [PubMed] [Google Scholar]
- 6. Hao W, Daniels T, Pipeleers DG, et al. . Radioimmunoassay for glutamic acid decarboxylase-65. Diabetes Technol Ther. 1999;1:13–20. [DOI] [PubMed] [Google Scholar]
- 7. Schlosser M, Hahmann J, Ziegler B, Augstein P, Ziegler M. Sensitive monoclonal antibody-based sandwich ELISA for determination of the diabetes-associated autoantigen glutamic acid decarboxylase GAD65. J Immunoassay. 1997;18:289–307. [DOI] [PubMed] [Google Scholar]
- 8. Waldrop MA, Suckow AT, Hall TR, Hampe CS, Marcovina SM, Chessler SD. A highly sensitive immunoassay resistant to autoantibody interference for detection of the diabetes-associated autoantigen glutamic acid decarboxylase 65 in blood and other biological samples. Diabetes Technol Ther. 2006;8:207–218. [DOI] [PubMed] [Google Scholar]
- 9. Rui M, Hampe CS, Wang C, et al. . Species and epitope specificity of two 65 kDa glutamate decarboxylase time-resolved fluorometric immunoassays. J Immunol Methods. 2007;319:133–143. [DOI] [PubMed] [Google Scholar]
- 10. Waldrop MA, Suckow AT, Marcovina SM, Chessler SD. Release of glutamate decarboxylase-65 into the circulation by injured pancreatic islet β-cells. Endocrinology. 2007;148:4572–4578. [DOI] [PubMed] [Google Scholar]
- 11. Shapiro AM, Hao EG, Lakey JR, et al. . Novel approaches toward early diagnosis of islet allograft rejection. Transplantation. 2001;71:1709–1718. [DOI] [PubMed] [Google Scholar]
- 12. Schlosser M, Walschus U, Kloting I, Walther R. Determination of glutamic acid decarboxylase (GAD65) in pancreatic islets and its in vitro and in vivo degradation kinetics in serum using a highly sensitive enzyme immunoassay. Dis Markers. 2008;24:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piemonti L, Guidotti LG, Battaglia M. Modulation of early inflammatory reactions to promote engraftment and function of transplanted pancreatic islets in autoimmune diabetes. Adv Exp Med Biol. 2010;654:725–747. [DOI] [PubMed] [Google Scholar]
- 14. Emamaullee JA, Shapiro AM. Interventional strategies to prevent β-cell apoptosis in islet transplantation. Diabetes. 2006;55:1907–1914. [DOI] [PubMed] [Google Scholar]
- 15. Eich T, Eriksson O, Lundgren T. Visualization of early engraftment in clinical islet transplantation by positron-emission tomography. N Engl J Med. 2007;356:2754–2755. [DOI] [PubMed] [Google Scholar]
- 16. Eriksson O, Eich T, Sundin A, et al. . Positron emission tomography in clinical islet transplantation. Am J Transplant. 2009;9:2816–2824. [DOI] [PubMed] [Google Scholar]
- 17. Pipeleers DG, in't Veld PA, Van de Winkel M, Maes E, Schuit FC, Gepts W. A new in vitro model for the study of pancreatic A and B cells. Endocrinology. 1985;117:806–816. [DOI] [PubMed] [Google Scholar]
- 18. Keymeulen B, Gillard P, Mathieu C, et al. . Correlation between β-cell mass and glycemic control in type 1 diabetic recipients of islet cell graft. Proc Natl Acad Sci USA. 2006;103:17444–17449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ling Z, Pipeleers DG. Prolonged exposure of human β-cells to elevated glucose levels results in sustained cellular activation leading to a loss of glucose regulation. J Clin Invest. 1996;98:2805–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eizirik DL, Pipeleers DG, Ling Z, Welsh N, Hellerstrom C, Andersson A. Major species differences between humans and rodents in the susceptibility to pancreatic β-cell injury. Proc Natl Acad Sci USA. 1994;91:9253–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang C, Ling Z, Pipeleers D. Comparison of cellular and medium insulin and GABA content as markers for living β-cells. Am J Physiol Endocrinol Metab. 2005;288:E307–313. [DOI] [PubMed] [Google Scholar]
- 22. Hilbrands R, Huurman VA, Gillard P, et al. . Differences in baseline lymphocyte counts and autoreactivity are associated with differences in outcome of islet cell transplantation in type 1 diabetic patients. Diabetes. 2009;58:2267–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Movahedi B, Keymeulen B, Lauwers MH, Goes E, Cools N, Delvaux G. Laparoscopic approach for human islet transplantation into a defined liver segment in type-1 diabetic patients. Transpl Int. 2003;16:186–190. [DOI] [PubMed] [Google Scholar]
- 24. Maleux G, Gillard P, Keymeulen B, et al. . Feasibility, safety, and efficacy of percutaneous transhepatic injection of β-cell grafts. J Vasc Interv Radiol. 2005;16:1693–1697. [DOI] [PubMed] [Google Scholar]
- 25. Motulsky HJ, Brown RE. Detecting outliers when fitting data with nonlinear regression—a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics. 2006;7:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoorens A, Pipeleers D. Nicotinamide protects human β-cells against chemically-induced necrosis, but not against cytokine-induced apoptosis. Diabetologia. 1999;42:55–59. [DOI] [PubMed] [Google Scholar]
- 27. Frank H, Gray SJ. The determination of plasma volume in man with radioactive chromic chloride. J Clin Invest. 1953;32:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beuneu C, Vosters O, Movahedi B, et al. . Human pancreatic duct cells exert tissue factor-dependent procoagulant activity: relevance to islet transplantation. Diabetes. 2004;53:1407–1411. [DOI] [PubMed] [Google Scholar]
- 29. Street CN, Lakey JR, Shapiro AM, et al. . Islet graft assessment in the Edmonton Protocol: implications for predicting long-term clinical outcome. Diabetes. 2004;53:3107–3114. [DOI] [PubMed] [Google Scholar]
- 30. Bellin MD, Kandaswamy R, Parkey J, et al. . Prolonged insulin independence after islet allotransplants in recipients with type 1 diabetes. Am J Transplant. 2008;8:2463–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Froud T, Baidal DA, Faradji R, et al. . Islet transplantation with alemtuzumab induction and calcineurin-free maintenance immunosuppression results in improved short- and long-term outcomes. Transplantation. 2008;86:1695–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Remaley AT, Wilding P. Macroenzymes: biochemical characterization, clinical significance, and laboratory detection. Clin Chem. 1989;35:2261–2270. [PubMed] [Google Scholar]
- 33. Jiang L, Brackeva B, Stange G, et al. . LC-MS/MS identification of doublecortin as abundant β-cell-selective protein discharged by damaged β-cells in vitro. J Proteomics. 2013;80:268–280. [DOI] [PubMed] [Google Scholar]
- 34. Jiang L, Brackeva B, Ling Z, et al. . Potential of protein phosphatase inhibitor 1 as biomarker of pancreatic β-cell injury in vitro and in vivo. Diabetes. 2013;62:2683–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Erener S, Mojibian M, Fox JK, Denroche HC, Kieffer TJ. Circulating miR-375 as a biomarker of β-cell death and diabetes in mice. Endocrinology. 2013;154:603–608. [DOI] [PubMed] [Google Scholar]
- 36. Akirav EM, Lebastchi J, Galvan EM, et al. . Detection of β-cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci USA. 2011;108:19018–19023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Husseiny MI, Kuroda A, Kaye AN, Nair I, Kandeel F, Ferreri K. Development of a quantitative methylation-specific polymerase chain reaction method for monitoring β-cell death in type 1 diabetes. PLoS One. 2012;7:e47942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fisher MM, Perez Chumbiauca CN, Mather KJ, Mirmira RG, Tersey SA. Detection of islet beta-cell death in vivo by multiplex PCR analysis of differentially methylated DNA. Endocrinology. 2013;154:3476–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Husseiny MI, Kaye A, Zebadua E, Kandeel F, Ferreri K. Tissue-specific methylation of human insulin gene and PCR assay for monitoring β-cell death. PLoS One. 2014;9:e94591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Freudenberg JA, Bembas K, Greene MI, Zhang H. Non-invasive, ultra-sensitive, high-throughput assays to quantify rare biomarkers in the blood. Methods. 2008;46:33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]