

Abstract
Context:
Germline mutations in the phosphatase and tensin homolog (PTEN) tumor suppressor gene are found in the majority of patients with Cowden syndrome (CS), who have an increased risk of breast, thyroid, and endometrial cancer. According to our current understanding of genetic changes in the PTEN gene and the resultant phenotypic features of CS, pancreatic neuroendocrine tumors (NETs) are not considered part of the clinical spectrum of CS.
Case description:
We report a unique case of an advanced NET of the pancreas in a patient with CS. The germline DNA sequencing confirmed the clinical diagnosis of CS and revealed a PTEN mutation c.697C→T (p.R233*) causing a premature stop codon in exon 7. The tumor DNA sequencing showed no loss of heterozygosity or any copy number changes and no other deleterious genetic alterations, including those commonly mutated in sporadic pancreatic NETs: MEN1, ATRX, DAXX, TP53, and genes involved in the mammalian target of rapamycin pathway. In addition, the high-throughput transcriptome analyzed by RNA-seq did not reveal any missed genetic alterations, aberrant splicing variants, gene fusions, or gene expression alterations. The immunohistochemical staining of the tumor for PTEN revealed an abnormal, uniformly strong cytoplasmic staining of tumor cells with virtually absent nuclear staining.
Conclusion:
The results from genetic testing and histopathological techniques used to confirm CS diagnosis and characterize this unusual tumor tempted us to believe that in this case, the pancreatic NET was not a sporadic malignancy that occurred by coincidence, but rather represented a new entity in the spectrum of malignancies associated with CS.
The phosphatase and tensin homolog (PTEN) gene on chromosome 10 encodes a protein with lipid phosphatase function and is a known tumor suppressor gene that negatively regulates the phosphatidylinositol 3-kinase-AKT and mammalian target of rapamycin (mTOR) signaling pathways (1). Germline mutations in the PTEN gene have been linked to Cowden syndrome (CS) and several other related disorders including Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, and Proteus syndrome, collectively known as PTEN hamartoma tumor syndrome (PHTS) (1–3). CS, the principal PHTS, is an autosomal dominant disorder characterized by macrocephaly and an increased risk of benign (hamartomas of the skin and colon) and malignant (breast, thyroid, endometrial, and renal) tumors (4, 5).
Although CS shares common genetic etiology and several clinical features with the other PTEN-related disorders, the rarity of phenotypic heterogeneity and the variable clinical presentation pose a significant diagnostic and management challenge. Since the initial proposed diagnostic criteria, there have been several consensus statements and revisions to encompass the growing list of CS clinical features (2, 6–9). It is therefore important to thoroughly document and report genetic and phenotypic features of CS, which will help to expand our knowledge and contribute to a better understanding, characterization, and management of this complex disorder and ultimately impact the outcome of patients with PHTS by early diagnosis and treatment (10).
Here, we report the genotype-phenotype correlation of a unique case of advanced, progressive, pancreatic neuroendocrine tumor (NET) as an unusual manifestation and a presenting feature in a patient with CS.
Case Report
A 51-year-old gentleman presented with a 1-week history of jaundice and increasing upper abdominal pain. His rich family history and past medical history of macrocephaly, multiple intestinal polyps, skin hamartomas, lipomas, and thyroidectomy for papillary thyroid cancer at age 38 years were highly suggestive of the clinical diagnosis of CS (Figure 1, A and B).
Figure 1. Pedigree and diagnostic criteria for CS.

A, Patient's (proband) family tree in four generations. The patient's three children are 5 to 11 years of age. B, Revised operational diagnostic criteria for CS (7).
At the initial evaluation, the patient had icterus, upper abdominal pain, no lymphadenopathy, and stigmata of CS including macrocephaly and facial papillomatous papules. The remainder of his physical examination was unremarkable. A computed tomography (CT) scan of the chest, abdomen, and pelvis at that time revealed a large (9 × 8 × 4 cm) ill-defined mass of the head and neck of the pancreas, invading the celiac axis; encasing the celiac artery, superior mesenteric, and splenic arteries and veins; and compressing the portal vein confluence. There were several enlarged locoregional lymph nodes. He underwent endoscopic ultrasound-guided fine-needle aspiration of the pancreatic mass and endoscopic retrograde cholangiopancreatography for biliary stent placement and biliary decompression. The cytopathological features of fine-needle aspiration specimen and positive immunohistochemical staining for chromogranin A and synaptophysin were consistent with a tumor of neuroendocrine cell origin, stage uT4N1Mx determined by endoscopic sonography. The patient underwent exploratory laparotomy with a planned Whipple procedure for complete tumor resection; however, this proved not to be feasible, and debulking of the tumor invading the celiac area with a bypass hepaticojejunostomy and gastrojejunostomy was performed. The pathological evaluation of the tumor showed a well-differentiated, World Health Organization grade 1 NET with a Ki-67 < 2% and involved margins. Immunohistochemistry was positive for chromogranin A and synaptophysin (Figure 2). The patient recovered from surgery well and, within 1 year after the procedure, underwent tumor ablation by NanoKnife technique, which was not successful. He had two cycles of chemotherapy with streptozocin and doxorubicin without any response. The chemotherapy regimen was switched to capecitabine and temozolomide; however, the therapy was halted because he developed a profound thrombocytopenia necessitating multiple transfusions of blood products. Follow-up CT and magnetic resonance imaging showed that disease had progressed and the primary tumor size increased from 9 to 12 cm in its greatest dimension.
Figure 2. Tumor immunohistochemistry.

A, Hematoxylin and eosin-stained section of pancreatic NET (20 × magnification). B, Immunohistochemistry for chromogranin A (20 × magnification; brown color). C, Immunohistochemistry for synaptophysin (20 × magnification; brown color). D, Immunohistochemistry for PTEN showing strong cytoplasmic staining of tumor cells (brown color) with absent nuclear staining.
The patient underwent genetic counseling and was referred to the Endocrine Oncology Branch, National Cancer Institute, for further evaluation and screening for enrollment in a clinical trial of mutation targeting therapy with sunitinib (a polytyrosine kinase inhibitor) or everolimus (an mTOR inhibitor), which are among the few available therapeutic options for patients with advanced, progressive, unresectable, low- or intermediate-grade pancreatic NETs (ClinicalTrials.gov Identifier: NCT02315625) (11). Recommendations were also made for genetic counseling and consideration for genetic testing of the patient's family members who meet the diagnostic criteria for CS; interested family members will be evaluated and followed up. The patient's workup included histopathological characterization, grading, immunohistochemistry, and tumor genotyping by next-generation sequencing and anatomic and functional imaging including CT and 68-Gallium DOTATATE-PET/CT (ClinicalTrials.gov Identifier: NCT01967537) (Figure 3). The results from the laboratory workup for biomarkers were consistent with a nonfunctional pancreatic NET, but with a chromogranin A level of 436 ng/mL (normal level ≤ 93 ng/mL), slightly elevated 5-hydroxyindoleacetic acid to 12 mg/24 hours (normal level ≤ 8 mg/24 h), and normal gastrin (50 pg/mL), neuron specific enolase (6.2 ng/mL), glucagon (57 pg/mL), vasointestinal peptide (28 pg/mL), and pancreatic polypeptide (<40 pg/mL).
Figure 3. Imaging studies of the pancreatic NET.
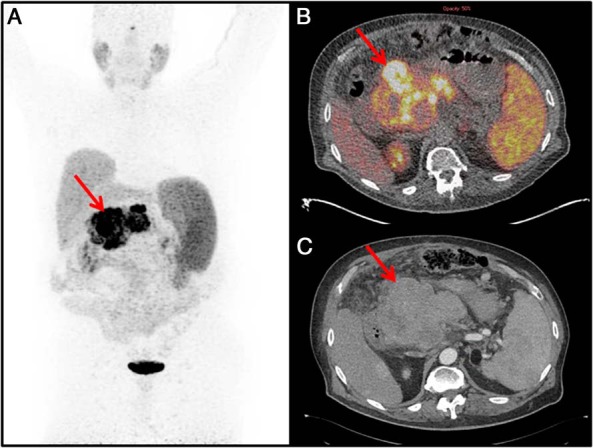
A, 68Ga-DOTATATE PET maximal intensity projection image showing a large central lesion in the abdomen (red arrow). B, 68Ga-DOTATATE PET/CT image showing the pancreatic head mass with a maximum standardized uptake value of 58 (red arrow). C, Arterial phase CT showing corresponding mass (red arrow).
As part of the screening process for the mutation targeting trial, tumor DNA was sequenced, and variants were analyzed relative to peripheral blood DNA in 500 genes associated with a broad spectrum of cancers. The gene panel coding region was sequenced to an average coverage of more than 100 ×. Blood DNA served as a germline reference to gauge somatic mutation status. Sequencing of whole transcriptome (RNA-seq) was used as an additional control and for validation (Supplemental Figure 1).
Results of the germline sequencing revealed a PTEN, nonsense mutation c.697C→T (p.R233*) causing a premature stop codon in exon 7, which encodes a portion of the C2 domain of the PTEN protein. This finding confirmed the clinical diagnosis of CS because up to 80–85% of patients who meet the diagnostic criteria carry a germline mutation in the PTEN gene (8, 12). The tumor DNA sequencing did not show loss of heterozygosity (LOH) or any copy number changes, and there were no other deleterious genetic alterations in the rest of the genes in the panel, including those commonly mutated in sporadic pancreatic NETs: MEN1, ATRX, DAXX, TP53, and genes involved in the mTOR pathway (13). In addition, the high-throughput transcriptome analyzed by RNA-seq did not reveal any missed genetic alterations, aberrant splicing variants, gene fusions, or gene expression alterations. Specifically, there were no large structural rearrangements in the PTEN gene or mutations or deletions in the PTEN promoter region that could have resulted in reduced PTEN expression. We further sought to find a possible effect on PTEN protein expression or an aberrant subcellular compartmentalization of the protein because it has recently been shown that normal pancreatic islets exhibit predominant nuclear PTEN expression, whereas malignant cells have lower nuclear protein levels (10, 14, 15). Consistent with the results of these previous studies, the immunohistochemical staining of the tumor tissue for PTEN revealed uniformly strong cytoplasmic staining of tumor cells with virtually absent nuclear staining (Figure 2). This finding hinted at a possible causative association of PTEN germline mutation with the pancreatic NET.
The results from genetic testing and histopathological techniques used to confirm CS diagnosis and characterize this unusual tumor tempted us to believe that in this case the pancreatic NET was not a sporadic malignancy that occurred by coincidence, but rather represented a new entity in the spectrum of malignancies associated with CS.
Discussion
CS is an autosomal dominant, multisystem disorder of the PHTS spectrum associated with germline mutations in the PTEN tumor suppressor gene located on 10q23.3 (3). Patients with CS have characteristic macrocephaly, benign mucocutaneous lesions, and increased risk of breast, thyroid, endometrial, colorectal, and renal cancers. The clinical diagnosis of CS is made when an individual meets the revised operational diagnostic criteria adopted by the National Comprehensive Cancer Network (Figure 1) (7). Once the diagnosis of CS is established, genetic counseling should be offered, and testing for germline PTEN mutation can be undertaken. Although any individual phenotypic feature of CS is nonspecific and can often be seen in the general population, our patient had a personal and family history highly suggestive of CS, which was confirmed by the presence of a nonsense (p.R233*) germline mutation in exon 7 of the PTEN gene.
However, according to our current stage of knowledge, pancreatic NETs are not considered part of the clinical spectrum of CS because they have not thus far been described in patients with this disorder. There were several possible scenarios that could explain, at least in part, how these two seemingly distinct entities could have coincided. First, we contemplated that, by virtue of the tumor suppressor paradigm, the pancreatic NET could have occurred by chance due to a second hit somatic LOH of the wild-type 10q23 region. This possibility was supported by the fact that the 10q23 region and PTEN tumor suppressor gene are among the most frequent targets for LOH and point mutations identified in a variety of human malignancies including pancreatic, brain, breast, endometrial, and prostate cancers (6, 12, 13, 16). The second option was that it occurred coincidentally as a sporadic, nonfamilial cancer due to mutation(s) in other genes frequently associated with sporadic pancreatic NETs. This scenario was supported by the findings of a large-scale whole exome sequencing study of sporadic pancreatic NETs showing that the majority (nearly 90%) of these tumors harbor genetic alterations in MEN1, ATRX, DAXX, TP53, and genes in mTOR pathway (13).
However, in our case, the tumor tissue DNA sequencing did not identify LOH, and there was no copy alteration in PTEN; neither did it reveal other deleterious genetic alterations in the rest of the genes in the panel including those commonly mutated in sporadic pancreatic NETs. In addition, the analysis of RNA-seq data did not reveal any missed genetic alterations, aberrant splicing variants, gene fusions, or gene expression alterations.
The third possibility was that this unusual tumor was actually part of another familial cancer syndrome associated with pancreatic NETs such as Von Hippel-Lindau, multiple endocrine neoplasia type 1 (MEN1), neurofibromatosis type 1, or tuberous sclerosis. This possibility was supported by the fact that besides the NET of the pancreas, the differential diagnosis of mucocutaneous lesions, which are the primary and pathognomonic feature of CS, includes neurofibromatosis type 1, MEN1, and tuberous sclerosis (1). However, no other germline alterations were found that could explain the occurrence of this unusual pancreatic NET and link it to another heritable cancer syndrome.
The notion that, for a true loss-of-function effect to occur, loss of both functional copies is required in many, but not all, tumor suppressor genes may help to explain how this unique pancreatic NET has occurred in our CS patient in the absence of a biallelic PTEN event (16, 17). It is possible that germline inactivation of one of the PTEN alleles may be sufficient for a tumor-promoting effect by itself or in combination with disease-modifying alterations in other genes not covered by the gene panel used in this study. In this line of thought, it has recently been demonstrated that catalytically inactive cancer-associated PTEN mutants heterodimerize with the wild-type PTEN and constrain its phosphatase activity in a dominant-negative manner (17). Furthermore, the possibility for a post-translational epigenetic modification and resultant inactivation of the second allele or an aberrant subcellular compartmentalization of PTEN should also be considered (10, 14). In this regard, it has been shown that normal pancreatic islets exhibit homogenous predominant nuclear PTEN expression, whereas malignant cells have lower nuclear PTEN levels (14, 15). In our case, we found that tumor cells exhibited a robust cytoplasmic PTEN localization with practically absent nuclear PTEN staining. Taken together, in light of evidence from the literature reviewed above, our findings enable us to speculate that the p.R233* mutation in one of the alleles was, perhaps, sufficient to cause an abnormal cytoplasmic subcellular compartmentalization or exert a dominant-negative effect over wild-type PTEN protein, leading to the development of the pancreatic NET.
Analysis of PTEN germline alterations in one of the largest series of CS patients with identified pathogenic mutations (n = 290) showed that the majority (61%) were nonsense (32%) and missense (29%) mutations (18). There were mutation hotspots clustered primarily in exon 5 encoding the phosphatase domain and exon 7 corresponding to the portion of the lipid-binding C2 domain. Of these, the p.R233* mutation was identified in 10 patients with breast, endometrial, colon, thyroid, and renal cancers. Somatic PTEN alterations and LOH are commonly identified in a variety of sporadic tumors that are also common in CS such as breast, endometrial, and thyroid cancers, but also in cancers of the prostate, central nervous system, lung, liver, melanoma, and pancreas, including pancreatic NETs. According to the Catalogue of Somatic Mutations in Cancer database, of 3383 unique cancer samples with mutations, the p.R233* PTEN mutation was commonly identified in endometrial (n = 62), central nervous system (n = 16), and colon cancer (n = 11), but it has not been described in pancreatic NETs.
Given the growing accessibility of tumor profiling at the genetic, molecular, and cellular level, the diagnosis, prognosis, and development of new therapeutic strategies for patients with CS in the future will increasingly rely on understanding and integrating the effects of genetic changes in the PTEN gene with the resultant diverse and expanding syndromic phenotype. Therefore, thorough documentation, characterization, and reporting of cases of CS with unique clinical feature and outlining genotype-phenotype correlation with the underlying PTEN genetic alterations is important for promoting our understanding of this complex disorder and improving management options for the patients and their families.
In conclusion, our findings lead us to believe that in this case, the pancreatic NET might not represent a sporadic, coincidental tumor, but rather a new feature in the spectrum of malignancies associated with CS.
Acknowledgments
This work was supported by the National Cancer Institute (NCT02315625).
Disclosure Summary: The authors have nothing to declare.
Funding Statement
This work was supported by the National Cancer Institute (NCT02315625).
Footnotes
- CS
- Cowden syndrome
- CT
- computed tomography
- LOH
- loss of heterozygosity
- MEN1
- multiple endocrine neoplasia type 1
- mTOR
- mammalian target of rapamycin
- NET
- neuroendocrine tumor
- PHTS
- PTEN hamartoma tumor syndrome
- PTEN
- phosphatase and tensin homolog.
References
- 1. Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009;11(10):687–694. [DOI] [PubMed] [Google Scholar]
- 2. Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Couns. 2009;18(1):13–27. [DOI] [PubMed] [Google Scholar]
- 3. Liaw D, Marsh DJ, Li J, et al. . Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16(1):64–67. [DOI] [PubMed] [Google Scholar]
- 4. Marsh DJ, Dahia PL, Coulon V, et al. . Allelic imbalance, including deletion of PTEN/MMACI, at the Cowden disease locus on 10q22–23, in hamartomas from patients with Cowden syndrome and germline PTEN mutation. Genes Chromosomes Cancer. 1998;21(1):61–69. [DOI] [PubMed] [Google Scholar]
- 5. Marsh DJ, Kum JB, Lunetta KL, et al. . PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8(8):1461–1472. [DOI] [PubMed] [Google Scholar]
- 6. Steck PA, Pershouse MA, Jasser SA, et al. . Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15(4):356–362. [DOI] [PubMed] [Google Scholar]
- 7. Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013;105(21):1607–1616. [DOI] [PubMed] [Google Scholar]
- 8. Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41(5):323–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salem OS, Steck WD. Cowden's disease (multiple hamartoma and neoplasia syndrome). A case report and review of the English literature. J Am Acad Dermatol. 1983;8(5):686–696. [DOI] [PubMed] [Google Scholar]
- 10. Orloff MS, Eng C. Genetic and phenotypic heterogeneity in the PTEN hamartoma tumour syndrome. Oncogene. 2008;27(41):5387–5397. [DOI] [PubMed] [Google Scholar]
- 11. Neychev V, Steinberg SM, Cottle-Delisle C, et al. . Mutation-targeted therapy with sunitinib or everolimus in patients with advanced low-grade or intermediate-grade neuroendocrine tumours of the gastrointestinal tract and pancreas with or without cytoreductive surgery: protocol for a phase II clinical trial [published online May 19, 2015]. BMJ Open. doi:10.1136/bmjopen-2015–008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eng C. Genetics of Cowden syndrome: through the looking glass of oncology. Int J Oncol. 1998;12(3):701–710. [DOI] [PubMed] [Google Scholar]
- 13. Jiao Y, Shi C, Edil BH, et al. . DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perren A, Komminoth P, Saremaslani P, et al. . Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol. 2000;157(4):1097–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ginn-Pease ME, Eng C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0–G1 in MCF-7 cells. Cancer Res. 2003;63(2):282–286. [PubMed] [Google Scholar]
- 16. Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22(14):2954–2963. [DOI] [PubMed] [Google Scholar]
- 17. Papa A, Wan L, Bonora M, et al. . Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157(3):595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan MH, Mester J, Peterson C, et al. . A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011;88(1):42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]