

SUMMARY
Alterations in distal regulatory elements that control gene expression underlie many diseases, including cancer. Epigenomic analyses of normal and diseased cells have produced correlative predictions for connections between dysregulated enhancers and target genes involved in pathogenesis. However, with few exceptions, these predicted cis-regulatory circuits remain untested. Here, we dissect cis-regulatory circuits that lead to overexpression of NEK6, a mitosis-associated kinase, in human B cell lymphoma. We find that only a minor subset of predicted enhancers is required for NEK6 expression. Indeed, an annotated super-enhancer is dispensable for NEK6 overexpression and for maintaining the architecture of a B cell-specific regulatory hub. A CTCF cluster serves as a chromatin and architectural boundary to block communication of the NEK6 regulatory hub with neighboring genes. Our findings emphasize that validation of predicted cis-regulatory circuits and super-enhancers is needed to prioritize transcriptional control elements as therapeutic targets.
eTOC BLURB
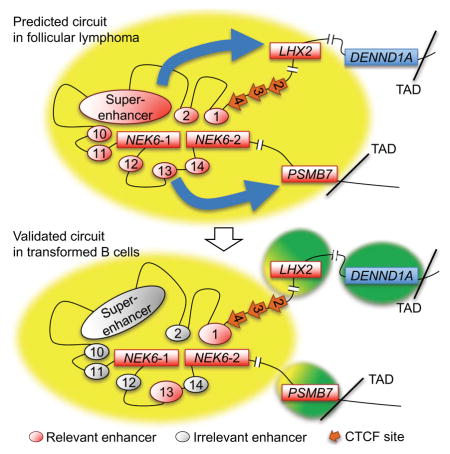
Huang et al. functionally dissect cis-regulatory circuits associated with NEK6, a mitotic kinase overexpressed in B cell lymphoma. Only a subset of predicted enhancers and CTCF sites cooperatively construct the regulatory hub of NEK6. A super-enhancer is completely dispensable for maintaining NEK6 expression and architecture in transformed B cells.
INTRODUCTION
Cell identity and function rely on stringently controlled programs of gene expression, perturbations of which underlie diseases, including autoimmunity and cancer. Genome-wide association studies have revealed that most pathogenic changes in gene expression are linked to variants in regulatory elements rather than coding sequences (Maurano et al., 2012). A dissection of cis-regulatory circuits controlling transcriptomes in normal and diseased cells remains an important objective. Most cis-regulatory circuits are composed of gene-proximal promoters and distal enhancers, which serve as conduits for transcription factors (TFs) and communicate with each other via physical contact, forming a series of loops in nuclear chromatin (Bulger and Groudine, 2011).
Conventional enhancers (CEs), both active and poised, can be identified in the genome as nucleosome-free regions. The activity level of each CE is revealed by the density of certain histone modifications, prototypically histone H3 acetylated at lysine 27 (H3K27ac) (Bulger and Groudine, 2011). Recent epigenome analyses have revealed a new class of regulatory regions, coined super-enhancers (SEs) (Whyte et al., 2013), which are characterized by broad stretches of H3K27ac. Most SEs are dense clusters of highly active CEs, which bind lineage-restricted TFs. Indeed, SEs normally co-localize with a limited set of genes that are most essential for cell identity and function. The acquisition or amplification of SEs near oncogenes contributes to several classes of cancer (Hnisz et al., 2013; Mansour et al., 2014). SEs are also enriched for disease-associated sequence variants, some of which presumably disrupt TF binding sites to alter SE function and expression of its associated gene(s) (Hnisz et al., 2013; Koues et al., 2016). However, contributions of SEs to gene expression programs have been mostly assumed from correlative chromatin profiling, rather than by direct testing (Proudhon et al., 2016). Furthermore, it remains controversial whether SEs represent a new paradigm in transcriptional regulation, or merely clusters of CEs that additively promote transcription (Dukler et al., 2016; Hay et al., 2016).
In addition to cis-regulatory elements, gene expression programs are significantly influenced by chromosome architecture, which facilitates or impairs promoter-enhancer contacts. The architecture of mammalian genomes is compartmentalized into topologically associated domains (TADs), which are highly conserved among cell types and species (Dixon et al., 2012). Loci within each TAD interact with one another, but are largely cordoned off from neighboring TADs. Each of these architectural building blocks is subdivided into structures called sub-TADs or contact domains, which are composed of loops between CTCF binding elements (structural loops) or between promoters and enhancers (regulatory loops). At a biochemical level, structural loops form via dimeric interactions between CTCF proteins bound in a convergent orientation at two distinct sites and are stabilized by association with the ring-like cohesin complex (Ghirlando and Felsenfeld, 2016; Rao et al., 2014). The bases of many structural loops serve as boundary elements that partition active and inactive chromatin domains within TADs and limit inappropriate interactions of regulatory elements with neighboring genes (Hnisz et al., 2016a; Ong and Corces, 2014). In keeping with their structural determinants, contact domains, unlike TADs, may vary significantly between cell types, developmental stages, or activation status (Dixon et al., 2016). Indeed, key questions remain about how intra-TAD architectures form and change during cellular differentiation and transformation. Answers to these fundamental questions will not only impact our understanding of basic gene regulatory mechanisms, but also the etiology of many diseases. A substantial subset of disease-associated SNPs and genomic alterations disrupt CTCF sites, breaking architectural borders, allowing inappropriate communication between enhancers and alternative genes (Lupiáñez et al., 2015; Hnisz et al., 2016).
Similarly, a deeper understanding of the regulatory determinants that underlie oncogenic gene expression programs remains a basic mission of cancer research (Sur and Taipale, 2016). Pathogenic expression programs have been characterized for many cancers, including various types of B cell lymphoma (BCL) (Jiang et al., 2016; Morin et al., 2010). A common class of BCL, termed follicular lymphoma (FL), is incurable. Most FLs exhibit an indolent clinical course, but often transform to a more aggressive cancer, termed diffuse large BCL (DLBCL) (Lenz and Staudt, 2010). Recently, we showed that pathogenic gene expression programs in FL are coordinated by a common set of TFs that, in turn, augment or attenuate activities of their target enhancers when compared with normal B cell counterparts, termed centrocytes (CCs) (Koues et al., 2015). Integrative transcriptome and epigenome analyses revealed a blueprint of pathogenic cis-regulatory circuits associated with FL, which predicted connections between distal enhancers and promoters of dysregulated genes. Similar correlation-based circuitries governing gene expression have been constructed for many normal and transformed cell types (Thurman et al., 2012), revealing a new collection of potential targets for epigenetic therapeutics. However, the validity of predicted circuits remains largely untested at the functional level. This gap is particularly important given that a majority of predicted cis-regulatory circuits consist of multiple enhancers connected to a single gene or, conversely, multiple genes connected to a single enhancer (Thurman et al., 2012).
Here, we functionally dissect a predicted cis-regulatory circuit for the mitosis-associated kinase, NEK6, which is commonly overexpressed in BCL (Mareschal et al., 2015). We find that only a subset of CEs, predicted by correlative algorithms to regulate NEK6 in BCL, is required to maintain its elevated expression. Strikingly, a B cell-specific super-enhancer is completely dispensable for NEK6 expression and maintenance of a regulatory hub that co-localizes its promoter with many distal CEs. A cluster of CTCF sites at one border of the NEK6 contact domain serves as a chromatin and architectural boundary to minimize the functional impact of its regulatory hub with neighboring genes. Our study not only provides insights into how NEK6 expression is regulated in normal and pathogenic B cells, but also emphasizes the need to rigorously test predictions, based solely on chromatin landscapes, regarding cis-regulatory circuits and super-enhancer function.
RESULTS
The NEK6 Cis-Regulatory Circuit Distinguishes FL Subsets
Very few correlation-based predictions for cis-regulatory circuits in normal or transformed cells have been validated functionally by targeted engineering of control elements within their native chromosomal context (Sur and Taipale, 2016). To rigorously test a manageable set of predictions, we prioritized pathogenic cis-regulatory circuits associated with CC transformation into FL (Koues et al., 2015). Prioritization of differentially expressed genes and their corresponding regulomes was tiered for recurrence of pathogenic enhancers in FL samples, altered levels of gene expression, relevant TF binding, and gene function (Fig. S1A, Supplemental Experimental Procedures, Table S1). The scheme yielded seven regulatory clusters and accompanying genes, which we considered to be of high priority for functional dissection (Table S2). Each of the seven regions consists of multiple enhancers and potential target genes, which renders comprehensive analysis of all prioritized circuits unwieldy. From the seven, we selected a region spanning NEK6 and several neighboring genes for in depth functional studies, based on multiple criteria. We first tested enhancer activities using luciferase reporters for a series of regulatory elements from the seven surviving regions, each of which displays augmented H3K27ac in FL compared with CC. A regulatory element in the NEK6 region (CE1) displays the most robust enhancer activity in both an EBV-transformed B cell line (GM12878) and a human BCL line (Farage, Fig. S1B). Moreover, NEK6, a central gene in the identified circuit, encodes a serine/threonine kinase that mediates mitotic progression, is overexpressed in many cancers, and is essential for sustained growth of tumors derived from numerous tissues (Fry et al., 2012).
With regard to B cell oncogenesis, NEK6 expression distinguishes the two known subtypes of DLBCL, exhibiting elevated expression in germinal center (GC-) compared with the activated B cell (ABC-) subtype (Mareschal et al., 2015). Epigenome analyses revealed that FL also segregates into two analogous classes (Koues et al., 2015), called subtype 1 (GC-like) and 2 (ABC-like). Strikingly, NEK6 expression is significantly elevated in subtype 1 FL, further highlighting its similarity to GC-DLBCL (Fig. 1A). One final criterion in selecting the NEK6 region for further study is its rich regulatory landscape, which seemingly consists of multiple enhancers augmented in BCL and a series of potential architectural elements (see below). Thus, we suspected that analysis of NEK6 cis-regulatory circuits would provide insights into enhancer and architectural elements important for cell type-, lymphoma-, or FL subtype-specific expression of this mitosis-associated kinase.
Figure 1. The NEK6 Regulatory Landscape in Normal and Transformed Cells.

(A) Expression levels of NEK6 in primary human cells. Each dot represents normalized microarray signals for a purified B cell sample from independent healthy volunteers or FL biopsies (CC: tonsillar centrocytes, TsB: unfractionated tonsillar B cells). Statistical tests were performed for subtype 1 or 2 FL versus other cell types. Only significant differences are shown for clarity (unpaired t-test with Welch’s correction): ***p<0.005, and ****p<0.001.
(B) Scheme depicting genes and regulatory elements in the NEK6 neighborhood. Red circles represent CEs that are FAIRE- and H3K27ac-positive in at least two FL samples from previously published data (Koues et al. 2015). Orange arrowheads depict CSs, as well as their orientations, as identified by chromatin profiling. UCSC Genome Browser views are shown for FAIRE- and H3K27ac ChIP-seq data from FL and CC samples (Koues et al. 2015), as well as DNase-seq, H3K27ac, CTCF and RAD21 ChIP-seq data in GM12878 (GM) and Jurkat cell lines (ENCODE). All sequencing data are presented as reads per million mapped reads.
(C) Rank order of increasing H3K27ac enrichment at enhancers in the indicated cell types. SEs were called using ROSE, with the NEK6-associated SE highlighted.
(D) NEK6 transcripts in the indicated cell types measured by RT-qPCR. Results represent the mean ± SEM of three independent experiments. Statistical significance (unpaired t-test with Welch’s correction): *p<0.05, ***p<0.005, and ****p<0.001.
(E) Expression levels of NEK6 neighboring genes in primary B cell samples, as measured by microarray. Each dot represents an independent sample. Statistical significance (unpaired t-test with Welch’s correction): *p<0.05, and ***p<0.005.
See also Figure S1.
The NEK6 Regulatory Landscape
To identify the collection of distal architectural and regulatory elements that contribute to elevated NEK6 expression in BCL, we leveraged data from public databases (ENCODE Project Consortium, 2012; Koues et al., 2015). Nucleosome-depleted regions demarcate more than a dozen active or poised elements spread over a 500 kb region encompassing NEK6 and its neighboring genes (Fig. 1B, FAIRE/DNase-seq). Several of these regions are bound by architectural factors, CTCF and RAD21, in GM12878, suggesting they may serve as structural or boundary elements (CTCF sites, CS1-7). NEK6 has two annotated transcription start sites (TSSs), which are both active in human B cells and GM12878 (Fig. S1C). H3K27ac peaks coincide with 14 nucleosome-depleted regions in FL samples, indicating positions of active conventional enhancers (CE1-14). Importantly, many of these enhancers exhibit a higher density of H3K27ac in FL compared with normal CC counterparts, suggesting they are hyperactive in transformed B cells. A subset of active enhancers (CE3-9) is clustered in a region −63 to −40 kb upstream of NEK6, which is designated as a super-enhancer (SE1) in both FL and CC samples using the ROSE algorithm (Lovén et al., 2013; Whyte et al., 2013) to analyze H3K27ac ChIP-seq data (Fig. 1C). When compared with other cell types, the activities of CE1, CE10, and SE1 are primarily restricted to B cells (Fig. S1D). Another conventional enhancer region, CE13-14, is also active in a subset of other cell types that express NEK6. These epigenome analyses suggest that CE1, CE10, SE1 and, perhaps, CE13-14 are critical enhancers for driving high levels of NEK6 expression in activated or transformed B cells.
Extensive genetic manipulations are required to dissect the NEK6 regulome; however, this approach is currently infeasible using primary human B cells. As such, we identified a tractable cell model that mirrors the NEK6 chromatin landscape in primary FL. As shown in Fig. 1B, the transformed human B cell line, GM12878, meets this criterion, while the human T lymphocyte cell line, Jurkat, exhibits a chromatin landscape largely devoid of active regulatory elements near NEK6, thus providing a negative control. In addition to recapitulating patterns of active enhancers in primary B and FL cells, the CE3-9 region is classified as an SE in GM12878 (Fig. 1C). NEK6 expression in GM12878 is comparable to levels observed in tonsillar B cells, the majority of which are activated, whereas NEK6 transcripts are nearly undetectable in Jurkat (Fig. 1D).
In addition to NEK6, two neighboring genes, LHX2 and PSMB7, are predicted to connect with many of the B cell-restricted enhancers in FL using a gene circuitry algorithm (Koues et al., 2015). LHX2 is a TF involved in the differentiation of developing lymphoid and neural cell precursors and is a putative oncogene for pancreatic tumors (Zhou et al., 2014). PSMB7 is a proteasome subunit that was identified as a biomarker for breast and colon cancers (Munkácsy et al., 2010). As shown in Fig. 1E, expression of these two genes, but not the more distal DENND1A, are modestly elevated in FL and/or tonsillar B cells compared with human CCs. All of these genes are expressed at varying levels in GM12878 (Fig. S1E). As such, functional dissection of the NEK6 cis-regulatory circuit can be achieved using GM12878, which recapitulates prominent features of the FL regulome.
Spatial Convergence of NEK6 Distal Regulatory Elements
Proper control of gene expression requires direct contact of distal regulatory elements with their target promoters. Many cell type-specific contacts between enhancers and promoters are confined within TADs and further restricted by boundary elements to minimize inappropriate enhancer-promoter communication. To elucidate the NEK6 interactome within its chromosomal neighborhood, we analyzed publicly available Hi-C data for a ~2 Mb region in GM12878 (Fig. 2A) (Rao et al., 2014). Based on interactomes conserved among cell types, the TAD containing NEK6 spans ~1 Mb encompassing DENND1A, LHX2, NEK6 and PSMB7. In GM12878, this region also contains several sub-TADs, one of which includes NEK6, spanning from the DENND1A promoter to PSMB7 (~500 kb). Within the sub-TAD, there is a robust contact domain spanning from the cluster of upstream CTCF sites (CS2-4) to the downstream NEK6 promoters (TSS1-2). More focal contacts are observed between both NEK6 promoters and pockets of upstream regulatory elements, especially with CE1 and SE1. Hi-C data revealed associations of the NEK6 locus with PSMB7 and, to a lesser extent, with LHX2, suggesting a potential mechanism for their elevated expression in FL. Finally, NEK6 is flanked by two sets of CTCF sites pointing in convergent orientations, a trio located approximately 130 kb upstream of TSS1 (CS2-4) and a pair located in a NEK6 intron (CS5) and near the PSMB7 promoter (CS6). The convergent orientation favors loop formation between CTCF regions (Ghirlando and Felsenfeld, 2016; Rao et al., 2014), perhaps spatially sequestering the NEK6 regulome.
Figure 2. The NEK6 Regulatory Hub.

(A) Hi-C data for the NEK6 region in GM12878, as visualized in Juicebox (Rao et al. 2014). The intensity of each pixel represents relative normalized numbers of contact between corresponding regions, for which red and blue represent enriched or depleted interaction frequencies, respectively. Knight and Ruiz normalization (balanced) is applied to remove locus-specific biases. The observed over expected (O/E) signal is displayed to account for a higher number of interactions with closer regions due to one-dimensional proximity (Rao et al. 2014). Several chromatin structures and contact points are highlighted with black boxes. In the left panel, genes within the NEK6-TAD are colored red and remaining genes are colored blue.
(B) Interaction frequencies, as measured by 3C-qPCR, for NEK6 TSS1 (top) and CE1 (bottom) viewpoints in GM12878 (NEK6 expressed) and Jurkat (NEK6 silent). Results represent the mean ± SEM of three independent experiments. Statistical significance (unpaired t-test with Welch’s correction): *p<0.05.
See also Figure S2.
To determine whether this regulatory architecture is cell type-specific, we performed 3C assays in GM12878 (NEK6+) and Jurkat (NEK6−), which directly probes interactions between a given viewpoint and selected regions of the NEK6 chromosomal neighborhood. As shown in Fig. 2B, a viewpoint spanning TSS1 interacts with upstream regulatory regions and with TSS2 at significantly higher frequencies in GM12878 compared with Jurkat. Peak TSS1 associations are with the CTCF cluster (CS2-4), CE1, CE2, and sites within SE1. To further validate the NEK6 interactome, we assayed a number of complementary viewpoints. Interactions with the distal CE1 element are significantly higher throughout the NEK6 sub-TAD in GM12878 compared with Jurkat. The enhanced CE1-PSMB7 contacts were confirmed using a PSMB7 promoter viewpoint (Fig. S2A). Coupled with 3C assays using viewpoints in SE1 (Fig. S2B, C), TSS2 (Fig. S2D), and the CTCF cluster (Fig. S2E), we conclude that the upstream region of NEK6 folds into a cell type-specific regulatory conformation, forming a hub for enhancers, promoters, and CTCF sites, which likely drives higher levels of NEK6 expression in activated B cells.
Conventional Enhancers Augment NEK6 Expression in Transformed B Cells
Our ultimate goal is to test predictions for key components of the cis-regulatory circuit associated with elevated NEK6 expression in transformed B cells. Chromatin profiling and interactome analyses revealed over a dozen enhancer elements that could potentially augment NEK6 expression in FL. To prioritize functional analyses, we first measured enhancer activities for each candidate regulatory element in GM12878 and Jurkat (Fig. 3A, Fig. S3A). In addition to the robust, GM12878-specific enhancer activity of CE1, four other elements augment luciferase expression from SV40 promoter-driven reporters. These include two regions in SE1 (CE5 and 9), the CE10 region upstream of TSS1 and the CE13 region upstream of TSS2. Despite its significant levels of interaction with NEK6 promoters (Fig. 2B), CE2 lacks enhancer activity in GM12878, which is consistent with minimal deposition of H3K27ac over this region (Fig. 1B). The activity status of CEs was bolstered by ChIP-seq data from GM12878 (ENCODE Project Consortium, 2012), which reveals significant peaks for EP300 and TFs important in B cell biology, including EBF1, OCT2, PU.1, PAX5, RELA and TCF3 (Fig. S3B). In contrast, CE2 lacks significant binding by any of these factors. These functional data led us to first focus on the role of three CEs located outside of SE1, which had the most robust activities in GM12878 (CE1, CE10 and CE13).
Figure 3. CEs Potentiate NEK6 in Transformed B Cells.

(A) Luciferase reporter assays for 14 putative CEs near NEK6. Enhancer activities were measured transiently in GM12878 or Jurkat cells and calculated relative to an SV40 promoter-only reporter construct. Human IGH enhancer was included as a positive control. Results show the mean ± SEM of at least four independent experiments in GM12878, and at least two in Jurkat.
(B and C) NEK6 transcripts, as measured by RT-qPCR, in different GM12878-derived CRISPR deletion subclones with the indicated genotypes or Jurkat cells, as a negative control. Each dot represents the Jurkat cell line or a unique subclone of GM12878, reported as the average of two independent RNA preparations, reverse transcription, and qPCR assays, the latter performed in triplicate. Statistical significance (unpaired t-test with Welch’s correction): *p<0.05, **p<0.01, and ***p<0.005.
(D) H3K27ac ChIP assays in GM12878-derived subclones harboring deletions of CE13 (left) or CE1 (right). ChIP-DNA was analyzed by qPCR with primers in or adjacent to indicated CEs. ChIP assays with a non-specific IgG antibody are shown as controls. For panels D and E, each bar represents the mean ± SEM of two subclones, each of which includes two independent experiments. Statistical significance (unpaired t-test with Welch’s correction): *p<0.05.
(E) Interaction frequencies, as measured by 3C-qPCR, in deletion subclones of CE13 (left) and CE1 (right) for the NEK6 TSS1 viewpoint.
(F) UCSC Genome Browser views of interaction profiles, as measured by 4C-seq, for CE1 wild-type and deletion subclones using CE1 and NEK6-TSS1 as anchors. For each viewpoint, the average counts per HindIII fragment normalized by DESeq2 are shown for three wild-type (red), and three CS2-4 deletion lines (green). A plot for differential signal between deletion and wild-type samples (Del-WT) is displayed below. None of the differences are statistically significant (DESeq2). The deleted CE1 region is shown as a yellow rectangle.
See also Figure S3.
To test the contributions of selected CEs to NEK6 expression, we individually deleted each enhancer from its endogenous site in GM12878 using CRISPR/Cas9 technology (Table S3). Deletion of CE13, which is proximal to TSS2, produces a modest, but significant decrease in NEK6 expression when compared with subclones retaining the enhancer on both alleles (Fig. 3B). Ablation of CE10 has no significant impact on NEK6 expression, despite its enhancer activity in luciferase assays. Importantly, NEK6 expression is attenuated substantially in subclones lacking the most distal enhancer, CE1, located 120 kb from TSS1. Consistently, NEK6 protein levels are dramatically reduced in CE1−/− subclones as measured by western blotting (Fig. S3C). The effects of each enhancer deletion are indistinguishable for transcripts derived from either TSS1 or TSS2 (Fig. S3D). Moreover, neither the CE1 nor the CE13 enhancer deletion impacts expression of neighboring LHX2 and PSMB7 genes (Fig. S3E). These data suggest that CE1 and CE13 both contribute to augmented NEK6 expression in transformed B cells. Indeed, compound deletion of both elements further diminishes NEK6 mRNA and protein expression (Fig. 3C, Fig. S3C). We conclude that two conventional enhancers, positioned outside of the large super-enhancer, additively potentiate NEK6 expression in GM12878.
To probe the effects of enhancer deletions on NEK6 chromatin and interaction landscapes, we analyzed subclones using ChIP and 3C, respectively. Deletion of CE13 reduces H3K27ac to near background levels at an adjacent region, verifying removal of the core enhancer (Fig. 3D). H3K27ac levels in CE13−/− mutants are unaffected at all other NEK6 enhancers tested. In sharp contrast, deletion of CE1 leads to significant reductions in H3K27ac not only at an adjacent region, but also at many locations within SE1 and other enhancers that associate with CE1. These data suggest that CE1 is a dominant element in sculpting the active epigenetic landscape near NEK6, perhaps through spatial interactions that form its regulatory hub. In this regard, the TSS1 interactome is unaffected by deletion of either CE13 or CE1 (Fig. 3E). Likewise, CE1 deletion does not alter long-range interactions between this region and downstream regulatory elements, including the TSSs (Fig. S3F). However, deletion of CE13 slightly boosts associations of CE1 with downstream enhancers, as well as NEK6 TSSs (Fig. S3G). This finding suggests that CE13 may partially compete with CE1 for association with TSSs and other elements of the regulatory hub. When CE13 is deleted, there may be a compensatory increase in CE1 interactions.
To further test whether the dominant CE1 element is dispensable for maintaining the NEK6 interactome, we performed 4C-seq on GM12878, as well as three independent CE1−/− and two wild-type subclones. Genome-wide interactome data probed from CE1 and TSS1 viewpoints show that CE1 deletion subclones have no significant differences for interactions with regions between CS2 and downstream of TSS2 (Fig. 3F and Fig. S3H), validating our 3C findings. These data indicate that maintenance of the NEK6 regulatory hub, which includes the distal CTCF cluster, CE1, SE1, CE13, and TSSs, is independent of the dominant conventional enhancer, CE1. However, this element contributes significantly to the maintenance of active chromatin marks at other CEs in the regulatory hub, boosting NEK6 expression in GM12878.
The NEK6 Super-Enhancer Is a Bystander
Super-enhancers are thought to be dominant regulatory elements for genes controlling cell identity, major cellular functions and, in some cases, oncogenesis (Hnisz et al., 2013). Our chromatin analysis identified SE1, a 23 kb region located between CE1 and the TSSs, as a B cell-specific NEK6 super-enhancer. Although two conventional enhancers (CE1 and CE13) contribute to NEK6 expression, a substantial level of transcripts remains following their deletion, further implicating SE1 as an important regulatory element. To test this directly, we deleted the entire SE1 region from both alleles of GM12878 using CRISPR/Cas9. Surprisingly, multiple independent clones lacking SE1 consistently express NEK6 mRNA at modestly higher levels when compared with subclones retaining an SE1+/+ configuration (Fig. 4A). Removal of SE1 also enhances or has minimal impact on NEK6 protein expression (Fig. S3C). ChIP analysis revealed a depletion of H3K27ac neighboring the deleted SE1, confirming removal of the super-enhancer (Fig. 4B). However, SE1 deletion does not impact H3K27ac levels at other tested CEs. Moreover, compound deletion of SE1 on one allele of CE1−/− clones has no significant impact on NEK6 expression (Fig. 4A).
Figure 4. SE1 Is a Dispensable Element in the NEK6 Regulome.

(A) NEK6 transcripts measured by RT-qPCR of SE1 deletion subclones. Each dot represents a unique subclone, which is reported as the average of two independent experiments. See Fig. 3B and C for details. For panels A–D, statistical significance (unpaired t-test with Welch’s correction): *p<0.05.
(B) H3K27ac ChIP assays in SE1 deletion subclones. See Fig. 3D for details. For panels B and C, each bar represents the mean ± SEM of two subclones, each of which includes two independent experiments.
(C) Interaction frequencies, as measured by 3C-qPCR, in SE1 deletion subclones for NEK6 TSS1 (left) and CE1 (right) viewpoints.
(D) LHX2 and PSMB7 transcripts measured by RT-qPCR in SE1 deletion subclones. Each dot represents a unique subclone, which is reported as the average of two independent experiments.
(E) Expression profile for all genes located within 5 Mb of SE1, as measured by RNA-seq, in SE1 wild-type and deletion subclones of GM12878. Average logCPM indicates the average expression level of each gene among three wild-type and three deletion subclones, reported as log2 read counts per million mapped reads. Log(SE1 Del/WT) represents the log2 fold-change of each gene between the average CPM of deletion versus wild-type subclones. Blue lines denote two-fold differences.
See also Figure S4.
One potential explanation for enhanced NEK6 expression following SE1 removal is that CE1 resides 23 kb closer to its promoters. However, this would imply that SE1 itself does not contribute fundamentally to NEK6 expression. To explore the impact of SE1 on the NEK6 regulatory hub, we performed 3C. As shown in Fig. 4C, SE1 deletion potentiates interactions between TSS1 and more distal elements (CE1 and CE2). The SE1−/− clones also show enhanced associations between TSS1 and more proximal regulatory regions (CE10 and TSS2), whose linear distances are unaffected by SE1 deletion. These data suggest that SE1 has a modest inhibitory impact on the frequency of enhancer associations in the NEK6 regulatory hub, as well as overall expression of this gene in GM12878.
An alternative explanation for the lack of SE1 regulatory function is that removal of critical enhancer elements drop NEK6 levels below a threshold required for GM12878 proliferation or survival. To test this possibility, we depleted NEK6 using several independent shRNAs. Reduced levels of NEK6 protein (20–30% normal, Fig. S4A, B) have no detectable impact on either proliferation or survival of GM12878 (Fig. S4C, D). The lack of a biological phenotype may also stem from expression of NEK7 in these cells, a closely related kinase with significant functional overlap (Fry et al., 2012). These data indicate that selective pressure from reduced NEK6 levels cannot reasonably explain the lack of a significant expression phenotype in SE1-deficient cells.
Although SE1 is dispensable for NEK6 expression in GM12878, it remains possible that this broad regulatory region may target another gene in its chromosomal neighborhood. Focused RT-qPCR analysis of SE1−/− clones revealed no significant change in PSMB7 expression (Fig. 4D). Similar to its effect on NEK6, SE1 deletion modestly enhances levels of LHX2 transcripts. To explore potential SE1 roles on a more global level, we analyzed three independent GM12878 subclones with SE1+/+ or SE1−/− genotypes using RNA-seq. SE1 deletion does not significantly change steady-state expression of any gene located within 5 Mb (Fig. 4E). On the transcriptome level, six genes are significantly increased or decreased in SE1−/− clones compared with their wild-type counterparts (Fig. S4E). The six genes are located on five different chromosomes; however, published promoter-capture Hi-C data reveal no significant inter-chromosomal interactions between any of the gene promoters and SE1 in GM12878 (Mifsud et al., 2015). We conclude that SE1, although clearly assigned as a super-enhancer using current algorithms, has no identifiable regulatory impact for maintaining expression of its nearest neighbors or any gene in a large chromosomal swath centered on NEK6.
A CTCF Cluster Establishes the NEK6 Contact Domain but Not the Regulatory Hub
Our functional data clearly demonstrate that two conventional enhancers, CE1 and CE13, additively increase NEK6 expression in transformed B cells. The more distal of these two elements, CE1, requires long-range looping (>120 kb) to communicate with NEK6 promoters. Architectural elements, largely consisting of CTCF sites, are common mediators of long-range looping that facilitate enhancer contact with gene promoters. Moreover, some CTCF sites serve as boundary elements to compartmentalize chromatin domains and inhibit inappropriate communication between enhancers and other neighboring genes (Ghirlando and Felsenfeld, 2016). CE1 is flanked by a cluster of CTCF sites positioned at one border of a robust contact domain containing NEK6. All three sites in this cluster are oriented convergently with a pair of downstream CTCF sites, located in a NEK6 intron (CS5) and near the PSMB7 promoter (CS6). The convergent orientation favors intermolecular CTCF interactions, which could form loops to cordon off NEK6-associated enhancers from other genes in the TAD. To explore architectural logic in the NEK6 cis-regulatory circuit, we deleted a region spanning all three sites in the upstream CTCF cluster (CS2-4). Minimal CTCF binding is detected at sites flanking CS2-4 following its deletion, compared with wild-type loci (Fig. 5A), whereas CTCF ChIP signals are unaffected at CS5 and CS6. NEK6 expression is reduced ~20% in subclones harboring the CS2-4 deletion on both alleles (Fig. 5B). In contrast, LHX2 expression is enhanced ~60% in knock-out subclones, while expression of the two other genes in this TAD, DENND1A and PSMB7, remains unchanged.
Figure 5. CS2-4 Serves as a Chromatin and Architectural Boundary for the NEK6 Regulatory Hub.

(A) CTCF ChIP assays in CS2-4 deletion subclones. ChIP-DNA was analyzed by qPCR using primers within or adjacent to indicated CSs. Each bar represents the mean ± SEM of two subclones, each of which includes two independent experiments. ChIP assays with a non-specific IgG antibody were performed as specificity controls. For panels A–E and G, statistical significance (unpaired t-test with Welch’s correction): *p<0.05, and **p<0.01.
(B) Transcript abundance of genes in the NEK6-TAD, as measured by RT-qPCR, for CS2-4 deletion subclones. Each dot represents a unique subclone, which is reported as an average of two independent experiments. See Fig. 3B and C for details.
(C) H3K27ac ChIP assays in C2-4 deletion subclones. See Fig. 3D for details. Each bar represents the mean ± SEM of two subclones, each of which includes two independent experiments.
(D and E) Interaction frequencies, as measured by 3C-qPCR, in CS2-4 deletion subclones for CE1, NEK6 TSS1 (D), and the LHX2 promoter (E) viewpoints. Each dot in (D) or bar in (E) represents the mean ± SEM of two subclones, each of which includes two independent experiments.
(F) UCSC Genome Browser views of interaction profiles, as measured by 4C-seq, for CS2-4 wild-type and deletion subclones using CE1 and NEK6-TSS1 as anchors. For each viewpoint, the average reads per HindIII fragment normalized by DESeq2 are shown for three wild-type (red), and three CS2-4 deletion lines (green). Reads located within the deleted CS2-4 region (yellow rectangle) are removed from all samples. Percentages of total normalized reads are displayed above each sample for regions upstream and downstream of CS2-4 deletion, as marked by double-headed arrow lines. For each viewpoint, a plot for differential signal between deletion and wild-type samples in natural log scale, ln (Del-WT), is displayed below. Statistical significance (generalized linear model adjusted by Benjamini-Hochberg procedure): p<0.05, are denoted by green or red asterisks for interactions that are increased or decreased in CS2-4 mutants, respectively.
(G) Interaction frequencies, as measured by 3C-qPCR, in CS2-4 deletion subclones for CS0 (left), CS5 (middle), and CS6 (right) viewpoints. Each bar represents the mean ± SEM of two subclones, each of which includes two independent experiments.
See also Figure S5.
These data suggest that CS2-4 serves as a boundary element to prevent the spread of active chromatin from NEK6 to LHX2, or to minimize long-range interactions between NEK6 enhancers and LHX2, or both (Ghirlando and Felsenfeld, 2016; Ong and Corces, 2014). To test the first possibility, we measured H3K27ac densities at sites in the NEK6 contact domain and adjacent LHX2 regions (Fig. 5C). Consistent with a role for CS2-4 as a chromatin boundary, its deletion permits H3K27ac spreading upstream of CE1 into the LHX2 locus. The CS2-4 deletion had an opposite effect on H3K27ac densities within the NEK6 contact domain, which are significantly reduced, and accompanied by an increase in the H3K27me3 modification (Fig. S5A). Thus, perturbed patterns of chromatin modifications correlate well with altered gene expression upon deletion of the 5′ CTCF cluster, supporting its functional assignment as a boundary element.
To determine whether CS2-4 also serves as a spatial boundary, precluding communication between NEK6 enhancers and other promoters, we performed 3C on subclones with wild-type and CS2-4−/− genotypes. As expected, mutant subclones generate no 3C signal for interactions between TSS1 and the deleted CS3 region (Fig. 5D). All other interactions between TSS1 and NEK6 regulatory elements are unaffected by the CS2-4 deletion. In contrast, TSS1 interactions with the LHX2 and DENND1A promoters, located further upstream in the sub-TAD, are significantly increased in mutant subclones. A similar enhancement of upstream interactions is observed for the CE1 element with LHX2 but not DENND1A, which correlates with the differential impacts of CS2-4 deletion on expression levels. Conversely, CE1 associations are decreased with downstream regions, including CS5 and the PSMB7 promoter. The enhanced interactions with LHX2 were confirmed using a complementary viewpoint corresponding to its promoter (Fig. 5E).
To support these findings, we performed 4C-seq on GM12878, as well as independent CS2-4−/− and wild-type subclones (Fig. 5F, Fig. S5B and C). Genome-wide interactome data probed from TSS1 and CE1 viewpoints reveal that, in general, CS2-4−/− subclones have more robust associations with upstream regions in the sub-TAD, reaching to the DENND1A promoter, as reflected in percent total normalized reads (Fig. 5F) (Guo et al., 2015). In contrast, interactions within the NEK6 contact domain itself are slightly attenuated following CS2-4 deletion (diminished percent normalized reads in Fig. 5F, Fig. S5B). In addition, 4C-seq data identify several interactions that differ significantly between CS2-4−/− and control clones. Deletion of the CTCF cluster significantly augments interactions between CE1 and several regions upstream (Fig. 5F, green asterisks), as well as with the LHX2 promoter, although the latter does not attain statistical significance in 4C data. Conversely, multiple interactions of CE1 with downstream regions in the NEK6 gene body and PSMB7 promoter region are significantly diminished following CS2-4 removal (Fig. 5F, red asterisks), consistent with our 3C data (Fig. 5D). Similarly, upon CS2-4 deletion, TSS1 has significantly elevated associations with the DENND1A and LHX2 promoters (Fig. 5F).
A potential explanation for the latter finding is that new contact loops may be formed between NEK6-proximal CTCF sites (e.g., CS5) and the properly oriented CTCF site upstream of the deleted CS2-4 region. A CTCF site located between the DENND1A promoter and LHX2, designated as CS0, has the same orientation as those deleted from the CS2-4 cluster (Fig. 5F). Indeed, 3C analyses indicate that the CS2-4 deletion enhances CS0-CS5 interactions, whereas CS0-CS6 crosslinking remains unaffected (Fig. 5G). The architectural remodeling of CTCF interactions, which may place the NEK6 gene in closer proximity to LHX2 and DENND1A, was confirmed using the complementary CS5 viewpoint (Fig. 5G). Together, these data indicate that CS2-4 contributes modestly to establishing the regulatory hub between NEK6 promoters and enhancers. Instead, this CTCF cluster predominantly functions as a chromatin and architectural boundary, minimizing the impact of the NEK6 regulatory hub on neighboring genes in its TAD.
DISCUSSION
Developmental and cell type-specific regulation of genes is orchestrated by changes in TF expression, enhancer activation, and alterations in chromatin landscapes, including architecture. Deciphering the contributions of each process to gene regulation is especially important given that a vast majority of disease-associated changes in the genome affect expression levels rather than coding potentials (Maurano et al., 2012). A prerequisite for understanding cis-regulatory circuits that govern normal or pathogenic gene expression is the profiling of enhancers and their contacts in distinct cell types. This milestone has largely been achieved in several hematologic malignancies and normal cellular counterparts (Chapuy et al., 2013; Koues et al., 2015). Based on chromatin and architectural profiles, pattern-based algorithms have been used to predict key regulatory connections between enhancers and their target genes. However, there is a critical need to test predicted circuits using reductionist, genetic approaches.
In this study, we dissected cis-regulatory circuits within a chromosomal neighborhood spanning at least three genes overexpressed in human BCL. Importantly, many predictions from pattern-based algorithms for NEK6 were not substantiated when tested directly. The predicted circuitry for pathogenic NEK6 expression involved at least a dozen enhancers with augmented H3K27ac loads in FL versus normal B cells. All of the CEs, including those comprising a super-enhancer, directly contact the NEK6 promoter in transformed B cells, further strengthening their predicted contributions to its elevated expression in BCL. Instead, we find that the NEK6 regulome is dominated by two conventional enhancers – one located near the TSSs (CE13), and a second, more powerful enhancer (CE1), located ~100 kb upstream. Although some of the predicted enhancers for NEK6 bind an overlapping set of factors, CE1 exhibits higher loads of TF binding than other enhancers (Fig. S3B), potentially explaining its dominant regulatory function. CE13 has lower levels of bound TFs and enhancer activity in luciferase assays, yet its proximity to TSSs may elevate its role in NEK6 regulation. The remaining CEs and, surprisingly, the super-enhancer, are all dispensable for NEK6 expression in transformed B cells, despite correlative changes in epigenetic and architectural landscapes. Thus, our study underscores the pressing need to hone predicted circuitry through rigorous testing. Although tedious, the emergence of high throughput methods for genetic dissection of TFs, enhancers, and chromosome architecture will speed achievement of this goal.
We suspect several potential reasons for disconnects between predictive algorithms and direct validation of cis-regulatory circuits. First, as shown here for NEK6, a dominant enhancer can affect the chromatin profile of other regulatory elements in its interactome. Deletion of CE1 attenuated H3K27ac loads on other CEs spread throughout the NEK6 region. Thus, increased CE1 activity in BCL likely augments H3K27ac on other elements in the regulatory hub, even if they do not contribute substantially to enhanced gene expression. Second, we cannot rule out that some CEs function as “back-up” elements to partially sustain NEK6 expression if CE1 activity is destroyed. This may be true for CE13, which contributes modestly to NEK6 expression in the absence of CE1. However, SE1 does not appear to have such a back-up role since deletion of the entire region or its composite CEs have no significant effect on NEK6 expression, whether CE1 is present or not.
The most surprising and significant finding from our study is that a clearly established SE has no discernable impact on the expression of NEK6 or any other gene on its chromosome. This finding is especially notable given the building dogma that SEs are a collection of key elements controlling high-level expression of genes critical for cell identity and function, as well as oncogenesis (Lovén et al., 2013). Not only does this finding underscore the need for functional evaluation of SEs in many cell types, but it also brings to light a third potential explanation for discrepancies between predicted and validated cis-regulatory circuits. Although the SE and a subset of other CEs are dispensable for NEK6 expression, these elements may be required earlier in B cell development or transformation to initially activate or augment transcription of this kinase gene. After these key activation events, SE1 or other CEs may become dispensable, with CE1 primarily maintaining elevated levels of NEK6 expression. These issues are currently intractable in primary human B cells, but may be approached in future studies by deletion of analogous regulatory regions for mouse NEK6. Notwithstanding, our findings indicate that at least a subset of SEs associated with oncogenesis would not be priority targets for current epigenetic-based therapeutic strategies to squelch expression of associated genes (Lovén et al., 2013).
A second surprise to emerge from our studies concerned determinants for regulatory architecture of the NEK6 chromosomal neighborhood. We found that most enhancers in this region converge spatially to form a regulatory hub with NEK6 promoters and flanking CTCF clusters. Although CE1 is the dominant NEK6 enhancer, its deletion does not significantly affect maintenance of the regulatory hub. Likewise, deletion of CS2-4 has only a modest impact on spatial interactions within this hub. These findings suggest several intriguing possibilities for architectural determinants of regulatory hubs, which await future dissection, including: (1) direct CE1-promoter interactions are redundant, structurally, with CS2-4 looping to downstream CTCF sites, (2) another element, excluding SE1 and CE1, is the key determinant for initiating regulatory hub formation, or (3) once the NEK6 sub-TAD is decorated with active histone modifications, homotypic chromatin interactions drive close association of the promoter with regional enhancers (Lieberman-Aiden et al., 2009). Nevertheless, our study identifies important dual roles for CS2-4 as a chromatin and architectural boundary, impairing the spread of active chromatin and enhancer interactions upstream of NEK6 into LHX2. Thus, many CTCF sites or clusters predicted to be important for formation of architectural loops may be more critical in establishing or maintaining borders of regulatory domains.
Our findings will also inform future studies to determine how NEK6 contributes to B lymphomagenesis. Despite consistent overexpression of the mitosis-associated kinase in BCL, NEK6 depletion had no detectable impact on viability or proliferation of transformed human B cells, including complete NEK6 knockout in two BCL lines (data not shown). In contrast, NEK6 knockdown in other cancer models significantly attenuated cell growth (Fry et al., 2012). We suspect that, in BCL, partial functional overlap with the closely related kinase, NEK7, may explain the lack of cellular phenotype. Indeed, NEK7 is overexpressed in primary cells derived from BCL biopsies compared with their normal counterparts (Koues et al., 2015). Human NEK6 and NEK7 loci appear to be partial duplicates of one another since both are flanked upstream by additional LHX and DENND genes. However, unlike NEK6, the NEK7 locus is devoid of chromatin hallmarks for active distal enhancers in B lymphocytes, FL, or other cell types (ENCODE Project Consortium, 2012; Koues et al., 2015). These correlative data suggest that NEK family kinases are essential components of the program for lymphomagenesis, requiring transformed B cells to augment NEK6 as a complement, or a back-up, to NEK7 overexpression, or vice versa. Thus, our dissection of the NEK6 regulome will be an important starting point to test such requirements in the germinal center program and oncogenic conversion to BCL.
EXPERIMENTAL PROCEDURES
Details for prioritization, 3C, 4C-seq, RT-PCR, RNA-seq, western blotting and NEK6 knockdown experiments are in Supplemental Experimental Procedures.
Luciferase Assay
Candidate enhancers (~800bp) were PCR amplified (Table S4) and cloned into SV40 promoter-driven pGL3 plasmid (Promega). Reporters were transfected into GM12878 and Farage (Roche 06366236001), or electroporated into Jurkat.
SE Calling
H3K27ac ChIP-seq data for primary B cells (Koues et al., 2015) and GM12878 (ENCODE Project Consortium, 2012) were aligned to the reference human genome (hg19) with Bowtie2 (Langmead et al. 2012). Peaks were called using MACS, and SEs were called using ROSE under default settings (Lovén et al., 2013; Whyte et al., 2013).
3C and 4C-seq
3C and 4C-seq assays were performed as described previously (Hagège et al., 2007; Majumder et al., 2015; Splinter et al., 2012) using strategies detailed in Supplemental Experimental Procedures. Primers and probes are shown in Table S4. 4C-seq statistics are shown in Table S5.
Chromatin Immunoprecipitation
ChIP assays were performed as described previously (Koues et al., 2015) using the following antibodies: 1 μg anti-H3K27ac (ab4729), 1 μg anti-H3K27me3 (ab6002), 8 μl anti-CTCF (Cell Signaling 2899) and anti-rabbit IgG (sc2027). ChIP DNA was analyzed with SYBR qPCR assays using primers listed in Table S4. Statistical analysis was performed using Prism.
CRISPR-Mediated Deletion
107 GM12878 cells or engineered subclones were electroporated with hCas9 plasmid (Addgene 41815), expression plasmids for two gRNAs targeting sequences that flank the region to be deleted, and a plasmid encoding hCD4. hCD4+ cells were purified 24 h post-transfection using magnetic beads (StemCell Technologies 18052), passaged for ~7 days, subcloned by limiting dilution, and screened for deletions using multiple independent primer pairs outside and inside of the gRNA target sites. gRNA sequences are shown in Table S3. Most gRNAs were cloned into the Addgene vector 41824, while gRNAs for CE13 were cloned into pKLV-U6gRNA(BbsI)-PGKpuro2ABFP (Addgene 50946). PCR primers for screening deletions are provided in Table S3. PCR products spanning deletion sites were purified and Sanger sequenced (Table S3). All molecular analyses were performed on sibling subclones corresponding to parental and mutant genotypes in the same experiment to avoid complications that might arise from drifts in bulk GM12878 cultures and experimental variations.
Supplementary Material
HIGHLIGHTS.
NEK6, a mitotic kinase gene, is overexpressed in specific types of B cell lymphoma
Super-enhancer is dispensable for elevated NEK6 expression in transformed B cells
Genome engineering reveals regulatory elements for NEK6 cis-regulatory circuits
Need for rigorous testing of predicted cis-regulatory circuits and super-enhancers
Acknowledgments
This work was supported by NIH grants CA156690, CA188286 (J.E.P. and E.M.O.), AI122726, AI115734 (E.M.O.), as well as WU-ICTS (TR000448) and Siteman Cancer Center (CA91842) grants. We thank Patrick Collins for helpful comments and the Genome Technology Access Center for assistance with –omics analyses.
Footnotes
Accession Numbers.
The accession number for raw reads and processed files for RNA-seq and 4C-seq datasets is GEO: GSE87323.
AUTHOR CONTRIBUTIONS
E.M.O., J.E.P., Y.H. and O.I.K. conceptualized the study and designed experiments. E.M.O. supervised all aspects of the project. Experiments and data analyses were performed by all authors. The manuscript was written by E.M.O. and Y.H. with input from J.E.P.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MGM, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Gorkin DU, Ren B. Chromatin Domains: The Unit of Chromosome Organization. Mol Cell. 2016;62:668–680. doi: 10.1016/j.molcel.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukler N, Gulko B, Huang YF, Siepel A. Is a super-enhancer greater than the sum of its parts? Nat Genet. 2016;49:2–3. doi: 10.1038/ng.3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry AM, O’Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. J Cell Sci. 2012;125:4423–4433. doi: 10.1242/jcs.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlando R, Felsenfeld G. CTCF: making the right connections. Genes Dev. 2016;30:881–891. doi: 10.1101/gad.277863.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell. 2015;162:900–910. doi: 10.1016/j.cell.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagège H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, Forné T. Quantitative analysis of chromosome conformation capture assays (3C-qPCR) Nat Protoc. 2007;2:1722–1733. doi: 10.1038/nprot.2007.243. [DOI] [PubMed] [Google Scholar]
- Hay D, Hughes JR, Babbs C, Davies JOJ, Graham BJ, Hanssen LLP, Kassouf MT, Oudelaar AM, Sharpe JA, Suciu MC, et al. Genetic dissection of the α-globin super-enhancer in vivo. Nat Genet. 2016;48:895–903. doi: 10.1038/ng.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Day DS, Young RA. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell. 2016a;167:1188–1200. doi: 10.1016/j.cell.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016b;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Dominguez PM, Melnick AM. The many layers of epigenetic dysfunction in B-cell lymphomas. Curr Opin Hematol. 2016;23:377–384. doi: 10.1097/MOH.0000000000000249. [DOI] [PubMed] [Google Scholar]
- Koues OI, Kowalewski RA, Chang LW, Pyfrom SC, Schmidt JA, Luo H, Sandoval LE, Hughes TB, Bednarski JJ, Cashen AF, et al. Enhancer Sequence Variants and Transcription-Factor Deregulation Synergize to Construct Pathogenic Regulatory Circuits in B-Cell Lymphoma. Immunity. 2015;42:186–198. doi: 10.1016/j.immuni.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koues OI, Collins PL, Cella M, Robinette ML, Porter SI, Pyfrom SC, Payton JE, Colonna M, Oltz EM. Distinct Gene Regulatory Pathways for Human Innate versus Adaptive Lymphoid Cells. Cell. 2016;165:1134–1146. doi: 10.1016/j.cell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010;362:1417–1429. doi: 10.1056/NEJMra0807082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder K, Rupp LJ, Yang-Iott KS, Koues OI, Kyle KE, Bassing CH, Oltz EM. Domain-Specific and Stage-Intrinsic Changes in Tcrb Conformation during Thymocyte Development. J Immunol. 2015;195:1262–1272. doi: 10.4049/jimmunol.1500692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareschal S, Ruminy P, Bagacean C, Marchand V, Cornic M, Jais J-P, Figeac M, Picquenot J-M, Molina TJ, Fest T, et al. Accurate Classification of Germinal Center B-Cell-Like/Activated B-Cell-Like Diffuse Large B-Cell Lymphoma Using a Simple and Rapid Reverse Transcriptase-Multiplex Ligation-Dependent Probe Amplification Assay: A CALYM Study. J Mol Diagn. 2015 doi: 10.1016/j.jmoldx.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. 2015;47:598–606. doi: 10.1038/ng.3286. [DOI] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkácsy G, Abdul-Ghani R, Mihály Z, Tegze B, Tchernitsa O, Surowiak P, Schäfer R, Györffy B. PSMB7 is associated with anthracycline resistance and is a prognostic biomarker in breast cancer. Br J Cancer. 2010;102:361–368. doi: 10.1038/sj.bjc.6605478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudhon C, Snetkova V, Raviram R, Lobry C, Badri S, Jiang T, Hao B, Trimarchi T, Kluger Y, Aifantis I, et al. Active and Inactive Enhancers Cooperate to Exert Localized and Long-Range Control of Gene Regulation. Cell Rep. 2016;15:2159–2169. doi: 10.1016/j.celrep.2016.04.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E, de Wit E, van de Werken HJG, Klous P, de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods. 2012;58:221–230. doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16:483–493. doi: 10.1038/nrc.2016.62. [DOI] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Gou S, Xiong J, Wu H, Wang C, Liu T. Oncogenicity of LHX2 in pancreatic ductal adenocarcinoma. Mol Biol Rep. 2014;41:8163–8167. doi: 10.1007/s11033-014-3716-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.