

Abstract
Purpose of review
Recent clinical studies and management guidelines for the treatment of the organic acidopathies methylmalonic acidemia (MMA) and propionic acidemia (PA) address the scope of interventions to maximize health and quality of life. Unfortunately, these disorders continue to cause significant morbidity and mortality due to acute and chronic systemic and end-organ injury.
Recent findings
Dietary management with medical foods has been a mainstay of therapy for decades, yet well controlled patients can manifest growth, development, cardiac, ophthalmological, renal and neurological complications. Patients with organic acidopathies suffer metabolic brain injury which targets specific regions of the basal ganglia in a distinctive pattern, and these injuries may occur even with optimal management during metabolic stress. Liver transplantation has improved quality of life and metabolic stability, yet transplantation in this population does not entirely prevent brain injury or the development of optic neuropathy and cardiac disease.
Summary
Management guidelines should identify necessary screening for patients with MMA and PA, and improve anticipatory management of progressive end-organ disease. Liver transplantation improves overall metabolic control, but injury to non-regenerative tissues may not be mitigated. Continued use of medical foods in these patients requires prospective studies to demonstrate evidence of benefit in a controlled manner.
Keywords: methylmalonic acidemia, propionic acidemia, brain injury, liver transplantation, medical foods
Introduction
Methylmalonic acidemia (MMA) and propionic acidemia (PA) are rare, autosomal recessive, multisystemic inborn errors of branched chain amino acid metabolism which cause significant morbidity and mortality in infancy and childhood, and, for survivors, significant debilitating end-organ damage into adulthood. MMA and PA, as organic acidopathies (OAs), result in defective mitochondrial metabolism of coenzyme A (coA)-activated carboxylic acids, which are largely derived from the metabolism of branched-chain amino acids, odd-chain fatty acids, and cholesterol. Clinical features of OAs may occur due to accumulation of toxic metabolites, altered mitochondrial energy metabolism, carnitine depletion, and coenzyme A sequestration. Acute illness may be associated with metabolic acidosis, acute alterations of consciousness or encephalopathy, anorexia, and nausea and vomiting [1, 2]. Chronic complications include poor growth, movement disorders, progressive spastic quadraparesis, epilepsy, cardiac dysfunction (PA>MMA), progressive renal disease (MMA), osteopenia/osteoporosis, vision loss (MMA>PA), and functional immunodeficiency [1, 3–35]. Recent investigation into the pathophysiology of the end-organ effects seen in patients with OAs has improved screening for disease related complications, and recent treatment recommendations are the first steps toward standardization of care [36].
Clinical Presentation, Diagnosis, and Management
PA and MMA classically present in a term neonate within the first 3 days of life, who feeds poorly, is pancytopenic, becomes progressively encephalopathic, and ultimately progresses to coma and death without prompt identification and management [22, 37–44]. The differential diagnosis in this age group includes sepsis, hypoxic-ischemic encephalopathy, drug intoxication (from maternal exposure before and/or during delivery), and other inborn errors of metabolism. Sick neonates who appear septic or encephalopathic with an anion gap metabolic acidosis, ketoacidosis, lactic acidosis, hyperammonemia, and/or hypoglycemia should be stabilized expectantly with reversal of catabolism using dextrose containing fluids with a glucose infusion rate of 6–8 mg/kg/min, with intralipids at 2–3 grams/kg/day, while removing all source of protein from the infant. Workup for intoxication-type inborn errors of metabolism (IEM) should occur immediately upon clinical indication and include blood gas (non-capillary), comprehensive metabolic panel, complete blood count, blood culture, urinalysis (specifically for urine ketones, which should be negative in a healthy newborn without and IEM), lactate, ammonia, urine organic acids, plasma amino acids, and an acylcarnitine profile [45].
Once the infant is acutely stabilized and diagnosed, lifelong aggressive management by metabolic physicians remains essential. Despite management based on best practices, including dietary protein restriction, carnitine supplementation, and the use of drugs to modulate ammonia, these patients frequently experience acute metabolic decompensation during acute illness or other stressors such as surgical or interventional procedures [1, 45–58]. In older patients with the classical OAs, acute decompensation events continue, frequently due viral illness or surgical procedures. Frequently, these patients will have significant complications due their disease, discussed below.
Patients with milder variants of isolated MMA (mut−, or the cobalamin disorders Cobalamin A (cblA) or B (cblB), which result in deficiency of the adenosylcobalamin cofactor for MUT) or PA may not present until later in infancy, childhood, or adolescence. While the definition of early versus late onset remains controversial within the OAs, Heringer et al classify late-onset disease as any patient in whom the first clinical symptoms occur outside of the neonatal (first 30 days of life) period, although some patients may not present until much later in life [59]. In patients who were not diagnosed by expanded newborn metabolic screening (NMS), Heringer and colleagues report that for MMA and PA, the median age at onset of clinical symptoms was 6–8 days in the early-onset group, while in the late-onset group, the median age at diagnosis was 210–348 days. Approximately half of the non-NMS MMA and PA cohort were classified as late-onset, but most of these diagnoses occurred during infancy [59]. In MMA, these childhood and adolescent-onset patients may present with chronic renal failure, and evaluation for MMA should occur in all patients who present with progressive proximal tubular renal dysfunction [60–64]. Late-stage presentation of PA may include seemingly isolated cardiomyopathy, while patients with PA and MMA may present with progressive spastic quadraparesis, progressive movement disorder, or vision loss [6, 7, 14, 32, 33, 35, 58, 65–71]. Some patients who self-restrict protein due limited protein tolerance may present later in life with metabolic decompensation or metabolic stroke following a surgical or interventional procedure where fasting for several hours is required. Additional complications of later-onset MMA and PA are similar to those with classical disease and are discussed below.
Diagnosis
Diagnosis typically occurs during an initial decompensation event in the neonatal period, which may resemble neonatal sepsis and present with poor feeding, vomiting, lethargy, and progression to coma and death without prompt and effective therapy. Diagnosis is based on clinical presentation and laboratory analysis, metabolic acidosis, ketoacidosis, lactic acidosis, hyperammonemia, hypoglycemia, pancytopenia, and elevated C3 acylcarnitines and organic acids in the urine. Metabolites elevated in PA include elevated plasma propionylcarnitine, glycine, and alanine, and elevated urinary 3-OH-propionate and methylcitrate [72–85]. In MMA, elevations of plasma propionylcarnitine, glycine, and alanine coupled with elevation of urinary methylmalonic, 3-OH-propionic, and methylcitric acids provide the diagnosis [72–78, 86–88].
Some infants, however, are detected based on NMS prior to a decompensation event. A recent study of organic acidemia outcomes using compiled data from the European registry and network for intoxication type metabolic diseases (E-IMD) demonstrates that, for infants diagnosed with OAs on newborn screening, 52% with cobalamin non-responsive MMA, 67% with cobalamin responsive MMA, and 49% with PA were asymptomatic at 8 days of life [59]. OA patients detected by expanded NMS are somewhat more likely to have normal development of motor milestones and less likely to have a movement disorder, although movement disorders and metabolic brain injury may occur at any age, and the median age of subjects in this study were under 10 years of age [59]. Furthermore, other neurological and neurocognitive outcomes were not explored, and thus, in spite of earlier detection, these patients are likely to develop some degree of neurological sequela of disease, and other end-organ sequelae have not been analyzed in this cohort.
Defects in other genes within the propionate catabolism pathway or mitochondrial disorders may also result in excretion of methylmalonic acid in the urine [89, 90]. These disorders are rarer than classical isolated MMA, often manifest other biochemically diagnostic markers in urine or plasma to suggest the diagnosis, and will not be addressed further in this review.
Biochemical Perturbations in PA and MMA
Mutations in the PCCA and PCCB genes cause PA by generating defective or absent propionyl-CoA Carboxylase (PCC), which is biochemically upstream of MUT [91–130]. Isolated MMA is caused by mutations in the MUT gene encoding methylmalonyl-CoA mutase (mut) or the genes encoding the enzymes responsible for the generation of 5-deoxyadenosylcobalamin (AdoCbl) cofactor of Mut (MMAA and MMAB) (Fig. 1)[131–148]. The subtypes of MMA are based on complementation subclasses and include mut, cblA, and cblB, based on the enzymatic cause of the condition[149]. Mut deficiency may be further divided into subclasses based on the degree of enzymatic activity of the mutase enzyme, designated mut0, for enzymes with null activity, and mut− for enzymes with reduced or minimal activity[141]. Other causes of isolated MMA include much rarer deficiencies in other enzymes within the propionate catabolism pathway or in other components of mitochondrial function.
Figure 1.
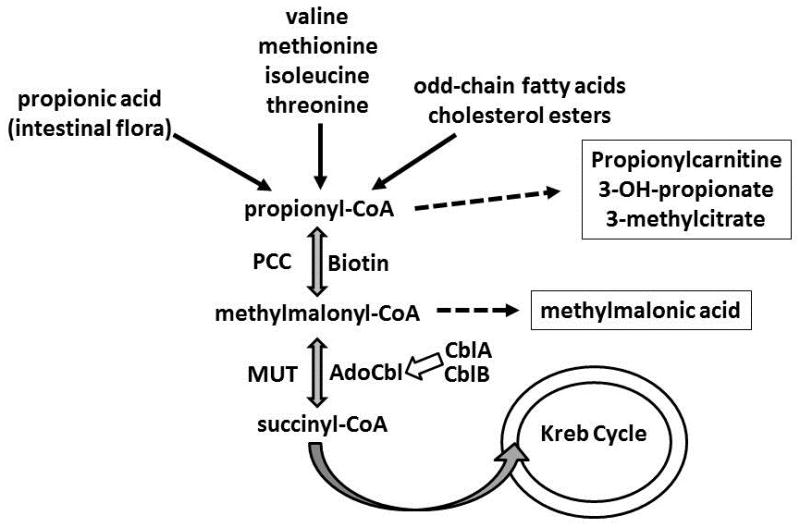
Biochemical Pathway of Propionate Metabolism. For simplification, the methylmalonyl-CoA epimerase enzyme step has been removed from the pathway diagram. AdoCbl: adenosylcobalamin, CblA: Cobalamin A, CblB: Cobalamin B, MUT: methylmalonyl-CoA mutase, PCC: propionyl coA carboxylase.
With normal enzymatic function, propiogenic precursors are converted sequentially from propionyl-CoA to methylmalonyl-CoA to succinyl-CoA, which is subsequently metabolized within the TCA cycle [131, 132, 150–152]. This complex role of PCC and MUT within mitochondrial energy metabolism mirrors the biochemical and clinical findings associated with OA disease [153–159]. Approximately two-thirds of normal propiogenic load is generated from dietary intake and muscle turnover, while around one-third naturally originates from gut bacterial sources [160, 161]. During decompensation, acidosis in these patients occurs due to accumulation of organic acids and ketoacids, while lactic acidosis also occurs, particularly in severe decompensation or with severe secondary mitochondrial dysfunction [61, 162–166]. Accumulation of propionyl-CoA, and to some extent methylmalonyl-CoA, results in secondary inhibition of N-acetylglutamate synthase (NAGS), causing secondary hyperammonemia in these patients [167–170]. Generation of propionylcarnitine moieties may result in cardiac arrhythmias and cardiomyopathy due to secondary carnitine deficiency [29, 171].
Complications and Management of MMA and PA
Because normal mitochondrial function requires sufficient energy production through the citric acid cycle and oxidative phosphorylation, MMA and PA result in multi-systemic chronic disease, particularly in the highly energetic organs such as brain, heart, kidney, and eye. End-organ injury occurs due to both primary toxicity of both the accumulating primary and secondary metabolites and deficiency of succinyl-CoA resulting in Kreb cycle and oxidative phosphorylation dysfunction. Periods of acute illness frequently chronically worsen the patient’s basal condition due to increased energetic dysfunction.
Dietary Management
In the well state, OA patients are typically maintained on a protein-limited diet, or if enteral gastric feedings are required, combinations of standard, age-appropriate enteral formulas with formulas specially-designed for OA patients may be employed. While protein restriction is more aggressive in patients with other inborn errors of metabolism, in OAs dietary protein intake should target the recommended daily allowance for protein (0.8 grams protein/kg body weight), unless differences based on the individual patient response require lower or higher concentrations. Patients with spasticity or severe choreoathetosis may require additional protein nutrition for their increased energetic demand, and other patients with brittle, difficult to manage disease may require less whole protein and, based on clinician preference, addition of medical foods or formulas [12, 53, 172–179]. The primary dietary goal in OA patients should remain prevention of catabolism and allow normal growth, without causing obesity. Thus, providing sufficient protein, preferably from natural protein sources with as little amino acid-deficient protein from medical foods as possible, is preferred. Recently, dietary analysis of a large cohort of patients has revealed that patients with MMA typically tolerate the recommended daily allowance of protein. However, many of these patients were receiving a large proportion of protein from propiogenic-deficient sources and were also noted to have poor growth in height and weight, elevated leucine levels, and low levels of isoleucine and valine, often requiring specific amino acid supplementation [180]. Prior patients on such diets developed severe amino acid deficiencies [176, 178]. Thus, the use of medical foods deficient in propiogenic precursors appears to result in branched chain amino acid deficiencies and may worsen outcomes. Therefore, coordination with a metabolic dietician is strongly recommended to ensure that nutritional and amino acid deficiencies are prevented, and future prospective studies on the safety and efficacy of medical foods for OAs should occur to ensure that iatrogenic secondary effects are prevented in this already vulnerable population.
Medical Management
Patients with PA and MMA require carnitine supplementation to prevent secondary carnitine deficiency (L-carnitine, enterally administered at 50–100mg/kg/day), and those patients with B12 responsive MMA, usually cblA disease, should receive daily injections of hydroxocobalamin (1 mg, intramuscularly every day) [12, 57, 144, 145, 181, 182]. Some patients are treated with cycles of enteral antibiotics (metronidazole) to reduce the burden of propiogenic gut flora [36, 161, 173, 183]. Some brittle patients with chronic hyperammonemia may be treated orally with sodium benzoate or sodium phenylbutyrate (Buphenyl) at 10 grams/m2/day, with careful monitoring of amino acid levels and electrolytes [12, 36]. This is not a standardized practice, but may be instituted by a metabolic physician based on professional experience and provider preference.
Acute Metabolic Decompensation
Patients with OAs may become very ill from otherwise mild viral illnesses, and other events that cause physical or emotional stress may trigger catabolism, including surgical procedures, labor and childbirth, and abrupt changes in nutritional status. Aggressive acute management of intercurrent illness and mitigation of other stressors must be undertaken to limit the degree of decompensation and sequelae from these events. Limited reports indicate that patients with PA and MMA may not maintain sufficient humoral immune response to combat viral infection [184, 185]. Reversal of catabolism, promotion of anabolism and identification and treatment of the underlying precipitating etiology are paramount to management of the acute decompensation.
With intercurrent illness, PA and MMA patients typically present with nausea and vomiting, worsening anorexia, and encephalopathy, with laboratory studies demonstrating metabolic acidosis, ketonuria, hyperammonemia, pancytopenia, and electrolyte disturbances [1, 22, 45, 86]. These decompensation events typically present with decreased oral intake or enteral feeding intolerance, vomiting, and altered mental status or lethargy. Without aggressive reversal of catabolism with intravenous dextrose-containing fluids (typically 10–12.5% dextrose in normal saline with appropriate electrolyte additives at 150% of maintenance, unless renal function demands different electrolyte composition or volume), patients with OAs may die or suffer severe metabolic brain injury [22, 36, 45]. Some providers choose to employ intralipid in addition to dextrose fluids for additional caloric support [36, 45]. In addition to reversal of catabolism, more aggressive metabolic therapies are employed during decompensation, including ammonia scavenging with sodium phenylacetate-sodium benzoate (Ammonul, intravenous preferably via central line, variable dosing based on age/weight) or disinhibiton of urea cycle function by N-carbamylglutamate (Carbaglu, oral, 100 mg/kg bolus followed by 25–62 mg/kg every 6 hours, currently under investigational status with the United States Food and Drug Administration) [36, 45, 186–189]. The ammonia scavengers allow conjugation of amino acids to the scavenger compounds to bypass the urea cycle and permit excretion [39, 159, 190, 191]. Inhibition of carbamylphosphate synthetase I (CPS1), a urea cycle enzyme, by accumulating metabolites in OAs causes secondary hyperammonemia in these disorders [167]. N-carbamylglutamate, an N-acetylglutamate analogue, allosterically activates CPS1 and inhibits the secondary effects of the propionate metabolites on CPS1 [36, 45, 189, 192–195]. Once catabolism has been reversed and acidosis corrected, complete nutrition should be reinitiated as soon as possible, preferably via the enteral route. Once the precipitating source is identified and treated or managed, the patient may be transitioned back to standard diet.
Chronic Management and Screening Recommendations
Optimal management of individuals with MMA and PA includes careful dietary management and regular screening for known complications of the OAs [12, 22, 36, 57]. Table 1 catalogues the most common complications associated with PA and MMA and the recommendations for screening and management. Metabolic “strokes”, frequently indicated by significant acute mental status changes or new or worsening abnormal movements, require immediate laboratory and imaging evaluation and reversal of catabolism [12, 17, 22, 34, 65–67, 196–198]. Once dextrose-containing intravenous fluids and treatment of the underlying precipitant are initiated, MRI and magnetic resonance spectroscopy may be performed to evaluate the location and extent of evolving injury [17, 66, 199–202]. Movement disorders or spastic quadra- or paraparesis, potential sequelae of metabolic strokes, should be managed in collaboration with a neurologist and/or physiatrist to allow for optimal function and minimum disability. If spasticity or movement disorder worsens and limits the patient’s ability to perform activities of daily living, referral to speech, physical, and/or occupational therapy should occur [203].
Table 1.
Chronic Manifestations and Management of PA and MMA
| Organ System | Manifestations | PA | MMA | Evaluation and Management |
|---|---|---|---|---|
|
| ||||
| Constitutional | Failure to thrive | ++ | ++ | Consider need for gastrostomy tube placement to permit sufficient caloric intake. |
| Anorexia | ++ | ++ | ||
| Feeding difficulty | ++ | ++ | ||
|
| ||||
| CNS | Movement Disorder Spastic quadra-/para-paresis (progressive) | ++ | ++ | Neurological evaluation and treatment of movement disorders and spasticity. |
| Metabolic “stroke” involving basal ganglia | ++ | ++ | MRI with spectroscopy to evaluate prior injury and acute/evolving injury in symptomatic patients. Reversal of catabolism. |
|
| Variable intellectual disability | ++ | + | Ensure appropriate legal documentation in place for power of attorney, guardianship, etc. as indicated based on level of functioning. | |
|
| ||||
| Ophthalmological | Optic nerve atrophy Retinal degeneration | + | ++ | Routine ophthalmological evaluation and treatment at regular intervals. |
|
| ||||
| Gastrointestinal | Pancreatitis | ++ | + | Monitoring of pancreatic enzyme levels with illness and adjustment of feeding paradigm as indicated. |
|
| ||||
| Renal | Tubulointerstitial nephritis | − | ++ | Routine screening of clinically available markers for renal disease. GFR preferable. |
| Chronic Progressive Renal Failure | + | ++ | Avoid/limit/renally dose nephrotoxic medications in patients with evidence of declining renal function. | |
| End Stage Renal Disease | + | ++ | Evaluation for organic acidopathies in patients with renal failure of unknown cause with suggestive history. | |
|
| ||||
| Cardiovascular | Arrhythmias, including prolonged QTc | ++ | + | Telemetry for arrhythmias and prolongation of QT interval. |
| ++ | + | Cardiology evaluation with echocardiography and EKG yearly or with symptoms. | ||
| Cardiomyopathy | ++ | + | ||
| Heart Failure | + | − | Concurrent management with cardiology for cardiac pathology. | |
|
| ||||
| Skeletal | Osteopenia/Osteoporosis | + | + | Routine screening with DXA every 5 years. |
| Pathological Fractures | + | Routine (yearly) monitoring, consider addition of bisphosphonates. | ||
Patients with PA and, less commonly, MMA may develop life-threatening cardiac arrhythmias, particularly prolonged QTc, or cardiomyopathy, with or without carnitine deficiency [4, 12, 28, 204, 205]. All individuals should be routinely screened with echocardiography and EKG yearly, during admissions for illness (EKG), or with cardiac symptoms [12, 36]. Medications that prolong the QT interval should be used with caution in OA patients and avoided to the extent possible in patients with known cardiac disease. All patients who are hospitalized for metabolic decompensation or due to an invasive or surgical procedure should be placed on telemetry to monitor for life-threatening arrhythmias during these crises.
The significant ophthalmological effects of MMA and PA require close collaboration with an ophthalmologist comfortable with managing the retinal and optic nerve degeneration associated with associated with these diseases [6, 7, 14, 35, 70, 206–209]. While effective ophthalmic therapies remain elusive, careful monitoring for vision loss and provision of support services are vital for maintaining patient function. As ophthalmological innovations occur for retinal and optic nerve disease, MMA and PA patients with access to an ophthalmologist may benefit from trials with new devices and therapies.
MMA frequently, and PA rarely, result in progressive, severe renal disease, often requiring transplant, [8, 61–64, 210–216]. Patients with MMA and PA should be carefully screened with laboratory markers of renal function including BUN and creatinine, which are frequently near normal until late stage disease, as well as calculated creatinine clearance or glomerular filtration rates, and cystatin C. Other markers of renal function, including erythropoietin and 1,25-hydroxy vitamin D in the setting of appropriate vitamin D intake/supplementation may indicate additional investigation for worsening renal disease. Patients with indications of declining renal function require referral to a nephrologist for further evaluation and management, as dialysis and/or renal transplantation may become necessary, particularly in adolescents and adults with MMA. Nephrotoxic medications should also be avoided or limited in these patients.
Although the specific mechanisms associated with bone health in OAs remain incompletely investigated, patients with PA and MMA are at significantly increased risk for osteopenia or osteoporosis that their age-matched peers, with and without renal disease [22, 41]. DXA scan evaluation should be performed routinely starting at age 5, the earliest age for which height, race, and gender adjustment norms exist, and radiographic evaluation for fractures in patients presenting with pain should always be considered. Various pathologies including vertebral fusion anomalies, vertebral compression and fractures, as well as generalized osteopenia, have been observed. Therapeutic intervention should address morbidity associated with such low BMD, and the use of bisphosphonates versus calcitriol to target anti-resorption versus anabolic measures to support increased deposition is dependent on the individual patient’s findings (BMD, bone age, parathyroid hormone, 1,25-OH and 25-OH vitamin D levels, sex hormone production), and even bone biopsy should be considered for responsiveness to bisphosphonates prior to their initiation. If pathological fractures or significant osteopenia/osteoporosis are detected on screening, more robust monitoring for bone density and response to interventions are indicated. Currently, further studies are required to determine the role of medical food use, renal disease, or other contributing factors such as immobility or metabolic fragility in the development of low bone mineral density.
Transplant
Some individuals have received liver transplants to treat PA and liver, kidney, or combined liver-kidney to metabolically stabilize MMA and/or address the chronic renal failure associated with MMA disease [12, 18, 24, 60, 196, 201, 217–239]. Liver transplantation has become an increasingly popular treatment choice for children with more severe or brittle disease, and some adults undergo isolated kidney transplant for MMA-related end-stage renal disease. For adolescents, however, combined liver and kidney transplant has also been employed in MMA. Liver transplantation does improve the metabolic stability of brittle OA patients, and some sequelae may be mitigated, including cardiomyopathy. One center has claimed that liver transplantation is more cost-effective than dietary therapy alone [228]. Not all sequelae of PA and MMA may be prevented by liver transplantation; some liver-transplanted patients have developed metabolic stroke after transplant and others have had progressive vision loss due to optic atrophy as well [18, 231, 232, 236, 239, 240]. Further studies on the long-term outcomes of transplantation and changes to the natural history of disease are indicated.
Conclusions
For patients with PA and MMA who survive their initial decompensation episode, significant morbidity remains a lifelong threat. Better therapies for these disorders remain elusive, but a critical mass of patients has now contributed to our understanding of the natural history of these diseases. Most importantly, recently proposed diagnostic and management guidelines have emerged from worldwide experts in these disorders and should improve management of these patients. Dietary management of the OAs should receive additional scrutiny in the near future with prospective, controlled studies to demonstrate the optimal circumstances for the use of medical foods and improvements in formulations of these foods to prevent iatrogenic morbidity. Better understanding of the pathophysiology of metabolic brain injury in the OAs must occur to permit further drug targeting for neuroprotection and recovery from metabolic insult. Finally, the use of orthotopic liver transplantation to improve metabolic stability in patients with OAs is burgeoning and will likely change the natural history of these disorders, yet we must ensure our patients’ families understand the limitations of this therapy, including ongoing risk to the CNS compartment following transplantation and the inherent risks of liver transplantation.
Key Points.
Diagnostic and management guidelines for PA and MMA are emerging and may improve long-term care.
Prospective, controlled studies are needed to support the use of medical foods and their formulations to limit iatrogenic morbidity in PA and MMA.
The pathophysiology of metabolic brain injury in the OAs requires further elucidation to permit future drug targeting for neuroprotection and recovery.
Orthotopic liver transplantation improves metabolic stability in patients with OAs and will likely change the natural history of these disorders, but is not curative.
Acknowledgments
Funding Sources: Child Health Research Career Development Award (Fraser), Intramural Research Funding – NHGRI (Venditti)
Financial support and sponsorship
This work was supported by the Intramural Research Program of the National Human Genome Research Institute (CPV) and the Child Health Research Career Development Award (K12) via Children’s National Medical Center (JLF).
Footnotes
Conflicts of interest
The authors have no conflicts of interest to report.
References
- 1.Matsui SM, Mahoney MJ, Rosenberg LE. The natural history of the inherited methylmalonic acidemias. The New England journal of medicine. 1983;308(15):857–61. doi: 10.1056/NEJM198304143081501. [DOI] [PubMed] [Google Scholar]
- 2.Henriquez H, el Din A, Ozand PT, et al. Emergency presentations of patients with methylmalonic acidemia, propionic acidemia and branched chain amino acidemia (MSUD) Brain & development. 1994;16(Suppl):86–93. doi: 10.1016/0387-7604(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura M, Tokura Y. Methylmalonic aciduria presenting with recurrent multiple molluscum contagiosum lesions. Dermato-endocrinology. 2010;2(2):60–1. doi: 10.4161/derm.2.2.13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prada CE, Al Jasmi F, Kirk EP, et al. Cardiac disease in methylmalonic acidemia. The Journal of pediatrics. 2011;159(5):862–4. doi: 10.1016/j.jpeds.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Vatanavicharn N, Champattanachai V, Liammongkolkul S, et al. Clinical and molecular findings in Thai patients with isolated methylmalonic acidemia. Molecular genetics and metabolism. 2012;106(4):424–9. doi: 10.1016/j.ymgme.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Pinar-Sueiro S, Martinez-Fernandez R, Lage-Medina S, et al. Optic neuropathy in methylmalonic acidemia: the role of neuroprotection. Journal of inherited metabolic disease. 2010;33(Suppl 3):S199–203. doi: 10.1007/s10545-010-9084-8. [DOI] [PubMed] [Google Scholar]
- 7.Williams ZR, Hurley PE, Altiparmak UE, et al. Late onset optic neuropathy in methylmalonic and propionic acidemia. American journal of ophthalmology. 2009;147(5):929–33. doi: 10.1016/j.ajo.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 8.Baumgarter ER, Viardot C. Long-term follow-up of 77 patients with isolated methylmalonic acidaemia. Journal of inherited metabolic disease. 1995;18(2):138–42. doi: 10.1007/BF00711749. [DOI] [PubMed] [Google Scholar]
- 9.Kanaumi T, Takashima S, Hirose S, et al. Neuropathology of methylmalonic acidemia in a child. Pediatric neurology. 2006;34(2):156–9. doi: 10.1016/j.pediatrneurol.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Lee NC, Chien YH, Peng SF, et al. Brain damage by mild metabolic derangements in methylmalonic acidemia. Pediatric neurology. 2008;39(5):325–9. doi: 10.1016/j.pediatrneurol.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 11.Azar MR, Shakiba M, Tafreshi RI, et al. Molecular genetics and metabolism. 92. United States: 2007. Heart failure in a patient with methylmalonic acidemia; p. 188. [DOI] [PubMed] [Google Scholar]
- 12.Sutton VR, Chapman KA, Gropman AL, et al. Chronic management and health supervision of individuals with propionic acidemia. Molecular genetics and metabolism. 2012;105(1):26–33. doi: 10.1016/j.ymgme.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 13.Haberlandt E, Canestrini C, Brunner-Krainz M, et al. Epilepsy in patients with propionic acidemia. Neuropediatrics. 2009;40(3):120–5. doi: 10.1055/s-0029-1243167. [DOI] [PubMed] [Google Scholar]
- 14.Nyhan WL, Bay C, Beyer EW, et al. Neurologic nonmetabolic presentation of propionic acidemia. Archives of neurology. 1999;56(9):1143–7. doi: 10.1001/archneur.56.9.1143. [DOI] [PubMed] [Google Scholar]
- 15.Bergman AJ, Van der Knaap MS, Smeitink JA, et al. Magnetic resonance imaging and spectroscopy of the brain in propionic acidemia: clinical and biochemical considerations. Pediatric research. 1996;40(3):404–9. doi: 10.1203/00006450-199609000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Rafique M. Propionic acidaemia: demographic characteristics and complications. Journal of pediatric endocrinology & metabolism : JPEM. 2013;26(5–6):497–501. doi: 10.1515/jpem-2013-0031. [DOI] [PubMed] [Google Scholar]
- 17.Scholl-Burgi S, Haberlandt E, Gotwald T, et al. Stroke-like episodes in propionic acidemia caused by central focal metabolic decompensation. Neuropediatrics. 2009;40(2):76–81. doi: 10.1055/s-0029-1231065. [DOI] [PubMed] [Google Scholar]
- 18*.Charbit-Henrion F, Lacaille F, McKiernan P, et al. Early and late complications after liver transplantation for propionic acidemia in children: a two centers study. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015;15(3):786–91. doi: 10.1111/ajt.13027. This study details the risks of liver transplantation in PA. [DOI] [PubMed] [Google Scholar]
- 19.Laemmle A, Balmer C, Doell C, et al. Propionic acidemia in a previously healthy adolescent with acute onset of dilated cardiomyopathy. European journal of pediatrics. 2014;173(7):971–4. doi: 10.1007/s00431-014-2359-6. [DOI] [PubMed] [Google Scholar]
- 20.Baruteau J, Hargreaves I, Krywawych S, et al. Successful reversal of propionic acidaemia associated cardiomyopathy: evidence for low myocardial coenzyme Q10 status and secondary mitochondrial dysfunction as an underlying pathophysiological mechanism. Mitochondrion. 2014;17:150–6. doi: 10.1016/j.mito.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Grünert SC, Müllerleile S, De Silva L, et al. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet journal of rare diseases. 2013;8:6. doi: 10.1186/1750-1172-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pena L, Franks J, Chapman KA, et al. Natural history of propionic acidemia. Molecular genetics and metabolism. 2012;105(1):5–9. doi: 10.1016/j.ymgme.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 23.Pena L, Burton BK. Survey of health status and complications among propionic acidemia patients. American journal of medical genetics Part A. 2012;158a(7):1641–6. doi: 10.1002/ajmg.a.35387. [DOI] [PubMed] [Google Scholar]
- 24.Romano S, Valayannopoulos V, Touati G, et al. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. The Journal of pediatrics. 2010;156(1):128–34. doi: 10.1016/j.jpeds.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Iwashima S, Ishikawa T, Ohzeki T, et al. Delayed enhancement cardiac magnetic resonance imaging in propionic acidemia. Pediatric cardiology. 2010;31(6):884–6. doi: 10.1007/s00246-010-9723-8. [DOI] [PubMed] [Google Scholar]
- 26.Sato S, Kasahara M, Fukuda A, et al. Liver transplantation in a patient with propionic acidemia requiring extra corporeal membrane oxygenation during severe metabolic decompensation. Pediatric transplantation. 2009;13(6):790–3. doi: 10.1111/j.1399-3046.2008.01029.x. [DOI] [PubMed] [Google Scholar]
- 27.Mizuguchi K, Hoshino H, Nagasawa T, et al. Extracorporeal membrane oxygenation in a patient with propionic acidaemia: a therapeutic option for cardiac failure. Journal of inherited metabolic disease. 2009;32(Suppl 1):S37–40. doi: 10.1007/s10545-009-1029-8. [DOI] [PubMed] [Google Scholar]
- 28.Baumgartner D, Scholl-Burgi S, Sass JO, et al. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. The Journal of pediatrics. 2007;150(2):192–7. 7.e1. doi: 10.1016/j.jpeds.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 29.Mardach R, Verity MA, Cederbaum SD. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Molecular genetics and metabolism. 2005;85(4):286–90. doi: 10.1016/j.ymgme.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Vernon HJ, Bagnasco S, Hamosh A, et al. Chronic kidney disease in an adult with propionic acidemia. JIMD reports. 2014;12:5–10. doi: 10.1007/8904_2013_237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lam C, Desviat LR, Perez-Cerda C, et al. 45-Year-old female with propionic acidemia, renal failure, and premature ovarian failure; late complications of propionic acidemia? Molecular genetics and metabolism. 2011;103(4):338–40. doi: 10.1016/j.ymgme.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 32*.Martinez Alvarez L, Jameson E, Parry NR, et al. Optic neuropathy in methylmalonic acidemia and propionic acidemia. The British journal of ophthalmology. 2016;100(1):98–104. doi: 10.1136/bjophthalmol-2015-306798. This study reports on the optic neuropathy of MMA and PA, a later effect of these disorders. [DOI] [PubMed] [Google Scholar]
- 33.Arias C, Raimann E, Peredo P, et al. Propionic acidemia and optic neuropathy: a report of two cases. JIMD reports. 2014;12:1–4. doi: 10.1007/8904_2013_234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schreiber J, Chapman KA, Summar ML, et al. Neurologic considerations in propionic acidemia. Molecular genetics and metabolism. 2012;105(1):10–5. doi: 10.1016/j.ymgme.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Ianchulev T, Kolin T, Moseley K, et al. Optic nerve atrophy in propionic acidemia. Ophthalmology. 2003;110(9):1850–4. doi: 10.1016/S0161-6420(03)00573-6. [DOI] [PubMed] [Google Scholar]
- 36**.Baumgartner MR, Horster F, Dionisi-Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet journal of rare diseases. 2014;9:130. doi: 10.1186/s13023-014-0130-8. While outside of the review period, this reference cites the first ever guidelines for treatment of the OA disorders. Despite limited evidence basis to most recommendations, this work should promote standardization of care for patients with OAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krizova A, Herath JC. Death of a Neonate With a Negative Autopsy and Ketoacidosis: A Case Report of Propionic Acidemia. The American journal of forensic medicine and pathology. 2015;36(3):193–5. doi: 10.1097/PAF.0000000000000156. [DOI] [PubMed] [Google Scholar]
- 38.Kasapkara CS, Akar M, Yuruk Yildirim ZN, et al. Severe renal failure and hyperammonemia in a newborn with propionic acidemia: effects of treatment on the clinical course. Renal failure. 2014;36(3):451–2. doi: 10.3109/0886022X.2013.865484. [DOI] [PubMed] [Google Scholar]
- 39.Deodato F, Boenzi S, Santorelli FM, et al. Methylmalonic and propionic aciduria. American journal of medical genetics Part C, Seminars in medical genetics. 2006;142c(2):104–12. doi: 10.1002/ajmg.c.30090. [DOI] [PubMed] [Google Scholar]
- 40.Matern D, Seydewitz HH, Lehnert W, et al. Primary treatment of propionic acidemia complicated by acute thiamine deficiency. The Journal of pediatrics. 1996;129(5):758–60. doi: 10.1016/s0022-3476(96)70162-2. [DOI] [PubMed] [Google Scholar]
- 41.North KN, Korson MS, Gopal YR, et al. Neonatal-onset propionic acidemia: neurologic and developmental profiles, and implications for management. The Journal of pediatrics. 1995;126(6):916–22. doi: 10.1016/s0022-3476(95)70208-3. [DOI] [PubMed] [Google Scholar]
- 42.Baumgartner ER, Bachmann C, Brechbuhler T, et al. Acute neonatal nonketotic hyperglycinemia: normal propionate and methylmalonate metabolism. Pediatric research. 1975;9(7):559–64. doi: 10.1203/00006450-197507000-00001. [DOI] [PubMed] [Google Scholar]
- 43.van der Meer SB, Spaapen LJ, Fowler B, et al. Prenatal treatment of a patient with vitamin B12-responsive methylmalonic acidemia. The Journal of pediatrics. 1990;117(6):923–6. doi: 10.1016/s0022-3476(05)80138-6. [DOI] [PubMed] [Google Scholar]
- 44.Childs B, Nyhan WL, Borden M, et al. Idiopathic hyperglycinemia and hyperglycinuria: a new disorder of amino acid metabolism. I. Pediatrics. 1961;27:522–38. [PubMed] [Google Scholar]
- 45.Chapman KA, Gropman A, MacLeod E, et al. Acute management of propionic acidemia. Molecular genetics and metabolism. 2012;105(1):16–25. doi: 10.1016/j.ymgme.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindblad B, Lindblad BS, Olin P, et al. Methylmalonic acidemia. A disorder associated with acidosis, hyperglycinemia, and hyperlactatemia. Acta paediatrica Scandinavica. 1968;57(5):417–24. doi: 10.1111/j.1651-2227.1968.tb07314.x. [DOI] [PubMed] [Google Scholar]
- 47.Morrow G, 3rd, Barness LA, Auerbach VH, et al. Observations on the coexistence of methylmalonic acidemia and glycinemia. The Journal of pediatrics. 1969;74(5):680–90. doi: 10.1016/s0022-3476(69)80130-7. [DOI] [PubMed] [Google Scholar]
- 48.Morrow G, 3rd, Barness LA, Cardinale GJ, et al. Congenital methylmalonic acidemia: enzymatic evidence for two forms of the disease. Proceedings of the National Academy of Sciences of the United States of America. 1969;63(1):191–7. doi: 10.1073/pnas.63.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baumgartner ER, Bachmann C, Wick H. Congenital methylmalonic acidemia: a variant of the B12 ‘non-responsive’ form with evidence for reduced affinity of methylmalonyl-CoA mutase for its B12-coenzyme. Enzyme. 1976;21(6):553–67. doi: 10.1159/000458907. [DOI] [PubMed] [Google Scholar]
- 50.Matsuda I, Terashima T, Yamamoto J, et al. Methylmalonic acidemia. European journal of pediatrics. 1978;128(3):181–6. doi: 10.1007/BF00444303. [DOI] [PubMed] [Google Scholar]
- 51.Morrow G, 3rd, Burkel GM. Long-term management of a patient with vitamin B12-responsive methylmalonic acidemia. The Journal of pediatrics. 1980;96(3 Pt 1):425–6. doi: 10.1016/s0022-3476(80)80687-1. [DOI] [PubMed] [Google Scholar]
- 52.Cathelineau L, Briand P, Ogier H, et al. Occurrence of hyperammonemia in the course of 17 cases of methylmalonic acidemia. The Journal of pediatrics. 1981;99(2):279–80. doi: 10.1016/s0022-3476(81)80478-7. [DOI] [PubMed] [Google Scholar]
- 53.Satoh T, Narisawa K, Igarashi Y, et al. Dietary therapy in two patients with vitamin B12-unresponsive methylmalonic acidemia. European journal of pediatrics. 1981;135(3):305–12. doi: 10.1007/BF00442109. [DOI] [PubMed] [Google Scholar]
- 54.Zwickler T, Riderer A, Haege G, et al. Usefulness of biochemical parameters in decision-making on the start of emergency treatment in patients with propionic acidemia. Journal of inherited metabolic disease. 2014;37(1):31–7. doi: 10.1007/s10545-013-9621-3. [DOI] [PubMed] [Google Scholar]
- 55.Zwickler T, Riderer A, Haege G, et al. Usefulness of biochemical parameters in decision-making on the start of emergency treatment in patients with propionic acidemia. Journal of inherited metabolic disease. 2013 doi: 10.1007/s10545-013-9621-3. [DOI] [PubMed] [Google Scholar]
- 56.Nizon M, Ottolenghi C, Valayannopoulos V, et al. Long-term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet journal of rare diseases. 2013;8:148. doi: 10.1186/1750-1172-8-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chapman KA, Summar ML. Propionic acidemia consensus conference summary. Molecular genetics and metabolism. 2012;105(1):3–4. doi: 10.1016/j.ymgme.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 58.Surtees RA, Matthews EE, Leonard JV. Neurologic outcome of propionic acidemia. Pediatric neurology. 1992;8(5):333–7. doi: 10.1016/0887-8994(92)90085-d. [DOI] [PubMed] [Google Scholar]
- 59*.Heringer J, Valayannopoulos V, Lund AM, et al. Impact of age at onset and newborn screening on outcome in organic acidurias. Journal of inherited metabolic disease. 2016;39(3):341–53. doi: 10.1007/s10545-015-9907-8. This study is the first large study evaluating differences in outcomes in patients with OAs diagnosed by NMS versus selective diagnostic testing. [DOI] [PubMed] [Google Scholar]
- 60.Lubrano R, Scoppi P, Barsotti P, et al. Kidney transplantation in a girl with methylmalonic acidemia and end stage renal failure. Pediatric nephrology (Berlin, Germany) 2001;16(11):848–51. doi: 10.1007/s004670100688. [DOI] [PubMed] [Google Scholar]
- 61.Manoli I, Sysol JR, Li L, et al. Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(33):13552–7. doi: 10.1073/pnas.1302764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Molteni KH, Oberley TD, Wolff JA, et al. Progressive renal insufficiency in methylmalonic acidemia. Pediatric nephrology (Berlin, Germany) 1991;5(3):323–6. doi: 10.1007/BF00867492. [DOI] [PubMed] [Google Scholar]
- 63.Rutledge SL, Geraghty M, Mroczek E, et al. Tubulointerstitial nephritis in methylmalonic acidemia. Pediatric nephrology (Berlin, Germany) 1993;7(1):81–2. doi: 10.1007/BF00861581. [DOI] [PubMed] [Google Scholar]
- 64.Wolff JA, Strom C, Griswold W, et al. Proximal renal tubular acidosis in methylmalonic acidemia. Journal of neurogenetics. 1985;2(1):31–9. doi: 10.3109/01677068509100141. [DOI] [PubMed] [Google Scholar]
- 65.Heidenreich R, Natowicz M, Hainline BE, et al. Acute extrapyramidal syndrome in methylmalonic acidemia: “metabolic stroke” involving the globus pallidus. The Journal of pediatrics. 1988;113(6):1022–7. doi: 10.1016/s0022-3476(88)80574-2. [DOI] [PubMed] [Google Scholar]
- 66.Rodan LH, Mishra N, Yau I, et al. Expanding the Spectrum of Methylmalonic Acid-Induced Pallidal Stroke: First Reported Case of Metabolic Globus Pallidus Stroke in Transcobalamin II Deficiency. JIMD reports. 2013;11:7–11. doi: 10.1007/8904_2013_215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson GN, Christodoulou J, Danks DM. Metabolic stroke in methylmalonic acidemia. The Journal of pediatrics. 1989;115(3):499–500. doi: 10.1016/s0022-3476(89)80867-4. [DOI] [PubMed] [Google Scholar]
- 68.Chakraborti S, Hasegawa H, Lumsden DE, et al. Bilateral subthalamic nucleus deep brain stimulation for refractory total body dystonia secondary to metabolic autopallidotomy in a 4-year-old boy with infantile methylmalonic acidemia: case report. Journal of neurosurgery Pediatrics. 2013;12(4):374–9. doi: 10.3171/2013.7.PEDS1350. [DOI] [PubMed] [Google Scholar]
- 69.Prada CE, Villamizar-Schiller IT. Globus pallidus involvement as initial presentation of methylmalonic acidemia. Movement disorders : official journal of the Movement Disorder Society. 2014;29(7):870. doi: 10.1002/mds.25890. [DOI] [PubMed] [Google Scholar]
- 70.Traber G, Baumgartner MR, Schwarz U, et al. Subacute bilateral visual loss in methylmalonic acidemia. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society. 2011;31(4):344–6. doi: 10.1097/WNO.0b013e31822db480. [DOI] [PubMed] [Google Scholar]
- 71.Sethi KD, Ray R, Roesel RA, et al. Adult-onset chorea and dementia with propionic acidemia. Neurology. 1989;39(10):1343–5. doi: 10.1212/wnl.39.10.1343. [DOI] [PubMed] [Google Scholar]
- 72.Rashed MS, Ozand PT, Bucknall MP, et al. Diagnosis of inborn errors of metabolism from blood spots by acylcarnitines and amino acids profiling using automated electrospray tandem mass spectrometry. Pediatric research. 1995;38(3):324–31. doi: 10.1203/00006450-199509000-00009. [DOI] [PubMed] [Google Scholar]
- 73.Stanley CA, Berry GT, Bennett MJ, et al. Renal handling of carnitine in secondary carnitine deficiency disorders. Pediatric research. 1993;34(1):89–97. doi: 10.1203/00006450-199307000-00021. [DOI] [PubMed] [Google Scholar]
- 74.Poorthuis BJ, Jille-Vlckova T, Onkenhout W. Determination of acylcarnitines in urine of patients with inborn errors of metabolism using high-performance liquid chromatography after derivatization with 4′-bromophenacylbromide. Clinica chimica acta; international journal of clinical chemistry. 1993;216(1–2):53–61. doi: 10.1016/0009-8981(93)90138-t. [DOI] [PubMed] [Google Scholar]
- 75.Kurczynski TW, Hoppel CL, Goldblatt PJ, et al. Metabolic studies of carnitine in a child with propionic acidemia. Pediatric research. 1989;26(1):63–6. doi: 10.1203/00006450-198907000-00018. [DOI] [PubMed] [Google Scholar]
- 76.Dugan RE, Schmidt MJ, Hoganson GE, et al. High-performance liquid chromatography of coenzyme A esters formed by transesterification of short-chain acylcarnitines: diagnosis of acidemias by urinary analysis. Analytical biochemistry. 1987;160(2):275–80. doi: 10.1016/0003-2697(87)90047-9. [DOI] [PubMed] [Google Scholar]
- 77.Roe CR, Millington DS, Maltby DA, et al. L-carnitine enhances excretion of propionyl coenzyme A as propionylcarnitine in propionic acidemia. The Journal of clinical investigation. 1984;73(6):1785–8. doi: 10.1172/JCI111387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Donato S, Rimoldi M, Garavaglia B, et al. Propionylcarnitine excretion in propionic and methylmalonic acidurias: a cause of carnitine deficiency. Clinica chimica acta; international journal of clinical chemistry. 1984;139(1):13–21. doi: 10.1016/0009-8981(84)90187-6. [DOI] [PubMed] [Google Scholar]
- 79.Matsumoto I, Kuhara T. A new chemical diagnostic method for inborn errors of metabolism by mass spectrometry-rapid, practical, and simultaneous urinary metabolites analysis. Mass spectrometry reviews. 1996;15(1):43–57. doi: 10.1002/(SICI)1098-2787(1996)15:1<43::AID-MAS3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 80.Kuhara T, Shinka T, Matsuo M, et al. Increased excretion of lactate, glutarate, 3-hydroxyisovalerate and 3-methylglutaconate during clinical episodes of propionic acidemia. Clinica chimica acta; international journal of clinical chemistry. 1982;123(1–2):101–9. doi: 10.1016/0009-8981(82)90118-8. [DOI] [PubMed] [Google Scholar]
- 81.Al-Hassnan ZN, Boyadjiev SA, Praphanphoj V, et al. The relationship of plasma glutamine to ammonium and of glycine to acid-base balance in propionic acidaemia. Journal of inherited metabolic disease. 2003;26(1):89–91. doi: 10.1023/a:1024048118294. [DOI] [PubMed] [Google Scholar]
- 82.Shimizu N, Yamaguchi S, Orii T. A study of urinary metabolites in patients with dicarboxylic aciduria for differential diagnosis. Acta paediatrica Japonica; Overseas edition. 1994;36(2):139–45. doi: 10.1111/j.1442-200x.1994.tb03149.x. [DOI] [PubMed] [Google Scholar]
- 83.Hayasaka K, Narisawa K, Satoh T, et al. Glycine cleavage system in ketotic hyperglycinemia: a reduction of H-protein activity. Pediatric research. 1982;16(1):5–7. doi: 10.1203/00006450-198201001-00002. [DOI] [PubMed] [Google Scholar]
- 84.Wadlington WB, Kilroy A, Ando T, et al. Hyperglycinemia and propionyl coA carboxylase deficiency and episodic severe illness without consistent ketosis. The Journal of pediatrics. 1975;86(5):707–12. doi: 10.1016/s0022-3476(75)80354-4. [DOI] [PubMed] [Google Scholar]
- 85.Shih VE, Mandell R, Levy HL, et al. Free amino acids in extracts of cultured skin fibroblasts from patients with various amino acid metabolic disorders. Clinical genetics. 1975;7(5):421–5. doi: 10.1111/j.1399-0004.1975.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 86.Zwickler T, Haege G, Riderer A, et al. Metabolic decompensation in methylmalonic aciduria: which biochemical parameters are discriminative? Journal of inherited metabolic disease. 2012;35(5):797–806. doi: 10.1007/s10545-011-9426-1. [DOI] [PubMed] [Google Scholar]
- 87.Tu WJ, Dai F, Wang XY, et al. Liquid chromatography-tandem mass spectrometry for analysis of acylcarnitines in dried blood specimens collected at autopsy from neonatal intensive care unit. Chinese medical sciences journal = Chung-kuo i hsueh k’o hsueh tsa chih/Chinese Academy of Medical Sciences. 2010;25(2):109–14. doi: 10.1016/s1001-9294(10)60032-6. [DOI] [PubMed] [Google Scholar]
- 88.Maeda Y, Ito T, Suzuki A, et al. Simultaneous quantification of acylcarnitine isomers containing dicarboxylic acylcarnitines in human serum and urine by high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid communications in mass spectrometry : RCM. 2007;21(5):799–806. doi: 10.1002/rcm.2905. [DOI] [PubMed] [Google Scholar]
- 89.Alfares A, Nunez LD, Al-Thihli K, et al. Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. Journal of medical genetics. 2011;48(9):602–5. doi: 10.1136/jmedgenet-2011-100230. [DOI] [PubMed] [Google Scholar]
- 90.Sloan JL, Johnston JJ, Manoli I, et al. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat Genet. 2011;43(9):883–6. doi: 10.1038/ng.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stojiljkovic M, Klaassen K, Djordjevic M, et al. Molecular and phenotypic characteristics of seven novel mutations causing branched-chain organic acidurias. Clinical genetics. 2016 doi: 10.1111/cge.12751. [DOI] [PubMed] [Google Scholar]
- 92.Gupta D, Bijarnia-Mahay S, Kohli S, et al. Seventeen Novel Mutations in PCCA and PCCB Genes in Indian Propionic Acidemia Patients, and Their Outcomes. Genetic testing and molecular biomarkers. 2016;20(7):373–82. doi: 10.1089/gtmb.2016.0017. [DOI] [PubMed] [Google Scholar]
- 93.Porntaveetus T, Srichomthong C, Suphapeetiporn K, et al. A novel PCCB mutation in a Thai patient with propionic acidemia identified by exome sequencing. Human genome variation. 2015;2:15033. doi: 10.1038/hgv.2015.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chiu YH, Liu YN, Liao WL, et al. Two frequent mutations associated with the classic form of propionic acidemia in Taiwan. Biochemical genetics. 2014;52(9–10):415–29. doi: 10.1007/s10528-014-9657-6. [DOI] [PubMed] [Google Scholar]
- 95.Gallego-Villar L, Perez-Cerda C, Perez B, et al. Functional characterization of novel genotypes and cellular oxidative stress studies in propionic acidemia. Journal of inherited metabolic disease. 2013;36(5):731–40. doi: 10.1007/s10545-012-9545-3. [DOI] [PubMed] [Google Scholar]
- 96.Kraus JP, Spector E, Venezia S, et al. Mutation analysis in 54 propionic acidemia patients. Journal of inherited metabolic disease. 2012;35(1):51–63. doi: 10.1007/s10545-011-9399-0. [DOI] [PubMed] [Google Scholar]
- 97.Perez B, Angaroni C, Sanchez-Alcudia R, et al. The molecular landscape of propionic acidemia and methylmalonic aciduria in Latin America. Journal of inherited metabolic disease. 2010;33(Suppl 2):S307–14. doi: 10.1007/s10545-010-9116-4. [DOI] [PubMed] [Google Scholar]
- 98.Desviat LR, Sanchez-Alcudia R, Perez B, et al. High frequency of large genomic deletions in the PCCA gene causing propionic acidemia. Molecular genetics and metabolism. 2009;96(4):171–6. doi: 10.1016/j.ymgme.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 99.Kaya N, Al-Owain M, Albakheet A, et al. Array comparative genomic hybridization (aCGH) reveals the largest novel deletion in PCCA found in a Saudi family with propionic acidemia. European journal of medical genetics. 2008;51(6):558–65. doi: 10.1016/j.ejmg.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 100.Desviat LR, Clavero S, Perez-Cerda C, et al. New splicing mutations in propionic acidemia. Journal of human genetics. 2006;51(11):992–7. doi: 10.1007/s10038-006-0068-3. [DOI] [PubMed] [Google Scholar]
- 101.Rodriguez-Pombo P, Perez-Cerda C, Perez B, et al. Towards a model to explain the intragenic complementation in the heteromultimeric protein propionyl-CoA carboxylase. Biochimica et biophysica acta. 2005;1740(3):489–98. doi: 10.1016/j.bbadis.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 102.Yang X, Sakamoto O, Matsubara Y, et al. Mutation spectrum of the PCCA and PCCB genes in Japanese patients with propionic acidemia. Molecular genetics and metabolism. 2004;81(4):335–42. doi: 10.1016/j.ymgme.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 103.Desviat LR, Perez B, Perez-Cerda C, et al. Propionic acidemia: mutation update and functional and structural effects of the variant alleles. Molecular genetics and metabolism. 2004;83(1–2):28–37. doi: 10.1016/j.ymgme.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 104.Clavero S, Perez B, Rincon A, et al. Qualitative and quantitative analysis of the effect of splicing mutations in propionic acidemia underlying non-severe phenotypes. Human genetics. 2004;115(3):239–47. doi: 10.1007/s00439-004-1147-1. [DOI] [PubMed] [Google Scholar]
- 105.Perez-Cerda C, Clavero S, Perez B, et al. Functional analysis of PCCB mutations causing propionic acidemia based on expression studies in deficient human skin fibroblasts. Biochimica et biophysica acta. 2003;1638(1):43–9. doi: 10.1016/s0925-4439(03)00039-5. [DOI] [PubMed] [Google Scholar]
- 106.Perez B, Desviat LR, Rodriguez-Pombo P, et al. Propionic acidemia: identification of twenty-four novel mutations in Europe and North America. Molecular genetics and metabolism. 2003;78(1):59–67. doi: 10.1016/s1096-7192(02)00197-x. [DOI] [PubMed] [Google Scholar]
- 107.Yorifuji T, Kawai M, Muroi J, et al. Unexpectedly high prevalence of the mild form of propionic acidemia in Japan: presence of a common mutation and possible clinical implications. Human genetics. 2002;111(2):161–5. doi: 10.1007/s00439-002-0761-z. [DOI] [PubMed] [Google Scholar]
- 108.Kim SN, Ryu KH, Lee EH, et al. Molecular analysis of PCCB gene in Korean patients with propionic acidemia. Molecular genetics and metabolism. 2002;77(3):209–16. doi: 10.1016/s1096-7192(02)00139-7. [DOI] [PubMed] [Google Scholar]
- 109.Clavero S, Martinez MA, Perez B, et al. Functional characterization of PCCA mutations causing propionic acidemia. Biochimica et biophysica acta. 2002;1588(2):119–25. doi: 10.1016/s0925-4439(02)00155-2. [DOI] [PubMed] [Google Scholar]
- 110.Muro S, Perez B, Desviat LR, et al. Effect of PCCB gene mutations on the heteromeric and homomeric assembly of propionyl-CoA carboxylase. Molecular genetics and metabolism. 2001;74(4):476–83. doi: 10.1006/mgme.2001.3254. [DOI] [PubMed] [Google Scholar]
- 111.Campeau E, Desviat LR, Leclerc D, et al. Structure of the PCCA gene and distribution of mutations causing propionic acidemia. Molecular genetics and metabolism. 2001;74(1–2):238–47. doi: 10.1006/mgme.2001.3210. [DOI] [PubMed] [Google Scholar]
- 112.Ravn K, Chloupkova M, Christensen E, et al. High incidence of propionic acidemia in greenland is due to a prevalent mutation, 1540insCCC, in the gene for the beta-subunit of propionyl CoA carboxylase. American journal of human genetics. 2000;67(1):203–6. doi: 10.1086/302971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chloupkova M, Ravn K, Schwartz M, et al. Changes in the carboxyl terminus of the beta subunit of human propionyl-CoA carboxylase affect the oligomer assembly and catalysis: expression and characterization of seven patient-derived mutant forms of PCC in Escherichia coli. Molecular genetics and metabolism. 2000;71(4):623–32. doi: 10.1006/mgme.2000.3097. [DOI] [PubMed] [Google Scholar]
- 114.Ugarte M, Perez-Cerda C, Rodriguez-Pombo P, et al. Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Human mutation. 1999;14(4):275–82. doi: 10.1002/(SICI)1098-1004(199910)14:4<275::AID-HUMU1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 115.Richard E, Desviat LR, Perez B, et al. Genetic heterogeneity in propionic acidemia patients with alpha-subunit defects. Identification of five novel mutations, one of them causing instability of the protein. Biochimica et biophysica acta. 1999;1453(3):351–8. doi: 10.1016/s0925-4439(99)00008-3. [DOI] [PubMed] [Google Scholar]
- 116.Muro S, Rodriguez-Pombo P, Perez B, et al. Identification of novel mutations in the PCCB gene in European propionic acidemia patients. Mutation in brief no. 253. Online. Human mutation. 1999;14(1):89–90. doi: 10.1002/(SICI)1098-1004(1999)14:1<89::AID-HUMU18>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 117.Campeau E, Dupuis L, Leon-Del-Rio A, et al. Coding sequence mutations in the alpha subunit of propionyl-CoA carboxylase in patients with propionic acidemia. Molecular genetics and metabolism. 1999;67(1):11–22. doi: 10.1006/mgme.1999.2850. [DOI] [PubMed] [Google Scholar]
- 118.Campeau E, Dupuis L, Leclerc D, et al. Detection of a normally rare transcript in propionic acidemia patients with mRNA destabilizing mutations in the PCCA gene. Human molecular genetics. 1999;8(1):107–13. doi: 10.1093/hmg/8.1.107. [DOI] [PubMed] [Google Scholar]
- 119.Rodriguez-Pombo P, Hoenicka J, Muro S, et al. Human propionyl-CoA carboxylase beta subunit gene: exon-intron definition and mutation spectrum in Spanish and Latin American propionic acidemia patients. American journal of human genetics. 1998;63(2):360–9. doi: 10.1086/301970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Richard E, Desviat LR, Perez B, et al. Three novel splice mutations in the PCCA gene causing identical exon skipping in propionic acidemia patients. Human genetics. 1997;101(1):93–6. doi: 10.1007/s004390050593. [DOI] [PubMed] [Google Scholar]
- 121.Gravel RA, Akerman BR, Lamhonwah AM, et al. Mutations participating in interallelic complementation in propionic acidemia. American journal of human genetics. 1994;55(1):51–8. [PMC free article] [PubMed] [Google Scholar]
- 122.Ohura T, Ogasawara M, Ikeda H, et al. The molecular defect in propionic acidemia: exon skipping caused by an 8-bp deletion from an intron in the PCCB allele. Human genetics. 1993;92(4):397–402. doi: 10.1007/BF01247343. [DOI] [PubMed] [Google Scholar]
- 123.Ohura T, Miyabayashi S, Narisawa K, et al. Genetic heterogeneity of propionic acidemia: analysis of 15 Japanese patients. Human genetics. 1991;87(1):41–4. doi: 10.1007/BF01213089. [DOI] [PubMed] [Google Scholar]
- 124.Tahara T, Kraus JP, Rosenberg LE. An unusual insertion/deletion in the gene encoding the beta-subunit of propionyl-CoA carboxylase is a frequent mutation in Caucasian propionic acidemia. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(4):1372–6. doi: 10.1073/pnas.87.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lamhonwah AM, Troxel CE, Schuster S, et al. Two distinct mutations at the same site in the PCCB gene in propionic acidemia. Genomics. 1990;8(2):249–54. doi: 10.1016/0888-7543(90)90279-4. [DOI] [PubMed] [Google Scholar]
- 126.Ohura T, Kraus JP, Rosenberg LE. Unequal synthesis and differential degradation of propionyl CoA carboxylase subunits in cells from normal and propionic acidemia patients. American journal of human genetics. 1989;45(1):33–40. [PMC free article] [PubMed] [Google Scholar]
- 127.Lamhonwah AM, Barankiewicz TJ, Willard HF, et al. Isolation of cDNA clones coding for the alpha and beta chains of human propionyl-CoA carboxylase: chromosomal assignments and DNA polymorphisms associated with PCCA and PCCB genes. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(13):4864–8. doi: 10.1073/pnas.83.13.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McKeon C, Eanes RZ, Wolf B. Biochemical characterization of propionyl CoA carboxylase deficiency: heterogeneity within a single genetic complementation group. Biochemical genetics. 1982;20(1–2):77–94. doi: 10.1007/BF00484937. [DOI] [PubMed] [Google Scholar]
- 129.Kidd JR, Wolf B, Hsia E, et al. Genetics of propionic acidemia in a Mennonite-Amish kindred. American journal of human genetics. 1980;32(2):236–45. [PMC free article] [PubMed] [Google Scholar]
- 130.Hsia YE, Scully KJ, Rosenberg LE. Human propionyl CoA carboxylase: some properties of the partially purified enzyme in fibroblasts from controls and patients with propionic acidemia. Pediatric research. 1979;13(6):746–51. doi: 10.1203/00006450-197906000-00005. [DOI] [PubMed] [Google Scholar]
- 131.Morrow G, 3rd, Mahoney MJ, Mathews C, et al. Studies of methylmalonyl coenzyme A carbonylmutase activity in methylmalonic acidemia. I. Correlation of clinical, hepatic, and fibroblast data. Pediatric research. 1975;9(8):641–4. doi: 10.1203/00006450-197508000-00006. [DOI] [PubMed] [Google Scholar]
- 132.Morrow G, 3rd, Lebowitz J. Studies of methylmalonyl-coenzyme A carbonylmutase activity in methylmalonic acidemia. II. In vitro binding kinetics with adenosylcobalamin. Biochemical medicine. 1976;15(3):241–5. doi: 10.1016/0006-2944(76)90054-5. [DOI] [PubMed] [Google Scholar]
- 133.Morrow G, 3rd, Revsin B, Clark R, et al. A new variant of methylmalonic acidemia-defective coenzyme-apoenzyme binding in cultured fibroblasts. Clinica chimica acta; international journal of clinical chemistry. 1978;85(1):67–72. doi: 10.1016/0009-8981(78)90102-x. [DOI] [PubMed] [Google Scholar]
- 134.Whelan DT, Ryan E, Spate M, et al. Methylmalonic acidemia: 6 years’ clinical experience with two variants unresponsive to vitamin B12 therapy. Canadian Medical Association journal. 1979;120(10):1230–5. [PMC free article] [PubMed] [Google Scholar]
- 135.Ledley FD, Lumetta M, Nguyen PN, et al. Molecular cloning of L-methylmalonyl-CoA mutase: gene transfer and analysis of mut cell lines. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(10):3518–21. doi: 10.1073/pnas.85.10.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ledley FD, Lumetta MR, Zoghbi HY, et al. Mapping of human methylmalonyl CoA mutase (MUT) locus on chromosome 6. American journal of human genetics. 1988;42(6):839–46. [PMC free article] [PubMed] [Google Scholar]
- 137.Jansen R, Ledley FD. Heterozygous mutations at the mut locus in fibroblasts with mut0 methylmalonic acidemia identified by polymerase-chain-reaction cDNA cloning. American journal of human genetics. 1990;47(5):808–14. [PMC free article] [PubMed] [Google Scholar]
- 138.Ledley FD, Crane AM, Lumetta M. Heterogeneous alleles and expression of methylmalonyl CoA mutase in mut methylmalonic acidemia. American journal of human genetics. 1990;46(3):539–47. [PMC free article] [PubMed] [Google Scholar]
- 139.Crane AM, Ledley FD. Clustering of mutations in methylmalonyl CoA mutase associated with mut- methylmalonic acidemia. American journal of human genetics. 1994;55(1):42–50. [PMC free article] [PubMed] [Google Scholar]
- 140.Ogasawara M, Matsubara Y, Mikami H, et al. Identification of two novel mutations in the methylmalonyl-CoA mutase gene with decreased levels of mutant mRNA in methylmalonic acidemia. Human molecular genetics. 1994;3(6):867–72. doi: 10.1093/hmg/3.6.867. [DOI] [PubMed] [Google Scholar]
- 141.Ledley FD, Rosenblatt DS. Mutations in mut methylmalonic acidemia: clinical and enzymatic correlations. Human mutation. 1997;9(1):1–6. doi: 10.1002/(SICI)1098-1004(1997)9:1<1::AID-HUMU1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 142.Mikami H, Ogasawara M, Matsubara Y, et al. Molecular analysis of methylmalonyl-CoA mutase deficiency: identification of three missense mutations in mut0 patients. Journal of human genetics. 1999;44(1):35–9. doi: 10.1007/s100380050103. [DOI] [PubMed] [Google Scholar]
- 143.Acquaviva C, Benoist JF, Callebaut I, et al. N219Y, a new frequent mutation among mut(degree) forms of methylmalonic acidemia in Caucasian patients. European journal of human genetics : EJHG. 2001;9(8):577–82. doi: 10.1038/sj.ejhg.5200675. [DOI] [PubMed] [Google Scholar]
- 144.Dobson CM, Wai T, Leclerc D, et al. Identification of the gene responsible for the cblA complementation group of vitamin B12-responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(24):15554–9. doi: 10.1073/pnas.242614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang X, Sakamoto O, Matsubara Y, et al. Mutation analysis of the MMAA and MMAB genes in Japanese patients with vitamin B(12)-responsive methylmalonic acidemia: identification of a prevalent MMAA mutation. Molecular genetics and metabolism. 2004;82(4):329–33. doi: 10.1016/j.ymgme.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 146.Acquaviva C, Benoist JF, Pereira S, et al. Molecular basis of methylmalonyl-CoA mutase apoenzyme defect in 40 European patients affected by mut(o) and mut- forms of methylmalonic acidemia: identification of 29 novel mutations in the MUT gene. Human mutation. 2005;25(2):167–76. doi: 10.1002/humu.20128. [DOI] [PubMed] [Google Scholar]
- 147.Worgan LC, Niles K, Tirone JC, et al. Spectrum of mutations in mut methylmalonic acidemia and identification of a common Hispanic mutation and haplotype. Human mutation. 2006;27(1):31–43. doi: 10.1002/humu.20258. [DOI] [PubMed] [Google Scholar]
- 148.Martinez MA, Rincon A, Desviat LR, et al. Genetic analysis of three genes causing isolated methylmalonic acidemia: identification of 21 novel allelic variants. Molecular genetics and metabolism. 2005;84(4):317–25. doi: 10.1016/j.ymgme.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 149.Coude FX, Grimber G, Parvy P, et al. Characterization of enzymatic deficiencies of branched chain amino-acid catabolism in human fibroblasts by genetic complementation. Biochemical and biophysical research communications. 1983;114(1):175–82. doi: 10.1016/0006-291x(83)91610-8. [DOI] [PubMed] [Google Scholar]
- 150.Cannata JJ, Focesi A, Jr, Mazumder R, et al. METABOLISM OF PROPIONIC ACID IN ANIMAL TISSUES. XII. PROPERTIES OF MAMMALIAN METHYLMALONYL COENZYME A MUTASE. The Journal of biological chemistry. 1965;240:3249–57. [PubMed] [Google Scholar]
- 151.Sprecher M, Clark MJ, Sprinson DB. The absolute configuration of methylmalonyl coenzyme A and stereochemistry of the methymalonyl coenzyme A mutase reaction. The Journal of biological chemistry. 1966;241(4):872–7. [PubMed] [Google Scholar]
- 152.Whitaker TR, Giorgio AJ. A direct radioassay of methylmalonyl-coenzyme A mutase using enzymatically synthesized DL-(3–14 C)methylmalonyl-CoA. Analytical biochemistry. 1973;52(2):522–32. doi: 10.1016/0003-2697(73)90057-2. [DOI] [PubMed] [Google Scholar]
- 153.Wajner M, Goodman SI. Disruption of mitochondrial homeostasis in organic acidurias: insights from human and animal studies. Journal of bioenergetics and biomembranes. 2011;43(1):31–8. doi: 10.1007/s10863-011-9324-0. [DOI] [PubMed] [Google Scholar]
- 154.Fragaki K, Cano A, Benoist JF, et al. Fatal heart failure associated with CoQ10 and multiple OXPHOS deficiency in a child with propionic acidemia. Mitochondrion. 2011;11(3):533–6. doi: 10.1016/j.mito.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 155.Brusque AM, Borba Rosa R, Schuck PF, et al. Inhibition of the mitochondrial respiratory chain complex activities in rat cerebral cortex by methylmalonic acid. Neurochemistry international. 2002;40(7):593–601. doi: 10.1016/s0197-0186(01)00130-9. [DOI] [PubMed] [Google Scholar]
- 156.Scholl-Burgi S, Sass JO, Zschocke J, et al. Amino acid metabolism in patients with propionic acidaemia. Journal of inherited metabolic disease. 2012;35(1):65–70. doi: 10.1007/s10545-010-9245-9. [DOI] [PubMed] [Google Scholar]
- 157.de Keyzer Y, Valayannopoulos V, Benoist JF, et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatric research. 2009;66(1):91–5. doi: 10.1203/PDR.0b013e3181a7c270. [DOI] [PubMed] [Google Scholar]
- 158.Knerr I, Weinhold N, Vockley J, et al. Advances and challenges in the treatment of branched-chain amino/keto acid metabolic defects. Journal of inherited metabolic disease. 2012;35(1):29–40. doi: 10.1007/s10545-010-9269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Filipowicz HR, Ernst SL, Ashurst CL, et al. Metabolic changes associated with hyperammonemia in patients with propionic acidemia. Molecular genetics and metabolism. 2006;88(2):123–30. doi: 10.1016/j.ymgme.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 160.Prasad C, Nurko S, Borovoy J, et al. The importance of gut motility in the metabolic control of propionic acidemia. The Journal of pediatrics. 2004;144(4):532–5. doi: 10.1016/j.jpeds.2003.12.044. [DOI] [PubMed] [Google Scholar]
- 161.Thompson GN, Walter JH, Bresson JL, et al. Sources of propionate in inborn errors of propionate metabolism. Metabolism: clinical and experimental. 1990;39(11):1133–7. doi: 10.1016/0026-0495(90)90084-p. [DOI] [PubMed] [Google Scholar]
- 162.Richard E, Monteoliva L, Juarez S, et al. Quantitative analysis of mitochondrial protein expression in methylmalonic acidemia by two-dimensional difference gel electrophoresis. Journal of proteome research. 2006;5(7):1602–10. doi: 10.1021/pr050481r. [DOI] [PubMed] [Google Scholar]
- 163.Chandler RJ, Zerfas PM, Shanske S, et al. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2009;23(4):1252–61. doi: 10.1096/fj.08-121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Murphy GE, Lowekamp BC, Zerfas PM, et al. Ion-abrasion scanning electron microscopy reveals distorted liver mitochondrial morphology in murine methylmalonic acidemia. Journal of structural biology. 2010;171(2):125–32. doi: 10.1016/j.jsb.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Melo DR, Kowaltowski AJ, Wajner M, et al. Mitochondrial energy metabolism in neurodegeneration associated with methylmalonic acidemia. Journal of bioenergetics and biomembranes. 2011;43(1):39–46. doi: 10.1007/s10863-011-9330-2. [DOI] [PubMed] [Google Scholar]
- 166.Melo DR, Mirandola SR, Assuncao NA, et al. Methylmalonate impairs mitochondrial respiration supported by NADH-linked substrates: involvement of mitochondrial glutamate metabolism. Journal of neuroscience research. 2012;90(6):1190–9. doi: 10.1002/jnr.23020. [DOI] [PubMed] [Google Scholar]
- 167.Coude FX, Sweetman L, Nyhan WL. Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemia. The Journal of clinical investigation. 1979;64(6):1544–51. doi: 10.1172/JCI109614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dercksen M, LIJ, Duran M, et al. Inhibition of N-acetylglutamate synthase by various monocarboxylic and dicarboxylic short-chain coenzyme A esters and the production of alternative glutamate esters. Biochimica et biophysica acta. 2014;1842(12 Pt A):2510–6. doi: 10.1016/j.bbadis.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 169.Stewart PM, Walser M. Failure of the normal ureagenic response to amino acids in organic acid-loaded rats. Proposed mechanism for the hyperammonemia of propionic and methylmalonic acidemia. The Journal of clinical investigation. 1980;66(3):484–92. doi: 10.1172/JCI109879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Glasgow AM, Chase HP. Effect of propionic acid on fatty acid oxidation and ureagenesis. Pediatric research. 1976;10(7):683–6. doi: 10.1203/00006450-197607000-00010. [DOI] [PubMed] [Google Scholar]
- 171.Sugiyama N, Kidouchi K, Kobayashi M, et al. Carnitine deficiency in inherited organic acid disorders and Reye syndrome. Acta paediatrica Japonica; Overseas edition. 1990;32(4):410–6. doi: 10.1111/j.1442-200x.1990.tb00854.x. [DOI] [PubMed] [Google Scholar]
- 172.Nyhan WL, Fawcett N, Ando T, et al. Response to dietary therapy in B 12 unresponsive methylmalonic acidemia. Pediatrics. 1973;51(3):539–48. [PubMed] [Google Scholar]
- 173.van der Meer SB, Poggi F, Spada M, et al. Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. The Journal of pediatrics. 1994;125(6 Pt 1):903–8. doi: 10.1016/s0022-3476(05)82005-0. [DOI] [PubMed] [Google Scholar]
- 174.Hauser NS, Manoli I, Graf JC, et al. Variable dietary management of methylmalonic acidemia: metabolic and energetic correlations. The American journal of clinical nutrition. 2011;93(1):47–56. doi: 10.3945/ajcn.110.004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lane TN, Spraker MK, Parker SS. Propionic acidemia manifesting with low isoleucine generalized exfoliative dermatosis. Pediatric dermatology. 2007;24(5):508–10. doi: 10.1111/j.1525-1470.2007.00505.x. [DOI] [PubMed] [Google Scholar]
- 176.Yannicelli S, Acosta PB, Velazquez A, et al. Improved growth and nutrition status in children with methylmalonic or propionic acidemia fed an elemental medical food. Molecular genetics and metabolism. 2003;80(1–2):181–8. doi: 10.1016/j.ymgme.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 177.Thomas E. A study of the response to protein-modified diets for propionic acidemia in twelve patients. Brain & development. 1994;16(Suppl):58–63. doi: 10.1016/0387-7604(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 178.De Raeve L, De Meirleir L, Ramet J, et al. Acrodermatitis enteropathica-like cutaneous lesions in organic aciduria. The Journal of pediatrics. 1994;124(3):416–20. doi: 10.1016/s0022-3476(94)70364-7. [DOI] [PubMed] [Google Scholar]
- 179.Satoh T, Narisawa K, Tazawa Y, et al. Dietary therapy in a girl with propionic acidemia: supplement with leucine resulted in catch up growth. The Tohoku journal of experimental medicine. 1983;139(4):411–5. doi: 10.1620/tjem.139.411. [DOI] [PubMed] [Google Scholar]
- 180*.Manoli I, Myles JG, Sloan JL, et al. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonic acidemias. Genetics in medicine : official journal of the American College of Medical Genetics. 2016;18(4):386–95. doi: 10.1038/gim.2015.102. This work calls into question the safety of medical foods in the OAs and asserts the need for further study regarding their use. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Solomon LR. Oral pharmacologic doses of cobalamin may not be as effective as parenteral cobalamin therapy in reversing hyperhomocystinemia and methylmalonic acidemia in apparently normal subjects. Clinical and laboratory haematology. 2006;28(4):275–8. doi: 10.1111/j.1365-2257.2006.00783.x. [DOI] [PubMed] [Google Scholar]
- 182.Ampola MG, Mahoney MJ, Nakamura E, et al. Prenatal therapy of a patient with vitamin-B12-responsive methylmalonic acidemia. The New England journal of medicine. 1975;293(7):313–7. doi: 10.1056/NEJM197508142930701. [DOI] [PubMed] [Google Scholar]
- 183.Rafique M. Emerging trends in management of propionic acidemia. Arquivos brasileiros de endocrinologia e metabologia. 2014;58(3):237–42. doi: 10.1590/0004-2730000002821. [DOI] [PubMed] [Google Scholar]
- 184.Raby RB, Ward JC, Herrod HG. Propionic acidaemia and immunodeficiency. Journal of inherited metabolic disease. 1994;17(2):250–1. doi: 10.1007/BF00711631. [DOI] [PubMed] [Google Scholar]
- 185*.Harrington EA, Sloan JL, Manoli I, et al. Neutralizing Antibodies Against Adeno-Associated Viral Capsids in Patients with mut Methylmalonic Acidemia. Human gene therapy. 2016;27(5):345–53. doi: 10.1089/hum.2015.092. This work highlights the immunological aspects of future gene-therapy studies, but it also indicates that patients with MMA lack the typical immune response to common environmental viruses, such as AAVs, lending additional credence to the immunodeficiency associated with OAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Schwahn BC, Pieterse L, Bisset WM, et al. Biochemical efficacy of N-carbamylglutamate in neonatal severe hyperammonaemia due to propionic acidaemia. European journal of pediatrics. 2010;169(1):133–4. doi: 10.1007/s00431-009-1036-7. [DOI] [PubMed] [Google Scholar]
- 187.Filippi L, Gozzini E, Fiorini P, et al. N-carbamylglutamate in emergency management of hyperammonemia in neonatal acute onset propionic and methylmalonic aciduria. Neonatology. 2010;97(3):286–90. doi: 10.1159/000255168. [DOI] [PubMed] [Google Scholar]
- 188.Ah Mew N, McCarter R, Daikhin Y, et al. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics. 2010;126(1):e208–14. doi: 10.1542/peds.2010-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tuchman M, Caldovic L, Daikhin Y, et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatric research. 2008;64(2):213–7. doi: 10.1203/PDR.0b013e318179454b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ierardi-Curto L, Kaplan P, Saitta S, et al. The glutamine paradox in a neonate with propionic acidaemia and severe hyperammonaemia. Journal of inherited metabolic disease. 2000;23(1):85–6. doi: 10.1023/a:1005659132147. [DOI] [PubMed] [Google Scholar]
- 191.Walter JH, Wraith JE, Cleary MA. Absence of acidosis in the initial presentation of propionic acidaemia. Archives of disease in childhood Fetal and neonatal edition. 1995;72(3):F197–9. doi: 10.1136/fn.72.3.f197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192*.Valayannopoulos V, Baruteau J, Delgado MB, et al. Carglumic acid enhances rapid ammonia detoxification in classical organic acidurias with a favourable risk-benefit profile: a retrospective observational study. Orphanet journal of rare diseases. 2016;11:32. doi: 10.1186/s13023-016-0406-2. This is the most recent study to characterize the effects of N-carbamylphosphate on ammona levels in patients with OAs, suggesting that this drug may be an good treatment choice mild hyperammonemia in OAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Abacan M, Boneh A. Use of carglumic acid in the treatment of hyperammonaemia during metabolic decompensation of patients with propionic acidaemia. Molecular genetics and metabolism. 2013;109(4):397–401. doi: 10.1016/j.ymgme.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 194.Kasapkara CS, Ezgu FS, Okur I, et al. N-carbamylglutamate treatment for acute neonatal hyperammonemia in isovaleric acidemia. European journal of pediatrics. 2011;170(6):799–801. doi: 10.1007/s00431-010-1362-9. [DOI] [PubMed] [Google Scholar]
- 195.Jones S, Reed CA, Vijay S, et al. N-carbamylglutamate for neonatal hyperammonaemia in propionic acidaemia. Journal of inherited metabolic disease. 2008;31(Suppl 2):S219–22. doi: 10.1007/s10545-008-0777-1. [DOI] [PubMed] [Google Scholar]
- 196.Chakrapani A, Sivakumar P, McKiernan PJ, et al. Metabolic stroke in methylmalonic acidemia five years after liver transplantation. The Journal of pediatrics. 2002;140(2):261–3. doi: 10.1067/mpd.2002.121698. [DOI] [PubMed] [Google Scholar]
- 197.Kalidas K, Behrouz R. Inherited metabolic disorders and cerebral infarction. Expert review of neurotherapeutics. 2008;8(11):1731–41. doi: 10.1586/14737175.8.11.1731. [DOI] [PubMed] [Google Scholar]
- 198.Haas RH, Marsden DL, Capistrano-Estrada S, et al. Acute basal ganglia infarction in propionic acidemia. Journal of child neurology. 1995;10(1):18–22. doi: 10.1177/088307389501000104. [DOI] [PubMed] [Google Scholar]
- 199*.Baker EH, Sloan JL, Hauser NS, et al. MRI characteristics of globus pallidus infarcts in isolated methylmalonic acidemia. AJNR American journal of neuroradiology. 2015;36(1):194–201. doi: 10.3174/ajnr.A4087. While outside of the review period, this study provides the first, and thus far only, radiographic analysis of evolution and progression of metabolic brain injury of MMA with novel classification system for correlating with phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kandel A, Amatya SK, Yeh EA. Reversible diffusion weighted imaging changes in propionic acidemia. Journal of child neurology. 2013;28(1):128–31. doi: 10.1177/0883073812441057. [DOI] [PubMed] [Google Scholar]
- 201.Davison JE, Davies NP, Wilson M, et al. MR spectroscopy-based brain metabolite profiling in propionic acidaemia: metabolic changes in the basal ganglia during acute decompensation and effect of liver transplantation. Orphanet journal of rare diseases. 2011;6:19. doi: 10.1186/1750-1172-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chemelli AP, Schocke M, Sperl W, et al. Magnetic resonance spectroscopy (MRS) in five patients with treated propionic acidemia. Journal of magnetic resonance imaging : JMRI. 2000;11(6):596–600. doi: 10.1002/1522-2586(200006)11:6<596::aid-jmri4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 203*.Ktena YP, Paul SM, Hauser NS, et al. Delineating the spectrum of impairments, disabilities, and rehabilitation needs in methylmalonic acidemia (MMA) American journal of medical genetics Part A. 2015;167A(9):2075–84. doi: 10.1002/ajmg.a.37127. This study characterizes the neuro-functional impairments of individuals with MMA and highlights the importance of wrap-around therapies to support patients with OAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chao PW, Chang WK, Lai IW, et al. Acute life-threatening arrhythmias caused by severe hyperkalemia after induction of anesthesia in an infant with methylmalonic acidemia. Journal of the Chinese Medical Association : JCMA. 2012;75(5):243–5. doi: 10.1016/j.jcma.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 205*.Bodi I, Grunert SC, Becker N, et al. Mechanisms of acquired long QT syndrome in patients with propionic academia. Heart rhythm : the official journal of the Heart Rhythm Society. 2016;13(6):1335–45. doi: 10.1016/j.hrthm.2016.02.003. This study delineates pathophysiological mechanisms associated with long QTc in PA patients. [DOI] [PubMed] [Google Scholar]
- 206.Tsina EK, Marsden DL, Hansen RM, et al. Maculopathy and retinal degeneration in cobalamin C methylmalonic aciduria and homocystinuria. Archives of ophthalmology. 2005;123(8):1143–6. doi: 10.1001/archopht.123.8.1143. [DOI] [PubMed] [Google Scholar]
- 207.Gerth C, Morel CF, Feigenbaum A, et al. Ocular phenotype in patients with methylmalonic aciduria and homocystinuria, cobalamin C type. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus/American Association for Pediatric Ophthalmology and Strabismus. 2008;12(6):591–6. doi: 10.1016/j.jaapos.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 208.Martinelli D, Deodato F, Dionisi-Vici C. Cobalamin C defect: natural history, pathophysiology, and treatment. Journal of inherited metabolic disease. 2011;34(1):127–35. doi: 10.1007/s10545-010-9161-z. [DOI] [PubMed] [Google Scholar]
- 209.Aleman TS, Brodie F, Garvin C, et al. Retinal Structure in Cobalamin C Disease: Mechanistic and Therapeutic Implications. Ophthalmic genetics. 2014 doi: 10.3109/13816810.2014.885059. [DOI] [PubMed] [Google Scholar]
- 210.Paik KH, Lee JE, Jin DK. Successful dialysis in a boy with methylmalonic acidemia. Pediatric nephrology (Berlin, Germany) 2004;19(10):1180–1. doi: 10.1007/s00467-003-1409-5. [DOI] [PubMed] [Google Scholar]
- 211.Ha TS, Lee JS, Hong EJ. Delay of renal progression in methylmalonic acidemia using angiotensin II inhibition: a case report. Journal of nephrology. 2008;21(5):793–6. [PubMed] [Google Scholar]
- 212.Kruszka PS, Manoli I, Sloan JL, et al. Renal growth in isolated methylmalonic acidemia. Genetics in medicine : official journal of the American College of Medical Genetics. 2013;15(12):990–6. doi: 10.1038/gim.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Schuck PF, Alves L, Pettenuzzo LF, et al. Acute renal failure potentiates methylmalonate-induced oxidative stress in brain and kidney of rats. Free radical research. 2013;47(3):233–40. doi: 10.3109/10715762.2012.762771. [DOI] [PubMed] [Google Scholar]
- 214.Brunelli SM, Meyers KE, Guttenberg M, et al. Cobalamin C deficiency complicated by an atypical glomerulopathy. Pediatric nephrology (Berlin, Germany) 2002;17(10):800–3. doi: 10.1007/s00467-002-0895-1. [DOI] [PubMed] [Google Scholar]
- 215.Kind T, Levy J, Lee M, et al. Cobalamin C disease presenting as hemolytic-uremic syndrome in the neonatal period. Journal of pediatric hematology/oncology. 2002;24(4):327–9. doi: 10.1097/00043426-200205000-00023. [DOI] [PubMed] [Google Scholar]
- 216.Komhoff M, Roofthooft MT, Westra D, et al. Combined pulmonary hypertension and renal thrombotic microangiopathy in cobalamin C deficiency. Pediatrics. 2013;132(2):e540–4. doi: 10.1542/peds.2012-2581. [DOI] [PubMed] [Google Scholar]
- 217.van ’t Hoff WG, Dixon M, Taylor J, et al. Combined liver-kidney transplantation in methylmalonic acidemia. The Journal of pediatrics. 1998;132(6):1043–4. doi: 10.1016/s0022-3476(98)70407-x. [DOI] [PubMed] [Google Scholar]
- 218.Nyhan WL, Gargus JJ, Boyle K, et al. Progressive neurologic disability in methylmalonic acidemia despite transplantation of the liver. European journal of pediatrics. 2002;161(7):377–9. doi: 10.1007/s00431-002-0970-4. [DOI] [PubMed] [Google Scholar]
- 219.Hsui JY, Chien YH, Chu SY, et al. Living-related liver transplantation for methylmalonic acidemia: report of one case. Acta paediatrica Taiwanica = Taiwan er ke yi xue hui za zhi. 2003;44(3):171–3. [PubMed] [Google Scholar]
- 220.Huang HP, Chien YH, Huang LM, et al. Viral infections and prolonged fever after liver transplantation in young children with inborn errors of metabolism. Journal of the Formosan Medical Association = Taiwan yi zhi. 2005;104(9):623–9. [PubMed] [Google Scholar]
- 221.Lubrano R, Elli M, Rossi M, et al. Renal transplant in methylmalonic acidemia: could it be the best option? Report on a case at 10 years and review of the literature. Pediatric nephrology (Berlin, Germany) 2007;22(8):1209–14. doi: 10.1007/s00467-007-0460-z. [DOI] [PubMed] [Google Scholar]
- 222.Morioka D, Kasahara M, Horikawa R, et al. Efficacy of living donor liver transplantation for patients with methylmalonic acidemia. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2007;7(12):2782–7. doi: 10.1111/j.1600-6143.2007.01986.x. [DOI] [PubMed] [Google Scholar]
- 223.Chen PW, Hwu WL, Ho MC, et al. Stabilization of blood methylmalonic acid level in methylmalonic acidemia after liver transplantation. Pediatric transplantation. 2010;14(3):337–41. doi: 10.1111/j.1399-3046.2009.01227.x. [DOI] [PubMed] [Google Scholar]
- 224.Clothier JC, Chakrapani A, Preece MA, et al. Renal transplantation in a boy with methylmalonic acidaemia. Journal of inherited metabolic disease. 2011;34(3):695–700. doi: 10.1007/s10545-011-9303-y. [DOI] [PubMed] [Google Scholar]
- 225.Brassier A, Boyer O, Valayannopoulos V, et al. Renal transplantation in 4 patients with methylmalonic aciduria: a cell therapy for metabolic disease. Molecular genetics and metabolism. 2013;110(1–2):106–10. doi: 10.1016/j.ymgme.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 226.Lubrano R, Bellelli E, Gentile I, et al. Pregnancy in a methylmalonic acidemia patient with kidney transplantation: a case report. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2013;13(7):1918–22. doi: 10.1111/ajt.12282. [DOI] [PubMed] [Google Scholar]
- 227.Lubrano R, Perez B, Elli M. Methylmalonic acidemia and kidney transplantation. Pediatric nephrology (Berlin, Germany) 2013;28(10):2067–8. doi: 10.1007/s00467-013-2536-2. [DOI] [PubMed] [Google Scholar]
- 228*.Li M, Dick A, Montenovo M, et al. Cost-effectiveness of liver transplantation in methylmalonic and propionic acidemias. Liver transplantation : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2015;21(9):1208–18. doi: 10.1002/lt.24173. This study is the first to analyze cost-effectiveness of liver transplantation versus standard care in the treatment of OAs, suggesting that transplantation is more cost-effective than standard of care. [DOI] [PubMed] [Google Scholar]
- 229*.Arrizza C, De Gottardi A, Foglia E, et al. Reversal of cardiomyopathy in propionic acidemia after liver transplantation: a 10-year follow-up. Transplant international : official journal of the European Society for Organ Transplantation. 2015;28(12):1447–50. doi: 10.1111/tri.12677. This study investigated the longer term effects of liver transplantation on pre-existent cardiomypathy in PA and suggests that liver transplant may have a beneficial role treating the cardiomyopathy of PA. [DOI] [PubMed] [Google Scholar]
- 230.Nagao M, Tanaka T, Morii M, et al. Improved neurologic prognosis for a patient with propionic acidemia who received early living donor liver transplantation. Molecular genetics and metabolism. 2013;108(1):25–9. doi: 10.1016/j.ymgme.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 231.Hussein MH, Hashimoto T, Suzuki T, et al. Children undergoing liver transplantation for treatment of inherited metabolic diseases are prone to higher oxidative stress, complement activity and transforming growth factor-beta1. Annals of transplantation : quarterly of the Polish Transplantation Society. 2013;18:63–8. doi: 10.12659/AOT.883820. [DOI] [PubMed] [Google Scholar]
- 232.Vara R, Turner C, Mundy H, et al. Liver transplantation for propionic acidemia in children. Liver transplantation : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2011;17(6):661–7. doi: 10.1002/lt.22279. [DOI] [PubMed] [Google Scholar]
- 233.Rela M, Battula N, Madanur M, et al. Auxiliary liver transplantation for propionic acidemia: a 10-year follow-up. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2007;7(9):2200–3. doi: 10.1111/j.1600-6143.2007.01899.x. [DOI] [PubMed] [Google Scholar]
- 234.Barshes NR, Vanatta JM, Patel AJ, et al. Evaluation and management of patients with propionic acidemia undergoing liver transplantation: a comprehensive review. Pediatric transplantation. 2006;10(7):773–81. doi: 10.1111/j.1399-3046.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- 235.Morioka D, Takada Y, Kasahara M, et al. Living donor liver transplantation for noncirrhotic inheritable metabolic liver diseases: impact of the use of heterozygous donors. Transplantation. 2005;80(5):623–8. doi: 10.1097/01.tp.0000167995.46778.72. [DOI] [PubMed] [Google Scholar]
- 236.Morioka D, Kasahara M, Takada Y, et al. Living donor liver transplantation for pediatric patients with inheritable metabolic disorders. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2005;5(11):2754–63. doi: 10.1111/j.1600-6143.2005.01084.x. [DOI] [PubMed] [Google Scholar]
- 237.Meyburg J, Hoffmann GF. Liver transplantation for inborn errors of metabolism. Transplantation. 2005;80(1 Suppl):S135–7. doi: 10.1097/01.tp.0000186905.10088.e5. [DOI] [PubMed] [Google Scholar]
- 238.Yorifuji T, Kawai M, Mamada M, et al. Living-donor liver transplantation for propionic acidaemia. Journal of inherited metabolic disease. 2004;27(2):205–10. doi: 10.1023/B:BOLI.0000028778.54210.13. [DOI] [PubMed] [Google Scholar]
- 239.Kayler LK, Merion RM, Lee S, et al. Long-term survival after liver transplantation in children with metabolic disorders. Pediatric transplantation. 2002;6(4):295–300. doi: 10.1034/j.1399-3046.2002.02009.x. [DOI] [PubMed] [Google Scholar]
- 240.Vernon HJ, Sperati CJ, King JD, et al. A detailed analysis of methylmalonic acid kinetics during hemodialysis and after combined liver/kidney transplantation in a patient with mut (0) methylmalonic acidemia. Journal of inherited metabolic disease. 2014;37(6):899–907. doi: 10.1007/s10545-014-9730-7. [DOI] [PMC free article] [PubMed] [Google Scholar]