

Abstract
Pain and depression are frequently co-existent in clinical practice, yet the underlying mechanisms remain largely to be determined. Microglia activation and subsequent pro-inflammatory responses play a crucial role in the development of neuropathic pain and depression. The process of microglia polarization to the pro-inflammatory M1 or anti-inflammatory M2 phenotypes often occurs during neuroinflammation. However, it remains unclear whether M1/M2 microglia polarization is involved in the neuropathic pain induced by spared nerve injury (SNI). In the present study, the mechanical withdrawal threshold, forced swim test, sucrose preference test, and open field test were performed. The levels of microglia markers including ionized calcium-binding adaptor molecule 1 (Iba1), cluster of differentiation 11b (CD11b), M1 markers including CD68, inducible nitric oxide synthase (iNOS), interleukin-1β (IL-1β), IL-6, tumor necrosis factor-a (TNF-α), 8-hydroxy-2-deoxyguanosine (8-OH-dG), and M2 markers including CD206, arginase 1 (Arg1), IL-4 in the prefrontal cortex were determined on day 14 after SNI. The results showed that SNI produced mechanical allodynia and depressive-like behaviors, and also increased the expressions of microglia markers (Iba1, CD11b) and M1 markers (CD68, iNOS, IL-1β, TNF-α, and 8-OH-dG) in the prefrontal cortex. Notably, minocycline administration reversed these abnormalities. In addition, minocycline also promoted M2 microglia polarization as evidenced by up-regulation of CD206 and Arg1. In conclusion, data from our study suggest that SNI can lead to depression-like behaviors, while M1 polarization and consequent overproduction of pro-inflammatory cytokines plays a key role in the pathogenesis of neuropathic pain. The data furthermore indicate that modulation of inflammation by inhibition of M1 polarization could be a strategy for treatment of neuropathic pain, and might prevent the induction of neuropathic pain-induced depression symptoms.
Keywords: neuropathic pain, depression, M1/M2 microglia, minocycline, inflammation
Introduction
Pain and depression are frequently co-existent. It is likely that pain can induce depression, while depression may enhance pain perception (Bair et al., 2003; Zhou et al., 2015). Neuropathic pain, one of the types of chronic pain, is a pain arising as a consequence of a primary lesion or disease affecting the somatosensory system (Treede et al., 2008). It is estimated that almost 50% of patients with major depression suffer from chronic pain (Maletic and Raison, 2009) and the prevalence of depression is around 30% in patients with neuropathic pain (Radat et al., 2013). The comorbidity of chronic pain and depression causes an increased disability, poor treatment response and consequently heavy disease burden (Dworkin and Gitlin, 1991; Arnow et al., 2006). Despite the broad spectrum of causes, little is known with regard to the mechanisms underlying the comorbid relationship, and consequently, few available treatments exist.
Neuroinflammatory activation is tended to be regarded as a causal role in the development of nerve injury-induced neuropathic pain and depression (Walker et al., 2014). Moreover, drugs or methods down-regulating neuroinflammation could alleviate the pain symptoms and depressive behaviors (Zhang et al., 2015; Luchting et al., 2016). Hence, modulation of neuroimmune response has been proposed for treatment of many central nervous system (CNS) disorders with an inflammatory component, including the comorbidity of neuropathic pain and depression (Kim et al., 2012; Walker et al., 2014; Bruning et al., 2015).
Microglia are resident macrophage-like immune cells in the CNS and play a critical role in both physiological and pathological conditions, including restoring the homeostasis of the CNS and driving the neuroinflammatory response of neurodegenerative disorders, respectively (Cherry et al., 2014). There are mainly two states of microglia polarization, “classical activation” (M1) and “alternative activation” (M2) (Colton, 2009; Colton and Wilcock, 2010). M1 microglia promote pro-inflammatory responses with excess tumor necrosis factor (TNF-α), interleukin-1β (IL-1β), inducible nitric oxide synthase (iNOS), and reactive oxygen species (ROS) production (Le et al., 2001; Block et al., 2007), contributing to neural network dysfunction. By contrast, M2 microglia are associated with up-regulation of anti-inflammatory cytokines and play a vital role in restoring homeostasis, such as dampening inflammation and wound healing (Ponomarev et al., 2007). Thus, modulation of microglia activation, including M1/M2 microglia polarization, for therapeutic purposes might be obtained by inhibiting the pro-inflammatory reaction and simultaneously promoting the anti-inflammatory effect. However, the status of microglia polarization in spared nerve injury (SNI)-induced neuropathic pain and depression-like behaviors has not been reported yet.
Based on these findings, we hypothesized that SNI causes an increase in M1 polarization and of inflammation markers in the prefrontal cortex, which may help to explain why patients with neuropathic pain frequently suffer from depression. In this study, we showed that SNI provoked depression-like behaviors and SNI-induced neuropathic pain was affected by microglia M1 polarization and overproduction of pro-inflammatory cytokines. Importantly, minocycline, a semisynthetic second-generation tetracycline with the anti-inflammatory properties, improved neuropathic pain and depression-like behaviors and attenuated M1 microglia polarization. In addition, it is well established that the prefrontal cortex plays an important role in the pathogenesis of pain (Metz et al., 2009), depression (Pan et al., 2014), and pain-related depression (Baliki et al., 2006; Guida et al., 2015), we therefore chose this brain region for biochemical analysis in the present study.
Materials and methods
Animal husbandry
Male Sprague-Dawley rats (8 weeks old, 200–250 g) were purchased from the Animal Center of Jinling Hospital, Nanjing, China. Animals were grouped in 4–5 individuals per cage and were housed in in a standard condition (a 12-h light/dark cycle, room temperature of 22 ± 1°C and 50 ± 10% humidity, and food and water were available ad libitum) for 2 weeks before the start of tests. All procedures were approved by the Ethics Committee of Jinling Hospital, and were performed according to the Guideline for the Care and Use of Laboratory Animals from the National Institutes of Health. The flow chart of the study protocol was shown in Figure 1A.
Figure 1.

Behavioral tests including mechanical allodynia and depressive-like behaviors were performed. (A) Schematic timeline of the experimental procedure. The total distance (B) in the open field test was not significantly different among the four groups. (C) SNI elicited mechanical allodynia, while minocycline administration prevented SNI-induced decreases in mechanical withdrawal thresholds on days 7 and 14. (D) SNI increased the immobility time significantly in the forced swim test, while minocycline administration decreased the immobility time. (E) SNI reduced the sucrose consumption significantly and minocycline reversed this change. (F) No significant difference of the total liquid consumption was observed. The data are presented mean ± SEM (n = 12). *P < 0.05 compared to the Sham group, #P < 0.05 compared to the SNI group.
SNI surgery
SNI surgery was performed to induce neuropathic pain as previously described (Decosterd and Woolf, 2000). A rat was anesthetized by intraperitoneal (i.p.) injection of sodium pentobarbital (60 mg/kg) before surgery. The skin on the lateral surface of the right thigh of the rat was incised and the terminal branches of the sciatic nerve: the sural, common peroneal and tibial nerves were exposed by dissecting the biceps femoris muscle. The common peroneal and tibial nerves were tightly ligated with 4-0 silk at the point of trifurcation and then cut distal to the knot, followed by removing the distal nerve ends about 3–5 mm. The sural nerve was untouched during the surgery. For the sham-operated rats, the sciatic nerve was exposed in the same way but not ligated and cut.
Experiment design and drug administration
The doses of minocycline from 20 to 50 mg/kg/day have been demonstrated as effective in attenuating microglia activation in the brain (Ekdahl et al., 2003; Raghavendra et al., 2003a). Thus, minocycline hydrochloride (Sangon Biotech, China) at a dose of 40 mg/kg/day was administered orally by rats' drinking water and minocycline solution was prepared at the concentration of 1 mg/mL in the drinking water every day (Hinwood et al., 2012). Minocycline or tap water was available ad libitum for rats via the drinking water except during behavioral tests, starting 24 h before sham/SNI surgery and consecutive 15 days[4 groups: Sham (n = 24), Sham + Minocycline (Mino) (n = 24), SNI (n = 24), and SNI + Mino (n = 24)]. For each group, half of them were for behavioral tests and the remaining half of them were for biochemical tests. Mechanical withdrawal thresholds (MWT) were assessed 1 day before SNI and on days 7 and 14 after SNI. The forced swim test (FST), sucrose preference test (SPT), and open field test (OFT) were performed on day 14 after SNI.
Open field test
The open field test was carried out in a white opaque plastic chamber (100 × 100 × 40 cm, XR-XZ301; Shanghai Softmaze Information Technology Co., Ltd., Shanghai, China) as described in the previous study (Huang et al., 2015) with slight modification. Rats were gently placed in the center of the field, and the movement was recorded for 5 min with a video tracking system. Between subjects, 75% ethanol was used to clean the open field area.
Von frey test
Mechanical allodynia was detected by the Von Frey test. The Von Frey monofilament test was performed as previously described (Wang et al., 2011). Rats were individually placed into plexiglass chambers over a mesh table and habituated for 15 min prior to the test. Von Frey filaments were used to stimulate the lateral 1/3 of right paws of animals starting with the 2.0 g filament and subsequently with logarithmically incremental stiffness from 0.6 to 15.0 g 1 day before the surgery or on days 7 and 14 after the surgery. Five trials for each paw were detected at 5-min intervals and the force (g) applied was recorded. Sudden paw withdrawal, flinching, and paw licking were defined as positive response. An up–down method was used, and 50% withdrawal threshold was calculated as described previously (Chaplan et al., 1994).
Forced swim test
The forced swim test was performed as previously described with slight modification (Maeng et al., 2008). Rats were placed individually into Plexiglas cylinders (height, 65 cm; diameter, 30 cm) filled with 45 cm height of water, conditioned at 22–23°C. After 1-min habituation, the time when the rat kept floating passively or immobile in the water for a period of 5 min was recorded. After the test, subjects were dried with towel and then rest under a lamp for 30 min. The water was changed after each swim.
Sucrose preference test
The sucrose preference test was performed as described by previous studies (Willner et al., 1987; Sigwalt et al., 2011). Animals were acclimated to the test room for at least 20 min before the test. Two bottles (1% sucrose solution vs. tap water) were presented to rats individually for 24 h. Each rat had free access to both 1% sucrose solution bottle and a tap water bottle and continued for 24 h. Sucrose and tap water intakes were measured by volume before and after the test. The total liquid consumption was the sum of sucrose water and tap water consumption. The sucrose preference of each rat was expressed as the percentage of sucrose water consumption divided by the total water consumption.
Western blot
The prefrontal cortex were harvested after anesthetized with sodium pentobarbital (60 mg/kg, i.p.; Sigma, St Louise, MO, USA) on day 14 after surgery. The samples were placed on ice plate and homogenized in a lysis buffer containing 0.1% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 66 mM EDTA, 10 mMTris–HCl, (pH 7.4) with protease inhibitor cocktail. Homogenates were centrifuged at 4°Cat 12,000 rpm for 15 min and then the supernatants were collected. According to Bradford assay, the protein concentration was determined. Twenty micrograms of proteins were separated on 8–12% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), which was then blocked with 5% non-fat milk for 1 h at room temperature condition. Next the membranes were incubated with rabbit anti-CD11b (1:1,000; Abcam, Cambridge, UK), mouse anti-CD68 (1:1,000; Abcam, Cambridge, UK), rabbit anti-CD206 (1:500, proteintech, USA) and rabbit anti-β-tubulin (1:1,000; Bioworld, St. Louis Park, MN, USA) overnight at 4°C room. After washing in TBST (Tris Buffered Saline with Tween) for three times, the horseradish peroxidase-conjugated secondary antibodies (goat anti-rabbit and goat anti-mouse; Bioworld Technology, St. Louis Park, MN, USA) were diluted as 1:10,000 and the membranes were incubated for 1 h at room temperature. Chemiluminesence was used to detect the protein bands and Image J Software (Wayne Rasband, National Institute of Health, USA) was used to quantitate the band intensity.
Immunofluorescence
Rats were anesthetized with 2% sodium pentobarbital in saline (60 mg/kg, i.p.; Sigma, St Louise, MO, USA) and perfused with saline and then perfused with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) on day 14 after surgery. Brains were collected and fixed in the 4% PFA for 2 h and dehydrated in 30% sucrose at 4°C overnight. 10-mm-thick coronally sections of the prefrontal cortex were cut and pasted on glass slides. After blocking with 10% norm goat serum for 1 h at room temperature, the slices were incubated with primary antibodies: rabbit anti-CD11b (1:250; Abcam, Cambridge, UK), rabbit anti-Iba1 (1:500; Wako, Japan), mouse anti-CD206 (1:500; Biolegend, San Diego, USA), or mouse anti-8-hydroxy-20-deoxyguanosine (8-OH-dG; 1:200; Santa Cruz Biotechnology, Dallas, TX, USA) in 1% BSA at 4°C overnight. After washing with PBS for 3 × 5 min, the slices were exposed to the secondary antibodies Alexa fluor 488/549 goat anti-rabbit and Alexa fluor 488/549 goat anti-mouse (1:400; Bioworld Technology, St. Louis Park, MN, USA), and DAPI (1:1,000; Sigma, St. Louis, MO, USA) for 1 h at room temperature. A confocal microscope (Leica, TCS SP2, Germany) was used to capture the fluorescent images. The immunofluorescence intensity was calculated by Image J (Wayne Rasband, National Institute of Health, USA).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from the prefrontal cortex and then isolated with Trizol (Takara Bio Inc). HiScriptQ RT SuperMix (Vazyme, Nanjing, China) was used to synthesize cDNA. qRT-PCR was performed by the ABI StepOne™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with SYBR Green PCR Master Mix (Vazyme, Nanjing, China) and analyzed using StepOne Software v2.3 (Applied Biosystems). The sequences of primers used for qRT-PCR amplification are available in Table 1. The cycling conditions were 95°C for 5 min and 40 cycles of 95°C for 10 s followed by 60°C for 30 s. GAPDH was used to normalize gene expression data and relative gene expression levels were calculated by ΔΔCT method.
Table 1.
The primer sequences using qRT-PCR of microglia markers and the reference gene.
| Microglia markers (5′–3′) | |||
| CD11b | GAGTGTGATCCAGCTTGGTG | Arginase 1 | TCCGCTGACAACCAGCTCTG |
| GGACATATTCACAGCCTCTG | GTCCACATCTCGCAAGCCGA | ||
| CD68 | CCTTACGGACAGCTTACCTT | CD206 | CTGAACTGGCTACCAGGAAG |
| CCAGGTGAATTGCTGGAGAA | CAGGCAGTAGGCACATCACT | ||
| IL-1β | TTGCTTCCAAGCCCTTGACT | IL-4 | CCTTGCTGTCACCCTGTTCTG |
| CTCCACGGGCAAGACATAGG | TGCATGGAGTCCCTTTTTCTG | ||
| TNF-α | CATGAGCACGGAAAGCATGA | GAPDH | TGCCACTCAGAAGACTGTGG |
| CCACGAGCAGGAATGAGAAGA | TTCAGCTCTGGGATGACCTT | ||
| iNOS | CCTGGTGCAAGGGATCTTGG | ||
| GAGGGCTTGCCTGAGTGAGC | |||
Enzyme-linked immunosorbent assay (ELISA)
Pro-inflammatory cytokines of TNF-α, IL-1β, and IL-6 and anti-inflammatory cytokine of IL-4 in the prefrontal cortex were quantified by the ELISA. Rats were sacrificed to obtain the prefrontal cortex on day 14 after surgery. The tissue samples were weighed and homogenized. Homogenates were centrifuged at 4°C for 10 min at 2,500 rpm and then collected the supernatants. Bradford method was used to quantify the protein concentration. The protocol was provided by the manufacturer (R&D Systems, Minneapolis, MN, USA) and the concentration of these cytokines was calculated by the standard curves, which was performed with the Optical Density (OD) tested at 450 nm.
Statistical analysis
All analyses were performed by SPSS 17.0 software (version 17.0, IL, USA) and data are presented as mean ± SEM. Comparisons were assessed by one-way ANOVA followed by a Tukey test. Mechanical allodynia was analyzed by repeated measures two-way ANOVA followed by a Bonferroni test. Significant statistical difference was regarded as P < 0.05.
Results
SNI induced mechanical allodynia and depressive-like behaviors, which were attenuated by chronic minocycline administration
To evaluate the locomotor activity of rats, the open field test was performed. There was no significant difference in the total distance among the 4 groups (Figure 1B), suggesting neither SNI nor minocycline administration influenced the locomotor activity. Mechanical allodynia and depressive-like behaviors were used to verify the occurrence of neuropathic pain and depression, respectively. On day 7 or 14 after surgery, SNI induced increased mechanical withdrawal thresholds, which was reversed by chronic minocycline administration (Figure 1C, P < 0.05). The forced swim test and sucrose preference test were used to assess the depressive-like behaviors. The immobility time was longer and the sucrose consumption was less in the SNI rats than the sham-operated rats on day 14 after surgery, which were reversed by minocycline administration (Figures 1D,E, P < 0.05). There was no significant difference in the total liquid consumption among the 4 groups (Figure 1F). Moreover, there was no difference between the Sham group and the Sham + Mino group in the behavioral tests.
SNI induced microglia activation in the prefrontal cortex, which was attenuated by chronic minocycline administration
To evaluate whether SNI could induce microglia activation in the prefrontal cortex, microglia markers including CD11b and Iba-1 were detected by immunofluorescence. SNI up-regulated the expression of CD11b (Figures 2A,B, P < 0.05) and the number of Iba1-positive cells (Figures 2C,D, P < 0.05) in the prefrontal cortex, whereas minocycline administration alleviated these abnormalities (Figure 2). There was no difference between the Sham group and the Sham + Mino group in the expression of CD11b and Iba1.
Figure 2.

Immunofluorescence staining to detect microglial activation in the prefrontal cortex. (A) Representative images of CD11b (green) in the prefrontal cortex. (B) Minocycline significantly attenuated SNI-induced fluorescence intensity increase of CD11b in the prefrontal cortex. (Scale bar = 300 μm) (C) Representative images of Iba1 (green) in the prefrontal cortex. (D) Minocycline significantly attenuated SNI-induced fluorescence intensity increase of Iba1 in the prefrontal cortex. (Scale bar = 50 μm). The data are expressed as the mean ± SEM of 4 rats in each group. *P < 0.05 compared to the Sham group, #P < 0.05 compared to the SNI group. DAPI (4′,6-diamidino-2-phenylindole) staining is shown in blue.
SNI induced different changes in M1/M2 microglia markers in the prefrontal cortex, which were altered by chronic minocycline administration
To quantify microglial M1 and M2 polarization at the protein level, the expressions of CD11b (a microglia marker), CD68 (a M1 microglia marker) and CD206 (a M2 microglia maker) were measured by the Western blot. SNI increased the expressions of CD11b and CD68 but not CD206 in the prefrontal cortex (Figure 3, P < 0.05), which was blocked by minocycline administration. In addition, minocycline administration increased the expression of CD206 after SNI in the prefrontal cortex (Figure 3, P < 0.05). There was no difference between the Sham group and the Sham + Mino group in the expression of CD11b, CD68 and CD206.
Figure 3.

Western blot results of the prefrontal cortex. (A) A representative blot and quantitative analysis of CD11b in the prefrontal cortex. (B) A representative blot and quantitative analysis of CD68 in the prefrontal cortex. (C) A representative blot and quantitative analysis of CD206 in the prefrontal cortex. β-tubulin was included as control. The data are expressed as the mean ± SEM of 4 rats in each group. *P < 0.05 compared to the sham group, #P < 0.05 compared to the SNI group.
Chronic minocycline administration reduced SNI-induced microglia activation and elicited up-regulation of M2 microglia in the prefrontal cortex
Since the results of Western blot have shown the similar trend between microglia marker CD11b and M1 microglia marker CD68 but not M2 microglia marker CD206, we used double-immunofluorescence staining to detect the co-localization of M2 microglia marker CD206 and another microglia marker Iba1 in the prefrontal cortex thus to further observe the trend of M2 microglia on day 14 after SNI. The co-localization result showed SNI increased the expression of Iba1 in the prefrontal cortex (Figure 4, P < 0.05), but did not affect the expression of CD206. However, minocycline administration blocked the change of Iba1 (Figure 4, P < 0.05) and increased the expression of CD206 (Figure 4, P < 0.05). There was no difference between the Sham group and the Sham + Mino group in the expression of Iba1 and CD206.
Figure 4.
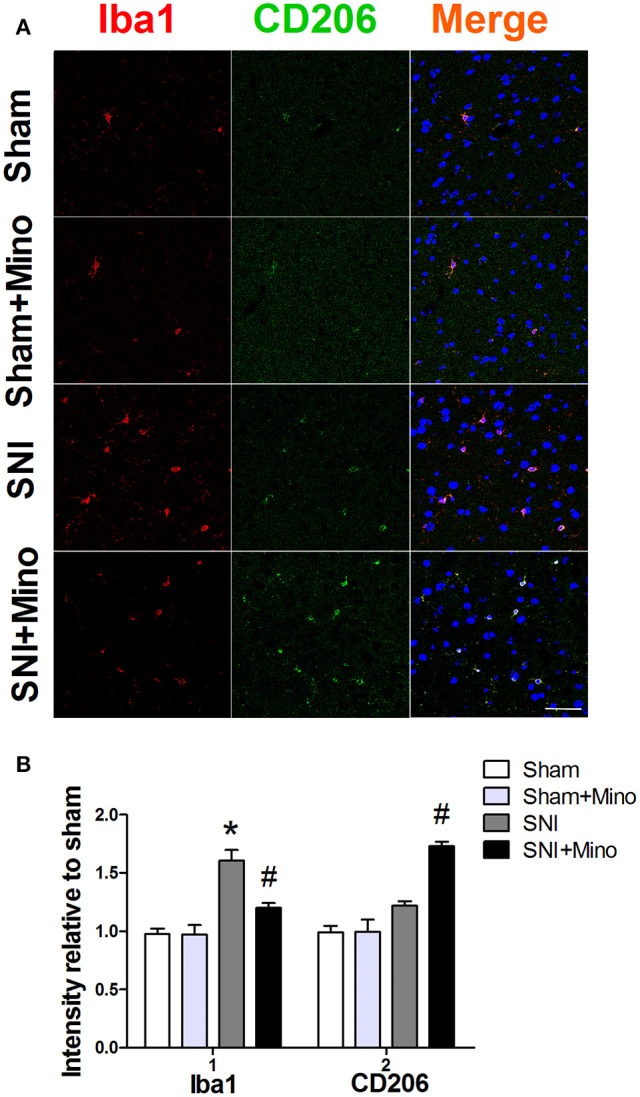
Double-immunofluorescence staining to detect co-localization of Iba1 and CD206 in the prefrontal cortex on day 14 post-SNI. (A) Representative images of Iba1 (red) and CD206 (green) in the prefrontal cortex. (Scale bar = 50 μm). (B) Quantification of Iba1 and CD206 fluorescence in the prefrontal cortex. The data are expressed as the mean ± SEM of 4 rats in each group. *P < 0.05 compared to the Sham group, #P < 0.05 compared to the SNI group. DAPI staining is shown in blue.
SNI alters the ratio between M1 and M2 polarization
To evaluate the alteration of M1/M2 microglia polarization at the genetic level, M1/M2 microglia markers were detected by qRT-PCR. SNI induced the increase of CD68 and iNOS (M1 markers; Figures 5A,B, P < 0.05) but did not affect CD206 and Arg1 (M2 markers; Figures 5C,D). However, minocycline administration rescued the increase of M1 microglia markers and increased M2 microglia markers (Figures 5A–D, P < 0.05). In addition, CD68 (M1 microglia marker)/CD206 (M2 microglia marker) ratio (Figure 5E, P < 0.05) was increased following SNI, whereas minocycline administration reversed the abnormalities, suggesting that minocycline redirected M1 activation to M2 activation. There was no difference between the Sham group and the Sham + Mino group in the expression of CD68, iNOS, CD206, Arg1, and CD68/CD206 ratio.
Figure 5.

mRNA expressions of M1/M2 microglia markers in the prefrontal cortex. Fold increase of M1 markers (CD68, iNOS) (A,B); Fold increase of M2 microglia markers (Arg1, CD206) (C,D). Fold increase of the ratio of CD68/CD206 (E). The data are expressed as mean ± SEM; n = 4 for each group; *P < 0.05 compared to the Sham group, #P < 0.05 compared to the SNI group.
SNI alters cytokine expression in the prefrontal cortex
To evaluate the changes of inflammatory cytokines at mRNA level, qRT-PCR was performed. SNI increased the mRNA level of pro-inflammatory cytokines (TNF-α and IL-1β; Figures 6A,B, P < 0.05) but did not affect the mRNA level of anti-inflammatory cytokine (IL-4; Figure 6C) in the prefrontal cortex. Minocycline administration reduced the mRNA level of pro-inflammatory cytokines (Figures 6A,B, P < 0.05) but failed to increase the mRNA level of anti-inflammatory cytokine (Figure 6C). In addition, IL-1β/IL-4 ratio was increased following SNI, whereas minocycline administration reversed this abnormity (Figure 6D, P < 0.05), suggesting that minocycline redirected pro-inflammatory state to anti-inflammatory state. There was no difference between the Sham group and the Sham + Mino group in the expression of TNF-α, IL-1β, IL-4, and IL-1β/IL-4 ratio.
Figure 6.

mRNA expressions of different inflammatory cytokines in the prefrontal cortex. Fold increase of pro-inflammatory cytokines (TNF-α, IL-1β) (A,B); Fold increase of M2 anti-inflammatory cytokine (IL-4) (C). Fold increase of the ratio of IL-1β/IL-4 (D). The data are expressed as mean ± SEM; n = 4 for each group; *P < 0.05 compared to the Sham group, #P < 0.05 compared to the SNI group.
SNI induced changes of pro- and anti-inflammatory cytokines in the prefrontal cortex, which were affected by chronic minocycline administration
To measure the levels of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) and anti-inflammatory cytokine (IL-4) in the prefrontal cortex, ELISA was applied. The levels of IL-1β and TNF-α were increased in the prefrontal cortex, whereas minocycline administration reversed the SNI-induced overproduction of TNF-α and IL-1β (Figures 7A,B, P < 0.05). In addition, there was no significant difference in IL-6 and IL-4 levels among the 4 groups (Figures 7C,D). Similarly, IL-1β/IL-4 ratio was increased following SNI, whereas minocycline administration reversed this change (Figure 7E, P < 0.05). There was no difference between the Sham group and the Sham + Mino group in the expression of TNF-α, IL-1β, IL-6, IL-4, and IL-1β/IL-4 ratio (Figure 7).
Figure 7.

ELISA analysis of different inflammatory cytokines in the prefrontal cortex. (A,B) SNI-induced increase in pro-inflammatory cytokines TNF-α and IL-1 β were alleviated by minocycline. (C) No significantly difference of IL-6 was found among the 4 groups. (D) There was no difference of IL-4 among the 4 groups. (E) SNI increased the ratio of IL-1β/IL-4 and minocycline decreased the ratio. The data are expressed as the mean ± SEM of 4 rats in each group. *P < 0.05 compared to the Sham group, #P < 0.05 compared to the SNI group.
SNI induced over-production of ROS in the brain, which was alleviated by minocycline
Since M1 microglia activation produces ROS apart from iNOS, we finally determined whether SNI could lead to augmented oxidative stress in microglia. For this purpose, 8-OH-dG immunostaining was used to detect cellular ROS. CD11b and 8-OH-dG staining were increased in the prefrontal cortex (Figure 8, P < 0.05), however, minocycline administration attenuated CD11b expression and ROS overproduction in the microglia in the prefrontal cortex on day 14 after SNI (Figure 8, P < 0.05). There was no difference between the Sham group and the Sham + Mino group in the expression of CD11b and 8-OH-dG (Figure 8).
Figure 8.
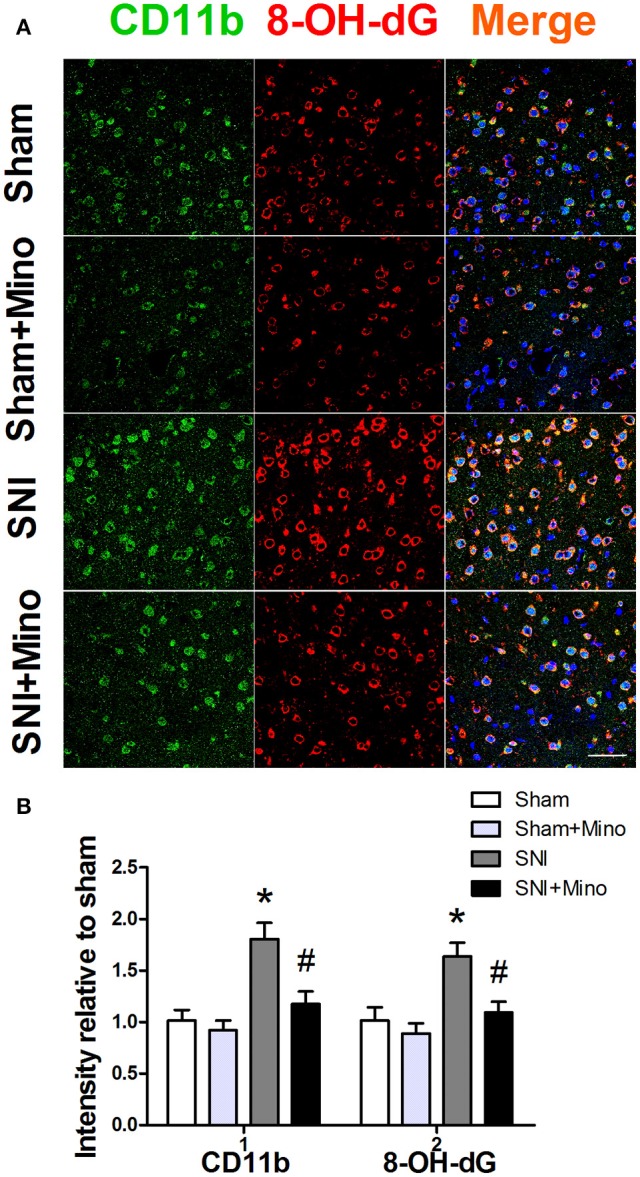
Immunofluorescence staining to detect ROS in the prefrontal cortex. (A) Representative images of CD11b (green) and 8-OH-dG (red) in the prefrontal cortex. (B) SNI significantly increased fluorescence intensity of CD11b and 8-OH-dG in the prefrontal cortex, which was attenuated by minocycline. (Scale bar = 50 μm). The data are expressed as the mean ± SEM of 4 rats in each group. *P < 0.05 compared to the Sham group, #P < 0.05 compared to the SNI group. DAPI staining is shown in blue.
Discussion
In the present study, we verified that SNI could induce the comorbidity of neuropathic pain and depression, M1 microglia polarization and amplified inflammatory cytokines as well as increased oxidative stress. Of note, we found that chronic minocycline administration reversed these abnormalities, probably mediated by inhibiting M1 microglia polarization and simultaneously promoting M2 microglia polarization. These findings may help to provide an avenue to treat neuropathic pain and a new explanation for the comorbidity of neuropathic pain and depression.
The comorbidity of chronic pain and depression has been well established in the literature (Bair et al., 2003; Miller and Cano, 2009). The common used models of comorbidity of neuropathic pain-induced depression include chronic constriction injury (Bennett and Xie, 1988), spinal nerve ligation (Kim and Chung, 1992), spinal nerve transection (Hu et al., 2010), and the SNI (Wang et al., 2011). In the present study, SNI-induced mechanical allodynia and depressive-like behaviors were confirmed by increased withdrawal threshold and immobility time and reduced sucrose preference, suggesting SNI induced the comorbidity of chronic pain and depression.
Previous studies have shown that the monoaminergic system, hypothalamic-pituitary-adrenal axis, neuroimmune responses, and neurotransmitters/neuromodulators are involved in the pathogenesis of the comorbidity of chronic pain and depression (Tsigos and Chrousos, 2002; Jasmin et al., 2003; Schatzberg, 2004; Andre et al., 2005; Gameiro et al., 2006; Walker et al., 2014), however the cellular mechanism underlying this comorbidity relationship remains largely to be elucidated. Accumulating evidence has suggested the key role of neuroinflammation mediated by microglia in linking neuropathic pain and depression (Burke et al., 2014; Fiore and Austin, 2016). It is reported that pro-inflammatory cytokines led to hyperalgesia (Schiavuzzo et al., 2015); meanwhile, pro-inflammatory cytokines also induced depressive-like behaviors in animals (Norman et al., 2010); In human studies, pro-inflammatory cytokines such as TNF-α and IL-1β are increased in the cerebrospinal fluid and blood in patients with chronic neuropathic pain (Alexander et al., 2005), suggesting a critical role of M1-mediated neuroinflammation in this disorder. Similar to peripheral macrophages, microglia are often classified into pro-inflammatory (M1) and alternatively activated (M2) phenotypes, in which the M1 phenotype was originally induced by lipopolysaccharide (LPS) or interferon (IFN)-γ stimulation, and the M2 phenotype by interleukin IL-4, IL-10, or IL-13 (Colton and Wilcock, 2010). It has been well documented that microglia M1/M2 polarization plays a pivotal role in controlling the balance between promotion and suppression of inflammation (Tao et al., 2016). In line with this notion, a recent study has shown that a transition to M1 phenotype is detrimental to neuroinflammation and neuropathic pain after SNI, whereas a failed M2 microglia response may contribute to the change from protective responses to harmful results because a failed inflammation control and decreased neuroprotective factors produced by microglia (Cherry et al., 2014). Thus, selective suppression of M1 and increase in the M2 polarization of microglia could be a potential strategy for treatment.
Minocycline, a commonly used inhibitor of microglia activation, has been proven to be neuroprotective in several animal models, such as experimental autoimmune encephalomyelitis (Howell et al., 2010), Parkinson's disease (Shan et al., 2011), and Alzheimer s disease (Malm et al., 2008). Minocycline also reduces the release of pro-inflammatory cytokines in the spinal cord and alleviates the neuropathic pain behaviors (Raghavendra et al., 2003b). In the present study, we studied the effects of minocycline on the neuropathic pain-induced depression and microglia M1/M2 markers in the prefrontal cortex and found that minocycline treatment attenuated the comorbidity by decreasing the expression of M1 microglia markers and associated pro-inflammatory cytokines and increasing the expression of the M2 microglia markers and associated anti-inflammatory cytokine. These results suggested minocycline modulated microglia polarization by boosting the M2 over the pro-inflammatory phenotype. In support with our results, it has been shown that minocycline has therapeutic effects on working memory deficit and depressive-like behaviors in these models (Hinwood et al., 2012; Majidi et al., 2016). However, another study showed that administration of minocycline attenuated the induction of M1 microglia markers but did not affect the transient enhancement of M2 microglia markers during the early pathogenesis phase (Kobayashi et al., 2013). A clear explanation for this discrepancy is still lacking because of our insufficient knowledge on the nature of microglia activation and the mechanisms of minocycline in controlling microglia in neuropathic pain models. However, our study did suggest that chronic minocycline administration differentially regulates the expression of M1/M2 microglia markers or relevant cytokines in the prefrontal cortex in an animal model of SNI.
There are some limitations in our study protocol. It should be mentioned that we only studied the prefrontal cortex here, which cannot represent the global brain expression of M1/M2 markers of microglia. However, we selected this brain region to study because one prior study has shown that the medial prefrontal cortex is an important region for the comorbidity of chronic pain and depression or anxiety (Burke et al., 2015). Consistently, the prefrontal cortex was found to involve in neuroimmune response in the olfactory bulbectomised model and following nerve injury (Apkarian et al., 2006; Myint et al., 2007; Borre et al., 2012), demonstrating the prefrontal cortex is implicated in the interaction between depression and pain. Second, it is important to measure the polarization markers at different time points, but we have detected only a time point. Longitudinal studies will be required in the future. Third, the negative results of serum IL-6 level may be the consequence of small sample sizes. Fourth, we cannot conclude that IL-4 is the primary M2-polarizing signal induced by chronic minocycline administration and a more extensive array analysis will be performed in the future to determine the hierarchy of M2 regulation.
In conclusion, our results suggested that SNI-induced neuropathic pain is associated with an enhanced pro-inflammatory M1/M2 microglia ratio. Notably, chronic minocycline administration alleviated the behavioral abnormities and selectively inhibited the microglia polarization to a pro-inflammatory state while attained an anti-inflammatory state after SNI. Our study provided additional evidence indicating the potential molecular mechanisms by which chronic minocycline administration can treat neuropathic pain and explained why patients with neuropathic pain frequently suffer from depression: because M1 polarization is a risk factor for depression. Therefore, future intervention should aim to prevent excessive microglia activities while retain their protective functions to treat the comorbidity of neuropathic pain and depression.
Author contributions
NX, ZZ, XT, and MZ conceived and designed the experiments. NX, XT, and WP performed the experiment. WP, ZX, and GZ supervised acquisition of results and analyzed the data. NX, MJ, and JY wrote and refined the article.
Funding
This study was supported by No. 81503053 from the National Natural Science Foundation of China.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Alexander G. M., van Rijn M. A., van Hilten J. J., Perreault M. J., Schwartzman R. J. (2005). Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. Pain 116, 213–219. 10.1016/j.pain.2005.04.013 [DOI] [PubMed] [Google Scholar]
- Andre J., Zeau B., Pohl M., Cesselin F., Benoliel J. J., Becker C. (2005). Involvement of cholecystokininergic systems in anxiety-induced hyperalgesia in male rats: behavioral and biochemical studies. J. Neurosci. 25, 7896–7904. 10.1523/jneurosci.0743-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian A. V., Lavarello S., Randolf A., Berra H. H., Chialvo D. R., Besedovsky H. O., et al. (2006). Expression of IL-1beta in supraspinal brain regions in rats with neuropathic pain. Neurosci. Lett. 407, 176–181. 10.1016/j.neulet.2006.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnow B. A., Hunkeler E. M., Blasey C. M., Lee J., Constantino M. J., Fireman B., et al. (2006). Comorbid depression, chronic pain, and disability in primary care. Psychosom. Med. 68, 262–268. 10.1097/01.psy.0000204851.15499.fc [DOI] [PubMed] [Google Scholar]
- Bair M. J., Robinson R. L., Katon W., Kroenke K. (2003). Depression and pain comorbidity: a literature review. Arch. Intern. Med. 163, 2433–2445. 10.1001/archinte.163.20.2433 [DOI] [PubMed] [Google Scholar]
- Baliki M. N., Chialvo D. R., Geha P. Y., Levy R. M., Harden R. N., Parrish T. B., et al. (2006). Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J. Neurosci. 26, 12165–12173. 10.1523/jneurosci.3576-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G. J., Xie Y. K. (1988). A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107. [DOI] [PubMed] [Google Scholar]
- Block M. L., Zecca L., Hong J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. 10.1038/nrn2038 [DOI] [PubMed] [Google Scholar]
- Borre Y., Sir V., de Kivit S., Westphal K. G., Olivier B., Oosting R. S. (2012). Minocycline restores spatial but not fear memory in olfactory bulbectomized rats. Eur. J. Pharmacol. 697, 59–64. 10.1016/j.ejphar.2012.09.005 [DOI] [PubMed] [Google Scholar]
- Bruning C. A., Martini F., Soares S. M., Sampaio T. B., Gai B. M., Duarte M. M., et al. (2015). m-Trifluoromethyl-diphenyl diselenide, a multi-target selenium compound, prevented mechanical allodynia and depressive-like behavior in a mouse comorbid pain and depression model. Prog. Neuropsychopharmacol. Biol. Psychiatry 63, 35–46. 10.1016/j.pnpbp.2015.05.011 [DOI] [PubMed] [Google Scholar]
- Burke N. N., Finn D. P., Roche M. (2015). Chronic administration of amitriptyline differentially alters neuropathic pain-related behaviour in the presence and absence of a depressive-like phenotype. Behav. Brain Res. 278, 193–201. 10.1016/j.bbr.2014.09.044 [DOI] [PubMed] [Google Scholar]
- Burke N. N., Kerr D. M., Moriarty O., Finn D. P., Roche M. (2014). Minocycline modulates neuropathic pain behaviour and cortical M1-M2 microglial gene expression in a rat model of depression. Brain Behav. Immun. 42, 147–156. 10.1016/j.bbi.2014.06.015 [DOI] [PubMed] [Google Scholar]
- Chaplan S. R., Bach F. W., Pogrel J. W., Chung J. M., Yaksh T. L. (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Cherry J. D., Olschowka J. A., O'Banion M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11:98. 10.1186/1742-2094-11-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton C. A. (2009). Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 4, 399–418. 10.1007/s11481-009-9164-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton C., Wilcock D. M. (2010). Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 9, 174–191. 10.2174/187152710791012053 [DOI] [PubMed] [Google Scholar]
- Decosterd I., Woolf C. J. (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. 10.1016/S0304-3959(00)00276-1 [DOI] [PubMed] [Google Scholar]
- Dworkin R. H., Gitlin M. J. (1991). Clinical aspects of depression in chronic pain patients. Clin. J. Pain 7, 79–94. [DOI] [PubMed] [Google Scholar]
- Ekdahl C. T., Claasen J. H., Bonde S., Kokaia Z., Lindvall O. (2003). Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. U.S.A. 100, 13632–13637. 10.1073/pnas.2234031100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore N. T., Austin P. J. (2016). Are the emergence of affective disturbances in neuropathic pain states contingent on supraspinal neuroinflammation? Brain Behav. Immun. 56, 397–411. 10.1016/j.bbi.2016.04.012 [DOI] [PubMed] [Google Scholar]
- Gameiro G. H., da Silva Andrade A., Nouer D. F., Ferraz de Arruda Veiga M. C. (2006). How may stressful experiences contribute to the development of temporomandibular disorders? Clin. Oral Investig. 10, 261–268. 10.1007/s00784-006-0064-1 [DOI] [PubMed] [Google Scholar]
- Guida F., Luongo L., Marmo F., Romano R., Iannotta M., Napolitano F., et al. (2015). Palmitoylethanolamide reduces pain-related behaviors and restores glutamatergic synapses homeostasis in the medial prefrontal cortex of neuropathic mice. Mol. Brain 8, 47. 10.1186/s13041-015-0139-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinwood M., Morandini J., Day T. A., Walker F. R. (2012). Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb. Cortex 22, 1442–1454. 10.1093/cercor/bhr229 [DOI] [PubMed] [Google Scholar]
- Howell O. W., Rundle J. L., Garg A., Komada M., Brophy P. J., Reynolds R. (2010). Activated microglia mediate axoglial disruption that contributes to axonal injury in multiple sclerosis. J. Neuropathol. Exp. Neurol. 69, 1017–1033. 10.1097/NEN.0b013e3181f3a5b1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Yang J., Hu Y., Wang Y., Li W. (2010). Amitriptyline rather than lornoxicam ameliorates neuropathic pain-induced deficits in abilities of spatial learning and memory. Eur. J. Anaesthesiol. 27, 162–168. 10.1097/EJA.0b013e328331a3d5 [DOI] [PubMed] [Google Scholar]
- Huang H., Zhao J., Jiang L., Xie Y., Xia Y., Lv R., et al. (2015). Paeoniflorin improves menopause depression in ovariectomized rats under chronic unpredictable mild stress. Int. J. Clin. Exp. Med. 8, 5103–5111. [PMC free article] [PubMed] [Google Scholar]
- Jasmin L., Rabkin S. D., Granato A., Boudah A., Ohara P. T. (2003). Analgesia and hyperalgesia from GABA-mediated modulation of the cerebral cortex. Nature 424, 316–320. 10.1038/nature01808 [DOI] [PubMed] [Google Scholar]
- Kim H., Chen L., Lim G., Sung B., Wang S., McCabe M. F., et al. (2012). Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Invest. 122, 2940–2954. 10.1172/jci61884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H., Chung J. M. (1992). An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50, 355–363. [DOI] [PubMed] [Google Scholar]
- Kobayashi K., Imagama S., Ohgomori T., Hirano K., Uchimura K., Sakamoto K., et al. (2013). Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 4, e525. 10.1038/cddis.2013.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le W., Rowe D., Xie W., Ortiz I., He Y., Appel S. H. (2001). Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson's disease. J. Neurosci. 21, 8447–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchting B., Heyn J., Woehrle T., Rachinger-Adam B., Kreth S., Hinske L. C., et al. (2016). Differential expression of P2X7 receptor and IL-1beta in nociceptive and neuropathic pain. J. Neuroinflammation 13, 100. 10.1186/s12974-016-0565-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S., Hunsberger J. G., Pearson B., Yuan P., Wang Y., Wei Y., et al. (2008). BAG1 plays a critical role in regulating recovery from both manic-like and depression-like behavioral impairments. Proc. Natl. Acad. Sci. U.S.A. 105, 8766–8771. 10.1073/pnas.0803736105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majidi J., Kosari-Nasab M., Salari A. A. (2016). Developmental minocycline treatment reverses the effects of neonatal immune activation on anxiety- and depression-like behaviors, hippocampal inflammation, and HPA axis activity in adult mice. Brain Res. Bull. 120, 1–13. 10.1016/j.brainresbull.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Maletic V., Raison C. L. (2009). Neurobiology of depression, fibromyalgia and neuropathic pain. Front. Biosci. (Landmark Ed.) 14, 5291–5338. 10.2741/3598 [DOI] [PubMed] [Google Scholar]
- Malm T. M., Magga J., Kuh G. F., Vatanen T., Koistinaho M., Koistinaho J. (2008). Minocycline reduces engraftment and activation of bone marrow-derived cells but sustains their phagocytic activity in a mouse model of Alzheimer's disease. Glia 56, 1767–1779. 10.1002/glia.20726 [DOI] [PubMed] [Google Scholar]
- Metz A. E., Yau H. J., Centeno M. V., Apkarian A. V., Martina M. (2009). Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 106, 2423–2428. 10.1073/pnas.0809897106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. R., Cano A. (2009). Comorbid chronic pain and depression: who is at risk? J. Pain 10, 619–627. 10.1016/j.jpain.2008.12.007 [DOI] [PubMed] [Google Scholar]
- Myint A. M., Steinbusch H. W., Goeghegan L., Luchtman D., Kim Y. K., Leonard B. E. (2007). Effect of the COX-2 inhibitor celecoxib on behavioural and immune changes in an olfactory bulbectomised rat model of depression. Neuroimmunomodulation 14, 65–71. 10.1159/000107420 [DOI] [PubMed] [Google Scholar]
- Norman G. J., Karelina K., Zhang N., Walton J. C., Morris J. S., Devries A. C. (2010). Stress and IL-1beta contribute to the development of depressive-like behavior following peripheral nerve injury. Mol. Psychiatry 15, 404–414. 10.1038/mp.2009.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y., Chen X. Y., Zhang Q. Y., Kong L. D. (2014). Microglial NLRP3 inflammasome activation mediates IL-1beta-related inflammation in prefrontal cortex of depressive rats. Brain Behav. Immun. 41, 90–100. 10.1016/j.bbi.2014.04.007 [DOI] [PubMed] [Google Scholar]
- Ponomarev E. D., Maresz K., Tan Y., Dittel B. N. (2007). CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 27, 10714–10721. 10.1523/jneurosci.1922-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radat F., Margot-Duclot A., Attal N. (2013). Psychiatric co-morbidities in patients with chronic peripheral neuropathic pain: a multicentre cohort study. Eur. J. Pain 17, 1547–1557. 10.1002/j.1532-2149.2013.00334.x [DOI] [PubMed] [Google Scholar]
- Raghavendra V., Tanga F., DeLeo J. A. (2003a). Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J. Pharmacol. Exp. Ther. 306, 624–630. 10.1124/jpet.103.052407 [DOI] [PubMed] [Google Scholar]
- Raghavendra V., Tanga F., Rutkowski M. D., DeLeo J. A. (2003b). Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain 104, 655–664. [DOI] [PubMed] [Google Scholar]
- Schatzberg A. F. (2004). The relationship of chronic pain and depression. J. Clin. Psychiatry 65(Suppl. 12), 3–4. [PubMed] [Google Scholar]
- Schiavuzzo J. G., Teixeira J. M., Melo B., da Silva dos Santos D. F., Jorge C. O., Oliveira-Fusaro M. C., et al. (2015). Muscle hyperalgesia induced by peripheral P2X3 receptors is modulated by inflammatory mediators. Neuroscience 285, 24–33. 10.1016/j.neuroscience.2014.11.020 [DOI] [PubMed] [Google Scholar]
- Shan S., Hong-Min T., Yi F., Jun-Peng G., Yue F., Yan-Hong T., et al. (2011). New evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson's disease. Neurobiol. Aging 32, 443–458. 10.1016/j.neurobiolaging.2009.03.004 [DOI] [PubMed] [Google Scholar]
- Sigwalt A. R., Budde H., Helmich I., Glaser V., Ghisoni K., Lanza S., et al. (2011). Molecular aspects involved in swimming exercise training reducing anhedonia in a rat model of depression. Neuroscience 192, 661–674. 10.1016/j.neuroscience.2011.05.075 [DOI] [PubMed] [Google Scholar]
- Tao Y., Li L., Jiang B., Feng Z., Yang L., Tang J., et al. (2016). Cannabinoid receptor-2 stimulation suppresses neuroinflammation by regulating microglial M1/M2 polarization through the cAMP/PKA pathway in an experimental GMH rat model. Brain Behav. Immun. 58, 118–129. 10.1016/j.bbi.2016.05.020 [DOI] [PubMed] [Google Scholar]
- Treede R. D., Jensen T. S., Campbell J. N., Cruccu G., Dostrovsky J. O., Griffin J. W., et al. (2008). Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 70, 1630–1635. 10.1212/01.wnl.0000282763.29778.59 [DOI] [PubMed] [Google Scholar]
- Tsigos C., Chrousos G. P. (2002). Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 53, 865–871. 10.1016/S0022-3999(02)00429-4 [DOI] [PubMed] [Google Scholar]
- Walker A. K., Kavelaars A., Heijnen C. J., Dantzer R. (2014). Neuroinflammation and comorbidity of pain and depression. Pharmacol. Rev. 66, 80–101. 10.1124/pr.113.008144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Goffer Y., Xu D., Tukey D. S., Shamir D. B., Eberle S. E., et al. (2011). A single subanesthetic dose of ketamine relieves depression-like behaviors induced by neuropathic pain in rats. Anesthesiology 115, 812–821. 10.1097/ALN.0b013e31822f16ae [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P., Towell A., Sampson D., Sophokleous S., Muscat R. (1987). Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl). 93, 358–364. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu L., Liu Y.-Z., Shen X.-L., Wu T.-Y., Zhang T., et al. (2015). NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int. J. Neuropsychopharmacol. 18:pyv006. 10.1093/ijnp/pyv006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Dantzer R., Budac D. P., Walker A. K., Mao-Ying Q. L., Lee A. W., et al. (2015). Peripheral indoleamine 2,3-dioxygenase 1 is required for comorbid depression-like behavior but does not contribute to neuropathic pain in mice. Brain Behav. Immun. 46, 147–153. 10.1016/j.bbi.2015.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]