

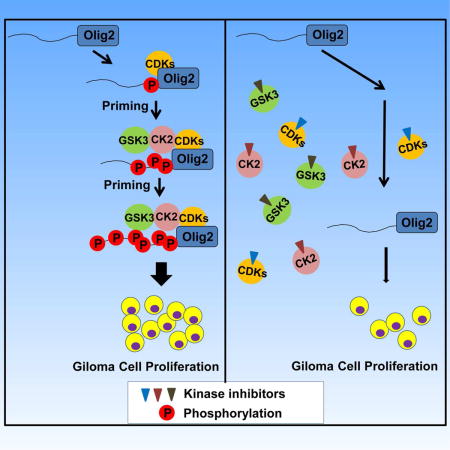
SUMMARY
During development of the vertebrate central nervous system, the bHLH transcription factor Olig2 sustains replication competence of progenitor cells that give rise to neurons and oligodendrocytes. A pathological counterpart of this developmental function is seen in human glioma wherein Olig2 is required for maintenance of stem-like cells that drive tumor growth. The mitogenic/gliomagenic functions of Olig2 are regulated by phosphorylation of a triple serine motif (S10, S13, and S14) in the amino terminus. Here we identify a set of three serine/threonine protein kinases (GSK3α/β, CK2 and CDK1/2) that are, collectively, both necessary and sufficient to phosphorylate the triple serine motif. We show that phosphorylation of the motif itself serves as a template to prime phosphorylation of additional serines and creates a highly charged “acid blob” in the amino terminus of Olig2. Finally, we show that small molecule inhibitors of this forward feeding phosphorylation cascade have potential as glioma therapeutics.
Graphical abstract
INTRODUCTION
A pivotal development in vertebrate evolution was the appearance of myelinating oligodendrocytes that enwrap neural axons in the central nervous system (CNS). By enabling saltatory conductivity of electrical impulses, oligodendrocytes enabled the vertebrate brain to grow large and complex. During CNS development, the basic helix-loop-helix (bHLH) transcription factor Olig2 plays two essential roles in formation of oligodendrocytes throughout the CNS. At late stages of CNS development, Olig2 instructs neural progenitors to exit the cell cycle and adopt an oligodendrocyte fate. However, at earlier stages of development, Olig2 actually opposes cell cycle exit and sustains replication competence so as to allow an adequate pool of oligodendrocyte progenitors to accumulate (Meijer et al., 2012).
Unfortunately, there is a pathological counterpart of this second function. Tumor initiating cells with stem-like properties have been isolated from a wide range of adult and pediatric astrocytomas (Galli et al., 2004; Hemmati et al., 2003; Ignatova et al., 2002; Singh et al., 2003). Irrespective of patient age or tumor grade, these stem-like cells are marked by Olig2 (Bouvier et al., 2003; Ligon et al., 2004; Ligon et al., 2007; Lu et al., 2001; Marie et al., 2001; Ohnishi et al., 2003). Beyond merely marking these stem-like cells, Olig2 is required for maintenance of the stem-like state and is essential for tumor formation from intracranial xenografts of human glioblastomas (Ligon et al., 2007; Mehta et al., 2011; Suva et al., 2014). To a large extent, the gliomagenic functions of Olig2 reflect an oppositional relationship with p53 functions (Mehta et al., 2011). Although p53 signaling is the most frequently mutated signaling axis in glioblastoma, the majority of glioblastomas retain at least one intact copy of the p53 gene (The Cancer Genome Atlas Research Network, 2008). Complete ablation of p53 in human gliomas or genetically relevant murine models of glioma eliminates the tumorigenic requirement for Olig2 (Mehta et al., 2011).
Several years ago, we showed that the mitogenic function of Olig2 in normal oligodendrocyte progenitors and the anti-p53 functions of Olig2 within stem-like, tumor initiating cells of glioma are regulated by phosphorylation of a triple serine motif in the Olig2 amino terminus at S10, S13, and S14. Phosphorylation of this motif is developmentally regulated and it is the phosphorylated form of Olig2 that has gliomagenic and anti-p53 functions (Sun et al., 2011). A more recent study demonstrates that this phosphorylation also regulates the switch from the proliferation to invasion in glioma cells (Singh et al., 2016). In studies summarized here, we use mass spectrometry, genetics and test tube biochemistry with synthetic peptides to identify a set of three protein kinases that are collectively both necessary and sufficient to phosphorylate the triple serine motif. We go on to show that the motif, when phosphorylated, serves as a template to prime phosphorylation of three adjacent serines thus creating a highly charged “acid blob” in the Olig2 amino terminus. Finally, we show that small molecule inhibitors of Olig2 protein kinases might have potential as glioma therapeutics.
RESULTS
Olig2 is Phosphorylated by GSK3 at S10
We interrogated the Olig2 triple serine motif and flanking amino acids using four different computer algorithms to identify candidate protein kinases for S10, S13 and S14 (Table S1). Small molecule inhibitors of the most frequent hits in this in silico screen were tested on Olig2-positive neural progenitor cells (NPCs) (Table S2). Lysates of the drug-treated cells were size fractionated by SDS-PAGE and immunoblotted with a phospho-specific antibody that recognizes Olig2 only when all three members of the triple serine motif are in a phosphorylated state (Sun et al., 2011). These procedures identified S10 as a potential substrate for the glycogen synthase kinase 3 (GSK3).
In mammals, two isoforms of GSK3 (α and β) share a high degree of homology, particularly in their kinase domain (Doble and Woodgett, 2003). As shown in Figures 1A and 1B, small molecule inhibitors of GSK3α/β and also lithium (a well-known GSK3α/β antagonist) suppress the phosphorylation of Olig2 in cultured mouse NPCs and also in low passage human glioma cells (the BT145 line) that have been maintained under conditions developed for neural progenitors so as to maintain “stemness” (see Experimental Procedures). GSK3α-null mice are viable (MacAulay et al., 2007), but GSK3β-null mice die at an early stage of embryonic development (Hoeflich et al., 2000). Accordingly, genetic validation of the GSK3 inhibitor data was achieved in NPC lines derived from informative intercrosses of GSKα-null and GSK3β conditional mice as shown schematically in Figure 1C. GSK3α/β double knockout cells do not survive (Figure 1C), and immunoblotting assays show no change in phospho-Olig2 levels within GSK3α homozygous null NPCs (Figures 1D and 1E). However, we observe significant reduction in phospho-Olig2 levels within GSK3α−/+; GSK3β−/− NPCs. Together these findings indicate that GSK3 function is essential for phosphorylation of Olig2 and that GSK3α and β function redundantly towards this end.
Figure 1. GSK3 phosphorylates Olig2 at S10.
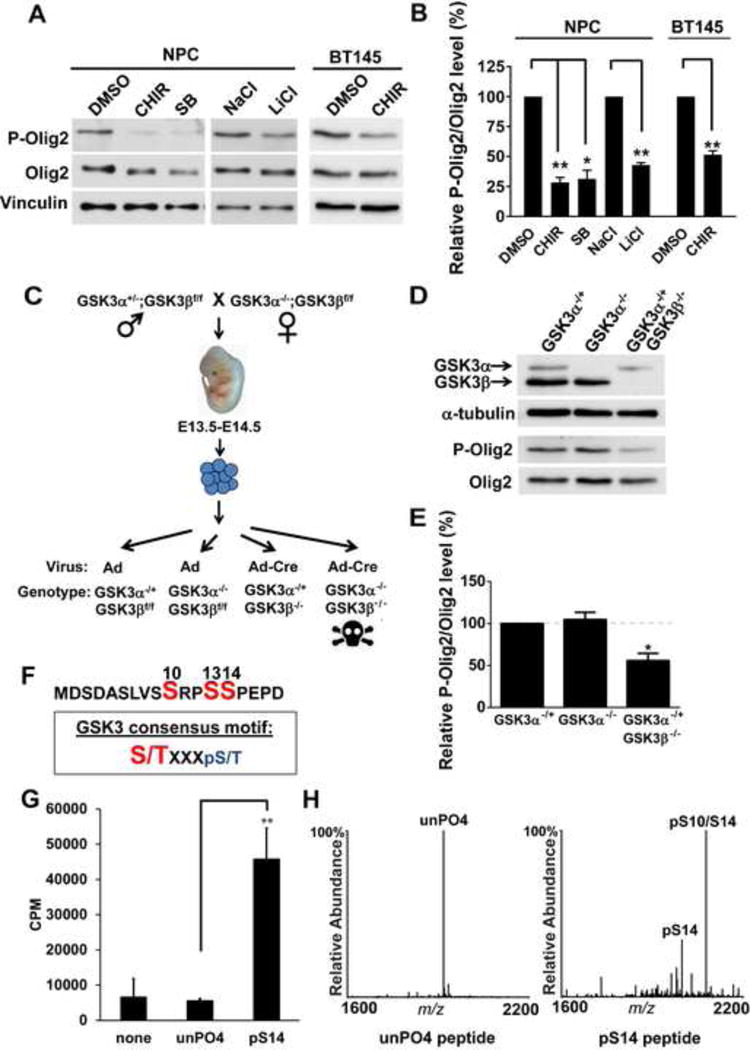
(A and B) Inhibition of GSK3 results in reduced Olig2 phosphorylation. Mouse NPCs and BT145 human glioma cells treated with GSK3 inhibitors, CHIR99021(CHIR), SB216763(SB) or LiCl for 4 hours show a decrease in P-Olig2 level, in comparison to control cells that were treated with either DMSO or NaCl. 5 μm CHIR99021, 10 μm SB216763, 10 mM of NaCl and 10 mM LiCl were used. Relative P-Olig2/Olig2 levels were quantified and compared between control and inhibitor-treated group. Data were analyzed by t-test and are represented as mean ± SEM. *p<0.05, **p<0.01, n=3. (C) A schematic diagram shows generation of mouse GSK3α- and β-knockout NPC lines. Control adeno or adeno-cre virus was introduced to generate GSK3α- or GSK3β-knockout NPC lines. (D and E) GSK3α and β function redundantly to phosphorylate Olig2. P-Olig2 levels were examined in GSK3α−/+, GSK3α −/−, and GSK3α−/+GSK3β−/− NPCs. Relative P-Olig2/Olig2 were quantified and compared between different groups. Data were analyzed by t-test and are represented as mean ± SEM. *p<0.05, n=3. (F) GSK3 consensus motif fits the S10 site upon phosphorylation at S14. S/T (highlighted with red), the kinase’s target Serine/Threonine residue; X, any amino acid; pS, phosphorylated serine. (G) in vitro kinase assay shows that GSK3 phosphorylates Olig2 N-terminal peptide, however, it requires a priming phosphorylation at S14. Synthetic Olig2 N-terminal peptides (a.a.1–18) were used, and both unphosphorylated Olig2 N-terminal peptide (unPO4) and phosphorylated peptide at S14 (pS14) were tested. Reactions without peptides (none) served as negative control. Data were analyzed by t-test and are represented as mean ± SEM. n=3; **p<0.01. (H) Mapping the GSK3 phosphorylation sites by mass spectrometry analysis. in vitro kinase reactions were analyzed by MALDI-MS and −MS/MS. The doubly phosphorylated peptide at S14 and S10 is indicated by pS14/S10.
To test whether Olig2 is directly phosphorylated by the GSK3s, we turned to in vitro kinase assays. As indicated in Figure 1F, the canonical substrates for GSK3α/β require a “priming” phospho-serine or phospho-threonine at +4 residue to the carboxyl side. In its phosphorylated state, S14 of Olig2 would serve this priming function. As shown in Figure 1G, recombinant GSK3β cannot phosphorylate an unmodified synthetic peptide corresponding to residues 1–18 of Olig2. However, an equivalent peptide with pre-phosphorylated S14 is an excellent substrate. Moreover, mass spectrometric analysis of the resulting products confirms that S10 is the enzymatic target of GSK3β in vitro. Besides singly phosphorylated S14, doubly phosphorylated peptides at S10 and S14 were detected when using phosphorylated S14 peptides as substrates (Figure 1H). Collectively, the data indicate that S10 of the Olig2 triple phospho-serine motif is phosphorylated by GSK3 and this requires pre-phosphorylation at S14.
Olig2 is Phosphorylated by Casein Kinase 2 (CK2) at S13
Computer algorithms identified S13 as a potential substrate for casein kinase 2 (CK2) (Table S1). CK2 functions as a tetramer composed of two catalytic subunits, CK2α and CK2α’, and two copies of an essential regulatory subunit, CK2β (Litchfield, 2003). As indicated in Figure 2A and Figure 2B, a small molecule inhibitor of CK2 (CX-4945) suppresses phosphorylation of Olig2 in murine NPCs and also in human glioma cells. Genetic validation of CK2 as an Olig2 kinase was achieved by using shRNA knockdown of CK2β as shown in Figures 2C and 2D.
Figure 2. CK2 phosphorylates Olig2 at S13.
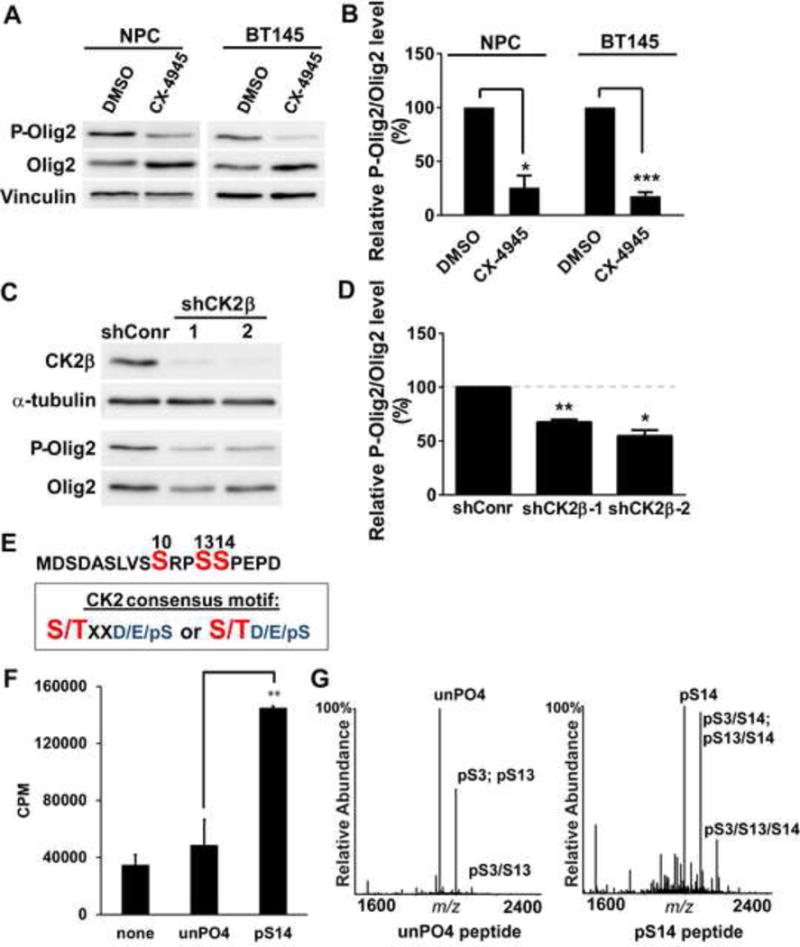
(A) Immunoblot assay. Mouse NPCs and BT145 human glioma cells were treated with the CK2 inhibitor CX-4945 (20 μM) for 4 hours. Cell lysates were size fractionated by polyacrylamide gel electrophoresis and immunoblotted with P-Olig2 and Olig2 antibodies. (B) Quantification of immunoblot assays. Relative P-Olig2/Olig2 levels were quantified and compared between control and inhibitor-treated group. Data were analyzed by t-test and are represented as mean ± SEM. *p<0.05 and ***p<0.001, n=3. (C) Genetic validation of inhibitor data. CK2β was knocked down in wild-type mouse NPCs by lentiviral vectors that express shRNAs targeting mCK2β. Cells that were stably transduced with lentivirus that encode non-target shRNA served as a control (shConr). (D) Quantification of knock-down data. Relative P-Olig2/Olig2 levels were quantified and compared between the knockdown group and control group. Data were analyzed by t-test and are represented as mean ± SEM. *p<0.05 and **p<0.01, n=3. (E) CK2 consensus motif fits S13 site. S/T (highlighted with red), the kinase’s target Serine/Threonine residue; X, could be any amino acid; pS, phosphorylated serine; D, aspartic acid; E, glutamic acid. (F) in vitro kinase assay demonstrates that CK2 phosphorylates the Olig2 N-terminal peptide, and this phosphorylation is facilitated by the phosphorylation of S14. Synthetic Olig2 N-terminal peptides (a.a.1–18) were generated without phosphorylation (unPO4) and with pre-phosphorylation at S14 (pS14). Reactions without peptides (none) served as negative control. Data were analyzed by t-test and are represented as mean ± SEM. n=3; **p<0.01. (G) Mapping CK2 phosphorylation sites by mass spectrometry analysis. in vitro kinase reactions were analyzed by MALDI-MS and −MS/MS. Peaks shown are pS3 (singly phosphorylated peptide at S3); pS13 (singly phosphorylated peptide at S13); pS3/S13 (doubly phosphorylated peptide at S3 and S13); pS3/S14 (doubly phosphorylated peptide at S3 and S14); pS13/S14 (doubly phosphorylated peptide at S13 and S14); pS3/S13/S14 (triply phosphorylated peptide at S3, S13 and S14).
Canonical substrates for CK2 require priming by negatively charged amino acids such as aspartic acid (D), glutamic acid (E) or a phosphoserine (pS), positioned at either +1 or +3 residues to the carboxyl side (Figure 2E) (Meggio and Pinna, 2003). The primary sequence of Olig2 suggests either E16 or phosphorylated S14 would prime S13 to be phosphorylated by CK2 and this prediction is borne out by in vitro kinase assays with recombinant CK2 showing: 1) that S13 is a direct enzymatic target for CK2, and 2) that CK2 phosphorylation of Olig2 at S13 is facilitated by pre-phosphorylation of S14 (Figures 2F and 2G). Of interest, mass spectrometric analysis of CK2-phosphorylated Olig2 peptides revealed that S3 is also a substrate for CK2, which is likely primed by aspartic acid (D) at +1 residue to the carboxyl side.
Cyclin-Dependent Kinases 1 and 2 Phosphorylate S14
As phosphorylation of the S10 and S13 sites was contingent upon prior phosphorylation of S14, we focused next on the identity of the S14 kinase. The S14 site precedes a proline residue, and phosphorylation of serine in this context is often mediated by so called “proline-directed kinases”. The known proline-directed kinases include mitogen-activated protein kinases (MAPKs) and cyclin dependent kinases (CDKs). We focused on these particular kinase groups because phosphorylation of Olig2 is observed only in cycling or replication competent cells (Sun et al., 2011).
As shown in Table S2, small molecule inhibitors of three major MAPKs, ERK1/2, p38MAPKs and JNK have no impact on the phosphorylation state of Olig2 in cycling NPCs. However, a subset of small molecule CDK antagonists suppresses Olig2 phosphorylation in murine NPCs and in the BT145 human glioma cell line. The active subset is confined to inhibitors that suppress the function of CDK1 and CDK2, whereas inhibitors active on other CDKs but not CDK1/2 show little or no activity on Olig2 phosphorylation (Table S2 and Figures 3A and 3B). In these assays, phosphorylation of retinoblastoma protein (RB) served as a positive control for all CDK antagonists. Accordingly, CDK1 and CDK2 were singled out for genetic and biochemical validation.
Figure 3. S14 of Olig2 is phosphorylated by CDK1/2.
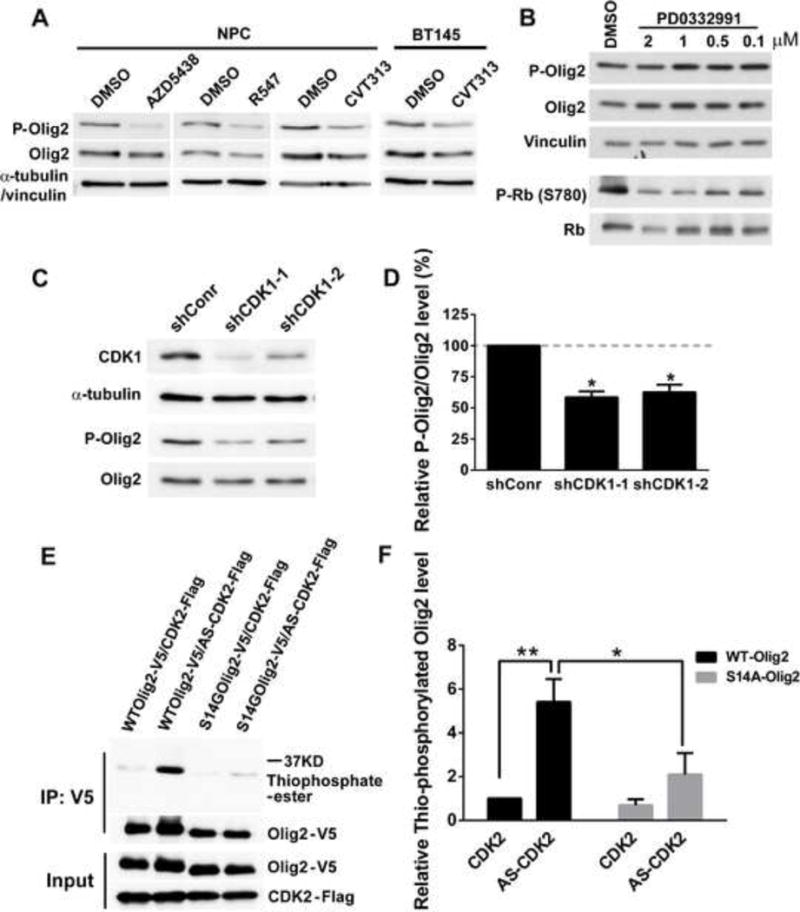
(A) Mouse NPCs and BT145 human glioma cells treated with CDK1/2 inhibitors show a decrease in P-Olig2 level. AZD5438, CDK1, 2 and 9 inhibitor; R547, CDK1, 2 and 4 inhibitor; CVT313, CDK1 and 2 inhibitor. P-Olig2 and Olig2 levels were examined 4 hr after AZD5438 and R547 treatment or 24 hr after CVT313 treatment in NPCs and 3 hr after CVT313 treatment in BT145 cells. 10 μm AZD5438, 5 μm R547, 10 μM of CVT313 were used. (B) Mouse NPCs treated with CDK4/6 inhibitor, PD0332991, show no obvious change on P-Olig2 level. P-Olig2 and Olig2 levels were examined 4 hr after PD0332991 treatment. (C) Genetic validation of CDK1/2 inhibitor data. Immunoblot assay shows that knock-down of CDK1 in CDK2-knockout NPCs decreases P-Olig2 level. CDK1 was acutely knocked down in CDK2-knockout NPCs by introducing AAV that expresses shRNA targeting mCDK1. Cells transduced with AAV that expresses non-target shRNA served as a control (shConr). The P-Olig2 and Olig2 levels were examined at 48 hr post viral transduction. (D) Quantification of knock-down data. Relative P-Olig2/Olig2 levels were quantified and compared between knockdown group and control group. Data are analyzed by t-test and are represented as mean ± SEM. *p<0.05; n=3. (E) An analog-sensitive kinase assay shows that CDK2 phosphorylates Olig2 at S14 site. (F) Quantification of analog-sensitive kinase assays. Thiophosphorylated Olig2 levels were quantified, normalized to total Olig2, and then compared between different groups. Data were analyzed by two-way ANOVA with Sidak posttest and are represented as mean ± SEM. *p<0.05, **p<0.01; n=4.
For genetic validation, we first generated CDK2-null NPC lines from E13.5 embryos of CDK2-knockout mice (Berthet et al., 2003). As indicated in Figures S1A and S1B, loss of CDK2 alone had no effect on Olig2 phosphorylation. Targeted disruption of CDK1 leads to arrest of embryonic development at a very early stage (Santamaria et al., 2007). We tried chronic CDK1 knockdown in NPCs by using shRNA targeting CDK1, however the CDK1-knockdown cell lines also fail to thrive (data not shown). We then used an adeno-associated virus (AAV) mediated shRNA for acute knockdown of CDK1 in either wild type or CDK2-null NPCs. As indicated in Figures 3C and 3D, acute shRNA knockdown of CDK1 within CDK2-null NPCs significantly reduced Olig2 phosphorylation, while acute knockdown of CDK1 alone in wild-type cells had no effect on Olig2 phosphorylation (Figure S1C and S1D). Together, these results indicate that CDK1 and CDK2 function redundantly for phosphorylation of Olig2.
Despite repeated efforts, we were unable to establish assay conditions wherein recombinant CDK1 or CDK2 would function on synthetic peptides in vitro, which might be due to the short length of Olig2 N-terminal peptide used in the assay. Accordingly, we used an in vivo phosphorylation assay with an analogue-sensitive mutant of CDK2 (AS-CDK2) to validate Olig2 as a direct substrate for CDK2. The ATP-binding pocket of this mutant CDK2 can utilize bulky adenine analogs, such as ATP-γ-S as a phosphate donor, therefore label CDK2 substrate with P-γ-S (Merrick et al., 2011). Expression vectors encoding wild-type CDK2 or this analogue-sensitive CDK2, together with V5 epitope tagged wild-type Olig2 or S14G Olig2 mutant were transfected into 293 cells that do not express endogenous Olig2. Incorporation of P-γ-S into wild type or mutant Olig2 was assessed by anti-thiophosphate ester antibody. As shown in Figures 3E and 3F, the analogue-sensitive mutant CDK2 can phosphorylate wild-type Olig2, whereas the phosphorylation level is reduced upon the mutation of S14 site. These data indicate that S14 is a direct substrate for CDK2.
Phosphorylation of the Triple Serine Motif Enables Formation of a Hexa-phosphoserine Acid Blob in the Amino Terminus of Olig2
During the course of this work, our procedures and protocols for mass spectrometric analysis of Olig2 were refined. Improved methodology enabled the detection of additional phosphoserine residues in the amino terminal end of Olig2 at S3, S6 and S9 (Figure 4A and Figure S2). However, as shown in Figure 4A (WT), peptide fragments with these additional phosphoserine residues were never observed without phosphorylations within the original S10, S13, and S14 sites.
Figure 4. Phosphorylation of the triple serine motif enables formation of a hexa-phosphoserine acid blob in the amino terminus of Olig2.

(A) Summary of mass spectrometric analyses on Olig2 N-terminal phosphorylated peptides detected in Olig2-null NPCs that were transduced with WT, TPN (S10A/S13A/S14G), DPN (S13A/S14G) or SPN (S14G) Olig2. Black font: phosphorylated peptides identified in previous study (Sun et al., 2011); Red font: newly identified phosphorylated peptides in WT-Olig2 sample; Blue font: contingent phospho peptides that are undetectable or barely detectable in WT-Olig2 sample. +, present; +/− detected but with low level; −, undetectable. (B) A sequentially priming phosphorylation cascade. Schematic diagram shows how the triple serine motif creates priming sites for additional phosphorylations at S3, S6 and S9 that create a hexaphosphate acid blob in the Olig2 amino terminus.
The relationship of S3, S6 and S9 to the phosphorylation state of the original triple serine motif was assessed in a set of “contingency” assays. Olig2-null NPCs were transduced with lentiviral expression vectors encoding Olig2 mutants with informative phospho-null substitutions at S10, S13 or S14. As shown in Figure 4A, ablation of the original triple serine motif completely eliminates phosphorylation at S3, S6 and S9. Phospho-null substitutions at S13 and S14 ablate the phosphorylation of S10 and likewise ablate phosphorylation of S6 and S9. Finally, a single S->G substitution at S14 almost completely eliminates phosphorylation at S10 and S13, although there is a low level of “breakthrough” phosphorylation events to the amino terminal side. This is consistent with the CK2 kinase assay, which suggests that the S13 and S3 can be phosphorylated even with the absence of S14 phosphorylation, although the efficiency is relatively low (Figure 2F and 2G).
The contingency relationships of S10 and S13 to the phosphorylation state of S14 are predicted by the priming requirements of GSK3 and CK2 (see Figures 1 and 2). Moreover, close scrutiny of the Olig2 amino acid sequence shows that S10 and S13, when phosphorylated, could themselves serve to prime S6 and S9 for phosphorylation by GSK3 and CK2. In addition, phosphorylation of S6 would facilitate CK2 phosphorylation on S3. Thus, as modeled in Figure 4B, phosphorylation of S14 by the cell cycle modulated kinases CDK1/2 initiates formation of a hexa-phosphate acidic domain in the amino terminus of Olig2.
Therapeutic potential for Small Molecule Inhibitors of Olig2 Phosphorylation
The phosphorylation state of Olig2 is developmentally regulated, being high in cycling neural progenitors and low in terminally differentiated, myelinating oligodendrocytes. Previous studies have shown that gliomagenic and anti-p53 functions of Olig2 are contingent upon phosphorylation of the triple serine motif (Sun et al., 2011). The functional relevance of this relationship to the pathobiology of human glioma is illustrated by an oppositional relationship between phospho-Olig2 and p53 status. As shown in Figure 5, the phospho-Olig2 content of gliomas with genetically intact p53 is more comparable to that of cycling neural progenitors than to myelinating oligodendrocytes and is significantly higher than that of p53-mutant gliomas.
Figure 5. Analysis P-Olig2 levels in a panel of gliomas cells with either wild-type p53 or mutant p53.
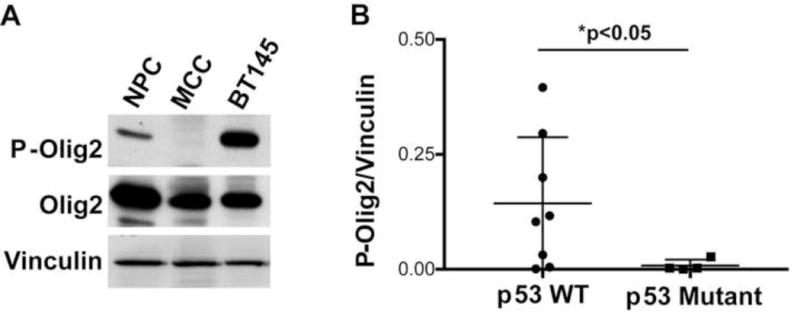
(A) Immunoblot of P-Olig2 in cycling neural progenitor cells (NPC), adult mouse corpus callosum (MCC) and low passage human glioma cells (BT145 line). (B) Glioma cell lines with intact p53 have significantly higher level of P-Olig2 than p53-mutant glioma cells. The relative P-Olig2/Vinculin levels were analyzed and compared in different glioma cell lines. Data were analyzed by Mann-Whitney test and represented as mean + SEM. n=8 p53 wild-type glioma cells lines (filled circles) and n=4 p53-mutant cell lines (filled boxes). For frame of reference, the P-Olig2/vinculin ratio of cycling mouse neural progenitor cells (open circle) was set at 1.0.
Are therapeutic opportunities for glioblastoma embedded in the kinases that phosphorylate the triple serine motif? To address this question, we turned to the genetically accessible murine NSCs. The anti-p53 functions of Olig2 manifest themselves in short term assays and are quantifiable by suppression of p21 expression. As shown in Figure 6, small molecule inhibitors of CDK1/2, CK2 and GSK3α/β all trigger elevated expression of p21. Although these small molecule inhibitors likely suppress the phosphorylation of multiple on-target and off-target protein substrates, the p21 responses shown in Figure 6 are largely rescued by expression of a triple phosphomimetic Olig2 (TPM; S10D/S13E/S14D), indicating that the drug effects on p21 expression are channeled, at least in part, through their action on Olig2.
Figure 6. Inhibition of Olig2 phosphorylation enhances p53 function.
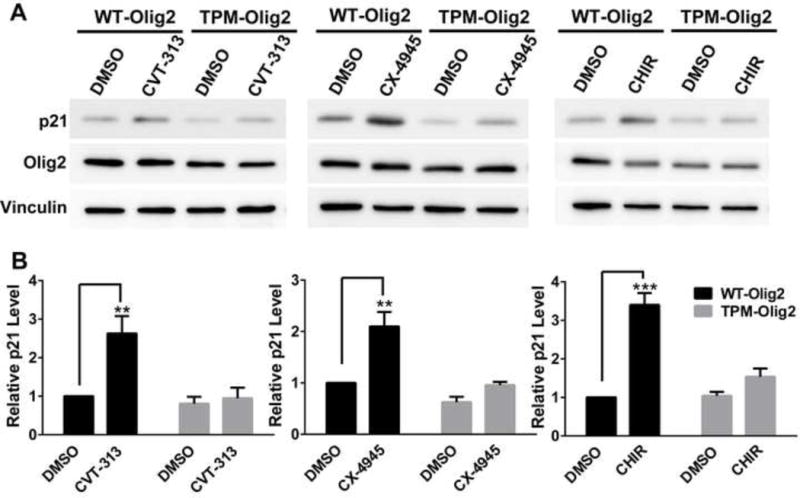
(A and B) Olig2 kinase inhibitors increase p21 level through inhibiting Olig2 phosphorylation. Olig2-null NPCs were transduced with retroviruses that express either WT-Olig2 or TPM-Olig2. p21 activation upon kinase inhibitor treatment was examined and compared between two NPC lines by western blot analysis. Cells were examined either after 24 hr treatment with 5 μM CVT313 (CDK1/2 inhibitor), or after 8 hr treatment with 10 μM CX-4945 (CK2 inhibitor) or 5 μM CHIR99021 (GSK3 inhibitor). Data were analyzed by two-way ANOVA with Sidak posttest. There is a significant difference between WT-Olig2 and TPM-Olig2 in response to Olig2 kinase inhibitors. Data are represented as mean ± SEM. **p<0.01, ***p<0.001; n=3.
To assess effects of the kinase inhibitors on cell proliferation, we used a preclinical murine model of pediatric glioma, a common Olig2-positive brain tumor of childhood (Bergthold et al., 2014). Activating mutations within the BRAF serine/threonine protein kinase (BRAFV600E) and homozygous deletion of Ink4a/ARF co-occur in a subset of WHO grades II–IV pediatric diffuse astrocytomas (Schiffman et al., 2010). Mouse NPCs bearing these two mutations are tumorigenic, and the resulting tumors recapture the histological characteristics and cellular markers (eg, Olig2, GFAP and Nestin) of human pediatric glioma (Huillard et al., 2012). We therefore utilized this mouse model to assess therapeutic potential of the triple serine motif as a drug target. Of the five small molecule kinase inhibitors studied here, the CK2 inhibitor CX-4945 scored second highest on the central nervous system multiparameter optimization (CNS MPO) algorithm for brain penetrance (Wager et al., 2010) (Table S3). Moreover, CX-4945 is the only one of the five drugs currently in clinical trials (Chon et al., 2015). Accordingly, our studies with this tumor model focused on CX-4945.
To address brain penetrance of CX-4945 we first monitored Olig2 phosphorylation in the brains of drug-treated mice. As shown in Figure 7A and 7B, CX-4945 treatment significantly reduces P-Olig2 positive cells in the brain of our glioma mouse model. Next, we sought to determine whether CX-4945 could inhibit cell proliferation in our tumor model by regulating P-Olig2 levels. This was addressed by using two knock-in lines, wherein the coding sequence of either wild-type Olig2 or triple phosphomimetic Olig2 (S10D/S13E/S14D, TPM-Olig2) was introduced into the Olig2 locus. As shown in Figure 7C and 7D, CX-4945 inhibits tumor cell proliferation, but the growth inhibition is rescued by the TPM-Olig2-variant. As noted for the studies on p21 expression (Figure 6), the TPM-Olig2 rescue of proliferation is only partial, presumably because the impact of a CK2 antagonist such as CX-4945 is not confined to Olig2.
Figure 7. Therapeutic potential of an Olig2 kinase antagonist in a murine model of pediatric low-grade astrocytoma.
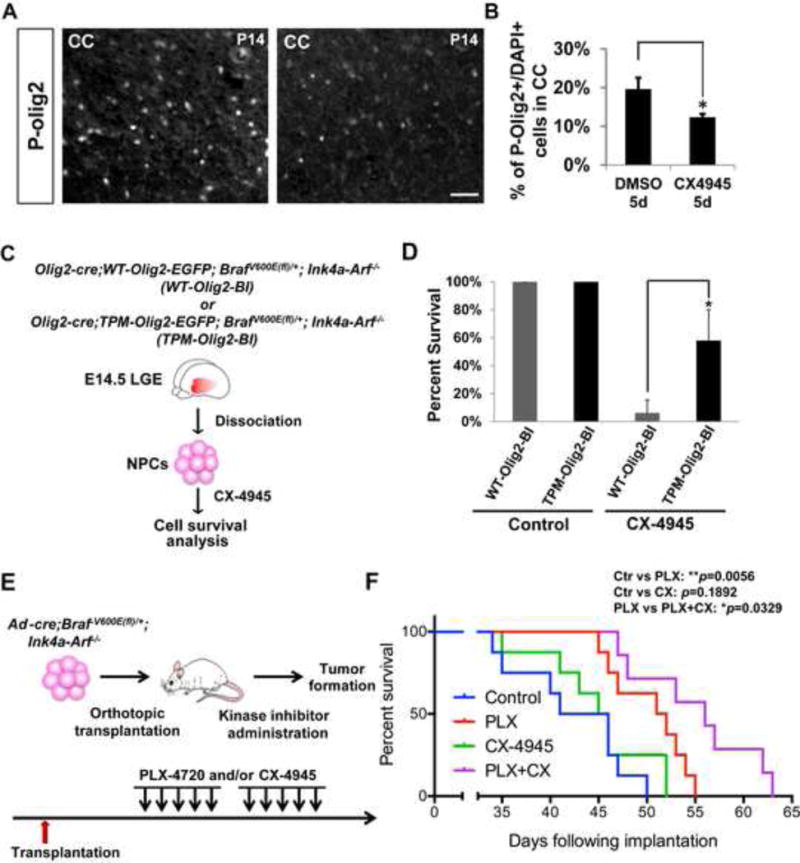
(A and B) CK2 inhibition decreases P-Olig2 level in vivo. Developing hGFAP-cre; BrafV600E(fl)/+; Ink4a-Arf−/− pups were treated with CK2 inhibitor CX-4945 25 mg/kg intraperitoneally for 5 days and were analyzed at P14. CC, Corpus Callosum. Scale bar, 50μm. Data are represented as mean ± SD. *p<0.05, n=3. (C) Schematic diagram shows generation of two BRAFV600E-transformed NPC lines in an Ink4a-Arf−/− background. The two lines express knock-in, epitope-tagged Olig2 wherein the triple serine motif is either wild type (WT-Olig2-BI) or triple phosphomimetic with the S10D/S13E/S14D substitutions (TPM-Olig2-BI). Of note, the Olig2-cre driver is used to activate expression of the BRAFV600E oncogene, while simultaneously disrupting another endogenous Olig2 allele (Schuller et al., 2008). The resulting mice exhibit early prenatal lethality, which precludes further study in postnatal pups. (D) Proliferation of the TPM-Olig2-BI line is partially resistant to the CK2 antagonist CX-4945. For proliferation assays, 5×104 cells were seeded at Day 0 and 1μM of CX-4945 was added at Day 1. The cell number was assessed at Day 4. Data were analyzed by t-test and represented as mean ± SD. *p<0.05, n=3. (E) Schematic diagram demonstrates orthotopic transplantation and the treatment regimen (see Experimental Procedures). (F) Combination of BRAF and CK2 inhibitors significantly improves survival in an orthotopic model of pediatric glioma. Kaplan-Meier graph is illustrated and Log-rank test was used to determine the survival differences between different treatment groups. *p<0.05, and **p<0.01; n=8.
With respect to tumor growth in vivo, previous studies have shown that PLX-4720 – a small molecule RAF inhibitor that is currently in clinical trials for BRAF mutant pediatric astrocytoma (Penman et al., 2015) – prolongs survival of mice that received intracranial grafts of the tumor cells (Bergthold et al., 2014). As shown in Figure 7E and 7F, CX-4945, as a single agent, had no statistically significant impact on survival of mice that were challenged with the tumor cells. However CX-4945 cooperated with PLX-4720 to give a survival benefit that exceeds that delivered by the RAF inhibitor alone. Collectively, these data suggest therapeutic potential for drugs that block phosphorylation of the triple serine motif, either as adjuvants to radiation and chemotherapy or in combination with other targeted therapeutics.
DISCUSSION
During development and also in disease, pro-mitogenic/anti-p53 functions of Olig2 are controlled by phosphorylation of a triple serine motif at the amino terminus of the protein (Mehta et al., 2011; Sun et al., 2011). Here we identify a set of three protein kinases that are both necessary and sufficient to phosphorylate the triple serine motif. We go on to show that in its phosphorylated state, the triple serine motif serves as a template that enables phosphorylation of an additional three serines including S9, S6 and S3, generating a hexa-phosphoserine acidic domain in the Olig2 amino terminus. A broad body of data documents the functional role of acidic domains (a.k.a. “acid blobs” or “negative noodles”) as facilitators of protein-protein interactions that enable transcription factor function (Banerjee and Kundu, 2003; Cross et al., 2011; Garcia-Rodriguez and Rao, 1998; Li and Botchan, 1993; Sigler, 1988). Protein alignment of Olig2 shows complete conservation of the six serines in the amino terminal acid blob among human, mouse, chicken, frog and fish (Meijer et al., 2012). Formation of the Olig2 acid blob is linked to the cell cycle by CDK1/2-mediated phosphorylation of S14.
The three kinases that phosphorylate the triple serine motif are broadly expressed and direct evidence of their developmental functions in formation of Olig2-dependent cell types is lacking. However, targeted disruption of the CK2 regulatory subunit CK2β in embryonic stem cells does recapitulate some features of the Olig2-null mouse including compromised neural progenitor cell proliferation and oligodendrogenesis (Huillard et al., 2010).
On a more practical note, a broad body of data suggests that Olig2 would be an attractive target for glioma therapeutics (Ligon et al., 2007; Mehta et al., 2011; Suva et al., 2014). Olig2-null mouse embryos develop to term but die within minutes following birth due to a total deficit of motor neurons (Lu et al., 2002; Zhou and Anderson, 2002). However, ablation of Olig2 in the postnatal mouse brain is survivable for extended periods of time (Cai et al., 2007). Moreover, as a glioma therapeutic, an Olig2 antagonist might be required only transiently as an adjuvant to standard of care therapeutics. Transcription factors per se are difficult targets for drug development because their interactions with DNA or co-regulator proteins tend to involve large surface areas. However, protein kinases lend themselves well to development of targeted therapeutics. Does the Olig2 acid blob constitute a therapeutic opportunity for glioma?
Data generated with small molecule inhibitors of the triple serine motif kinases can be correlated to a mutational analysis conducted by Sun et al. (Sun et al., 2011). Using site-directed mutagenesis, Sun et al. found that a triple phospho-null mutation (S10G/S13A/S14A) had a severe proliferation phenotype, and a double phospho-null mutation (S13A/S14G) had a partial proliferation phenotype whereas a single phospho-null substitution at S14 (S14G) had no discernable impact on cell proliferation. The lack of a phenotype with the S14G mutation is somewhat at odds with the pivotal role of S14 as “the first domino” to fall in a sequentially priming, forward feeding phosphorylation cascade as shown in Figure 4. However the negative result may be explained by the mass spectrometry data summarized in Figure 4A, which suggests a low level of “breakthrough” phosphorylation events to the amino terminal side of the S14G substitution. The partial loss of proliferative ability exhibited by the double phospho-null substitution at S13/S14 (Sun et al., 2011) is in accord with the Figure 4A data, showing that double substitution delivers a more complete suppression of phosphorylation events at S3, S6, S9 and S10. It should also be noted that the cells may adapt to chronic (i.e. genetic) loss of P-Olig2 in ways that cannot be achieved with acute, drug-induced loss.
Against this genetic backdrop, small molecule inhibitors targeted to any one of the three kinases that are essential to formation of the acid blob stimulate expression of p21 – a canonical p53 target gene – and this effect is channeled, at least in part, through phosphorylation of the Olig2 triple serine motif (Figure 6). Among these kinases inhibitors, the selective CK2 inhibitor, CX-4945 is orally available and has been on clinical trials for multiple solid tumors. Although it has not been tested clinically in glioma, CK2α is frequently overexpressed in glioblastoma, particularly in the classical subtype of glioma (50.7%) (Zheng et al., 2013). In preclinical studies, CK2 demonstrates an important role in regulating glioma cell viability and is necessary for glioma tumorigenesis in vivo. Inhibition of CK2 kinase by CX-4945 impairs glioma cells proliferation in vitro and suppresses human glioma growth in the xenograph model (Dixit et al., 2012; Nitta et al., 2015; Zheng et al., 2013). In our study, we further tested CX-4945 in a pediatric astrocytoma model. As shown in Figure 7, CX-4945 inhibits proliferation of Olig2-positive tumor cells and cooperates with a targeted BRAF antagonist to improve survival in a murine model of BRAF mutant pediatric astrocytoma. Collectively, the genetic and pharmacologic data suggest that small molecule inhibitors of the triple serine motif protein kinases could have therapeutic potential for malignant gliomas, either as standalone modalities or as adjuvants to radiotherapy, genotoxic drugs and targeted therapeutics.
EXPERIMENTAL PROCEDURES
Kinase Inhibitors
The serine kinase inhibitors used in this study were: AZD5438 (CDK1/2/9 inhibitor, Selleckchem), R547 (CDK1/2/4 inhibitor, Selleckchem), CVT313 (CDK1/2 inhibitor, EMD Millipore), PD0332991 (CDK4/6 inhibitor, Selleckchem), CX-4945 (CK2 inhibitor, Selleckchem or Pfizer), CHIR99021 (GSK3 inhibitor, Selleckchem), SB216763 (GSK3 inhibitor, Selleckchem), PLX-4720 (BRAFV600E inhibitor, Plexxikon) and LiCl (GSK3 inhibitor, Sigma). All the small molecule inhibitors were dissolved in DMSO and stock solutions were kept at −20°C, except LiCl which was dissolved in water and kept at room temperature.
NPC and Human Glioma cell Culture
Mouse NPCs were isolated from the lateral ganglionic eminence (LGE) of E13.5–14.5 embryos. All human glioma cell lines were provided by Dr. Ligon’s group (Brigham and Women’s Hospital) and were originally derived from Brigham and Woman’s Hospital patients undergoing surgery in accordance with IRB protocols. Both NPCs and human glioma cells were cultured in serum-free medium containing B27 and N2 supplements, epidermal growth factor (EGF, 20 ng/ml) and basic fibroblast growth factor (bFGF, 20 ng/ml). Cells were grown on laminin-coated plates for maintaining adherent monolayer culture for most assays, except for the experiments showed in Figure 5, where glioma cells were grown as neurospheres cultures.
Animals
All animal handling and procedures were performed according to UNC, DFCI, NWU Feinberg School of Medicine or UCSF guidelines under IACUC-approved protocols.
GSK3-null NPC Lines
To generate GSK3α/β-null NPC lines, we crossed GSK3α+/−; GSK3βfloxP/floxP male and GSK3α−/−; GSK3βfloxP/floxP female mice (Kim et al., 2009; MacAulay et al., 2007; Patel et al., 2008) and derived GSK3α+/−; GSK3βfloxP/floxP or GSK3α−/−; GSK3βfloxP/floxP NPCs from the lateral ganglionic eminence (LGE) of individual E13-14 embryos. To generate GSK3β-null NPC lines, we infected above cell lines with either adenovirus that expresses the Cre recombinase gene or adenovirus only. The ratio of virus to cells was 100:1. Transduced cells were cultured for at least three passages before examination to ensure complete recombination for GSK3β locus.
In Vitro Peptide Kinase Assay
Active kinases, GSK3β and CK2, were purchased from either Promega (GSK3β, V1991) or New England Biolabs (CK2, P6010L). Peptides that cover 18 amino acids of N-terminus of Olig2 protein were synthesized as either native peptides or phosphorylated peptides at S14 (Tufts Physiology Core, Boston). For kinase reactions, we mixed active kinase, Olig2 N-terminal peptides at a final concentration of 20 mM and 32P-labeled ATP (3000 Ci/mmol) in reaction buffer and incubated for 15 min at 30°C. Reactions were then blotted onto nitrocellulose membrane and washed three times with 150 mM phosphoric acid, dried and subjected to liquid scintillation counting. The products of cold peptide kinase assays were subjected to MALDI-MS and −MS/MS analyses to identify the phosphorylation site(s) (see supplemental materials and methods).
Analog-Sensitive Kinase Assay with CDK2
WT-CDK2 and AS-CDK2 (F80G) were expressed by the p3xFLAG-CMV (Sigma) vectors. WTOlig2-V5 and S14GOlig2-V5 were cloned into pcDNA backbone vectors. To test whether CDK2 can phosphorylate Olig2, we transfected 293 cells with WTOlig2-V5 or S14GOlig2-V5 with either WT-CDK2-Flag or AS-CDK2-Flag. In vivo phosphorylation assay was performed according to a previous protocol with slight modification (Banko et al., 2011) (see supplemental materials and methods).
Olig2 Protein Purification
As described previously, we have cloned full-length and different mutant forms of Olig2 into the pWZL-blast retrovirus vector (Sun et al., 2011). We generated mouse NPC lines that stably express either wild-type or different mutant forms of Olig2 by transducing Olig2-null NPC with the appropriate virus. Exogenous Olig2 proteins contain V5-tag at the C-terminus. For mass spectrometry analysis, we isolated the nuclear fraction and purified Olig2 by using Anti-V5 Agarose Affinity Gel (Sigma). The IP samples were then digested and subjected to nanoLC-ESI-MS analysis for phospho-Olig2 peptides.
Preclinical Murine Models of Pediatric Low-Grade Astrocytoma
BRAFV600E in the context of Ink4a-Arf−/− is a common genetic driver set for pediatric glioma (Huillard et al., 2012). BrafV600E(fl)/+ mice, Ink4a-Arf−/− mice and Olig2-cre mice have been described previously (Dankort et al., 2007; Schuller et al., 2008; Serrano et al., 1996). Two knock-in mouse lines that express either WT-Olig2 or TPM-Olig2 (S10D/S13E/S14D) fused to IRES-EGFP cassettes were newly generated through homologus recombination of the Olig2 locus in ES cells. Of note, Olig2-cre; BrafV600E(fl)/+; Ink4a-Arf−/− mice exhibit early prenatal lethality. However, by intercrossing above mouse lines (see schematic Figure 6C), we were able to generate Olig2-cre;WT-Olig2-EGFP;BrafV600E(fl)/+;Ink4a-Arf−/− and Olig2-cre;TPM-Olig2-EGFP;BrafV600E(fl)/+; Ink4a-Arf−/− embryos and isolate NPCs from E14.5 LGE. These Olig2 knock-in cell lines (labeled WT-Olig2-BI or TPM-Olig2-BI) were used for in vitro proliferation assays.
For in vivo drug tests, we used adenovirus-Cre to activate BRAFV600E within NPCs isolated from BrafV600E(fl)/+;Ink4a-Arf−/− embryos (see schematic Figure 6E). 2×105 cells were injected into the brains of 6-week old immunodeficient (nude) mice as previously described (Hashizume et al., 2010). Mice were randomly assigned into four cohorts and treated with 20mg/kg of PLX-4720 and/or 25mg/kg of CX-4945 by intraperitoneal injection for five consecutive days (Mon–Fri) for 2 weeks starting five days after transplantation. All mice were monitored every day for the development of symptoms related to tumor growth. Mice were euthanized when they exhibited symptoms indicative of significant impairment to neurological function.
Statistical Methods
Differential levels of P-Olig2 levels in glioma cell lines were analyzed by Mann-Whitney test. Two-way ANOVA with Sidak posttests were performed when analyzing the analog-sensitive kinase assays and when comparing the response of WT-Olig2 and TPM-Olig2 cell lines to Olig2 kinase inhibitors. The log-rank test was used to determine the survival differences between different treatment groups of mice and t-tests were used to analyze sample differences in other assays as described in the figure legends. The statistical values were obtained using GraphPad Prism.
Supplementary Material
Acknowledgments
The authors thank Philipp Kaldis for the CDK2-knockout mice and Martin McMahon for BrafV600E(fl)/+ mice. Nathanael Gray, Sara Buhrlage, Tinghu Zhang, Jinhua Wang and Per Hydbring contributed useful suggestions on kinase inhibitors. Expert technical assistance and advice was provided by Krister Barkovich, Sandra Chang and Emmanuelle Huillard. We thank the American Brain Tumor Association for postdoctoral fellowship support (A-C.T.) and AACR Anna D. Baker Fellowship in Basic Cancer Research (A.G.). This work was supported by NIH grants (NS057727 to C.D.S. and NS040511 to D.H.R.) and by funding from Dana-Farber Strategic Research Initiative (J.A.M.), Howard Hughes Medical Institute (D.H.R.) and A Cure for Kid’s Brain Tumor Foundation (C.D.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
J.Z. identified the kinases involved in phosphorylating Olig2, A-C.T. tested Olig2 kinases in vivo in pediatric glioma model cells, J.A.A. did the in vitro kinase assays, S.B.F., J.D.C. and J.A.M. performed the mass spectrometry experiments, A.G. set up the mouse tumor system, J.S.D. provided technical assistance, M.M-S. and W.D.S. provided GSK3 knockout embryos, W.M. and P.S. provided the analog-specific kinase system and helpful discussions, K.L.L offered the human glioma cell lines, Y.S. and J.A.A. examined and analyzed the P-Olig2 levels in different p53 wild-type and p53 mutant glioma cell lines, R.H. and C.D.J. performed orthotopic survival study, and J.Z., D.H.R and C.D.S. designed the experiments and wrote the paper.
References
- Banerjee S, Kundu TK. The acidic C-terminal domain and A-box of HMGB-1 regulates p53-mediated transcription. Nucleic Acids Res. 2003;31:3236–3247. doi: 10.1093/nar/gkg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villen J, Wang B, Kim SR, Sakamoto K, et al. Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Mol Cell. 2011;44:878–892. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergthold G, Bandopadhayay P, Bi WL, Ramkissoon L, Stiles C, Segal RA, Beroukhim R, Ligon KL, Grill J, Kieran MW. Pediatric low-grade gliomas: how modern biology reshapes the clinical field. Biochim Biophys Acta. 2014;1845:294–307. doi: 10.1016/j.bbcan.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Bouvier C, Bartoli C, Aguirre-Cruz L, Virard I, Colin C, Fernandez C, Gouvernet J, Figarella-Branger D. Shared oligodendrocyte lineage gene expression in gliomas and oligodendrocyte progenitor cells. J Neurosurg. 2003;99:344–350. doi: 10.3171/jns.2003.99.2.0344. [DOI] [PubMed] [Google Scholar]
- Cai J, Chen Y, Cai WH, Hurlock EC, Wu H, Kernie SG, Parada LF, Lu QR. A crucial role for Olig2 in white matter astrocyte development. Development. 2007;134:1887–1899. doi: 10.1242/dev.02847. [DOI] [PubMed] [Google Scholar]
- Chon HJ, Bae KJ, Lee Y, Kim J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front Pharmacol. 2015;6:70. doi: 10.3389/fphar.2015.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross B, Chen L, Cheng Q, Li B, Yuan ZM, Chen J. Inhibition of p53 DNA binding function by the MDM2 protein acidic domain. J Biol Chem. 2011;286:16018–16029. doi: 10.1074/jbc.M111.228981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–384. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFalpha)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012;3:e271. doi: 10.1038/cddis.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Garcia-Rodriguez C, Rao A. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP) J Exp Med. 1998;187:2031–2036. doi: 10.1084/jem.187.12.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume R, Ozawa T, Dinca EB, Banerjee A, Prados MD, James CD, Gupta N. A human brainstem glioma xenograft model enabled for bioluminescence imaging. J Neurooncol. 2010;96:151–159. doi: 10.1007/s11060-009-9954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Huillard E, Hashizume R, Phillips JJ, Griveau A, Ihrie RA, Aoki Y, Nicolaides T, Perry A, Waldman T, McMahon M, et al. Cooperative interactions of BRAFV600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc Natl Acad Sci U S A. 2012;109:8710–8715. doi: 10.1073/pnas.1117255109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huillard E, Ziercher L, Blond O, Wong M, Deloulme JC, Souchelnytskyi S, Baudier J, Cochet C, Buchou T. Disruption of CK2beta in embryonic neural stem cells compromises proliferation and oligodendrogenesis in the mouse telencephalon. Mol Cell Biol. 2010;30:2737–2749. doi: 10.1128/MCB.01566-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- Kim WY, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR, Snider WD. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009;12:1390–1397. doi: 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Botchan MR. The acidic transcriptional activation domains of VP16 and p53 bind the cellular replication protein A and stimulate in vitro BPV-1 DNA replication. Cell. 1993;73:1207–1221. doi: 10.1016/0092-8674(93)90649-b. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD, Rowitch DH. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol. 2004;63:499–509. doi: 10.1093/jnen/63.5.499. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Park JK, Noll E, Chan JA, Alberta J, Yuk D, Alzamora MG, Louis DN, Stiles CD, Rowitch DH, et al. Oligodendrocyte lineage genes (OLIG) as molecular markers for human glial brain tumors. Proc Natl Acad Sci U S A. 2001;98:10851–10856. doi: 10.1073/pnas.181340798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Marie Y, Sanson M, Mokhtari K, Leuraud P, Kujas M, Delattre JY, Poirier J, Zalc B, Hoang-Xuan K. OLIG2 as a specific marker of oligodendroglial tumour cells. Lancet. 2001;358:298–300. doi: 10.1016/S0140-6736(01)05499-X. [DOI] [PubMed] [Google Scholar]
- Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- Mehta S, Huillard E, Kesari S, Maire CL, Golebiowski D, Harrington EP, Alberta JA, Kane MF, Theisen M, Ligon KL, et al. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell. 2011;19:359–371. doi: 10.1016/j.ccr.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer DH, Kane MF, Mehta S, Liu H, Harrington E, Taylor CM, Stiles CD, Rowitch DH. Separated at birth? The functional and molecular divergence of OLIG1 and OLIG2. Nat Rev Neurosci. 2012;13:819–831. doi: 10.1038/nrn3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick KA, Wohlbold L, Zhang C, Allen JJ, Horiuchi D, Huskey NE, Goga A, Shokat KM, Fisher RP. Switching Cdk2 on or off with small molecules to reveal requirements in human cell proliferation. Mol Cell. 2011;42:624–636. doi: 10.1016/j.molcel.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta RT, Gholamin S, Feroze AH, Agarwal M, Cheshier SH, Mitra SS, Li G. Casein kinase 2alpha regulates glioblastoma brain tumor-initiating cell growth through the beta-catenin pathway. Oncogene. 2015;34:3688–3699. doi: 10.1038/onc.2014.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi A, Sawa H, Tsuda M, Sawamura Y, Itoh T, Iwasaki Y, Nagashima K. Expression of the oligodendroglial lineage-associated markers Olig1 and Olig2 in different types of human gliomas. J Neuropathol Exp Neurol. 2003;62:1052–1059. doi: 10.1093/jnen/62.10.1052. [DOI] [PubMed] [Google Scholar]
- Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314–6328. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penman CL, Faulkner C, Lowis SP, Kurian KM. Current Understanding of BRAF Alterations in Diagnosis, Prognosis, and Therapeutic Targeting in Pediatric Low-Grade Gliomas. Front Oncol. 2015;5:54. doi: 10.3389/fonc.2015.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, Fisher PG, Rowitch DH, Ford JM, Berger MS, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70:512–519. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Sigler PB. Transcriptional activation. Acid blobs and negative noodles. Nature. 1988;333:210–212. doi: 10.1038/333210a0. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Singh SK, Fiorelli R, Kupp R, Rajan S, Szeto E, Lo Cascio C, Maire CL, Sun Y, Alberta JA, Eschbacher JM, et al. Post-translational Modifications of OLIG2 Regulate Glioma Invasion through the TGF-beta Pathway. Cell Rep. 2016;16:950–966. doi: 10.1016/j.celrep.2016.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Meijer DH, Alberta JA, Mehta S, Kane MF, Tien AC, Fu H, Petryniak MA, Potter GB, Liu Z, et al. Phosphorylation state of Olig2 regulates proliferation of neural progenitors. Neuron. 2011;69:906–917. doi: 10.1016/j.neuron.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, McFarland BC, Drygin D, Yu H, Bellis SL, Kim H, Bredel M, Benveniste EN. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin Cancer Res. 2013;19:6484–6494. doi: 10.1158/1078-0432.CCR-13-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.