

ABSTRACT
Exon 16 of protein 4.1R encodes a spectrin/actin-binding peptide critical for erythrocyte membrane stability. Its expression during erythroid differentiation is regulated by alternative pre-mRNA splicing. A UUUUCCCCCC motif situated between the branch point and the 3′ splice site is crucial for inclusion. We show that the UUUU region and the last three C residues in this motif are necessary for the binding of splicing factors TIA1 and Pcbp1 and that these proteins appear to act in a collaborative manner to enhance exon 16 inclusion. This element also activates an internal exon when placed in a corresponding intronic position in a heterologous reporter. The impact of these two factors is further enhanced by high levels of RBM39, whose expression rises during erythroid differentiation as exon 16 inclusion increases. TIA1 and Pcbp1 associate in a complex containing RBM39, which interacts with U2AF65 and SF3b155 and promotes U2 snRNP recruitment to the branch point. Our results provide a mechanism for exon 16 3′ splice site activation in which a coordinated effort among TIA1, Pcbp1, and RBM39 stabilizes or increases U2 snRNP recruitment, enhances spliceosome A complex formation, and facilitates exon definition through RBM39-mediated splicing regulation.
KEYWORDS: alternative splicing, erythropoiesis, Pcbp1, protein 4.1R, RBM39, TIA1
INTRODUCTION
Alternative pre-mRNA splicing provides for the generation of numerous protein isoforms with diverse biological functions from a single gene (1–3). The excision of introns followed by the joining of exons is catalyzed by the spliceosome (4), which is assembled by the stepwise addition of discrete small nuclear ribonucleoprotein particles (U1, U2, U4, U5, and U6 snRNPs) and numerous accessory non-snRNP splicing factors (5, 6) on the pre-mRNA.
Splicing requires the presence of three critical sequence elements: the 5′ splice site (5′ ss), the 3′ splice site (3′ ss), and the branch point (BP) (7). The initial step is 5′ ss recognition by U1 snRNP and binding of SF1 and a U2 snRNP auxiliary factor (U2AF) to the 3′ ss. SF1 binds the branch point sequence (BPS), whereas the large subunit of U2AF (U2AF65) binds the polypyrimidine tract and the small U2AF subunit (U2AF35) binds the AG dinucleotide (8–12). These factors and additional proteins form the early complex E. It bridges the intron and brings the splice sites together. In the presence of ATP, U2AF recruits the U2 snRNP and converts the E complex to the A complex by interacting with U2AF65 and SF3b155 (13), by binding of U2 snRNP proteins at the BPS (14), and by base pairing of U2 snRNA with the BPS (15). U2 snRNP binding provides a platform for the tri-snRNP U4/U5/U6 and other factors. This fully assembled spliceosome supports a series of rearrangements via RNA-RNA and RNA-protein interactions that activate catalytic steps of cleavage, exon joining, and intron release (1, 2).
Splice site signals that define the 5′ ss and 3′ ss of an alternatively spliced exon are often weak, and the mammalian BP is an extremely degenerate motif (16). Splice site use is modulated by a complex interplay of positive and negative cis elements and trans-acting factors (1, 2) that govern alternative splicing. Splicing regulation occurs throughout the assembly pathway. Many factors enhance splicing by recruiting and/or stabilizing U1 snRNP to the 5′ ss or U2 snRNP to the 3′ ss. For example, T-cell intracellular antigen 1 (TIA1) regulates splicing by binding to U-rich motifs that are abundant in introns downstream of the 5′ ss in numerous alternatively spliced exons (17). TIA1 interacts with U1C, stabilizes the pre-mRNA U1 snRNP, and activates the 5′ ss of the target exon (18). Position- or context-dependent regulation by factors also occurs. RBFOX proteins bind to (U)GCAUG motifs, repressing splicing when located upstream of the exon (19) but activating splicing when located downstream (20–23).
Some factors have a direct or indirect role in the recruitment or assembly of spliceosomes. For example, the splicing-promoting activity of poly(C)-binding protein 1 (PCBP1) can arise either from direct binding to a C-rich polypyrimidine tract and interaction with U2 snRNP complex (24) or through interaction with RBM39 (also known as HCC1.3, Caper α, FSAP59, and RNPC2) (25). RBM39, a paralogue of U2AF65, interacts with U2AF65 and functions in the pre-mRNA splicing pathways of Saccharomyces cerevisiae, Drosophila, and humans (26–33). U2 snRNP-associated SF3b155 contains U2AF ligand motifs (ULM); it coordinates the recruitment of U2AF homology motifs (UHM) that contain RBM39 to the pre-mRNA 3′ ss (34). The net effect of RBM39 in pre-mRNA splicing is to recruit and/or stabilize U2 snRNP at the BP through these interactions (32–34).
The pre-mRNA binding sequences for many of the RNA binding proteins lack tight consensus motifs; cooperative recognition of these signals by multiple factors is required for efficient splicing. Examples include Fox-3 and PSF interacting to activate neural-cell-specific alternative splicing (35), ASD-2 and SUP-12 cooperatively switching alternative pre-mRNA splicing patterns of the ADF/cofilin gene in Caenorhabditis elegans (36), and RBFOX and SUP-12 sandwiching a G base to regulate tissue-specific splicing (37, 38).
The 80-kDa erythrocyte 4.1R is the prototype of a diverse array of protein 4.1R isoforms. Expression of its exon 16 (E16), which encodes peptides within the spectrin/actin-binding domain needed for the mechanical stability of the red cell membrane (39–41), is induced during late erythroid differentiation. Its absence results in hereditary elliptocytosis (42). We now report that UUUUCCCCCC, situated at bp −15 to −24 upstream of the 3′ ss, is vital for exon 16 splicing. TIA1 and Pcbp1 bind to the U and last three-C region, respectively, and activate exon 16 in a collaborative manner. TIA1 and Pcbp1 exert a notably more pronounced effect in cell types that express high levels of RBM39, whose expression increases markedly during late erythroid differentiation when exon 16 is induced. TIA1 interacts with Pcbp1 and then associates with RBM39 in complexes with U2AF65 and SF3B155. This favors U2 snRNP recruitment to the BP and formation of the spliceosome A complex. Our results suggest a potential molecular basis for 4.1R exon 16 3′ ss activation that requires coordination among TIA1, Pcbp1, and RBM39.
RESULTS
Two upstream pyrimidine-rich elements within the region of −1 to −25 are important for TIA1 responsiveness and exon 16 inclusion.
We (43) along with others (21, 44) previously showed that several cis elements mediate the erythroid differentiation-specific splicing in of 4.1R exon 16. In addition to factors identified earlier (21, 22, 44, 45), we found that TIA1 activates exon 16 inclusion on both endogenous 4.1R mRNA and an exon 16 minigene template. The wild-type (WT) exon 16 minigene replicated the endogenous splicing pattern with ∼12% exon 16 inclusion. Inclusion increased to ∼44% and 37% in response to TIA1 expression on endogenous 4.1R and exon 16 minigene pre-mRNA, respectively (Fig. 1C).
FIG 1.

TIA1 facilitates exon 16 inclusion through two pyrimidine-rich regions located between the branch point and the 3′ ss. (A) Schematic representation of the exon 16 minigene construct. Primers used in RT-PCR are indicated by arrows. (B) RT-PCR primers are minigene specific. MEL and HeLa cells were transiently transfected with the exon 16 minigene for 40 h; RNA isolated from transfected (+) and untransfected (−) cells was analyzed by RT-PCR. (C) TIA1 activates exon 16 splicing on both endogenous 4.1R and minigene pre-mRNA. The WT minigene was cotransfected with a vector (−) or HA-TIA1 (+) in MEL cells and analyzed for endogenous and exogenous exon 16 expression. E16 inclusion was calculated as the percentage of total RNA products containing exon 16. Averages and SDs were obtained for three independent experiments (n = 6), and results are presented at the bottom of each lane and as a bar graph. Anti-HA antibody detected HA-TIA1. β-Actin served as a loading control. (D) TIA1 effect depends on the U-rich (U-ISE) and C-rich (C-ISE) region upstream of exon 16. The top panel shows mutated minigene constructs with the replaced nucleotides indicated. The middle panel shows the effect of each mutation on exon 16 inclusion in the presence of vector (V), TIA1, or TIAR. Anti-HA antibody detected HA-TIA1 or HA-TIAR. As shown in the bottom panel, the C-rich region is critical for exon 16 inclusion even in the presence of a consensus 5′ ss. Minigenes with a consensus 5′ ss in the presence of the wild-type (AA) or mutated C-rich region (Xm/AA) were analyzed for exon 16 inclusion in response to TIA1. IB, immunoblotting.
TIA1 binds to U-rich sequences downstream of the 5′ ss and promotes U1 snRNP recruitment (18). Twelve putative binding motifs (46) span upstream and downstream introns of exon 16 (Fig. 1D, m1 to m11). Our analyses showed that, surprisingly, activation of exon 16 by TIA1 does not occur via these downstream sites. We thus expanded the search for TIA1-responsive elements (Fig. 1D). We knew that disrupting the upstream intronic sequence (−1 to −25) (GIa) resulted in complete exon 16 exclusion (43) and that altering two pyrimidine-rich elements within the region of −1 to −25, three Cs (−15 to −17) or four Us (−21 to −24), abolished exon 16 inclusion (Fig. 1D). The C residues in positions −15 to −17 are situated between the BP (−42) and the 3′ ss of exon 16. Fortunately, the Branch-site Analyzer Tool (http://ibis.tau.ac.il/ssat/BranchSite.htm) predicted that mutations there would reflect TIA1 effects since they would not impact the branch site score or change the branch site position.
We tested whether TIA1 or TIAR (TIA-1-related protein) could facilitate the splicing of these mutated minigenes. TIA1 exerted a strong enhancing effect on WT exon 16 inclusion (∼35%), but TIAR had a minimal effect (∼12%) (Fig. 1D). In m67, enhancing effects were also observed only for TIA1 (∼25%). Conversely, GIa, Xm, and Up minigenes exhibited complete exon 16 exclusion in the presence of TIA1 or TIAR (Fig. 1D). These results suggest that two cassettes upstream of the 3′ ss are required for TIA1's effects even though they do not match the TIA1 consensus. They are designated here as U-rich and C-rich intronic splicing enhancers (U-ISE and C-ISE, respectively).
Exon 16 possesses a weak 5′ ss. Reinforcing the site with a 5′ ss consensus sequence (AA) increases exon 16 inclusion to ∼95% (47). We analyzed the importance of the C-ISE on exon 16 inclusion in the presence of a consensus 5′ ss (Xm/AA). The AA construct with the consensus 5′ ss resulted in ∼90% exon 16 inclusion; TIA1 increased inclusion to ∼98% (Fig. 1D, bottom panel, lanes AA). The Xm/AA construct with the C-ISE mutation reduced exon 16 inclusion to ∼37% and abolished the TIA1 effect (Fig. 1D, lanes Xm/AA). C-ISE thus enhances exon 16 splicing with either a strong or a weak 5′ ss.
Two protein complexes assemble on the U-ISE and C-ISE regions.
Since TIA1 had not previously been shown to bind to C-rich sequences, we reasoned that a more complicated binding scenario occurred around the C-ISE region. We performed electrophoretic mobility shift assays (EMSAs) using probes consisting of positions −15 to −17 and flanking sequences derived from the WT or Xm minigene (yielding a wt or xm probe, respectively) (Fig. 2A). In HeLa cell nuclear extracts, wt probes shifted two bands; xm probes shifted only one band that migrated to the same position as that of the wt upper band (Fig. 2B). Intensity of the retarded bands increased when either probe was incubated with increasing amounts of extract (Fig. 2B).
FIG 2.

Two distinct RNA-protein complexes are formed on the U-ISE and C-ISE regions. (A) Wild type (wt) and C-mutated (xm) RNA probes used for EMSA. (B) wt probe formed two RNA-protein complexes; xm probe formed one complex in HeLa extracts. NE, nuclear extract (in microliters); B, RNA-protein complex; P, probe. (C and D) Specificity of the RNA-protein complexes analyzed by competition assay. A 2- to 50-fold molar excess of unlabeled wt or xm RNAs (comp, competitor) was added along with the biotinylated wt or xm probe. −, probe incubated without competitors. (E) HeLa cell nuclear extract lacking xm binding proteins (Δxm) do not form upper RNA complexes. Extracts were depleted by preincubation with streptavidin-Sepharose beads containing biotinylated xm probes. Depleted extracts were analyzed by EMSA with wt or xm probe in the absence (−) or presence of a 5-, 20-, or 50-fold molar excess of unlabeled competitors (comp). (F) Probes used to map the RNA-protein complex binding sites shown in panel G. (G) EMSA performed using the wt or mutated probe (up, u/d, dn, xm, and xup) and HeLa nuclear extracts. −, probe alone; B, RNA-protein complex; P, probe.
To assess the specificity of binding, competitor assays were performed with 1- to 25-fold molar excesses of unlabeled wt or xm RNAs. Both wt complexes were partially competed away by a 5-fold excess of wt and completely inhibited by a 25-fold excess (Fig. 2C). Only the upper band was diminished by the same concentrations of unlabeled xm RNAs (Fig. 2C). Both unlabeled wt and xm RNAs impeded the xm-retarded band (Fig. 2D). These results suggest that the lower wt RNA-protein interaction is specific to the last three-C sequence, while the upper RNA-protein interaction is similar to that of xm.
When EMSA was performed using nuclear extracts depleted by xm RNA, wt probes shifted only the lower band. This complex was competed away by an unlabeled wt probe at the same efficiency as that in undepleted nuclear extracts (Fig. 2E). The upper retarded band with the xm probe completely disappeared in xm-depleted nuclear extracts (Fig. 2E). These results demonstrate that the upper complexes in the wt are identical to the complexes formed on xm.
wt and xm probes share U-rich elements flanking the central C-rich region. The upstream U-ISE region is required for TIA1 enhancement. To test whether U-ISE is responsible for the upper complexes, EMSA was done using wt probes with upstream (up), downstream (dn), or both U mutations (u/d) (Fig. 2F). The up and u/d mutations exclusively retained the lower complexes (Fig. 2G, up and u/d). The dn mutations shifted two complexes with results similar to those of the wt (Fig. 2G, dn). The upper complex is thus upstream U specific. This was validated with the xup probe in which the upper U-rich region in the xm probe was mutated (Fig. 2F). xup also abolished the upper retarded band (Fig. 2G, xup). Additional studies showed that, among the upstream mutations, only the four Us immediately upstream of the Cs play a role (data not shown). Thus, binding at the upstream four Us likely accounts for the upper complex.
RNA affinity purification identifies proteins associated with the pyrimidine-rich elements.
To characterize the proteins associated with the U-ISE and C-ISE regions, we purified them from HeLa cell nuclear extracts using an RNA affinity column containing wt probes. Bound proteins were stepwise eluted and tested by EMSA. KCl at 0.5 M eluted the upper complex while the 1.25 M and 1.50 M KCl fractions contained the lower complex. The 0.75 M and 1.0 M KCl fractions retained both complexes (Fig. 3).
FIG 3.
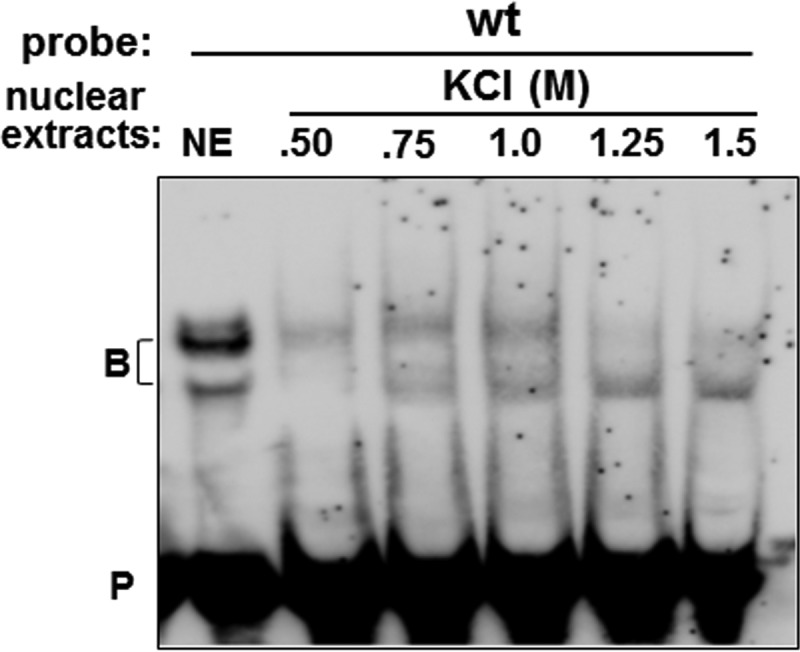
EMSAs of nuclear proteins fractionated by RNA affinity chromatography. HeLa cell nuclear extract was incubated with wt probes bound to streptavidin-Sepharose beads. Bound proteins were washed and eluted with increasing concentrations of KCl (see Materials and Methods). EMSA was performed using wt probes with 1 μl of nuclear extracts (NE) or 10 μl of the eluted KCl fractions. B, RNA-protein complex; P, probe.
The 1.0 M KCl eluate was sequenced by the Harvard Mass Spectrometry and Proteomics Facility. Among ∼200 proteins identified, many are associated with the 3′ ss but not the 5′ ss splicing machinery. Table 1 shows the representative proteins identified. Notably, U2AF subunits (U2AF35 and U2AF65), U2 snRNP-specific proteins (subunits of SF3A and SF3B), and BP binding SF1 are abundant. As anticipated, TIA1/TIAR and TIA1-interacting proteins (FUBP1, KHSRP, and FUBP3) are among the most prevalent species identified. A group of poly(C)-binding proteins (PCBPs; PCBP2, PCBP1, PCBP4, and hnRNPK) and PCBP1-interacting protein, RBM39, were also identified.
TABLE 1.
Representative proteins associated with the wt RNA probes
| Name or group | Associated protein(s) (accession no.) |
|---|---|
| Poly(C)-binding proteins | PCBP2 (Q15366; B4DLC0), PCBP1 (Q15365), PCBP4 (P57723), hnRNPK (highly similar; B4DUQ1) |
| PCBP-interacting protein | RBM39 (Q14498) |
| TIA1 | TIAL1 (Q01085), TIA1 (C9JTN7), p40-TIA1 (P31483) |
| TIA1-interacting proteins | FUBP1 (B4E0X8; Q96AE4), KHSRP/FUBP2 (Q92945), FUBP3 (Q96I24) |
| 3′ ss proteins | |
| U2AF | U2AF2/U2AF65 (P26368), U2AF1/U2AF35 (Q01081), U2AF1L4/U2AF26 (Q8WU68) |
| U2 snRNP-specific proteins | PUF60 (Q9UHX1; E9PQ56), SF3B2 (Q13435), SF3B1 (O75533), SF3B3 (Q15393), SF3B14 (Q9Y3B4), SF3B4 (Q15427), SF3B5 (Q9BWJ5), SF3A1 (Q15459), SF3A3 (Q12874), SF3A2 (Q15428), SNRPA1/U2-A′ (P09661), SNRPB2/U2-B″ (P08579) |
| U2 snRNP core/ring proteins | SNRPB/B&B′ (P14678), SNRPD2 (P62316), SNRPD1 (P62314), SNRPE (P62304), SNRPD3 (P62318), SNRPF (P62306) |
| Branch binding protein | SF1 (Q15637) |
| U2 snRNP-associated protein | U2SURP (O15042) |
TIA1 and Pcbp1 bind to U-ISE and C-ISE regions.
TIA1 and PCBPs have strong affinities for U-rich (17) and C-rich sequences (48), respectively. We thus asked whether a specific interaction existed between TIA1 or PCBP with a wt or xm probe.
TIA1/glutathione S-transferase (GST) and TIA1, but not GST, formed a complex with the wt or xm probe (Fig. 4B). Binding of TIA1 was further mapped to the upstream U-rich sequences; mutations in this region (Fig. 2F) abolished the complex (Fig. 4B, up). These results suggest that the RNA-TIA1 interaction occurs on sequences common to both the wt and xm probes; TIA1 binding also requires the upstream U-rich sequence. TIA1 possesses a Q-rich and three RRM domains (Fig. 4A). Only RRM2 formed a complex with the wt probe (Fig. 4C). Combinations of domains that included RRM2 also formed a complex (Fig. 4C). RRM2 of TIA1 is thus the domain that interacts with the U-rich region.
FIG 4.

TIA1 binds to the U-ISE region through the RRM2 domain. (A) Schematic diagram of TIA1 and subdomains analyzed by EMSA. Residue numbers and domains are indicated. (B) EMSA with wt, xm, or up probe (sequences are shown in Fig. 2A and F) and purified TIA1. The left panel shows results for Coomassie blue-stained TIA1/GST, TIA1, and GST. Middle and right panels show results for the wt, xm, or up probe incubated with TIA1/GST, TIA1, or GST and then analyzed by EMSA. (C) EMSA with purified individual or combined domains of TIA1. Coomassie blue-stained GST fusions of TIA1 domains and RNA-protein complexes formed on WT probes are shown, as indicated. B, RNA-protein complex; P, probe.
PCBP is a subfamily of KH domain-containing RNA binding proteins. PCBP1 and PCBP2 are predominantly nuclear with specific enrichment of PCBP1 in nuclear speckles, while PCBP3 and PCBP4 are restricted to the cytoplasm (49). Thus, we analyzed the interaction of Pcbp1 with the wt or xm probe.
Pcbp1/GST formed an intense upper and a faint lower band with the wt probes. Increased Pcbp1 significantly enhanced the intensity of the upper band and, to a lesser extent, the lower band (Fig. 5B, middle panel, wt). The xm probe formed a fainter shifted band that migrated similarly to the wt lower band (Fig. 5B, middle panel, xm). The upper complex assembled on the wt probes was competed away by a 10- or 25-fold molar excess of unlabeled wt but not xm RNA (Fig. 5B, right panel). The lower band was not competed away, suggesting nonspecific binding. These results suggest that Pcbp1 interacts with the wt probes specifically in region of the last three C residues. Of its three KH domains (Fig. 5A), the KHI domain was responsible for the binding (Fig. 5C). Thus, Pcbp1 binds to the last three-C region through its KHI domain.
FIG 5.

Pcbp1 binds to the C-ISE region through the KHI domain. (A) Schematic diagram of Pcbp1 and its domains. Residue numbers and domains are indicated. (B) EMSA with wt or xm probe (sequences are given in Fig. 2A) and purified Pcbp1. The left panel shows Coomassie blue staining for Pcbp1/GST. The middle panel shows results of an EMSA with wt or xm probe and nuclear extracts (NE) or increasing amounts of Pcbp1. −, probe alone. The right panel represents competition analyses employing a 10- or 25-fold molar excess of unlabeled wt or xm RNA. (C) EMSA with wt probe and purified individual KH domains of Pcbp1. The left panel shows Coomassie blue staining for GST fusions of Pcbp1 KH domains. The right panel shows RNA-Pcbp1 KH domain protein complexes formed on wt probes. B, RNA-protein complex; P, probe.
Coordination between TIA1 and Pcbp1 is critical for exon 16 splicing.
The Xm minigene with the three-C (−15 to −17) mutation abolished the effect of TIA1 on exon 16 splicing (Fig. 1D). TIA1 binds to the upstream U-rich region whereas Pcbp1 binds to the C-rich regions. We examined whether TIA1 and Pcbp1 and their respective binding sites are interdependent in their enhancing activities. The WT, C-mutated (Xm), or U-mutated (Up) exon 16 minigene (Fig. 6A) was cotransfected with TIA1, Pcbp1, or both TIA1 and Pcbp1 (TP) and analyzed for exon 16 inclusion.
FIG 6.

Combined TIA1 and Pcbp1 action on U-ISE and C-ISE is critical for exon 16 splicing. (A) Schematic diagram of the WT, Xm, and Up exon 16 minigenes showing replaced sequences at positions −15 to −24. (B and C) Splicing patterns of WT, Xm, or Up in the presence of vector (V), TIA1 (T), Pcbp1 (P), or both TIA1 and Pcbp1 (TP). MEL or HeLa cells were transfected with the indicated constructs and analyzed for exon 16 expression. E16 inclusion was calculated as the percentage of total RNA products containing exon 16. Averages and SDs were obtained for three independent experiments (n = 6), and results are presented at the bottom of each lane and as a bar graph. Anti-HA antibody detected HA-TIA1, and anti-Flag antibody detected Flag-Pcbp1. Actin served as a loading control.
In uninduced murine erythroleukemia (MEL) cells, exon 16 inclusion in the WT minigene increased from 10% in the absence of both factors to 37% with TIA1 and 25% with Pcbp1 (Fig. 6B, WT). However, no additional increase was noted when both proteins were coexpressed (Fig. 6B, WT, lane TP). Xm and Up reduced exon 16 inclusion to 4% and 0%, respectively, and completely abolished responsiveness to TIA1 and/or Pcbp1 (Fig. 6B, Xm and Up). These results suggest that the presence of both the U-ISE and C-ISE regions is necessary to elicit a positive effect from either TIA1 or Pcbp1 overexpression.
We next analyzed whether TIA1 and Pcbp1 exerted the same effects in different cell types. TIA1 or Pcbp1 alone enhanced exon 16 inclusion from ∼65% to 80% or 73%, respectively, in HeLa cells. In contrast to the MEL cell system, coexpression increased enhancement to ∼93% exon 16 inclusion (Fig. 6C, WT, lane TP). As in MEL cells, Xm and Up abolished the effect of either factor on exon 16 splicing (Fig. 6C, Xm and Up). These results further suggest that coordination between Pcbp1 and TIA1 is likely critical for exon 16 splicing. The differing results between HeLa and MEL cells led us to surmise that TIA1 and Pcbp1 might work with another factor(s), expressed in limiting amounts in uninduced MEL cells.
Erythroid differentiation stage- and cell-type-specific expression of Pcbp1-interacting protein RBM39.
The impact of Pcbp1 has been shown by other investigators to be mediated by other cell types through its interacting protein RBM39 (25). We thus analyzed the effect of RBM39 on exon 16 splicing regulation. Exon 16 increased from 12% to ∼25% when RBM39 was overexpressed in uninduced MEL cells (Fig. 7A). Since HeLa cells express higher levels of exon 16, we analyzed the effect of RBM39 depletion on exon 16 inclusion. We observed that when RBM39 was decreased by 80%, exon 16 inclusion was reduced from 78% to ∼57% (Fig. 7B). The effect depended on the presence of TIA1 and Pcbp1 binding sequences because mutation of either abolished the effect of RBM39 (Fig. 7C). An association among TIA1, Pcbp1, and RBM39 thus seems to be critical for exon 16 splicing regulation.
FIG 7.

RBM39 activates exon 16 splicing in a U-ISE- and C-ISE-dependent manner, and its expression levels correlate with exon 16 inclusion. (A) RBM39 facilitates exon 16 inclusion in MEL cells. The E16 WT minigene was cotransfected with vector (V), HA-TIA1 (TIA1), Flag-Pcbp1 (Pcbp1), or HA-RBM39 (RBM39) and analyzed for exon 16 inclusion. Average and SDs of expression levels obtained from three independent experiments (n = 6) are indicated at the bottom of each lane. In immunoblotting (IB), anti-HA antibody detected HA-TIA1 and HA-RBM39, and anti-Flag antibody detected Flag-Pcbp1. β-Actin served as a loading control. (B) RBM39 depletion reduced exon 16 inclusion in HeLa cells. RNA from HeLa cells treated with a control (−) or a RBM39 siRNA (+) and transfected with the E16 WT minigene was analyzed for exon 16 splicing. RBM39 knockdown was validated with an anti-RBM39 antibody. β-Actin served as a loading control. (C) Effect of RBM39 depends on the U-ISE and C-ISE regions. The E16 Xm or E16 Up minigene (Fig. 6A) cotransfected with a vector (V) or HA-RBM39 was analyzed for exon 16 inclusion. (D) RBM39 expression increases as exon 16 inclusion increases during MEL cell differentiation. Exon 16 splicing during DMSO induced MEL differentiation (top panel). Levels of endogenous TIA1, Pcbp1, and RBM39 are shown in the same set of samples (lower panel). (E) Endogenous and transfected proteins expressed in HeLa and MEL cells. Lysates from cells transfected with vector (−) or expression plasmids (+) were probed by Western blotting for the presence of endogenously (endo) and exogenously (exo) expressed TIA1, Pcbp1, and RBM39. (F) RBM39 does not interact with wt probes. Results of Coomassie blue staining for RBM39/GST are shown at left, and results of EMSAs with wt probes and nuclear extract (NE) or an increasing amount of RBM39 are shown at right. −, probe alone. (G) TIA1 interacts with Pcbp1 in a blot overlay assay. Results of Coomassie blue staining for BSA, actin, GST, U1C/GST, and Pcbp1/GST are shown at left. Note the purified U1C/GST fusion proteins (arrowhead) and contaminated host proteins (bracket). In the experiment shown on the right, a replicate of the gel was overlaid with purified TIA1/His and then subjected to Western blotting with an anti-His antibody.
We examined the expression patterns of TIA1, Pcbp1, and RBM39 during dimethyl sulfoxide (DMSO)-induced MEL cell differentiation. TIA1 expression slightly increased and then decreased, and Pcbp1 expression was unchanged throughout differentiation (Fig. 7D), but RBM39 expression drastically increased greater than 3-fold (Fig. 7D). Since an increased enhancing effect of combining TIA1 and Pcbp1 occurs only in HeLa and not in uninduced MEL cells, we further compared the expression levels of these proteins in these cells. While no differences in TIA1 and Pcbp1 expression levels were noted, RBM39 levels were approximately 2.5- to 4-fold higher in HeLa cells than in uninduced MEL cells (Fig. 7E). Exogenously expressed levels of TIA1 and Pcbp1 were comparable to endogenous protein expression levels in both HeLa and MEL cells. Conversely, exogenous levels of RBM39 were twice that of endogenous protein in MEL cells but only ∼50% that of endogenous protein in HeLa cells (Fig. 7E). Importantly, we observed no change in proliferation when MEL cells were transiently transfected with TIA1-, Pcbp1-, and RBM39-expressing plasmids. Similarly, doubling TIA1 and Pcbp1 or tripling RBM39 levels had no effect on erythroid differentiation (data not shown).
RBM39 enhanced exon 16 inclusion but did not interact directly with the wt probe (Fig. 7F). The effect of RBM39 is thus most likely mediated by interaction with Pcbp1.
TIA1-Pcbp1 interactions were analyzed in a blot overlay assay. TIA1-interacting protein U1C (18) was used as a positive control. Since U1C/GST was insoluble, we attempted to extract the fusion protein causing copurification of several low-molecular-weight bacterial proteins (Fig. 7G, left panel, bracket), but U1C and Pcbp1 were clearly recognized by TIA1 (Fig. 7G, right panel). Thus, TIA1 interacts with Pcbp1 in an RNA-independent manner.
The juxtaposed enhancers activate an internal exon in a neutral reporter system.
We examined the impact of U-ISE and C-ISE in the context of a neutral reporter, DUP4-1 (20). DUP4-1 supports a binary splicing pattern in which inclusion of the internal chimeric exon 2 (E2) requires addition of enhancer sequences (20).
DUP4-1 minigenes in which native sequences upstream of E2 (−15 to −28) were replaced with the corresponding exon 16 sequences (WT, Xm, or Up) are designated DUP-TP, DUP-Xm, and DUP-Up, respectively (Fig. 8A). DUP4-1 excluded E2 in MEL cells (Fig. 8C). Cotransfection with TIA1, Pcbp1, or both had no effect on E2 inclusion (Fig. 8C). DUP-TP supported ∼95% E2 inclusion with or without TIA1 or Pcbp1. DUP-Xm yielded only ∼7% inclusion, and DUP-Up resulted in 0%, with or without TIA1 or Pcbp1 (Fig. 8C). We obtained similar results in HeLa cells (Fig. 8D). These results suggest that the impact of U-ISE and C-ISE is not restricted to an exon 16 context since they are as essential in the DUP reporter.
FIG 8.

U-ISE and C-ISE sequences activate an internal exon when placed in a corresponding position in a heterologous reporter. (A) Schematic diagram of the DUP4-1 minigene and mutants with replaced sequences at positions −15 to −28. RT-PCR primers are indicated by arrows. (B) RT-PCR primers are minigene specific. MEL and HeLa cells were transiently transfected with the DUP-Xm minigene for 40 h; RNA from transfected (+) and untransfected (−) cells was analyzed by RT-PCR. (C and D) Splicing patterns of DUP4-1, DUP-TP, DUP-Xm, and DUP-Up in the presence of vector (V), HA-TIA1 (T), Flag-Pcbp1 (P), or both HA-TIA1 and Flag-Pcbp1 (TP). MEL or HeLa cells transfected with the indicated constructs were analyzed for the percentage of E2 inclusion. E2 inclusion was calculated as the percentage of total RNA products containing E2. Averages and SDs were calculated from three independent experiments (n = 6), and results are presented at the bottom of each lane and as a bar graph. Anti-HA and anti-FLAG antibodies detected HA-TIA1 and Flag-Pcbp1. β-Actin served as a loading control.
TIA1/Pcbp1-mediated splicing enhancement acts through the branch point.
Since TIA1 and Pcbp1 were identified along with many 3′ ss-associated factors in proteins assembled on the intronic splicing enhancer (Table 1), we reasoned that they might act through the 3′ ss and its obligatory signal, the BP.
E2 of DUP has a strong BPS (BPS, CACUGAC; consensus sequence, YNYYRAY). The BPS promotes base pairing with U2 snRNAs. Base replacements in the BPS impede splicing by disrupting this base pairing (50). To ascertain whether TIA1/Pcbp1 enhancement is BP dependent, we substituted sequences that are known to weaken the BPS (50, 51). Mutation mu4 (CACAGAC; the mutation is underlined) drastically reduces BP strength, while mu5 (CACUUAC) and mu7 (CACUGAG) cause intermediate impairment (Fig. 9A) (50). Each mutation impaired E2 inclusion in MEL cells, reducing it from 95% to 5% (mu4), 72% (mu5), and 58% (mu7) (Fig. 9B). We then asked whether TIA1 and/or Pcbp1 could enhance splicing on these constructs. Neither factor caused more than minimal enhancement with DUP-TP and mu4 (Fig. 9B), suggesting that neither has enhancing activity in the presence of a consensus or extremely weak BP. TIA1, Pcbp1, or both improved E2 splicing moderately with mu5 and mu7 (Fig. 9B). The enhancing action of these proteins was noticeable only when a weak BP was present. TIA1 and/or Pcbp1 binding thus seems to encourage recruitment of U2 snRNP by enhancing weak 3′ ss recognition.
FIG 9.

TIA1 and Pcbp1 mediate splicing enhancement via the branch point. (A) Branch point-weakened forms of DUP-TP minigenes were constructed by single nucleotide replacements at the positions indicated. (B) TIA1 and Pcbp1 facilitate E2 inclusion on weak branch points. MEL cells transfected with the indicated minigenes in the presence of vector (V), HA-TIA1 (T), Flag-Pcbp1 (P), or both HA-TIA1 and Flag-Pcbp1 (TP) were analyzed for E2 inclusion. E2 inclusion was calculated as the percentage of total RNA products containing E2. Averages and SDs were calculated from three independent experiments (n = 6), and results are presented at the bottom of each lane and as a bar graph. Anti-HA and anti-FLAG antibodies detected HA-TIA1 and Flag-Pcbp1, respectively. β-Actin served as a loading control. (C) TIA1 and Pcbp1 associate with RBM39 and 3′ ss factors U2AF65 and SF3b155. DMSO-induced MEL cell lysates immunoprecipitated (IP) with goat IgG (GIgG), anti-TIA1, anti-Pcbp1, rabbit IgG (RIgG), anti-U2AF65, anti-SF3b155, mouse IgG (MIgG), anti-Numb, or anti-GAPDH antibody, as indicated, were probed for their associated proteins.
TIA1 and Pcbp1 proteins form a complex with RBM39 and U2 snRNP-associated proteins.
Stable U2 snRNP recruitment to the BP requires interactions between SF3b155 and U2AF65. RBM39 might also play a role via its interactions with SF3b155 and U2AF65 (30, 32–34). We asked whether TIA1 and Pcbp1 participation in U2 snRNP recruitment involves associations with RBM39, SF3b155, and/or U2AF65. When immunoprecipitated with TIA1 or Pcbp1 antibodies, day 4 DMSO-induced MEL cell extracts (expressing high levels of RBM39) yielded RBM39, U2AF65, and SF3b155 in anti-TIA1 and anti-Pcbp1 precipitates. Reverse coimmunoprecipitation yielded TIA1, Pcbp1, and RBM39 in anti-SF3b155 and anti-U2AF65 precipitates (Fig. 9C). No coprecipitations were found with negative controls (Fig. 9C). We did not detect a clear association between U2AF35 and either TIA1 or Pcbp1. These results demonstrate that TIA1 and Pcbp1 associate with U2 snRNP via the U2AF65 subunit of U2AF.
TIA1 and Pcbp1 binding sequences promote recruitment of U2 snRNP and spliceosome A complex formation.
TIA1 and Pcbp1 strengthen a weak BP and associate with U2AF and U2 snRNP. They might then promote spliceosome A complex formation in this manner. To test this idea, we first truncated the downstream tandem repeat sequences of the DUP4-1, DUP-TP, DUP-Up, and DUP-Xm minigenes to form constructs (Fig. 10A) that allow splicing only on the upstream intron (designated dup4-1A, dup-tpA, dup-upA, and dup-xmA, respectively). This ensures that the transcript assembles into a single spliceosome that can be readily assayed.
FIG 10.

TIA1 and Pcbp1 binding sequences are required for U2 snRNP recruitment and spliceosome A complex formation. (A) Schematic representation of a single intron template containing exon 1 through exon 2 of DUP4-1 and its mutant derivatives. RNA substrates containing the wild type (dup4-1A), both TIA1 and Pcbp1 binding sequences (dup-tpA), a mutated TIA1 site (dup-upA), and a mutated Pcbp1 site (dup-xmA) are represented. (B) Psoralen-dependent cross-linked species detected after incubation of 32P-labeled RNA substrates in HeLa cell nuclear extracts. The identity of the cross-linked species was determined by hybridizing nuclear extracts to DNA complementary to positions 1 to 15 of U1 snRNA or 28 to 42 of U2 snRNA, followed by RNase H digestion before cross-linking. The positions of free substrates, U1 snRNA, and U2 snRNA cross-linked complexes are indicated. (C) Neither TIA1 nor Pcbp1 binding sequence affects E complex formation. RNA transcripts were incubated in ATP-depleted HeLa cell nuclear extracts and incubated at 0°C or 30°C for the indicated times, fractionated on 1.5% low-melting-point agarose gels, fixed, dried, and exposed to X-ray film. Prespliceosomal H and E complexes are indicated. (D) TIA1 and Pcbp1 binding sequences activate spliceosomal A complex formation. Transcripts were incubated in ATP-depleted (−ATP) or nondepleted (+ATP) HeLa cell nuclear extracts at 30°C for the indicated times and processed as described for panel C. Prespliceosomal H and E (H/E) complexes as well as A and B complexes are indicated. (E) Correlation between U2 snRNP recruitment/spliceosomal A complex formation and splicing efficiency. The top panel shows a schematic representation of the dup minigene construct. Primers used in RT-PCR are indicated by arrows. RT-PCR primers are minigene specific (middle panel). RNA was isolated from dup-tpA transfected (tp) and untransfected (−) cells after 40 h and analyzed by RT-PCR. dup4-1A and its mutant constructs were transfected into HeLa or MEL cells and analyzed for unspliced (E1-E2) and spliced (E12) products (bottom panel). Splicing efficiency was calculated as the percentage of total RNA products containing spliced E12. Averages and SDs were obtained for three independent experiments (n = 6), and results are presented at the bottom of each lane. CMV, cytomegalovirus.
BP recognition requires base pairing with bases 28 to 42 of U2 snRNA. We used psoralen-mediated UV cross-linking assays to detect U2 snRNP recruitment to various RNA substrates. dup-tpA produced two psoralen-dependent cross-linked bands (Fig. 10B). Both dup4-1A and dup-upA produced one band at the same position as that of the dup-tpA upper band. dup-xmA produced an intense upper and a very faint lower band at the same positions. We analyzed these cross-linked species by RNase H digestion with anti-snRNA-specific DNA oligonucleotides. Elimination of positions 1 to 15 of U1 snRNA inhibited cross-linking of the upper band with all substrates (Fig. 10B, U1). Eliminating positions 28 to 42 of U2 snRNA abolished the lower band with both dup-tpA and dup-xmA (Fig. 10B, U2). The upper cross-linked species is thus U1 snRNP specific while the lower species is U2 snRNP specific. The presence of both TIA1 and Pcbp1 binding sequences enhanced U2 snRNP recruitment while mutations in these binding sites drastically reduced U2 snRNP recruitment.
We next tested the effect of TIA1 and Pcbp1 binding on spliceosome formation. The initial formation of the E complex is ATP independent. Subsequent complexes A, B, and C are ATP dependent. In the E complex assay, the same patterns and quantities of H and E complexes were obtained with every substrate whether or not the U- or C-rich region was present (Fig. 10C). In ATP-depleted extracts, all transcripts assembled the E/H but not the A complex (Fig. 10D, −ATP lanes). When incubated in normal extracts, the dup4-1A construct assembled a faint A complex after 2 min, which increased in intensity at 5 min; an A complex and a very weak B complex formed after 25 min. The dup-tpA formed a robust A complex at 2 min, which increased at 5 min, and then proceeded to form both an A and B complex at 25 min (Fig. 10D). dup-upA formed no complexes under the same conditions, while dup-xmA exhibited A complex formation like that of dup4-1A (Fig. 10D). These results suggest that binding sites for both TIA1 and Pcbp1 are critical and specific for efficient spliceosome A complex formation. Loss of either site drastically reduced A complex formation.
We verified that there is a correlation between U2 recruitment/spliceosome A complex formation and actual splicing efficiency. We analyzed splicing in MEL and HeLa cells transfected with the single-intron construct dup4-1A and its derivatives. Minigenes with either a U-ISE or C-ISE mutation yielded only 0 to 8% spliced product (E12) (Fig. 10E). dup-tpA yielded spliced products in HeLa (56%) and MEL (34%) cells (Fig. 10E), a finding consistent with robust U2 recruitment and A spliceosome complex formation in dup-tpA in the presence of U-ISE and C-ISE. These results suggest that the U- and C-rich regions promote actual splicing efficiency.
RBM39 interacts with TIA1 and Pcbp1 to enhance spliceosome A complex formation.
To ask whether RBM39 participated in the enhancer effects of TIA1 and Pcbp1, we added increasing amounts of TIA1, Pcbp1, or RBM39 protein onto transcripts with different combinations of TIA1 and Pcbp1 binding sites and assessed spliceosome formation. dup-tpA responded to the addition of TIA1 and Pcbp1 in a dose-dependent manner, with stronger A and B complex formation (Fig. 11B). Similar results were observed for RBM39 but with a lower impact (Fig. 11C). Mutations in either TIA1 or Pcbp1 binding sites abolished the effect of TIA1, Pcbp1, or RBM39 (Fig. 11B and C). Thus, TIA1, Pcbp1, and RBM39 aided spliceosome assembly by binding to U-ISE and C-ISE.
FIG 11.

TIA1 and Pcbp1 activation of spliceosomal A complex formation is RBM39 dependent. (A) Purified TIA1/His and Pcbp1/GST proteins. (B and C) TIA1, Pcbp1, and RBM39 enhance spliceosome A complex formation on a dup-tpA substrate containing TIA1 and Pcbp1 binding sequences. TIA1 or Pcbp1 (B) or RBM39 (C) was added at increasing concentrations into each substrate, and complex formation was analyzed. Prespliceosomal E/H complexes as well as A and B complexes are indicated. −, probe alone. (D) Validation of RBM39 silencing. Lysates from HeLa cells treated with a control (Ctrl) or RBM39 siRNA (siRBM39-1 and siRBM39-2) were analyzed for RBM39 knockdown with an anti-RBM39 antibody. Anti-TIA1 and anti-Pcbp1 antibodies detected their respective proteins. β-Actin served as a loading control. (E) RBM39-dependent A complex formation. RBM39 silencing reduces the effect of TIA1 and Pcbp1 on A complex formation (left panel). TIA1 or Pcbp1 was added at increasing concentrations into RBM39-depleted HeLa cell nuclear extracts (ΔRBM39) with dup-tpA substrate and analyzed for complex formation. RBM39 reconstituted complex A formation when added back to depleted nuclear extracts (right panel). −, probe alone.
Since the effect of RBM39 on exon 16 splicing required the TIA1 and Pcbp1 binding sites (Fig. 7C), we further investigated whether RBM39 depletion would affect spliceosome A complex. When RBM39 was depleted by 90% in HeLa cell nuclear extracts while TIA1 and Pcbp1 were maintained at the same levels (Fig. 11D), the extracts not only failed to form an A complex but also showed no TIA1- or Pcbp1-mediated enhancement of A complex formation on dup-tpA (Fig. 11E). Adding back RBM39 rescued A complex formation in a dose-dependent manner (Fig. 11E). These results suggest that an association among TIA1, Pcbp1, and RBM39 is necessary for exon 16 splicing regulation.
Taken together, our data suggest a novel mechanism for exon 16 3′ ss activation by which concerted binding by TIA1 and Pcbp1 to the upstream intronic splicing enhancer UUUUCCCCCC in the presence of RBM39 stabilizes the pre-mRNA U2 snRNP complex and promotes spliceosomal A complex formation through their concerted association with U2AF and U2 snRNP (Fig. 12).
FIG 12.

Possible mechanism for TIA1- and Pcbp1-mediated recruitment of U2 snRNP to the branch point. TIA1 and Pcbp1 interact with each other and bind to the UUUUCCCCCC enhancer elements upstream of the target exon through its RRM2 and KHI domains, respectively. Without TIA1 and Pcbp1 binding, a weak branch point is poorly recognized by U2 snRNP. This prevents A complex formation and results in exon 16 exclusion. When present, TIA1 and Pcbp1 bind to their cognate enhancers and promote recruitment and stabilization of U2 snRNP to a weak branch point via interactions with RBM39, U2AF65, and SF3b155. This stimulates A complex formation and subsequent exon inclusion.
DISCUSSION
Regulated alternative pre-mRNA splicing depends on multiple cis elements and trans-acting factors that govern the recruitment of splicing machinery to cassette exons with weak splice site sequences. Although many splicing factors and their cognate RNA sites have been characterized, it has not been possible to delineate an alternative splicing “code,” in part because the same cis elements can act as binding sites for combinations of factors in a cooperative manner. Compilation of a comprehensive list of splicing factors and their target sites is needed to better comprehend splicing regulation. Our results show that TIA1 and Pcbp1 modulate 4.1R exon 16 splicing via a cooperative interaction with an upstream intronic enhancer located between the BP and the 3′ ss. When TIA1 and Pcbp1 concurrently bind to the UUUUCCCCCC element, they promote recognition of the BPS by U2 snRNP. This requires interaction with RBM39 and association with U2AF65 and SF3B155 proteins. This TIA1/Pcbp1-mediated regulation occurs at an early step of an ATP-dependent reaction in the splicing pathway. This interdependent interaction among TIA1/Pcbp1 and RBM39 appears to be an integral to the way that TIA1 and Pcbp1 regulate splicing activation via their interaction with an upstream intronic enhancer.
The TIA1 splicing factor family has been known to bind to U-rich motifs downstream of the target exon, thereby activating splicing by recruiting U1 snRNP to the weak 5′ ss (18). Despite the presence of numerous TIA1 putative binding motifs downstream of exon 16, TIA1 does not exert such a positive effect in our study. The coordinated effects of TIA1, Pcbp1, and RBM39 presented in this study point instead to a novel enhancing action upstream of exon 16.
Both TIA1 and TIAR are expressed in MEL cells. Only TIA1 exerts a positive effect on exon 16 inclusion. The RNA recognition motifs in TIA1 and TIAR have different RNA binding specificities (52) that may explain this differential activation. Alternatively, it could be that only TIA1 binds Pcbp1. Pcbp1 depends on RBM39 and also requires collaboration with TIA1. The effect of Pcbp1 could be mediated indirectly by interaction with RBM39 (25) or directly by binding to a C-rich polypyrimidine tract and interacting with U2 snRNP complex (24). This concept of splicing regulators acting in a context-dependent and position-sensitive mode is supported by a large collection of evidence in other systems (53, 54).
RBM39 mRNA expression increases during erythroid differentiation by 3.1-fold (http://www.cbil.upenn.edu/ErythronDB/) (55). Similarly, RBM39 protein expression increased in this study during induced MEL cell differentiation, while no significant changes in both TIA1 and Pcbp1 occurred. This rise in RBM39 expression thus suggests that upregulation of RBM39 probably facilitates exon 16 inclusion.
Our findings highlight the importance of the U-ISE and C-ISE elements for the splicing of exon 16 but do not imply that they completely control splicing. Database searches revealed the presence of UUUUYCYCCC motifs between the BP and the 3′ ss in several other alternatively spliced exons. It will be interesting to see if TIA1 and Pcbp1 exert similar effects on the splicing of these exons. Studying the effect of these elements and their associated binding factors in their native context, such as in a more extensive native segment of the splice acceptor region encompassing the BP, polypyrimidine track, and AG dinucleotide, should also add more insight.
Our current working model for protein 4.1R exon 16 splicing is that binding of TIA1/Pcbp1 to the upstream cis elements activates a 3′ ss through direct interaction with RBM39 and then connects to SF3b155 for 3′ ss recognition. While the exact molecular mechanisms remain to be clarified, these interactions appear to be critical for RBM39-mediated 3′ ss enhancement.
MATERIALS AND METHODS
Plasmid constructs.
All DNA constructs were made using standard cloning procedures and confirmed by sequencing. The exon 16 wild-type (WT) minigene and mutant constructs include a three-exon (exons 13, 16, and 17) splicing cassette with a consensus 5′ ss (AA) or mutations in upstream intronic sequences (GIa and Xm) as previously described (43). The double-mutation Xm/AA construct was created using a QuikChange II XL site-directed mutagenesis kit (Stratagene), replacing the TT at positions +3 and +4 of the exon 16 5′ ss in the Xm minigene with AA. The putative TIA1 binding site (TCTT or TTCT) (46) mutation constructs (E16 m1 to E16 m11) spanning 214 nucleotides (nt) upstream to 200 nt downstream of exon 16 were produced in which T bases were replaced with A bases. The E16 Up construct was made by replacing the upstream intronic UUUU sequence at −21 to −24 with GAAG.
The DUP minigene originally obtained from D. Black (20) and subcloned into pcDNA3 to form DUP4-1 was previously described (22). DUP exon 1 is β-globin exon 1, and exon 3 is β-globin exon 2. The internal second exon is a fusion between the first and second β-globin exons to make a 33-nt hybrid exon (E2). The DUP-TP minigene was constructed by replacing the sequence at positions −15 to −28 (GCCTATTGGTCTAT) upstream of the internal exon E2 with the corresponding intronic sequence UCUGUUUUCCCCCC from 4.1R exon 16. DUP-Up and DUP-Xm were generated by replacing −21 to −24 and −15 to −17 with GAAG and GGG, respectively, in the DUP-TP minigene.
Branch point mutants were constructed by single nucleotide substitutions within CACUGAC (−35 to −41) of DUP-TP at position 4, 5, or 7 with A, U, or G to form BP-mu4, BP-mu5, and BP-mu7, respectively.
For A/B complex assembly and psoralen cross-linking constructs, the NcoI fragment containing sequences spanning E1 and exon 2 of E2 were purified from their respective DUP minigene, amplified, and cloned into the NheI and EcoRI sites in pcDNA3.1(+) (Invitrogen) to form dup4-1A, dup-tpA, dup-upA, and dup-xmA. The substrates consist of 61 nt of the 3′ end of E1, 91 nt of the intron downstream of E1, 42 nt of the intron upstream of E2, and 12 nt of the 5′ end of E2 with different combinations of TIA1 and Pcbp1 binding sites.
TIA1 (GenBank accession number NM_022173) was amplified from human CD34+ RNA (Stem Cell Technologies, Inc.) with hTIA1-S (5′-ATGGCGGAGGGCGCCCAGCCGCAGCA-3′) and hTIA1-As (5′-TCAGTAGGGGGCAAATCGGCTGTAG-3′). TIAR (GenBank accession number NM_001033925) was amplified with hTIAR-S (5′-ATGATGGAAGACGACGGGCAGC-3′) and hTIAR-As (5′-TCACTGTGTTTGGTAACTTGCC-3′). The product was subsequently cloned in frame into the expression vector pcDNA3.1(+)-HA, a pcDNA3.1(+) derivative with three copies of the hemagglutinin (HA) tag inserted between the HindIII and BamHI sites. Full-length TIA1 (amino acids [aa] 1 to 385) and its domain deletion constructs RRM123 (aa 1 to 284), RRM12 (aa 1 to 207), RRM23 (aa 93 to 284), RRM1 (aa 1 to 92), RRM2 (aa 93 to 207), RRM3 (aa 208 to 284), Q (aa 285 to 385), RRM3Q (aa 208 to 385), and RRM23Q (aa 93 to 385) were cloned into pGEX6p1 to generate its respective expression constructs.
Pcbp1 cDNA (GenBank accession number NM_011865.3) was cloned into pcDNA3.1(+)-FLAG, a pcDNA3.1(+) derivative with three copies of the Flag tag inserted between the HindIII and BamHI sites, and pGEX6p3 for protein expression in mammalian and bacterial cells, respectively. Full-length Pcbp1 (aa 1 to 356) and its domain deletion constructs KHI (aa 1 to 88), KHII (aa 93 to 173), and KHIII (aa 273 to 356) were subsequently cloned into pGEX6p3 to generate its respective expression constructs. RBM39 (GenBank accession number BC030493) was cloned into pcDNA3.1(+)-HA and pGEX6p1 for protein expression.
The pSecT7His expression vector for secreted protein production was constructed by inserting mouse Igκ chain leader sequence (56) between the NheI and HindIII sites, T7 tag between the HindIII and BamHI sites, and 6×His tag between the XhoI and XbaI sites in pcDNA3.1(+). TIA1 was cloned in frame between the T7 and 6×His tag to form TIA1/pSecT7His for TIA1/His protein production.
Cell culture and transfection.
Murine erythroleukemia (MEL), HeLa, and HEK-293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate, and 10% fetal bovine serum (Sigma). MEL cells were induced to erythroid differentiation as described previously (22). Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For RBM39 depletion, HeLa was treated with a small interfering RNA (siRNA) with target sequences at positions 875 to 894 (GenBank accession number NM_184234.2) obtained from Thermo Fisher using Lipofectamine RNAiMAX transfection reagent. For His-tagged protein production, HEK-293 cells transfected with pSecT7His-based constructs were grown in advanced DMEM (Invitrogen) plus 2% fetal bovine serum for 4 to 6 days, and medium was collected for protein purification.
RT-PCR analyses.
Reverse transcription-PCR (RT-PCR) analysis of splicing products was performed using a limiting-cycle amplification protocol that obtains the PCR product within its linear range (43). RNAs from exon 16, DUP, or dup minigene transfected cells were reverse transcribed using the SP6 primer. Endogenous exon 16 detection used reverse transcription with Ex18-As (5′-TGATGGTCTTGGTCTCAGT-3′). PCRs were performed with Ex13-S (5′-AGAGCCCACAGAAGCATGGA-3′) and Ex17-As (5′-GTGTGTAGATAAGCCCTTGTCCCA-3′) for exon 16-based minigenes, with DUP-ex1-S (5′-AAGGTGAACGTGGATGAAGTTGGT-3′) and DUP-ex3-As (5′-ACAGATCCCCAAAGGACTCAAAGAAC-3′) for DUP-based minigenes, and with DUP-ex1-S and BGH_rev_primer for dup-based minigenes. The specificities of RT-PCR primers were verified by amplifying RNAs isolated from untransfected cells. Spliced products were fractionated on 2% agarose gels or 5% DNA polyacrylamide gels and quantified using analytic software from a ChemiImager 5500 system (Alpha Innotech Co.). Two transfections were performed with each construct in each experiment. Each experiment was repeated three times. Averages and standard deviations (SDs) were calculated from three independent experiments (n = 6). E16 inclusions were calculated as the percentage of exon 16 inclusion (+E16)/total products (set as 100%) in individual lanes.
Recombinant protein production.
GST, TIA1/GST, Pcbp1/GST, and RBM39/GST and their deletion derivatives were affinity purified by coupling to glutathione-Sepharose beads (GE Healthcare) according to the manufacturer's protocol. His-tagged proteins were purified from medium of HEK-293 cells transfected with pSecT7His-based constructs using an Ni-nitrilotriacetic acid (Ni-NTA) purification system under native conditions (Invitrogen). The collected medium was dialyzed in native purification buffer (50 mM NaH2PO4, pH 8.0, 500 mM NaCl), applied to Ni-NTA–agarose columns equilibrated with native purification buffer plus 10 mM imidazole, washed in purification buffer plus 20 mM imidazole, and eluted with native purification buffer plus 100 mM imidazole. Purified proteins were dialyzed and concentrated in buffer D (57).
Gel overlay assays.
Gel overlay assays were performed as previously described (58). Pcbp1/GST, U1C/GST (47), GST, bovine serum albumin (BSA), and actin were fractionated on 10% SDS-PAGE gels and either stained with Coomassie blue dye or electrotransferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore). The membrane was incubated in blocking buffer (50 mM Tris-HCl, pH 7.5, 140 mM NaCl, 0.1% Tween 20, 0.5% NP-40, 3% BSA, 0.5% gelatin, and 2 mM dithiothreitol [DTT]) for 12 h at 4°C and then overlaid with 3 μg/ml of TIA1/His in binding buffer (50 mM Tris-HCl, pH 7.5, 140 mM NaCl, 0.5% NP-40, 1% BSA, 0.25% gelatin, 2 mM ATP, and 2 mM DTT) for 3 h at 4°C. The membrane was washed in washing buffer (50 mM Tris-HCl, pH 7.5, 140 mM NaCl, 1.0% NP-40, and 2 mM DTT), followed by Western blotting using an anti-His antibody (Santa Cruz).
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed as described previously (59) with modifications. The biotinylated wt (UCUGUUUUCCCCCCUUUCAUUU), xm (UCUGUUUUCCCGGGUUUCAUUU), up (ACAGGAAGCCCCCCUUUCAUUU), dn (UCUGUUUUCCCCCCGAACAGAA), and u/d (ACAGGAAGCCCCCCGAACAGAA) RNA substrates were synthesized by Integrated DNA Technologies (Coralville, IA). RNA mobility shift was achieved by incubating 0.2 nM biotinylated RNA with nuclear extracts or purified proteins for 30 min at 4°C in 20 μl of binding buffer (10 mM Tris-HCl, 10 mM HEPES, 100 mM NaCl, 0.1% Triton X-100, 2 mM MgCl2, 1.5 mM DTT, pH 7.5, 7% glycerol). For competition assays, a molar excess of unlabeled competitor RNAs was added to the preincubation reaction mixture. Samples were fractionated on 5% polyacrylamide gels, transferred to Hybond-N+ nylon membranes (Amersham Biosciences), and detected using a LightShift Chemiluminescent EMSA kit (Pierce) according to the manufacturer's protocol.
Affinity chromatography purification of RNA binding proteins.
HeLa nuclear extracts were precleared for 30 min on streptavidin-Sepharose beads equilibrated with buffer D (57) and then incubated with streptavidin-Sepharose beads and the biotinylated RNA oligonucleotides (3 μg) for 60 min at 4°C. Bound complexes were washed, step eluted with increasing concentrations of KCl (0.5, 0.75, 1.0, 1.25, and 1.5 M), dialyzed in buffer D, and analyzed for binding proteins in RNA gel shift assays. Trichloroacetic acid (TCA) precipitates of eluates were fractionated in an Invitrogen NuPAGE gel and stained with colloidal blue (Invitrogen). The protein lanes were excised and washed with 50% acetonitrile in water. The identities of the proteins were established by the Harvard Mass Spectrometry and Proteomics Facility (Cambridge, MA).
Spliceosome assembly and psoralen cross-linking assays.
Pre-mRNA substrates for spliceosome assembly and psoralen-mediated UV cross-linking were produced from linearized plasmids by transcription with T7 RNA polymerase (Amersham Pharmacia Biotech) in the presence of a Ribo m7G cap analog (Promega) and [α-32P]NTP (PerkinElmer). Alternatively, gel-purified fragments containing the T7 promoter were used as templates for probe production.
HeLa cell nuclear extracts were prepared as described previously (57). Nuclear extract or extracts incubated at room temperature for 20 min to deplete ATP were used for spliceosomal complex E assembly (60, 61). For assembly of the A, B, and C complexes, 5 ng of 32P-labeled pre-mRNA transcripts was incubated at 30°C for 0, 2, 5, and 25 min in 25 μl of splicing reaction mixtures with untreated nuclear extracts (61). Where indicated (Fig. 11B and C), increasing amounts (20, 40, and 75 ng) of purified TIA1/His, Pcbp1/GST, or RBM39/GST protein was added to the reaction mixture with untreated or RBM39-depleted nuclear extracts and incubated for 2 min. The assembled RNA-protein complexes were fractionated in 1.5% low-melting-point agarose gels, fixed in 10% acetic acid and 10% methanol for 30 min, dried, and exposed to X-ray film (61).
Psoralen cross-linking reactions were carried out as described previously (62) using the dup4-1A, dup-tpA, dup-upA, and dup-xmA transcripts. RNase H-mediated inactivation of U1 snRNP or U2 snRNP was performed as described previously (63) by incubating the nuclear extract with RNase H and an oligodeoxynucleotide complementary to positions 1 to 15 of U1 snRNA or 28 to 42 of U2 snRNA before cross-linking. The cross-linked products were analyzed on 5% polyacrylamide gels containing 8.3 M urea and imaged using a PhosphorImager (Molecular Dynamics).
Western blot analysis.
Cell lysates or immunoprecipitates were fractionated by SDS-PAGE and electrotransferred onto a PVDF or nitrocellulose membrane. The detection of TIA1, Pcbp1, RBM39, U2AF65, SF3b155, Numb, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and actin was carried out by immunoblotting with anti-TIA1 (Santa Cruz), anti-Pcbp1 (T-18; Santa Cruz), anti-RBM39 (Bethyl Lab), anti-U2AF65 (Bethyl), anti-SF3b155 (Bethyl), anti-Numb (Developmental Studies Hybridoma Bank [DSHB]), anti-GAPDH (Sigma), and antiactin (Sigma) antibodies diluted in antibody enhancer diluent (Amresco, Inc.) and developed using an ECL detection kit (Amersham Pharmacia). VeriBlot secondary antibody (ab131366 or ab131368; Abcam) was used for coimmunoprecipitation and Western blot analyses. Exogenously expressed Flag-Pcbp1, HA-TIA1, His-TIA1, and HA-RBM39 proteins were detected with anti-FLAG (Sigma), anti-HA (H6908 [Sigma] or 3F10 [Roche]), and anti-His (Novagen/Sigma) antibodies. The same set of samples was sometimes probed with different antibodies on different dates by running and blotting new gels. Each membrane was probed with the desired antibody as well as an actin loading control to ensure that no pipetting error occurred.
ACKNOWLEDGMENTS
We thank D. L. Black (University of California, Los Angeles) for the DUP4-1 minigene. We thank Steve Papp for initial identification of TIA1 and Anyu Zhou for technical assistance.
This work was supported by NIH grant HL24385 (E.J.B.) and the Claudia Barr Award (S.-C.H.).
REFERENCES
- 1.Maniatis T, Tasic B. 2002. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 418:236–243. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- 2.Black DL. 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 3.Stetefeld J, Ruegg MA. 2005. Structural and functional diversity generated by alternative mRNA splicing. Trends Biochem Sci 30:515–521. doi: 10.1016/j.tibs.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Nilsen TW. 2002. The spliceosome: no assembly required? Mol Cell 9:8–9. doi: 10.1016/S1097-2765(02)00430-6. [DOI] [PubMed] [Google Scholar]
- 6.Jurica MS, Moore MJ. 2003. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell 12:5–14. doi: 10.1016/S1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 7.Reed R. 1989. The organization of 3′ splice-site sequences in mammalian introns. Genes Dev 3:2113–2123. doi: 10.1101/gad.3.12b.2113. [DOI] [PubMed] [Google Scholar]
- 8.Berglund JA, Abovich N, Rosbash M. 1998. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev 12:858–867. doi: 10.1101/gad.12.6.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berglund JA, Chua K, Abovich N, Reed R, Rosbash M. 1997. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 89:781–787. doi: 10.1016/S0092-8674(00)80261-5. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Luyten I, Bottomley M, Messias A, Houngninou-Molango S, Sprangers R, Zanier K, Krämer A, Sattler M. 2001. Structural basis for recognition of the intron branch site by splicing factor 1. Science 294:1098–1102. doi: 10.1126/science.1064719. [DOI] [PubMed] [Google Scholar]
- 11.Zamore PD, Patton JG, Green MR. 1992. Cloning and domain structure of the mammalian splicing factor U2AF. Nature 355:609–614. doi: 10.1038/355609a0. [DOI] [PubMed] [Google Scholar]
- 12.Moore M. 2000. Intron recognition comes of AGe. Nat Struct Biol 7:14–16. doi: 10.1038/71207. [DOI] [PubMed] [Google Scholar]
- 13.Gozani O, Potashkin J, Reed R. 1998. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol 18:4752–4760. doi: 10.1128/MCB.18.8.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Query CC, Strobel SA, Sharp PA. 1996. Three recognition events at the branch-site adenine. EMBO J 15:1392–1402. [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, Manley JL. 1989. Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev 3:1553–1561. doi: 10.1101/gad.3.10.1553. [DOI] [PubMed] [Google Scholar]
- 16.Gao K, Masuda A, Matsuura T, Ohno K. 2008. Human branch point consensus sequence is yUnAy. Nucleic Acids Res 36:2257–2267. doi: 10.1093/nar/gkn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aznarez I, Barash Y, Shai O, He D, Zielenski J, Tsui LC, Parkinson J, Frey BJ, Rommens JM, Blencowe BJ. 2008. A systematic analysis of intronic sequences downstream of 5′ splice sites reveals a widespread role for U-rich motifs and TIA1/TIAL1 proteins in alternative splicing regulation. Genome Res 18:1247–1258. doi: 10.1101/gr.073155.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Förch P, Puig O, Martínez C, Séraphin B, Valcárcel J. 2002. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J 21:6882–6892. doi: 10.1093/emboj/cdf668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou HL, Baraniak AP, Lou H. 2007. Role for Fox-1/Fox-2 in mediating the neuronal pathway of calcitonin/calcitonin gene-related peptide alternative RNA processing. Mol Cell Biol 27:830–841. doi: 10.1128/MCB.01015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Modafferi EF, Black DL. 1997. A complex intronic splicing enhancer from the c-src pre-mRNA activates inclusion of a heterologous exon. Mol Cell Biol 17:6537–6545. doi: 10.1128/MCB.17.11.6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ponthier JL, Schluepen C, Chen W, Lersch RA, Gee SL, Hou VC, Lo AJ, Short SA, Chasis JA, Winkelmann JC, Conboy JG. 2006. Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. J Biol Chem 281:12468–12474. doi: 10.1074/jbc.M511556200. [DOI] [PubMed] [Google Scholar]
- 22.Yang G, Huang SC, Wu JY, Benz EJ Jr. 2008. Regulated Fox-2 isoform expression mediates protein 4.1R splicing during erythroid differentiation. Blood 111:392–401. doi: 10.1182/blood-2007-01-068940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. 2009. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji X, Park JW, Bahrami-Samani E, Lin L, Duncan-Lewis C, Pherribo G, Xing Y, Liebhaber SA. 2016. αCP binding to a cytosine-rich subset of polypyrimidine tracts drives a novel pathway of cassette exon splicing in the mammalian transcriptome. Nucleic Acids Res 44:2283–2297. doi: 10.1093/nar/gkw088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng Q, Rayala SK, Gururaj AE, Talukder AH, O'Malley BW, Kumar R. 2007. Signaling-dependent and coordinated regulation of transcription, splicing, and translation resides in a single coregulator, PCBP1. Proc Natl Acad Sci U S A 104:5866–5871. doi: 10.1073/pnas.0701065104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behzadnia N, Golas MM, Hartmuth K, Sander B, Kastner B, Deckert J, Dube P, Will CL, Urlaub H, Stark H, Lührmann R. 2007. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J 26:1737–1748. doi: 10.1038/sj.emboj.7601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bessonov S, Anokhina M, Will CL, Urlaub H, Lührmann R. 2008. Isolation of an active step I spliceosome and composition of its RNP core. Nature 452:846–850. doi: 10.1038/nature06842. [DOI] [PubMed] [Google Scholar]
- 28.Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, Will CL, Urlaub H, Stark H, Lührmann R. 2010. Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceosome activation and first step catalysis. RNA 16:2384–2403. doi: 10.1261/rna.2456210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prigge JR, Iverson SV, Siders AM, Schmidt EE. 2009. Interactome for auxiliary splicing factor U2AF65 suggests diverse roles. Biochim Biophys Acta 1789:487–492. doi: 10.1016/j.bbagrm.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stepanyuk GA, Serrano P, Peralta E, Farr CL, Axelrod HL, Geralt M, Das D, Chiu HJ, Jaroszewski L, Deacon AM, Lesley SA, Elsliger MA, Godzik A, Wilson IA, Wüthrich K, Salomon DR, Williamson JR. 2016. UHM-ULM interactions in the RBM39-U2AF65 splicing-factor complex. Acta Crystallogr D Struct Biol 72:497–511. doi: 10.1107/S2059798316001248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. 2004. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci U S A 101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dowhan DH, Hong EP, Auboeuf D, Dennis AP, Wilson MM, Berget SM, O'Malley BW. 2005. Steroid hormone receptor coactivation and alternative RNA splicing by U2AF65-related proteins CAPERα and CAPERβ. Mol Cell 17:429–439. doi: 10.1016/j.molcel.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 33.Shao W, Kim HS, Cao Y, Xu YZ, Query CC. 2012. A U1-U2 snRNP interaction network during intron definition. Mol Cell Biol 32:470–478. doi: 10.1128/MCB.06234-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loerch S, Maucuer A, Manceau V, Green MR, Kielkopf CL. 2014. Cancer-relevant splicing factor CAPERα engages the essential splicing factor SF3b155 in a specific ternary complex. J Biol Chem 289:17325–17337. doi: 10.1074/jbc.M114.558825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim KK, Kim YC, Adelstein RS, Kawamoto S. 2011. Fox-3 and PSF interact to activate neural cell-specific alternative splicing. Nucleic Acids Res 39:3064–3078. doi: 10.1093/nar/gkq1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohno G, Ono K, Togo M, Watanabe Y, Ono S, Hagiwara M, Kuroyanagi H. 2012. Muscle-specific splicing factors ASD-2 and SUP-12 cooperatively switch alternative pre-mRNA processing patterns of the ADF/cofilin gene in Caenorhabditis elegans. PLoS Genet 8:e1002991. doi: 10.1371/journal.pgen.1002991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuroyanagi H, Ohno G, Mitani S, Hagiwara M. 2007. The Fox-1 family and SUP-12 coordinately regulate tissue-specific alternative splicing in vivo. Mol Cell Biol 27:8612–8621. doi: 10.1128/MCB.01508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuwasako K, Takahashi M, Unzai S, Tsuda K, Yoshikawa S, He F, Kobayashi N, Güntert P, Shirouzu M, Ito T, Tanaka A, Yokoyama S, Hagiwara M, Kuroyanagi H, Muto Y. 2014. RBFOX and SUP-12 sandwich a G base to cooperatively regulate tissue-specific splicing. Nat Struct Mol Biol 21:778–786. doi: 10.1038/nsmb.2870. [DOI] [PubMed] [Google Scholar]
- 39.Takakuwa Y, Tchernia G, Rossi M, Benabadji M, Mohandas N. 1986. Restoration of normal membrane stability to unstable protein 4.1-deficient erythrocyte membranes by incorporation of purified protein 4.1. J Clin Invest 78:80–85. doi: 10.1172/JCI112577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Discher D, Parra M, Conboy JG, Mohandas N. 1993. Mechanochemistry of the alternatively spliced spectrin-actin binding domain in membrane skeletal protein 4.1. J Biol Chem 268:7186–7195. [PubMed] [Google Scholar]
- 41.Horne WC, Huang SC, Becker PS, Tang TK, Benz EJ Jr. 1993. Tissue-specific alternative splicing of protein 4.1 inserts an exon necessary for formation of the ternary complex with erythrocyte spectrin and F-actin. Blood 82:2558–2563. [PubMed] [Google Scholar]
- 42.Tchernia G, Mohandas N, Shohet SB. 1981. Deficiency of skeletal membrane protein band 4.1 in homozygous hereditary elliptocytosis. Implications for erythrocyte membrane stability. J Clin Invest 68:454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deguillien M, Huang SC, Moriniere M, Dreumont N, Benz EJ Jr, Baklouti F. 2001. Multiple cis elements regulate an alternative splicing event at 4.1R pre-mRNA during erythroid differentiation. Blood 98:3809–3816. doi: 10.1182/blood.V98.13.3809. [DOI] [PubMed] [Google Scholar]
- 44.Hou VC, Lersch R, Gee SL, Ponthier JL, Lo AJ, Wu M, Turck CW, Koury M, Krainer AR, Mayeda A, Conboy JG. 2002. Decrease in hnRNP A/B expression during erythropoiesis mediates a pre-mRNA splicing switch. EMBO J 21:6195–6204. doi: 10.1093/emboj/cdf625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang G, Huang SC, Wu JY, Benz EJ Jr. 2005. An erythroid differentiation-specific splicing switch in protein 4.1R mediated by the interaction of SF2/ASF with an exonic splicing enhancer. Blood 105:2146–2153. doi: 10.1182/blood-2004-05-1757. [DOI] [PubMed] [Google Scholar]
- 46.Shukla S, Del Gatto-Konczak F, Breathnach R, Fisher SA. 2005. Competition of PTB with TIA proteins for binding to a U-rich cis-element determines tissue-specific splicing of the myosin phosphatase targeting subunit 1. RNA 11:1725–1736. doi: 10.1261/rna.7176605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang SC, Ou AC, Park J, Yu F, Yu B, Lee A, Yang G, Zhou A, Benz EJ Jr. 2012. RBFOX2 promotes protein 4.1R exon 16 selection via U1 snRNP recruitment. Mol Cell Biol 32:513–526. doi: 10.1128/MCB.06423-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makeyev AV, Liebhaber SA. 2002. The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA 8:265–278. doi: 10.1017/S1355838202024627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chkheidze AN, Liebhaber SA. 2003. A novel set of nuclear localization signals determine distributions of the αCP RNA-binding proteins. Mol Cell Biol 23:8405–8415. doi: 10.1128/MCB.23.23.8405-8415.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reed R, Maniatis T. 1988. The role of the mammalian branchpoint sequence in pre-mRNA splicing. Genes Dev 2:1268–1276. doi: 10.1101/gad.2.10.1268. [DOI] [PubMed] [Google Scholar]
- 51.Corvelo A, Hallegger M, Smith CW, Eyras E. 2010. Genome-wide association between branch point properties and alternative splicing. PLoS Comput Biol. 6:e1001016. doi: 10.1371/journal.pcbi.1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dember LM, Kim ND, Liu KQ, Anderson P. 1996. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J Biol Chem 271:2783–2788. doi: 10.1074/jbc.271.5.2783. [DOI] [PubMed] [Google Scholar]
- 53.Fu XD, Ares M Jr. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15:689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ke S, Chasin LA. 2011. Context-dependent splicing regulation: exon definition, co-occurring motif pairs and tissue specificity. RNA Biol 8:384–388. doi: 10.4161/rna.8.3.14458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kingsley PD, Greenfest-Allen E, Frame JM, Bushnell TP, Malik J, McGrath KE, Stoeckert CJ, Palis J. 2013. Ontogeny of erythroid gene expression. Blood 121:e5–. doi: 10.1182/blood-2012-04-422394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coloma MJ, Hastings A, Wims LA, Morrison SL. 1992. Novel vectors for the expression of antibody molecules using variable regions generated by polymerase chain reaction. J Immunol Methods 152:89–104. doi: 10.1016/0022-1759(92)90092-8. [DOI] [PubMed] [Google Scholar]
- 57.Dignam JD, Lebovitz RM, Roeder RG. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kontrogianni-Konstantopoulos A, Huang SC, Benz EJ Jr. 2000. A nonerythroid isoform of protein 4.1R interacts with components of the contractile apparatus in skeletal myofibers. Mol Biol Cell 11:3805–3817. doi: 10.1091/mbc.11.11.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang J, Lee MS, Watowich SJ, Gorenstein DG. 2006. Chemiluminescence-based electrophoretic mobility shift assay of RNA-protein interactions: application to binding of viral capsid proteins to RNA. J Virol Methods 131:155–159. doi: 10.1016/j.jviromet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 60.Michaud S, Reed R. 1993. A functional association between the 5′ and 3′ splice site is established in the earliest prespliceosome complex (E) in mammals. Genes Dev 7:1008–1020. doi: 10.1101/gad.7.6.1008. [DOI] [PubMed] [Google Scholar]
- 61.Das R, Reed R. 1999. Resolution of the mammalian E complex and the ATP-dependent spliceosomal complexes on native agarose mini-gels. RNA 5:1504–1508. doi: 10.1017/S1355838299991501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu H, Hasman RA, Young KM, Kedersha NL, Lou H. 2003. U1 snRNP-dependent function of TIAR in the regulation of alternative RNA processing of the human calcitonin/CGRP pre-mRNA. Mol Cell Biol 23:5959–5971. doi: 10.1128/MCB.23.17.5959-5971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tarn WY, Steitz JA. 1994. SR proteins can compensate for the loss of U1 snRNP functions in vitro. Genes Dev 8:2704–2717. doi: 10.1101/gad.8.22.2704. [DOI] [PubMed] [Google Scholar]