

ABSTRACT
Extracellular signal-regulated kinase 3 (ERK3) is an atypical mitogen-activated protein kinase (MAPK) whose regulatory mechanisms and biological functions remain superficially understood. Contrary to most protein kinases, ERK3 is a highly unstable protein that is subject to dynamic regulation by the ubiquitin-proteasome system. However, the effectors that control ERK3 ubiquitination and degradation are unknown. In this study, we carried out an unbiased functional loss-of-function screen of the human deubiquitinating enzyme (DUB) family and identified ubiquitin-specific protease 20 (USP20) as a novel ERK3 regulator. USP20 interacts with and deubiquitinates ERK3 both in vitro and in intact cells. The overexpression of USP20 results in the stabilization and accumulation of the ERK3 protein, whereas USP20 depletion reduces the levels of ERK3. We found that the expression levels of ERK3 correlate with those of USP20 in various cellular contexts. Importantly, we show that USP20 regulates actin cytoskeleton organization and cell migration in a manner dependent on ERK3 expression. Our results identify USP20 as a bona fide regulator of ERK3 stability and physiological functions.
KEYWORDS: deubiquitinating enzymes, ERK3, MAPKs
INTRODUCTION
Extracellular signal-regulated kinase 3 (ERK3) along with its paralogous kinase ERK4 define a distinct subfamily of atypical mitogen-activated protein kinases (MAPKs) (1). ERK3 is expressed ubiquitously in adult mammalian tissues, with the highest levels being found in the central nervous system, skeletal muscle, lung, thymus, and testis (2–4). ERK4 shows a more restricted expression profile and is predominantly detected in brain tissue (4). Much remains to be learned about the substrates and physiological functions of these signaling enzymes. Unlike classical MAPKs, such as ERK1/ERK2, cJun NH2-terminal kinases, and p38s, that phosphorylate a large spectrum of substrates, ERK3 and ERK4 appear to have a restricted substrate specificity. Their best-characterized and validated substrate is the protein kinase MAPK-activated protein kinase 5 (MK5) (5–8). Genetic invalidation studies in mice have revealed important functions of ERK3 in fetal growth, pulmonary maturation, thymocyte development, and neuromuscular control (9–12). Biochemical and cellular studies also suggest that ERK3 plays key roles in transcriptional control, cell adhesion, cell migration, and the DNA damage response (13–16). The physiological functions of ERK4 are unknown.
The activity of ERK3 and ERK4 is regulated in part by the phosphorylation of the Ser-Glu-Gly motif in the activation loop, which stimulates their intrinsic kinase activity and affinity for the substrate MK5 (17, 18). We and others have identified group I p21-activated kinases as ERK3/ERK4 activation loop kinases (19, 20). Of note, the activating phosphorylation of ERK3/ERK4 is not modulated by classical MAPK stimuli or by any other extracellular stimuli tested, suggesting that other regulatory mechanisms control their biological activity (17).
ERK3 is a short-lived protein with a half-life of 30 to 60 min that is constitutively degraded by the ubiquitin-proteasome system in proliferating cells (21). The turnover of ERK3 involves two degrons found in the N-terminal lobe of the kinase domain that are both necessary and sufficient to target ERK3 for proteasomal degradation. The phosphorylation of ERK3 in the C-terminal extension by cyclin-dependent kinase 1 (CDK1) leads to its transient accumulation in mitosis as a result of protein stabilization (22). ERK3 is also stabilized and accumulates with time during the myogenic differentiation of C2C12 myoblasts (21). These observations lend strong support to the importance of protein turnover in regulating the signaling functions of ERK3.
The effectors of the ubiquitin-proteasome system that control ERK3 protein degradation remain to be identified. In this study, we performed a comprehensive loss-of-function RNA interference (RNAi) screen of the human deubiquitinating enzymes (DUBs) that regulate ERK3 protein levels and identified ubiquitin-specific protease 20 (USP20) as the first ERK3 DUB. USP20 (also known as von Hippel-Lindau protein-interacting deubiquitinating enzyme 2) was first identified as a substrate of the cullin-RING ligase (CRL) family member CRL2VHL (23, 24). Subsequent work reported that it deubiquitinates hypoxia-inducible factor 1α, inducing its stabilization and therefore antagonizing CRL2VHL action (25). In addition to its role in hypoxia, USP20 has been implicated in the DNA damage response by promoting the deubiquitination and stabilization of Rad17 and claspin (26–28). Recent findings also uncovered a novel function of USP20 in the morphogenesis of granule neuron dendrites and axons (29). Here we report that USP20 binds to and directly deubiquitinates ERK3, resulting in the stabilization of the kinase. Importantly, USP20 was found to regulate the ERK3-dependent control of actin cytoskeleton dynamics and cell migration. Our results identify USP20 as a regulator of ERK3 biological functions.
RESULTS
The DUB USP20 regulates the protein levels of ERK3 by increasing its stability.
We used a systematic and unbiased approach to identify DUBs that regulate ERK3 protein levels. A genome-scale RNAi screen of the human DUB family was performed by using a library of SMARTpool small interfering RNAs (siRNAs) to individually deplete the 99 annotated DUBs in the human colon adenocarcinoma cell line HT-29. The expression of endogenous ERK3 protein was monitored by immunoblot analysis. From this screen, the silencing of 5 DUBs induced a decrease in ERK3 protein levels by at least 40% (Fig. 1A). One of these enzymes, USP20, was previously identified by a global proteomic analysis as a candidate ERK3-interacting protein (30). We therefore sought to determine whether USP20 regulates ERK3 expression and biological activity. We first examined the effect of ectopically overexpressing USP20 on ERK3 protein levels in 293T cells. The expression of increasing amounts of USP20 resulted in a dose-dependent increase in the levels of ERK3 protein, with a strong linear correlation (Pearson's r = 0.98) (Fig. 1B). To rule out any transcriptional effect of USP20 on ERK3 expression, we measured ERK3 mRNA expression levels by real-time quantitative PCR (qPCR). The overexpression of USP20 had no significant effect on ERK3 mRNA levels in these cells (Fig. 1C). We also tested the ability of USP20 to regulate the levels of ERK3 transcribed from an exogenous promoter. For these experiments, 293T cells were cotransfected with Myc6-tagged ERK3 and increasing amounts of USP20. As for endogenous ERK3, increasing the levels of USP20 led to a dose-dependent and linear increase of the ERK3 protein abundance (r = 0.99) (Fig. 1D). We also examined the contribution of the related DUB USP33, which shares 59% amino acid identity with USP20. However, the overexpression of USP33 did not result in the accumulation of ERK3 (data not shown).
FIG 1.

The DUB USP20 regulates ERK3 protein levels. (A) HT-29 cells were transfected in duplicate with individual SMARTpool siRNAs targeting 99 human DUBs. After 72 h, cell extracts were analyzed by immunoblotting with anti-ERK3 antibody and antitubulin, which was used as loading control. The blots were quantified by densitometry, and the mean expression value from two independent experiments was calculated. Results are expressed as the mean log2 fold change relative to the value for control siRNAs. Error bars indicate standard deviations. (B) 293T cells were transfected with increasing amounts of pCMV6-XL4-USP20 as indicated. After 48 h, cell lysates were analyzed by immunoblotting with anti-ERK3 antibody and anti-HSC70 as a loading control. (Top) Representative blot; (bottom) quantification of ERK3 expression levels performed in three independent experiments. Results are expressed as means ± standard deviations. (C) 293T cells were transfected with 2 μg of pCMV6-XL4-USP20. RNA was extracted 48 h after transfection, and the ERK3 mRNA level was quantified by real-time qPCR. RU, relative units. (D) 293T cells were cotransfected with pcDNA3-Myc6-ERK3 and increasing amounts of pCMV6-XL4-USP20 as indicated. The expression of ERK3 was analyzed by immunoblotting as described above for panel B. (E) HeLa cells were transfected with SMARTpool USP20 or nontarget (NT) siRNAs. After 48 h, the cells were exposed to pH 6.4 acidic medium or treated with 0.5 mM CoCl2 for 3 h. The expression of ERK3 was analyzed by immunoblotting as described above for panel B. Statistical significance was determined by two-way analysis of variance with a Bonferroni posttest using GraphPad Prism software version 5. (F) HeLa cells were transfected with SMARTpool USP20 siRNAs in the absence or presence of the pCMV6-XL4-USP20 plasmid. After 48 h, the cells were exposed to pH 6.4 acidic medium for 3 h, and ERK3 expression was analyzed by immunoblotting. Statistical significance was determined by one-way analysis of variance with a Bonferroni posttest. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
We further evaluated the effect of USP20 depletion on ERK3 protein levels in HeLa cells. For these experiments, we examined the impact of USP20 silencing in a cellular context associated with the accumulation of ERK3. Unpublished data from our laboratory and data from others (31) revealed that ERK3 expression is upregulated upon acidification of the medium or under hypoxic conditions. HeLa cells were transfected with USP20 siRNAs, and after 48 h, either the cells were treated with the hypoxia-mimetic agent CoCl2 or the culture medium was replaced with medium adjusted to pH 6.4. The depletion of USP20 significantly decreased ERK3 protein levels in cells exposed to CoCl2 or acidic medium (Fig. 1E). Finally, to rigorously validate that the inhibition of ERK3 expression was the result of USP20 depletion, we performed rescue experiments. The overexpression of USP20 completely rescued the decrease in ERK3 expression levels observed in HeLa cells transfected with USP20 siRNAs and exposed to acidic conditions (Fig. 1F).
USP20 silencing only modestly reduces ERK3 protein expression levels, which may reflect the involvement of multiple redundant ERK3 DUBs. To test this hypothesis, we examined the impact of the simultaneous depletion of USP20 with either USP13 or USP16, the two best hits in our DUB RNAi screen (Fig. 1A). In agreement with the results of the screen, the depletion of USP13 or USP16 alone modestly but significantly reduced the levels of ERK3 in HeLa cells cultured in acidic medium (Fig. 2). Importantly, the silencing of both USP13 and USP20 together resulted in a larger decrease in ERK3 expression levels, suggesting that the two DUBs cooperatively regulate ERK3 protein levels. No such additive effect was observed with USP16 siRNAs.
FIG 2.
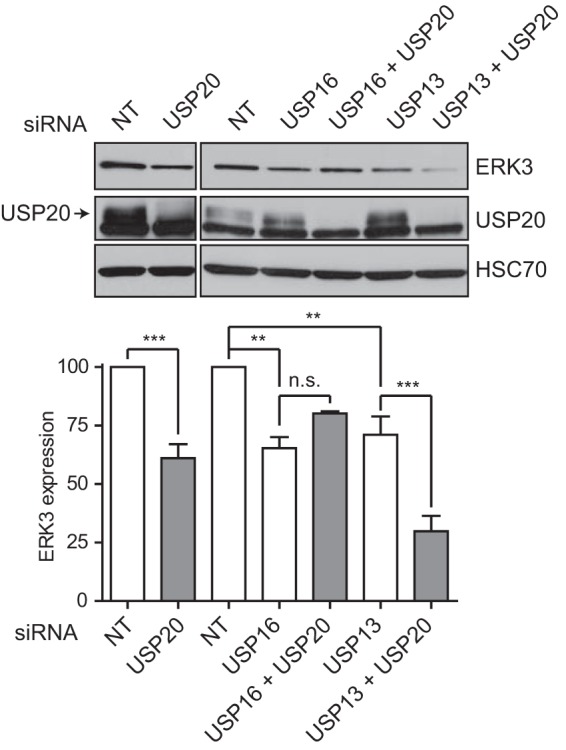
USP13 cooperates with USP20 to regulate ERK3 protein expression. HeLa cells were transfected with the indicated SMARTpool siRNAs. After 48 h, cells were exposed to pH 6.4 acidic medium for 3 h, and lysates were analyzed by immunoblotting with anti-ERK3 antibody. (Top) Representative immunoblot; (bottom) quantification of ERK3 levels performed in three independent experiments. Results are expressed as means ± standard deviations. Statistical significance was determined by one-way analysis of variance with a Bonferroni posttest. ***, P < 0.001; **, P < 0.01; *, P < 0.05; n.s., not significant.
The ERK3 expression level is controlled by protein turnover (21). To determine if USP20 affects the stability of ERK3, we measured the half-life of ERK3 by cycloheximide chase experiments. The overexpression of USP20 prolonged the half-life of endogenous ERK3 by 2.5-fold, consistent with its accumulation (Fig. 3A). Reciprocally, the depletion of USP20 by siRNAs significantly decreased the half-life of ERK3 in cells treated with CoCl2 or cultured in acidic medium (Fig. 3B and C). Together, these results demonstrate that USP20 regulates ERK3 expression by increasing its stability.
FIG 3.

USP20 stabilizes ERK3. (A) 293T cells were transiently transfected with an empty vector (Ctl) or pCMV6-XL4-USP20. After 48 h, the cells were treated with 100 μg/ml cycloheximide (CHX) for the indicated times. Endogenous ERK3 levels were analyzed by immunoblotting. (B and C) HeLa cells were transfected with SMARTpool USP20 siRNAs. After 48 h, the cells were treated with 0.5 mM CoCl2 (B) or switched to pH 6.4 culture medium (C) for 3 h. The half-life of endogenous ERK3 was measured by cycloheximide chase experiments. (Left) Representative immunoblot; (right) quantification of immunoblotting data performed in three independent experiments. Results are expressed as means ± standard deviations of relative ERK3 expression levels normalized to values for the 0-min time point. Degradation curves were fitted to a one-phase decay model.
USP20 interacts with and deubiquitinates ERK3.
Interestingly, ERK3 has been identified as a candidate USP20-interacting protein (DN score of 0.83, just below the threshold of a high-confidence interacting protein score) in a global proteomic analysis of the DUB interactome (30). To confirm the physical interaction of the two proteins, we performed coimmunoprecipitation experiments. Endogenous ERK3 was found to coimmunoprecipitate with endogenous USP20 in cells (Fig. 4A). The ERK3 protein is composed of an N-terminal kinase domain and a unique C-terminal extension. Previous studies have shown that ERK3 binds to the cell cycle regulators cyclin D3 (32) and Cdc14A (33) and to the cytoskeletal protein septin 7 (34) through its C-terminal extension. Therefore, we asked if the C-terminal region of ERK3 was necessary for its interaction with USP20. As shown in Fig. 4B, both full-length ERK3 or a deletion mutant comprising the first 365 amino acids of the protein [ERK3Δ(1–365)] formed a complex with endogenous USP20 in 293T cells. This suggests that the kinase domain of ERK3 is sufficient for its interaction with USP20.
FIG 4.
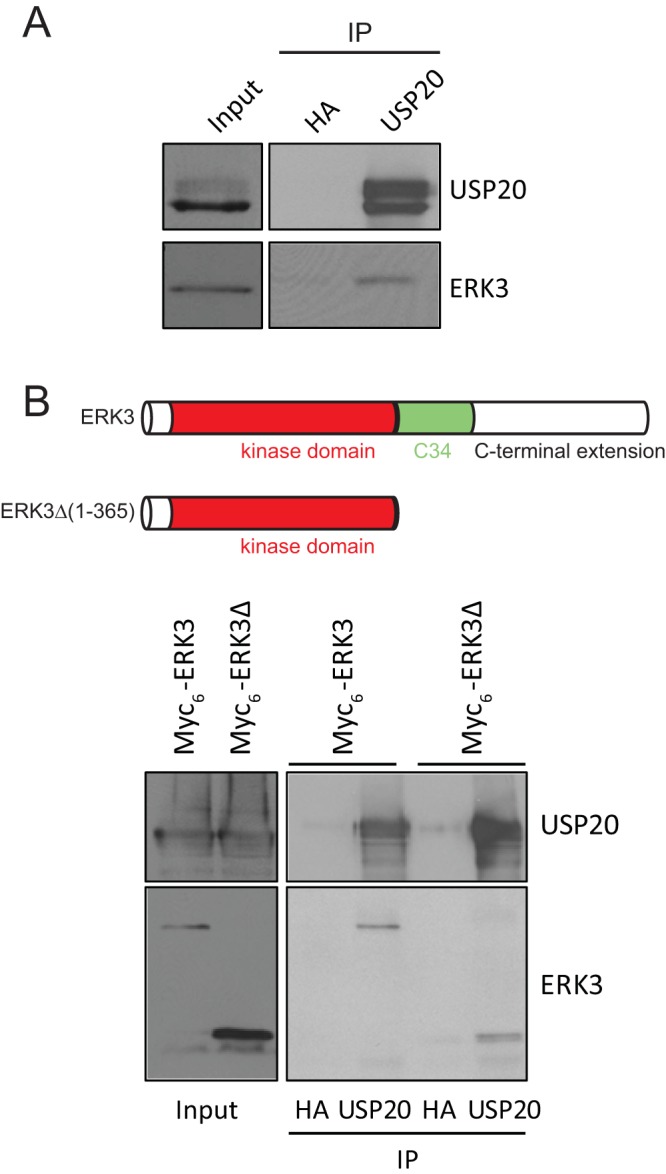
USP20 physically interacts with ERK3. (A) Lysates of exponentially proliferating 293T cells were subjected to immunoprecipitation (IP) with anti-USP20 or anti-HA antibody, which was used as a control. Immunoprecipitated proteins were analyzed by immunoblotting with the indicated antibodies. (B) 293T cells were transfected with full-length ERK3 or the truncated ERK3Δ(1–365) mutant comprising only the kinase domain. Cell lysates were immunoprecipitated with anti-USP20 or anti-HA antibody and analyzed by immunoblotting.
USP20 is a DUB, and so we investigated whether it regulates ERK3 stability and accumulation through deubiquitination. 293T cells were cotransfected with glutathione S-transferase (GST)-tagged ERK3 and hemagglutinin (HA)-ubiquitin, in the absence or presence of USP20, and the in vivo ubiquitination of ERK3 was analyzed by anti-HA immunoblotting after GST pulldown. We found that the expression of USP20 markedly reduces the ubiquitination of ERK3 in cells (Fig. 5A). To determine if ERK3 is a direct substrate of USP20, ubiquitinated ERK3 was isolated from 293T cells by GST pulldown and incubated with purified recombinant active USP20 for in vitro deubiquitination. Incubation with USP20 significantly decreased the levels of ubiquitination species of ERK3 (Fig. 5B). We conclude from these results that USP20 is a bona fide ERK3 DUB.
FIG 5.
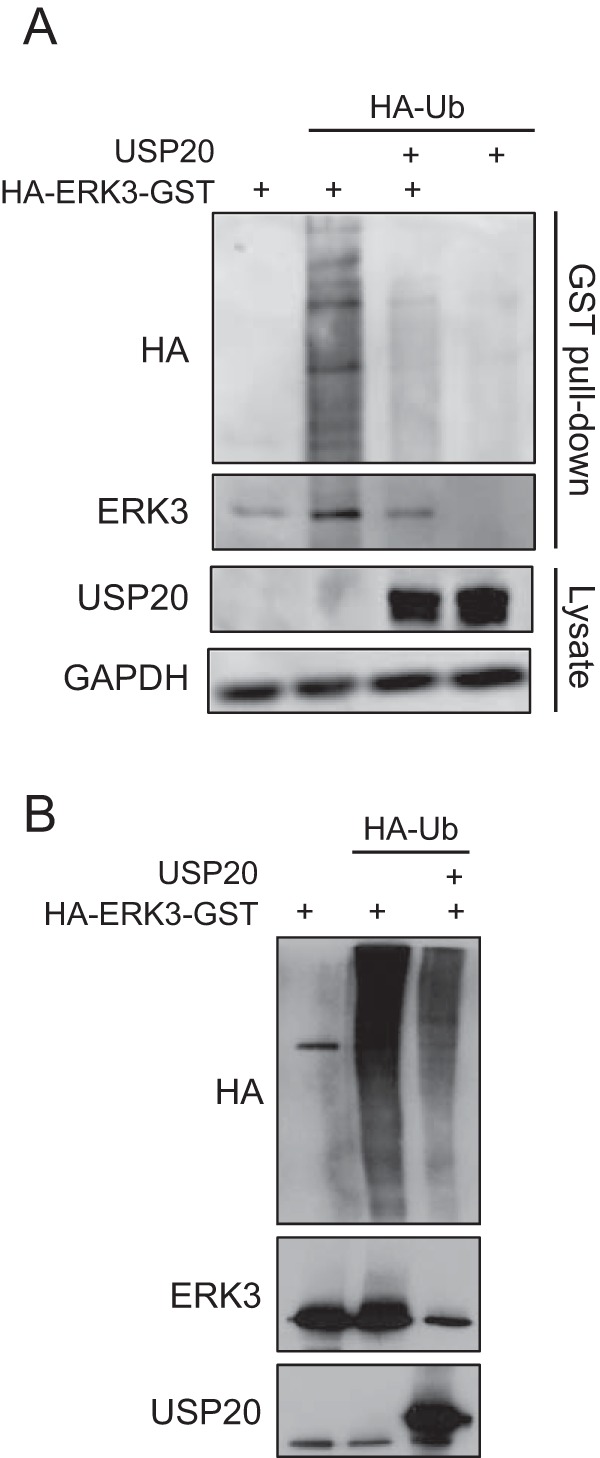
USP20 deubiquitinates ERK3 in vivo and in vitro. (A) 293T cells were transfected with pcDNA3-HA-ERK3-GST, pMT123-HA-Ubiquitin, and pCMV6-XL4-USP20 as indicated. After 48 h, the cells were lysed, and ERK3 was pulled down by using glutathione-agarose beads. Ubiquitination was analyzed by immunoblotting with anti-HA antibody. (B) Cells were processed as described above for panel A, and purified HA-ERK3-GST was incubated with purified recombinant USP20 in deubiquitination buffer for 2 h. Ubiquitination of ERK3 was assessed by immunoblotting with anti-HA antibody. Ub, ubiquitin.
USP20 deubiquitinates and stabilizes a lysine-less ERK3 mutant.
We previously reported that ectopically expressed ERK3 can be conjugated to ubiquitin via its free NH2 terminus and that a lysine-less mutant of ERK3 is degraded by the proteasome in a ubiquitin-dependent manner in cells (35). We therefore sought to determine if USP20 can regulate the ubiquitination and stability of a lysine-less ERK3 mutant. We first measured the half-life of wild-type (WT) or lysine-less (0K) ERK3Δ(1–365) coexpressed with USP20 in 293T cells. The overexpression of USP20 induced a similar increase of ERK3 protein stability for the WT and the lysine-less mutant (Fig. 6A). We next examined the effect of USP20 on the N-terminal ubiquitination of ERK3. WT HA-ERK3Δ(1–365) or the lysine-less mutant was cotransfected with HA-ubiquitin in the absence or presence of USP20, and the extent of ERK3 ubiquitination was analyzed by anti-HA immunoblotting. The overexpression of USP20 induced a marked decrease of the polyubiquitination of both the WT and lysine-less ERK3 proteins (Fig. 6B). These results indicate that USP20 is active on substrate proteins ubiquitinated at the free amino terminus.
FIG 6.
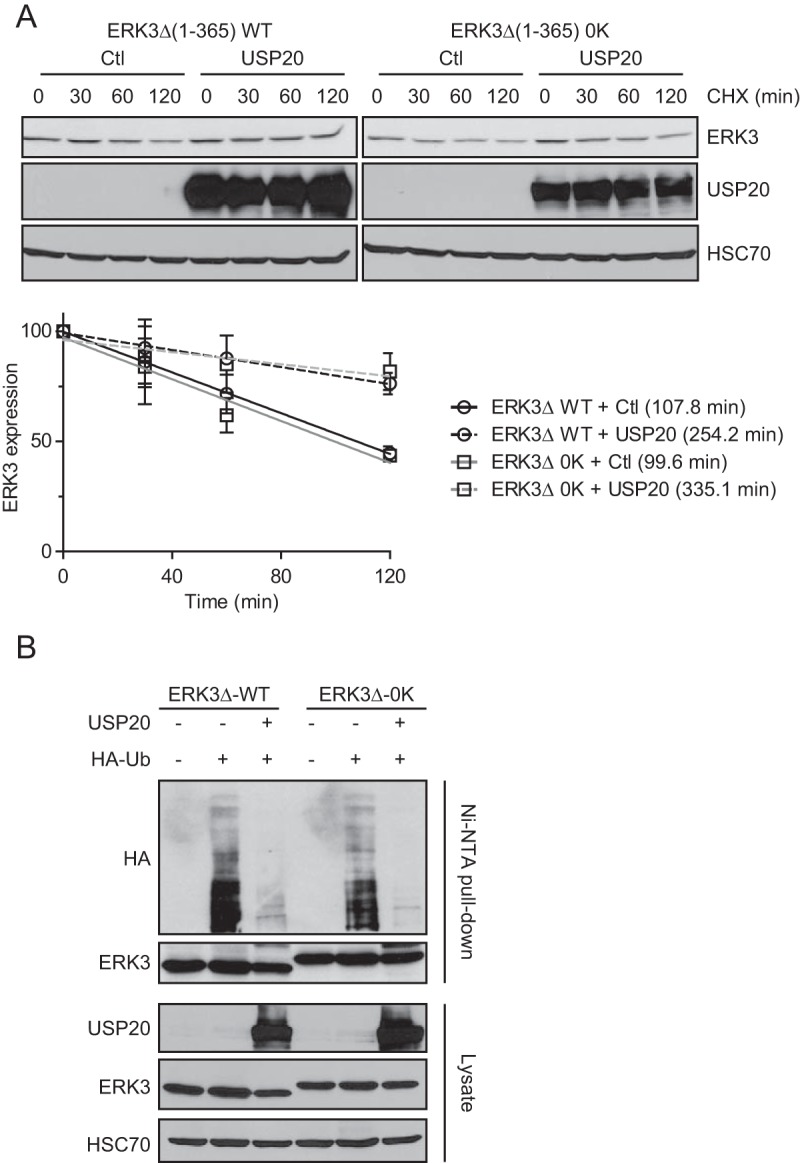
USP20 regulates the ubiquitination and stability of a lysine-less ERK3 mutant. (A) 293T cells were transfected with the WT or the lysine-less (0K) ERK3Δ(1–365) construct. After 48 h, the cells were treated with 100 μg/ml cycloheximide for the indicated times. Endogenous ERK3 levels were analyzed by immunoblotting. (Top) Representative immunoblots; (bottom) quantification of immunoblotting data performed in three independent experiments. Results are expressed as means ± standard deviations of relative ERK3 expression levels normalized to values for the 0-min time point. Degradation curves were fitted to a linear curve model. (B) 293T cells were transfected with pcDNA3-HA-ERK3Δ-His6 (WT or 0K), pMT123-HA-Ubiquitin, and pCMV6-XL4-USP20, as indicated. After 48 h, the cells were lysed, and ERK3 was pulled down with Ni-NTA–agarose beads. Ubiquitination was analyzed by immunoblotting with anti-HA antibody.
USP20 regulates ERK3 protein levels in a myogenic differentiation model.
We next studied the physiological importance of the USP20-dependent stabilization of ERK3. We previously reported that ERK3 accumulates as a result of protein stabilization during the myogenic differentiation of C2C12 myoblasts (21). Interestingly, we observed that USP20 is coexpressed with ERK3 in C2C12 cells and that its expression is markedly upregulated during myogenic differentiation with kinetics that parallel the accumulation of ERK3 (Fig. 7A). The overexpression of USP20 in C2C12 cells increased the levels of ERK3 protein both in proliferating cells and in cells undergoing myogenic differentiation (Fig. 7B). No change in ERK3 mRNA expression was observed under these experimental conditions (Fig. 7C). These results identify USP20 as a physiological regulator of ERK3 expression in skeletal muscle cells.
FIG 7.
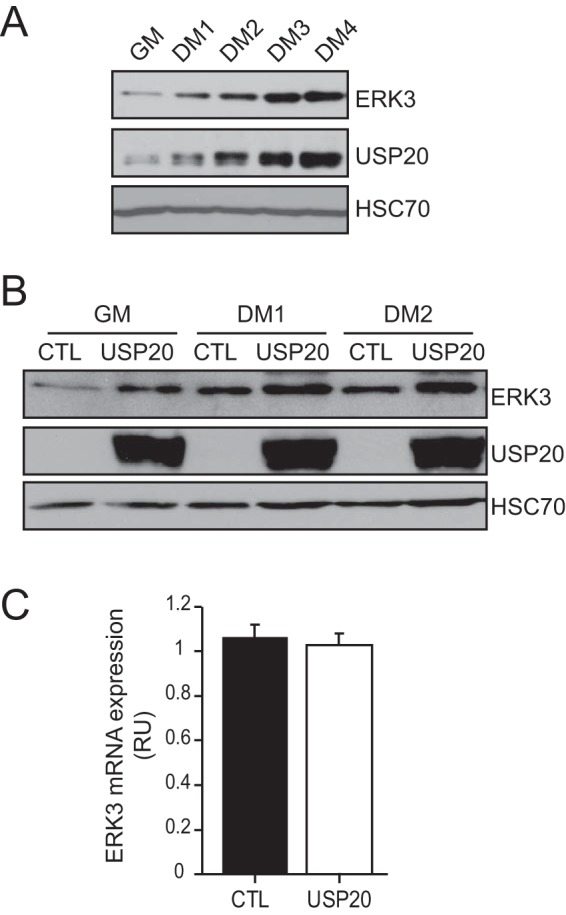
USP20 controls ERK3 protein levels during myogenic differentiation. (A) C2C12 myoblasts were cultured in growth medium (GM) or switched to differentiation medium (DM) for 4 days. The expression of ERK3 and USP20 was analyzed by immunoblotting. (B) C2C12 cells were transfected with an empty vector (Ctl) or pCMV6-XL4-USP20 and cultured as described above. The expression of ERK3 and USP20 was analyzed by immunoblotting. (C) C2C12 cells were transfected with an empty vector (Ctl) or pCMV6-XL4-USP20. After 48 h, the levels of ERK3 mRNA were quantified by real-time qPCR. Results are expressed as means ± standard deviations (n = 3).
USP20 regulates cell adhesion and migration through ERK3.
ERK3 has been proposed to regulate actin cytoskeleton dynamics and to promote cellular migration in various cell types (13–15). We therefore investigated the possible effect of USP20 on these cellular responses and the contribution of ERK3. In agreement with previously reported observations (15), the ectopic expression of ERK3 in HeLa cells caused a disorganization of the filamentous actin cytoskeleton, with a decrease in the number of stress fibers (Fig. 8A). The overexpression of USP20 similarly affected actin cytoskeleton organization, and this effect was reversed by the depletion of ERK3 (Fig. 8A). The alteration of actin cytoskeleton dynamics translated into a significantly reduced adhesive area in cells transfected with either ERK3 or USP20 (Fig. 8B). Again, the phenotype induced by USP20 was completely rescued by ERK3 silencing. Immunoblot analysis confirmed that ectopic USP20 expression upregulates ERK3 protein levels in HeLa cells (Fig. 8C). We next examined the impact of USP20 on cell migration. The overexpression of both ERK3 and USP20 enhanced the migration ability of HeLa cells in a wound-healing assay (Fig. 8D). Importantly, the effect of USP20 was abolished by RNAi depletion of ERK3 expression.
FIG 8.

USP20 regulates actin cytoskeleton dynamics and migration via ERK3. (A) HeLa cells were transfected with the indicated constructs in the absence or presence of SMARTpool ERK3 siRNAs. After 48 h, the cells were fixed, and actin filaments were stained with rhodamine-conjugated phalloidin. Actin cytoskeleton organization was visualized by epifluorescence microscopy. (B) The cellular adhesive area was quantified and is presented as a ratio of transfected (GFP-positive [GFP+])/untransfected (GFP-negative [GFP−]) cells. A minimum of 50 cells were counted, and the bar graph represents the means ± standard deviations of data from at least three independent experiments. (C and D) HeLa cells were infected with lentiviral vectors encoding GFP-ERK3 or USP20, and populations of transduced cells were selected with blasticidin. USP20-overexpressing cells were transfected with nontarget or SMARTpool ERK3 siRNAs. After 24 h, the cell lysates were analyzed for ERK3 expression by immunoblotting (C) and cells were plated at 90% confluence (D). Twenty-four hours after seeding, the confluent monolayer of cells was scraped with a sterile P200 tip. Cell migration was assessed by measuring the surface of the wound area 24 h later by phase-contrast microscopy. The bar graph represents the means ± standard deviations of data from at least three independent experiments. Statistical significance was determined by an unpaired t test. **, P < 0.01; *, P < 0.05.
We further explored the potential role of the USP20-ERK3 axis in regulating the migration of breast cancer epithelial cells. To determine if USP20 controls ERK3 levels in breast epithelial cells, we first measured the expression levels of ERK3 and USP20 in a panel of 9 immortalized and transformed breast epithelial cell lines. We observed a strong and statistically significant correlation between the expression levels of ERK3 and USP20 in these breast epithelial cell lines, suggesting an epistatic relationship (Fig. 9A). To test this hypothesis, we performed loss-of-function experiments in MCF7, T47D, and MCF10A cell lines, which all express high levels of USP20 and ERK3. The depletion of USP20 with siRNAs led to a decrease of ERK3 protein levels in all tested cell lines, although the difference did not reach statistical significance for MCF7 cells (Fig. 9B). We next analyzed the impact of USP20 and ERK3 on cell migration. Silencing of USP20 significantly impaired the migration of MCF7 and MCF10A cells in a wound-healing assay (Fig. 9C and D). This inhibitory effect was completely rescued by the coexpression of ERK3. These results indicate that the USP20-mediated stabilization of ERK3 promotes actin remodeling and cellular migration.
FIG 9.

The USP20-ERK3 axis regulates the migration of breast cancer cells. (A) The expression of ERK3 and USP20 was analyzed by immunoblotting with a panel of immortalized and transformed breast epithelial cell lines. Protein expression was quantified by densitometry, normalized by Ponceau S staining, and expressed as mean centered expression levels. (B) MCF7, MCF10A, and T47D cells were transfected with SMARTpool USP20 siRNAs. After 48 h, the cells were lysed, and ERK3 expression was analyzed by immunoblotting. (Left) Representative immunoblot; (right) densitometric quantification of immunoblotting data from three independent experiments. Results are expressed as means ± standard deviations. (C) MCF7 cells were infected with pLenti6-GFP or pLenti6-GFP-ERK3 in the absence or presence of SMARTpool USP20 siRNAs as indicated. After 24 h, the cells were plated at 90% confluence. Twenty-four hours after seeding, the cell monolayer was scratched, and cellular migration was analyzed by measuring the surface of the wound area after 24 and 48 h of recovery. Statistical significance was determined by two-way ANOVA with a Bonferroni posttest. (D) MCF10A cells stably infected with pLenti6-GFP or pLenti6-GFP-ERK3 were infected with two distinct shRNAs targeting USP20. After selection, wound-healing assays were performed as described above for panel C. Statistical significance was determined by one-way ANOVA with a Bonferroni posttest. **, P < 0.01; ****, P < 0.0001.
DISCUSSION
Unlike classical MAPKs and most protein kinases, which are stable proteins regulated mainly by phosphorylation and protein-protein interactions, ERK3 is a highly unstable protein kinase that is constitutively degraded by the ubiquitin-proteasome system (21, 35, 36). We have proposed that the biological activity of ERK3 is regulated, at least in part, by its cellular abundance through the control of ubiquitination and protein turnover (21). However, the regulatory pathways and enzymes that control ERK3 ubiquitination and degradation remain to be identified. Using an unbiased functional screening approach, we have identified the DUB USP20 as the first regulator of ERK3 stability. We show that USP20 physically interacts with and directly deubiquitinates ERK3 in vitro. Overexpression and loss-of-function experiments revealed that USP20 promotes the deubiquitination of ERK3 in vivo, resulting in the stabilization and accumulation of the protein. Importantly, we demonstrate that the USP20-dependent stabilization of ERK3 has physiological relevance.
Little is known about the regulation of ERK3 ubiquitination and degradation. The turnover rate of ERK3 is independent of activation loop phosphorylation and kinase activity, indicating that the two processes are uncoupled, at least in certain cellular contexts (21). Detailed biochemical analyses have revealed that the N-terminal kinase domain of ERK3 plays a key role in mediating its ubiquitination and degradation by the proteasome (21, 35). Interestingly, we showed that ERK3 can be targeted for degradation via an alternative mode of ubiquitination involving the conjugation of a polyubiquitin chain to the NH2 terminus of the protein (35). In this study, we show that USP20 binds to the N-terminal kinase domain of ERK3 and is able to deubiquitinate both WT and lysine-less ERK3 proteins in cells. We also reported that the addition of bulky tags (such as Myc6) to the N terminus of ERK3 inhibits its polyubiquitination and stabilizes the protein (35). The overexpression of USP20 leads to a further accumulation of ectopic Myc6-ERK3 (Fig. 1D), suggesting that multiple ubiquitination pathways control ERK3 turnover. This may prevent the accumulation of ERK3 to high levels that may be deleterious to the cell.
We observed that the overexpression of USP20 has a quantitatively greater impact on ERK3 stability than does USP20 depletion. This suggests the existence of additional DUBs for ERK3 whose relative functional importance may vary depending on the cell type and/or cellular context. Analysis of the DUB interaction landscape has documented that several key cellular regulators interact with multiple DUBs (30). Our RNAi screen of the human DUB family also suggests that other DUBs regulate ERK3 stability. Indeed, results of codepletion experiments indicate that USP13, one of the top hits in our screen, acts redundantly with USP20 in regulating ERK3 levels in HeLa cells. In contrast, no cooperative effect of USP16 and USP20 siRNAs was observed, suggesting that the two DUBs may act in the same pathway. Of note, HERC2, a regulator of USP20 expression and activity, has been shown to interact with USP16 and regulate its function (37). It will be interesting in future studies to investigate the individual functions of these DUBs and their redundancy with USP20.
In a previous study, we reported that ERK3 stability increases with time during myogenic differentiation of the model cell line C2C12, resulting in a marked upregulation of the protein (21). The observation that the expression of USP20 is upregulated during C2C12 differentiation, with kinetics similar to those of ERK3 accumulation, suggests that USP20 may act as a physiological regulator of ERK3 levels in skeletal muscle cells. In support of this idea, we found that the overexpression of USP20 increases ERK3 protein levels in C2C12 cells. Interestingly, large-scale transcriptional profiling studies revealed that the Usp20 gene is induced during C2C12 cell differentiation as well as in mouse embryonic fibroblasts in which myogenic differentiation is induced by the expression of the myogenic regulating factor MyoD (38, 39, 43). Therefore, it is tempting to speculate that a MyoD-USP20-ERK3 signaling pathway may play a role in skeletal muscle differentiation and physiology.
Recent studies have shown that ERK3 stimulates the migration of lung and breast cancer cell lines (13, 15). In agreement with these findings, we found that ERK3 overexpression increases the migration of HeLa cells. Importantly, we showed that the overexpression of USP20 phenocopies the effect of ERK3 by inducing actin cytoskeleton reorganization and promoting HeLa cell migration in an ERK3-dependent manner. Reciprocally, the silencing of USP20 expression markedly inhibits MCF7 and MCF10A cell migration, a phenotype that is completely rescued by the ectopic overexpression of ERK3. Our findings reveal novel roles of USP20 in actin cytoskeleton remodeling and cell migration and demonstrate the epistatic relationship of USP20 and ERK3 in regulating these cellular responses. Together, these results reinforce the importance of protein turnover in regulating the signaling functions of ERK3. The identification of the network of DUBs and E3 ligases controlling the stability of ERK3 will help elucidate the biological functions of this atypical kinase and its possible involvement in human disease.
MATERIALS AND METHODS
Reagents and antibodies.
Rhodamine-phalloidin was purchased from Molecular Probes. Cycloheximide and N-ethylmaleimide (NEM) were obtained from Sigma. SMARTpool siRNAs targeted to USP20 and ERK3 were obtained from Dharmacon. Recombinant USP20 protein was obtained from Abnova. The human ON-TARGETplus DUBs siRNA library was purchased from Dharmacon. The following The RNAi Consortium (TRC) lentiviral shRNA constructs for human USP20 were purchased from Sigma: TRCN0000007609 (shUSP20-A) and TRCN0000007611 (shUSP20-B).
The following commercial antibodies were obtained: anti-USP20 (catalog number A301-189A) from Bethyl Laboratories; anti-ERK3 (catalog number EP1720Y) from Abcam; anti-MK5 (catalog number M32220-050) from BD Biosciences; anti-phospho-MK5(T182) (catalog number 36-007) from Upstate Biotechnology; anti-HSP27 (catalog number 2402) and anti-phospho-HSP27(S82) (catalog number 2406) from Cell Signaling Technologies; and anti-HSC70 (catalog number sc-7298), anti-HA (catalog number sc-805), and anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (catalog number sc-25778) from Santa Cruz Biotechnology.
Plasmid constructs.
pCMV6-XL4-USP20 was obtained from Origene. Plasmids pcDNA3-Myc6-ERK3 for the WT and the truncated Δ1–365 mutant (0K mutant), pMT123-HA-Ubiquitin, and pcDNA3-HA-ERK3-GST were previously described (17, 35). pLenti6-USP20 and pLenti6-GFP-ERK3 were generated by subcloning the inserts from pCMV6-XL4-USP20 and pcDNA3-Myc6-ERK3, respectively, into the pLenti6 and pLenti6-GFP vectors. All mutations and PCR products were verified by DNA sequencing. Sequences of primers used for PCR and details about cloning strategies are available upon request.
Cell culture, transfections, and lentiviral infections.
293T, HeLa, MCF-7, hMEC 184A1, ZR-75-B, SKBR3, T47D, MDA-MB-231, Hs578T, MCF10A, and HT-29 cells were obtained from the American Type Culture Collection. The HCC1937 cell line was kindly provided by S. Mader (Université de Montréal). C2C12 myoblasts were obtained from J. F. Côté (Université de Montréal). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) or RPMI supplemented with 10% fetal bovine serum and antibiotics at 37°C in a 5% CO2 humidified atmosphere. Muscle differentiation of C2C12 myoblasts was induced by replacing growth medium (GM) (DMEM supplemented with 10% fetal bovine serum) with differentiation medium (DM) (DMEM supplemented with 2% horse serum) when cells reached 90% confluence. Cell lines were routinely tested for mycoplasma contamination.
293T cells were transiently transfected by using polyethylenimine. Other cell lines were transfected by using Lipofectamine 3000 (Invitrogen) according to the manufacturer's protocol. For siRNA transfections, cells were transfected with RNAiMAX (Invitrogen) according to the manufacturer's instructions. For lentiviral infections, 293T cells were transfected with pMD2/VSVG, pMDLg/pREE, pRSV/Rev, and the indicated pLenti6 constructs. After 48 h, virus-containing culture media were filtered and used to infect cells. Polyclonal populations of infected cells were obtained by selection with blasticidin. Short hairpin RNA (shRNA) lentiviral infections of MCF10A cells were performed as described above, and populations of infected cells were selected with puromycin.
Immunoblotting and immunoprecipitation.
Cell lysis, immunoprecipitation, and immunoblot analyses were performed as described previously (21, 40, 41). For immunoprecipitation experiments, 750 μg of lysate proteins was incubated with the indicated antibodies for 2 to 4 h at 4°C. Cycloheximide chase experiments were carried out as described previously (21). Protein levels were quantified by densitometry analysis of immunoblotting data. The ERK3 half-life was estimated by either one-phase decay or linear curve fitting. The choice between the two models was determined by the quality of the fit based on the R-squared value.
Deubiquitination assays.
For in vivo deubiquitination assays, 293T cells were transfected with expression vectors for HA-ERK3-GST or HA-ERK3(1–365)-His6 together with HA-ubiquitin. After 48 h, cells were treated with 20 μM the proteasome inhibitor MG132 for 16 h. The cells were lysed in lysis buffer supplemented with 10 mM NEM. Cell lysates (750 μg of protein) were then incubated for 2 h at 4°C with 20 μl of glutathione-agarose beads (GE Healthcare) for HA-ERK3-GST or Ni-nitrilotriacetic acid (NTA)-agarose beads (Qiagen) for HA-ERK3(1–365)-His6. Bound proteins were washed four times in lysis buffer and resolved by SDS-PAGE. Ubiquitin-containing conjugates were detected by immunoblotting with anti-HA antibody. For in vitro deubiquitination assays, HA-ERK3-GST was affinity purified from the cell lysate and then incubated with recombinant USP20 in deubiquitination buffer (50 mM Tris-HCl [pH 8.0], 50 mM NaCl, 1 mM EDTA, 10 mM dithiothreitol [DTT], 5% glycerol) for 2 h at 37°C. Ubiquitination of ERK3 was analyzed by anti-HA immunoblotting.
Migration assays.
Cell migration was assessed by using a wound-healing assay. The cells were grown as a single monolayer in 12-well plates and then wounded by using a 200-μl pipette tip. Phase-contrast microscopy images of the wound area were taken at time zero and at 24 and 48 h, and wound healing was estimated by measuring the areas of the wound by using ImageJ software.
Quantitative PCR analysis.
Real-time quantitative PCR was performed as previously described (42).
ACKNOWLEDGMENTS
We thank S. Mader and J. F. Côté for reagents and P.-L. Tanguay and J.-P. Guégan for critical comments.
This work was supported by grants from the Canadian Institutes for Health Research and the Natural Sciences and Engineering Research Council of Canada to S. Meloche. S. Meloche holds the Canada Research Chair in Cellular Signaling.
REFERENCES
- 1.Klinger S, Meloche S. 2012. Erk3 and Erk4, p 593–596. In Choi S. (ed), Encyclopedia of signaling molecules. Springer, New York, NY. [Google Scholar]
- 2.Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. 1991. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65:663–675. doi: 10.1016/0092-8674(91)90098-J. [DOI] [PubMed] [Google Scholar]
- 3.Turgeon B, Saba-El-Leil MK, Meloche S. 2000. Cloning and characterization of mouse extracellular-signal-regulated protein kinase 3 as a unique gene product of 100 kDa. Biochem J 346(Part 1):169–175. doi: 10.1042/bj3460169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rousseau J, Klinger S, Rachalski A, Turgeon B, Deleris P, Vigneault E, Poirier-Heon JF, Davoli MA, Mechawar N, El Mestikawy S, Cermakian N, Meloche S. 2010. Targeted inactivation of Mapk4 in mice reveals specific nonredundant functions of Erk3/Erk4 subfamily mitogen-activated protein kinases. Mol Cell Biol 30:5752–5763. doi: 10.1128/MCB.01147-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schumacher S, Laass K, Kant S, Shi Y, Visel A, Gruber AD, Kotlyarov A, Gaestel M. 2004. Scaffolding by ERK3 regulates MK5 in development. EMBO J 23:4770–4779. doi: 10.1038/sj.emboj.7600467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seternes OM, Mikalsen T, Johansen B, Michaelsen E, Armstrong CG, Morrice NA, Turgeon B, Meloche S, Moens U, Keyse SM. 2004. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J 23:4780–4791. doi: 10.1038/sj.emboj.7600489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aberg E, Perander M, Johansen B, Julien C, Meloche S, Keyse SM, Seternes OM. 2006. Regulation of MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAPK ERK4/MAPK4. J Biol Chem 281:35499–35510. doi: 10.1074/jbc.M606225200. [DOI] [PubMed] [Google Scholar]
- 8.Kant S, Schumacher S, Singh MK, Kispert A, Kotlyarov A, Gaestel M. 2006. Characterization of the atypical MAPK ERK4 and its activation of the MAPK-activated protein kinase MK5. J Biol Chem 281:35511–35519. doi: 10.1074/jbc.M606693200. [DOI] [PubMed] [Google Scholar]
- 9.Klinger S, Turgeon B, Levesque K, Wood GA, Aagaard-Tillery KM, Meloche S. 2009. Loss of Erk3 function in mice leads to intrauterine growth restriction, pulmonary immaturity, and neonatal lethality. Proc Natl Acad Sci U S A 106:16710–16715. doi: 10.1073/pnas.0900919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marquis M, Daudelin JF, Boulet S, Sirois J, Crain K, Mathien S, Turgeon B, Rousseau J, Meloche S, Labrecque N. 2014. The catalytic activity of the mitogen-activated protein kinase extracellular signal-regulated kinase 3 is required to sustain CD4+ CD8+ thymocyte survival. Mol Cell Biol 34:3374–3387. doi: 10.1128/MCB.01701-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sirois J, Daudelin JF, Boulet S, Marquis M, Meloche S, Labrecque N. 2015. The atypical MAPK ERK3 controls positive selection of thymocytes. Immunology 145:161–169. doi: 10.1111/imm.12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuevas Guaman M, Sbrana E, Shope C, Showalter L, Hu M, Meloche S, Aagaard K. 2014. Administration of antenatal glucocorticoids and postnatal surfactant ameliorates respiratory distress syndrome-associated neonatal lethality in Erk3(−/−) mouse pups. Pediatr Res 76:24–32. doi: 10.1038/pr.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long W, Foulds CE, Qin J, Liu J, Ding C, Lonard DM, Solis LM, Wistuba II, Qin J, Tsai SY, Tsai MJ, O'Malley BW. 2012. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J Clin Invest 122:1869–1880. doi: 10.1172/JCI61492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Bian K, Vallabhaneni S, Zhang B, Wu RC, O'Malley BW, Long W. 2014. ERK3 promotes endothelial cell functions by upregulating SRC-3/SP1-mediated VEGFR2 expression. J Cell Physiol 229:1529–1537. doi: 10.1002/jcp.24596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Mahdi R, Babteen N, Thillai K, Holt M, Johansen B, Wetting HL, Seternes OM, Wells CM. 2015. A novel role for atypical MAPK kinase ERK3 in regulating breast cancer cell morphology and migration. Cell Adh Migr 9:483–494. doi: 10.1080/19336918.2015.1112485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bian K, Muppani NR, Elkhadragy L, Wang W, Zhang C, Chen T, Jung S, Seternes OM, Long W. 2016. ERK3 regulates TDP2-mediated DNA damage response and chemoresistance in lung cancer cells. Oncotarget 7:6665–6675. doi: 10.18632/oncotarget.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deleris P, Rousseau J, Coulombe P, Rodier G, Tanguay PL, Meloche S. 2008. Activation loop phosphorylation of the atypical MAP kinases ERK3 and ERK4 is required for binding, activation and cytoplasmic relocalization of MK5. J Cell Physiol 217:778–788. doi: 10.1002/jcp.21560. [DOI] [PubMed] [Google Scholar]
- 18.Perander M, Aberg E, Johansen B, Dreyer B, Guldvik IJ, Outzen H, Keyse SM, Seternes OM. 2008. The Ser(186) phospho-acceptor site within ERK4 is essential for its ability to interact with and activate PRAK/MK5. Biochem J 411:613–622. doi: 10.1042/BJ20071369. [DOI] [PubMed] [Google Scholar]
- 19.Deleris P, Trost M, Topisirovic I, Tanguay PL, Borden KL, Thibault P, Meloche S. 2011. Activation loop phosphorylation of ERK3/ERK4 by group I p21-activated kinases (PAKs) defines a novel PAK-ERK3/4-MAPK-activated protein kinase 5 signaling pathway. J Biol Chem 286:6470–6478. doi: 10.1074/jbc.M110.181529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De la Mota-Peynado A, Chernoff J, Beeser A. 2011. Identification of the atypical MAPK Erk3 as a novel substrate for p21-activated kinase (Pak) activity. J Biol Chem 286:13603–13611. doi: 10.1074/jbc.M110.181743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coulombe P, Rodier G, Pelletier S, Pellerin J, Meloche S. 2003. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol Cell Biol 23:4542–4558. doi: 10.1128/MCB.23.13.4542-4558.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanguay PL, Rodier G, Meloche S. 2010. C-terminal domain phosphorylation of ERK3 controlled by Cdk1 and Cdc14 regulates its stability in mitosis. Biochem J 428:103–111. doi: 10.1042/BJ20091604. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Na X, Wang D, Schoen SR, Messing EM, Wu G. 2002. Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J Biol Chem 277:4656–4662. doi: 10.1074/jbc.M108269200. [DOI] [PubMed] [Google Scholar]
- 24.Li Z, Wang D, Na X, Schoen SR, Messing EM, Wu G. 2002. Identification of a deubiquitinating enzyme subfamily as substrates of the von Hippel-Lindau tumor suppressor. Biochem Biophys Res Commun 294:700–709. doi: 10.1016/S0006-291X(02)00534-X. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Wang D, Messing EM, Wu G. 2005. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha. EMBO Rep 6:373–378. doi: 10.1038/sj.embor.7400377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shanmugam I, Abbas M, Ayoub F, Mirabal S, Bsaili M, Caulder EK, Weinstock DM, Tomkinson AE, Hromas R, Shaheen M. 2014. Ubiquitin-specific peptidase 20 regulates Rad17 stability, checkpoint kinase 1 phosphorylation and DNA repair by homologous recombination. J Biol Chem 289:22739–22748. doi: 10.1074/jbc.M114.550459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan J, Luo K, Deng M, Li Y, Yin P, Gao B, Fang Y, Wu P, Liu T, Lou Z. 2014. HERC2-USP20 axis regulates DNA damage checkpoint through Claspin. Nucleic Acids Res 42:13110–13121. doi: 10.1093/nar/gku1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu M, Zhao H, Liao J, Xu X. 2014. HERC2/USP20 coordinates CHK1 activation by modulating CLASPIN stability. Nucleic Acids Res 42:13074–13081. doi: 10.1093/nar/gku978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anckar J, Bonni A. 2015. Regulation of neuronal morphogenesis and positioning by ubiquitin-specific proteases in the cerebellum. PLoS One 10:e0117076. doi: 10.1371/journal.pone.0117076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand F. 2012. Regulation and function of ERK3/MK5-mediated signaling. PhD thesis Hannover Medical School, Hannover, Germany. [Google Scholar]
- 32.Sun M, Wei Y, Yao L, Xie J, Chen X, Wang H, Jiang J, Gu J. 2006. Identification of extracellular signal-regulated kinase 3 as a new interaction partner of cyclin D3. Biochem Biophys Res Commun 340:209–214. doi: 10.1016/j.bbrc.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Hansen CA, Bartek J, Jensen S. 2008. A functional link between the human cell cycle-regulatory phosphatase Cdc14A and the atypical mitogen-activated kinase Erk3. Cell Cycle 7:325–334. doi: 10.4161/cc.7.3.5354. [DOI] [PubMed] [Google Scholar]
- 34.Brand F, Schumacher S, Kant S, Menon MB, Simon R, Turgeon B, Britsch S, Meloche S, Gaestel M, Kotlyarov A. 2012. The extracellular signal-regulated kinase 3 (mitogen-activated protein kinase 6 [MAPK6])-MAPK-activated protein kinase 5 signaling complex regulates septin function and dendrite morphology. Mol Cell Biol 32:2467–2478. doi: 10.1128/MCB.06633-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coulombe P, Rodier G, Bonneil E, Thibault P, Meloche S. 2004. N-terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol Cell Biol 24:6140–6150. doi: 10.1128/MCB.24.14.6140-6150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. 2008. Global protein stability profiling in mammalian cells. Science 322:918–923. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Yang H, Wang H. 2014. The histone H2A deubiquitinase USP16 interacts with HERC2 and fine-tunes cellular response to DNA damage. J Biol Chem 289:32883–32894. doi: 10.1074/jbc.M114.599605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao Y, Kumar RM, Penn BH, Berkes CA, Kooperberg C, Boyer LA, Young RA, Tapscott SJ. 2006. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J 25:502–511. doi: 10.1038/sj.emboj.7600958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Padova M, Caretti G, Zhao P, Hoffman EP, Sartorelli V. 2007. MyoD acetylation influences temporal patterns of skeletal muscle gene expression. J Biol Chem 282:37650–37659. doi: 10.1074/jbc.M707309200. [DOI] [PubMed] [Google Scholar]
- 40.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. 2001. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J 20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Servant MJ, Coulombe P, Turgeon B, Meloche S. 2000. Differential regulation of p27(Kip1) expression by mitogenic and hypertrophic factors: involvement of transcriptional and posttranscriptional mechanisms. J Cell Biol 148:543–556. doi: 10.1083/jcb.148.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voisin L, Saba-El-Leil MK, Julien C, Fremin C, Meloche S. 2010. Genetic demonstration of a redundant role of extracellular signal-regulated kinase 1 (ERK1) and ERK2 mitogen-activated protein kinases in promoting fibroblast proliferation. Mol Cell Biol 30:2918–2932. doi: 10.1128/MCB.00131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajan S, Chu Pham Dang H, Djambazian H, Zuzan H, Fedyshyn Y, Ketela T, Moffat J, Hudson TJ, Sladek R. 2012. Analysis of early C2C12 myogenesis identifies stably and differentially expressed transcriptional regulators whose knock-down inhibits myoblast differentiation. Physiol Genomics 44:183–197. doi: 10.1152/physiolgenomics.00093.2011. [DOI] [PubMed] [Google Scholar]