

Abstract
The transformation of vascular smooth muscle cells [VSMC] into foam cells leading to increased plaque size and decreased stability is a key, yet understudied step in atherogenesis. We reported that Interleukin-19 (IL-19), a novel, anti-inflammatory cytokine, attenuates atherosclerosis by anti-inflammatory effects on VSMC. In this work we report that IL-19 induces expression of miR133a, a muscle-specific miRNA, in VSMC. Although previously unreported, we report that miR133a can target and reduce mRNA abundance, mRNA stability, and protein expression of Low Density Lipoprotein Receptor Adaptor Protein 1, (LDLRAP1), an adaptor protein which functions to internalize the LDL receptor. Mutations in this gene lead to LDL receptor malfunction and cause the Autosomal Recessive Hypercholesterolemia (ARH) disorder in humans. Herein we show that IL-19 reduces lipid accumulation in VSMC, and LDLRAP1 expression and oxLDL uptake in a miR133a-dependent mechanism. We show that LDLRAP1 is expressed in plaque and neointimal VSMC of mouse and human injured arteries. Transfection of miR133a and LDLRAP1 siRNA into VSMC reduces their proliferation and uptake of oxLDL. miR133a is significantly increased in plasma from hyperlipidemic compared with normolipidemic patients. Expression of miR133a in IL-19 stimulated VSMC represents a previously unrecognized link between vascular lipid metabolism and inflammation, and may represent a therapeutic opportunity to combat vascular inflammatory diseases.
Keywords: Vascular Smooth muscle Cell, miR133a, LDLRAP1, cholesterol uptake
1.0 Introduction
Atherosclerotic vascular syndromes account for 50% of all mortality in the United States and is increasingly prevalent in the developing world. It is a considerable medical and socioeconomic problem contributing to mortality of many conditions including myocardial infarction, stroke, renal failure, and peripheral vascular disease. Most studies on atherosclerosis focus on the role of macrophages, but VSMC play an important, and understudied role in atherogenesis. Recent studies have implicated that as many as 70% of all cells in atherosclerotic lesions are SMC-derived [1,2]. Migration, proliferation, and synthesis of extracellular matrix by VSMC contribute to early formation of lesions [3]. Secretion of proliferative and inflammatory cytokines and immune modulators by VSMC promulgate autocrine activation of VSMC and recruitment of macrophage to the lesion in a paracrine manner [4]. Regulation of lipid uptake in VSMC is less studied and less understood than in macrophages, but nevertheless a crucial event in atherogenesis [5,6]. VSMC express receptors for lipid and can form foam cells, affecting the lipid content of the atherosclerotic plaque. Because they cannot egress from the plaque, uptake of excess lipid by medial and intimal VSMC lead to plaque progression, apoptosis, and eventual plaque instability [3,7]. Identification of a cytokine and its associated molecular effectors that can decrease uptake by VSMC has obvious therapeutic potential.
IL-19 was first described in 2001, and is a member of an IL-10 sub-family including IL-20, IL-22, and IL-24 [8]. IL-19 has been shown to polarize T-lymphocytes to the Th2 phenotype, and is thus considered to be anti-inflammatory [9,10]. We reported that IL-19 is expressed in EC and VSMC in injured, but not naive arteries, and in cytokine-stimulated, but not unstimulated cultured EC and VSMC[11,12]. We recently reported that when injected into LDLR−/− mice, IL-19 could reduce atherosclerosis by attenuating inflammatory gene expression in macrophage, VSMC, and EC [13,14].
MicroRNAs (miRNAs) are a class of small, non-coding RNA which regulate gene expression by binding to specific regions in the 3′UTR of target mRNAs to mediate them for translational repression or degradation. miRNA expression can be tissue specific and/or inducible by cytokines, and based on their structure and predicted mRNA targets, a number of miRNA have been predicted to modulate atherosclerosis [15–18]. A number of siRNAs have been implicated as important in maintenance of vascular homeostasis and lipoprotein uptake [16,17,19–21].
In a search to identify molecular mechanisms which mediate IL-19 anti-atherosclerotic effects, we determined that in cultured human VSMC, stimulation with IL-19 induced expression of miRNA133a, a muscle cell-specific miRNA. Because its expression is muscle-specific, most studies on miR133a focus on cardiac myocytes [22–27]. Much less is known about miR133a in VSMC. One report indicates that miR133a is known to regulate mouse VSMC phenotype, proliferation, and intimal hyperplasia [28], presumably by targeting Serum Response Factor (SRF) and SP1, transcription factors known to regulate VSMC phenotype [29]. In searching a public database (www.targetscan.org) [30], for potential miRNA133a targets, when sorted for Aggregate Pct, the highest scoring potential target (>99%) for miR133a is an mRNA coding for Low Density Lipoprotein Receptor Adaptor Protein 1 (LDLRAP1, transcript NM_015627), (also known as Autosomal Recessive Hypercholesterolemia, or ARH protein) with a total of 4 conserved miRNA133 recognition sites. LDLRAP1 is a cytosolic protein which interacts with the cytoplasmic tail of the LDL receptor and functions to internalize the LDL receptor when it engages with LDL [31,32]. Mutations in this gene lead to LDL receptor malfunction and cause the Autosomal Recessive Hypercholesterolemia [ARH] disorder in humans [33,34]. ARH patients have much lower clearance of LDL from the circulation compared to normal subjects and subsequently, increased atherosclerosis [35]. LDLRAP1 function has been posited to be tissue specific, as hepatocytes and lymphoblasts from ARH patients display impaired LDL internalization, but internalization is normal in fibroblasts from the same patients [36]. Although LDLRAP1 is expressed in VSMC, surprisingly, no investigation into a function for LDLRAP1 in lipid internalization, or any other function in VSMC has been reported.
The purpose of this study was to test the hypothesis that IL-19 can reduce oxLDL uptake in human VSMC by induction of miR133a and subsequent reduction in LDLRAP1 abundance. Confirmation of this hypothesis would be the first to implicate LDLRAP1 as a target for miR133a, show LDLRAP1 plays a role in VSMC lipid homeostasis, and demonstrate IL-19-driven miR133a expression and IL-19 itself as a link for two important physiological processes: anti-inflammation and vascular cell lipid homeostasis.
2.0 Results
2.1 IL-19 induces expression of miR133a
Although VSMC respond to cytokines by proliferation and phenotype modulation, cytokine induction of miR133a in VSMC has not been reported. To determine if IL-19 could induce miR133a expression, human VSMC were serum-starved for 24 hours then stimulated with IL-19 for various times. Figure 1A shows that miR133a is rapidly and transiently increased in response to IL-19, peaking at levels 14-fold above basal, between 2 and 6 hours post-stimulation. miR133a expression is reported to be muscle-specific, and accordingly, IL-19 was unable to induce miR133a expression in either endothelial cells or bone marrow-derived macrophage (BMDM). We did not observe expression of miR133b in IL-19 stimulated VSMC, which is consistent with reports suggesting that this miRNA is expressed only in skeletal muscle [37,38]. This is the first report to describe IL-19 induction of microRNA as well as and cytokine induction of miR133a.
Figure 1.

IL-19 significantly increases expression of miRNA133a. A. Human VSMC, EC, or mouse BMDM were treated with IL-19, RNA extracted at the indicated times, and miR133a abundance was detected by quantitative RT-PCR. * indicate P<0.05, ** P<0.01.
2.2 miR133a targets LDLRAP-1
In searching a public database [www.targetscan.org] for potential targets, the highest scoring miR133a target (>99%) when sorted for Aggregate Pct, is a transcript for a protein called Low Density Lipoprotein Receptor Adaptor Protein 1 (LDLRAP1, transcript NM_015627), (also known as Autosomal Recessive hypercholesterolemia, or ARH protein) with a total of 4 conserved miRNA133 recognition sites. The locations of complementary pairing sites to several areas of the 3′ region of LDLRAP1 mRNA are shown in Figure 2A.
Figure 2.

miR133a reduces LDLRAP1 expression. A. Targetscan analysis identifies seed regions of miR133a which target regions of LDLRAP1 mRNA. The locations of the miR133a complementary sites on human LDLRAP1 3′UTR are shown. B. Transfection of miR133a mimic at different concentrations reduce LDLRAP1 protein expression. Cell lysates were harvested 24 hours post-transfection and western blotted using the indicated antibodies. C. Densiometric quantitation of LDLRAP1 protein in VSMC transfected with different concentrations of miR133a mimic. D. Time course showing LDLRAP1 mRNA is significantly decreased after transfection with 25ng/ml miR133a mimic. RNA was extracted at the indicated times after transfection, and LDLRAP1 mRNA abundance detected by quantitative RT-PCR. E. Time course showing LDLRAP1 protein is significantly decreased after transfection with 25ng/ml miR133a mimic. Cell lysates were harvested at the indicated times post-transfection and western blotted using the indicated antibodies. F. Densiometric quantitation of LDLRAP1 protein in VSMC transfected with 25ng/ml miR133a mimic. Results from three independent experiments. G. miR133a significantly reduces LDLRAP1 mRNA stability. Human VSMC transfected with either miR133a or scrambled control were treated with Actinomycin D, and RNA extracted at the indicated times. H. miR133a targets and silences LDLRAP1 3′UTR. NIH 3T3 cells transfected with a luciferase reporter containing LDLRAP1 3′UTR and also miR133a or scrambled control. Asterisks indicate significant difference from control. * indicate P<0.05, *** P<0.001.
Several experiments were performed to confirm an association between miR133a expression and LDLRAP1 abundance. First, different concentrations of miR133a mimic was transfected into human VSMC to determine a dose response. Figures 2B and 2C show that both 25 and 100 ng/mL 133a mimic were able to significantly reduce LDLRAP1 protein expression, and 25 ng of miR133a mimic was used for all subsequent studies. Next, a time course of LDLRAP1 mRNA expression determined that LDLRAP1 mRNA abundance was reduced two hours post miR133a transfection (Figure 2D). LDLRAP1 protein abundance is also significantly decreased between 8 and 24 hours post-miR133a mimic transfection (Figure 2E,F.). One mechanism of miRNA function is reduction of mRNA stability [15]. miR133a-transfected hVSMC were stimulated with IL-19, then treated with the transcription inhibitor Actinomycin D. Figure 2G shows that that miR133a significantly reduces stability of LDLRAP1 mRNA. Lastly, NIH-3T3 cells were transfected with a luciferase reporter containing LDLRAP1 3′UTR and miR133a or scrambled control. NIH 3T3 fibroblast cells were used to avoid potential background of endogenous miR133a in muscle cells. Figure 2H shows that miR133a potently silences LDLRAP1 through interaction with the LDLRAP1 3′UTR. Together these data are the first to report targeting of LDLRAP1 by miR133a and demonstrate that miR133a can reduce LDLRAP1 mRNA and protein abundance in human VSMC.
2.3 LDLRAP1 is increased in injured arteries and stimulated VSMC
LDLRAP1 expression is uncharacterized in injured vasculature or activated VSMC, and nothing has been reported concerning its cytokine induction in any cell type. Several experiments were conducted to characterize LDLRAP1 expression in different models of vascular injury and in stimulated VSMC. First, using specific antibody and immunohistochemistry of mouse vascular tissue, we determined that LDLRAP1 is expressed preferentially in neointimal VSMC in ligated mouse carotid arteries [Figure 3A]. Development of neointima in this model is primarily VSMC-driven, and neointimal VSMC are in an activated, synthetic state. The LDLRAP1 promoter contains an NF-κB consensus site, suggesting inflammation-responsive expression. To determine if LDLRAP-1 expression can be induced in cultured human VSMC by inflammatory stimuli, TNFα (10ng/ml), a potent proinflammatory cytokine, was found to induce LDLRAP1 protein expression (Figure 3C). LDLRAP1 expression is also induced in VSMC by oxidized LDL (oxLDL) (50μg/ml), together supporting the concept that LDLRAP1 expression is an inflammation responsive gene in activated VSMC.
Figure 3.
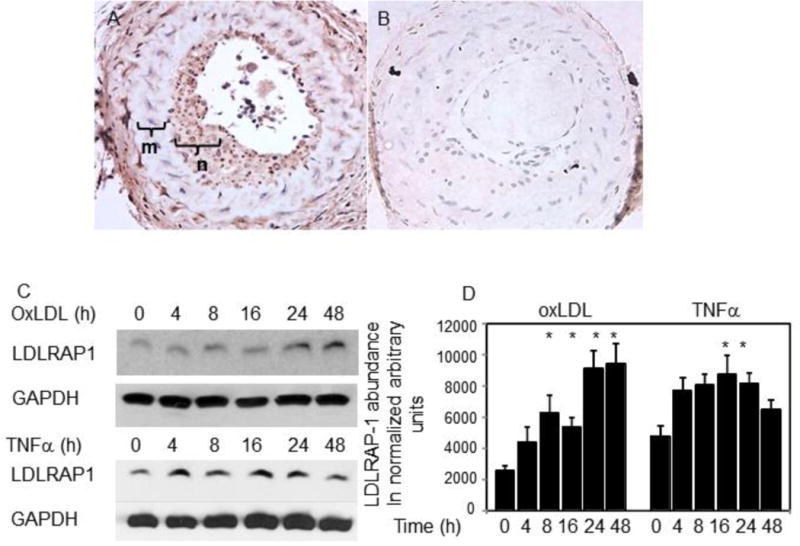
LDLRAP1 expression in activated VSMC and injured arteries. A. Increased LDLRAP1 expression localizes to neointima in ligated mouse carotid artery. Area marked “m” is the media, “n” is the neointima. B. Secondary IgG [negative control] staining of “A”. C. LDLRAP1 expression in oxLDL stimulated and TNFα-stimulated human VSMC. VSMC lysates were harvested at the indicated times post stimulation and western blotted using the indicated antibodies. D. Densiometric quantitation of LDLRAP1 protein expression in VSMC treated with oxLDL or TNFα. Asterisks indicate significant difference from unstimulated VSMC. * indicate P<0.05.
We isolated RNA from aortic arch from ApoE mice, and determined that miRNA133a expression is significantly decreased in mice fed a high-fat diet (HFD) compared with chow-fed mice [Figure 4A]. Correspondingly, LDLRAP1 mRNA is increased in aortic arch in HFD-fed mice (Figure 4B). RNA isolated from experiments in which LDLR−/− mice fed a HFD were injected with IL-19 [13] demonstrates significantly less LDLRAP1 mRNA expression compared with saline controls [Figure 4C]. Using LDLRAP1-specific antibody, we confirmed that LDLRAP1 protein is expressed in plaque from LDLR−/− mice fed an atherogenic diet (Figure 4D). Higher magnification shows that LDLRAP1 abundance is higher in those VSMC within the plaque and VSMC within the cap in atherosclerotic plaque regions, whereas LDLRAP1 is barely detectable in medial VSMC located on the opposite side of the aorta from the plaque (Figures 4 E,G). In human coronary arteries, LDLRAP1 expression is enhanced in myelofibrotic VSMC rich regions of atherosclerotic plaque compared with medial VSMC (Figure 4H). This is the first description and characterization of LDLRAP1 expression in vascular injury and VSMC in response to inflammatory stimuli.
Figure 4.

miR133a and LDLRAP1 expression in atherosclerosis tissue. A. miR133a expression is significantly increased in aortic arch from LDLR−/− mice fed a HFD compared with controls. B. LDLRAP1 mRNA expression is significantly increased in aortic arch from LDLR−/− mice fed a HFD compared with controls. C. LDLRAP1 mRNA is significantly reduced in aortic arch from LDLR−/− mice fed a HFD and also injected with IL-19 compared with saline controls. D. LDLRAP1 protein is expressed in plaque VSMC from LDLR−/− mice. E. higher magnification of “D” showing increased LDLRAP1 expression in medial and cap VSMC in proximity to the plaque. F. Secondary IgG [negative control] staining of “E”. G. higher magnification of “D” showing little to no LDLRAP1 expression in VSMC on the opposite side of the aorta from the plaque. H. higher LDLRAP1 expression in myelofibrotic plaque region compared with medial cells in human atherosclerotic plaque. I. Secondary IgG (negative control) staining of “H”. * indicate P<0.05.
2.4 IL-19 decreases LDLRAP-1 mRNA and protein abundance
To further link IL-19 stimulation with LDLRAP-1 abundance, human VSMC were serum-starved for 48 hours, then stimulated with IL-19. Figure 4A shows that IL-19 significantly decreases LDLRAP-1 mRNA 2 hours post-stimulation, which demonstrates similar kinetics to IL-19 induction of miR133a. IL-19 could also rapidly decrease LDLRAP1 protein abundance, with significant reduction beginning four hours post-stimulation (Figure 5B and C). It was important to determine if IL-19 reduction in LDLRAP1 expression was mediated by miR133a. Human VSMC were transfected with scrambled control, or LDLRAP1 anti-miR133a, serum starved then stimulated with IL-19. Figures 5D,E shows that IL-19 does not reduce LDLRAP1 protein abundance in VSMC transfected with anti-miR133a. This suggests that IL-19 reduction of LDLRAP1 is dependent upon miR133a.
Figure 5.
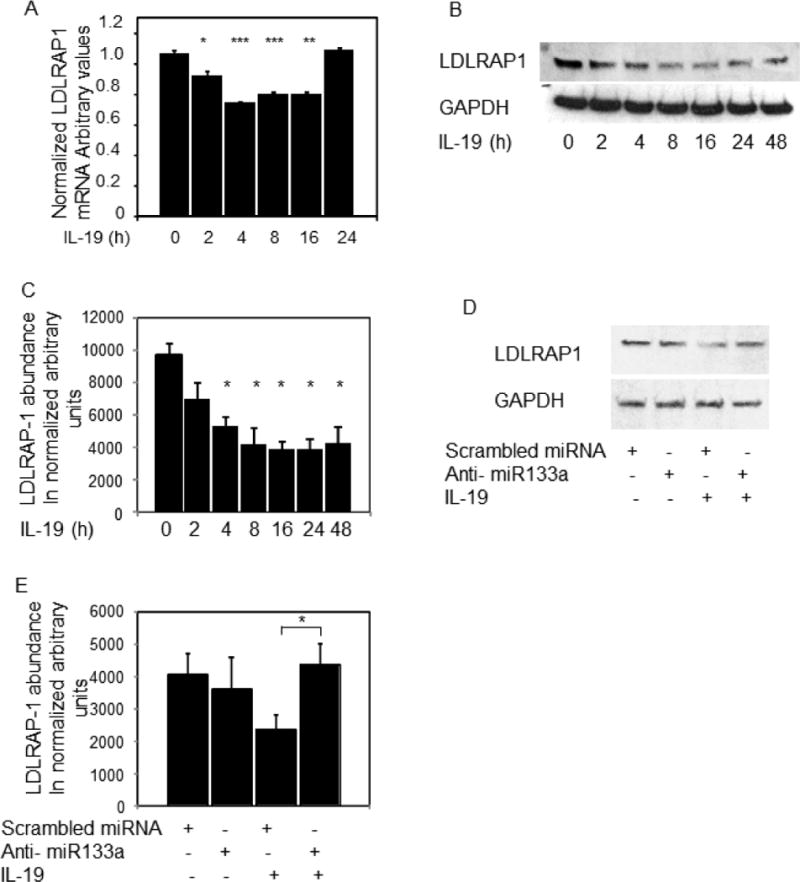
IL-19 reduces LDLRAP1 expression. A. LDLRAP1 mRNA expression is significantly reduced by IL-19. Human VSMC were treated with IL-19, RNA extracted at the indicated times, and LDLRAP1 mRNA abundance detected by quantitative RT-PCR. B. IL-19 significantly reduces LDLRAP1 protein expression. VSMC lysates were harvested at the indicated times post IL-19 stimulation and western blotted using the indicated antibodies. C. Densiometric quantitation of LDLRAP1 protein in VSMC treated with IL-19. Times from 4 to 48 hours were significantly lower compared with untreated cells. D. anti-miR133a prevents IL-19 from reducing LDLRAP1 protein abundance. 25ug anti-miR133a or scrambled control were transfected into human VSMC and stimulated with IL-19. E. densiometric quantitation of LDLRAP1 protein expression in VSMC transfected with anti-miR133a. Asterisk indicates significant reduction in the scrambled versus anti-miR transfected VSMC. * indicate P<0.05, **P<0.01, *** P<0.001.
2.5 IL-19 decreases cholesterol uptake in VSMC
One presumed function of LDLRAP1 in hepatocytes is internalization of cholesterol upon LDLR engagement [31,32], but nothing is reported for LDLRAP1 function in VSMC. To determine if IL-19 could decrease oxLDL uptake, human VSMC were serum starved for 24 hours, then stimulated with IL-19 for 16 hours, then incubated with 5ng/ml DiI oxLDL. Lipid uptake was quantitated by flow cytometry as we described [14]. Figure 6A shows that IL-19 treatment can significantly decrease oxLDL uptake by 39% percent (575.4+/−12.0 vs 356.6+/−19.4 mean fluorescence units, for untreated and IL-19-treated VSMC, respectively, P<0.001). VSMC from wild type mice were isolated and treated with Dil oxLDL as described for human, and oxLDL uptake quantitated by flow cytometry. Figure 6B shows that similar to human, addition of rIL-19 to mouse VSMC decreases lipid uptake 24% (913.7+/−28.1 vs 695.3+/− 21.74 for untreated and IL-19 treated, P<0.01). To strengthen the link between IL-19 and LDLRAP1 expression, aortic VSMC were isolated from IL-19−/− mice [39]. In contrast with wild type mice, IL-19−/− VSMC take up significantly more oxLDL compared with wild type VSMC (589.7+/−16.7 vs 1067.0+/− 48.6 for wild type and control, P<0.001) (Figure 6C). Correspondingly, significantly more LDLRAP-1 mRNA and protein is expressed in VSMC isolated from IL-19−/− VSMC compared with wild type (Figures 6D–6F).
Figure 6.
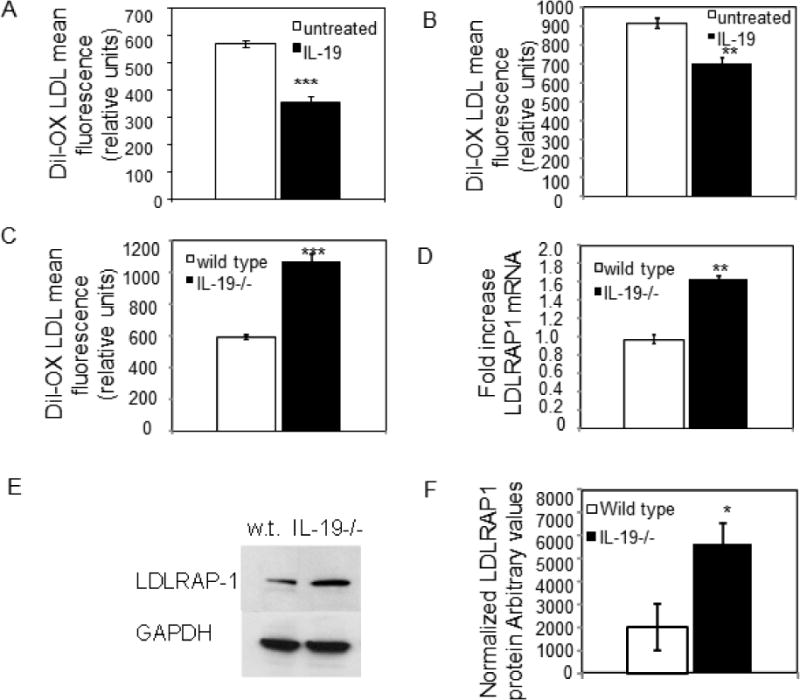
Reduction in uptake of fluorescently labeled oxLDL in VSMC by IL-19 and miR133a. A. Human VSMC were treated with IL-19 for 16 hours, then uptake quantitated by flow cytometry. B. mouse VSMC were treated with IL-19 for 16 hours, then uptake quantitated by flow cytometry. C. VSMC from IL-19−/− mice take up significantly more oxLDL than wild type VSMC. D. VSMC from IL-19−/− mice express significantly more LDLRAP1 mRNA than wild type VSMC. E. VSMC from IL-19−/− mice express significantly more LDLRAP1 protein than wild type VSMC. F. Densiometric quantitation of LDLRAP1 protein expression in IL-19−/− and wild-type VSMC. Asterisks indicate significant reduction in the treated versus control VSMC. * indicate P<0.05, **P<0.01, *** P<0.001.
2.6 miR133a, and LDLRAP1 knock down decreases cholesterol uptake in VSMC
To link miR133a expression with lipid uptake, miR133a was transfected into human VSMC, and oxLDL uptake quantitated. Figure 7A shows that miR133a mimic significantly reduces LDLRAP1 expression and also cholesterol internalization in VSMC (754.0+/−26.1 vs 524.5+/−10.0 mean fluorescence units, for scrambled and miR133a respectively, P<0.01). Importantly, in VSMC transfected with miR133a mimic IL-19 does not reduce abundance of LDLRAP1 protein or oxLDL internalization, further suggesting that IL-19 reduction in oxLDL internalization is mediated by miR133a. In a second experiment, human VSMC were transfected with LDLRAP1 siRNA or scrambled control. Figure 6B shows that knock down of LDLRAP1 reduces LDLRAP1 protein and also oxLDL internalization (840.5+/−20.8 vs 599.3+/−15.7 mean fluorescence units for scramble and LDLRAP1 siRNA, respectively, P<0.01). Similar to miR133a, addition of IL-19 to LDLRAP1 siRNA transfected VSMC does not reduce abundance of LDLRAP1 protein or oxLDL internalization, further suggesting that IL-19 reduction in oxLDL internalization is mediated by reduction in LDLRAP1.
Figure 7.

miR133a and LDLRAP1 regulate oxLDL uptake in human VSMC. Oxidized LDL uptake was quantitated by flow cytometry 48 hours after transfection. A. transfection of miR133a mimic significantly reduces oxLDL uptake in human VSMC. IL-19 does not further reduce this uptake. B. transfection of LDLRAP1 siRNA significantly reduces oxLDL uptake in human VSMC. IL-19 does not further reduce this uptake. C. anti-miR133a prevents IL-19 from reducing oxLDL uptake in human VSMC. Anti-miR133a also prevents IL-19 from reducing LDLRAP1 protein expression. D. Densiometric quantification of effect of anti-miR133a on LDLRAP1 expression shown in “C”. Asterisks indicate significant reduction in the treated versus control VSMC. * * indicate P<0.01.
To determine if IL-19 induced reduction of oxLDL uptake was mediated by miR133a, human VSMC were transfected with anti-miR133a and oxLDL uptake quantitated by flow cytometry. Figures7C, shows that IL-19 cannot reduce LDLRAP1 protein in VSMC transfected with anti-miR133a. Correspondingly, IL-19 cannot further reduce oxLDL uptake in VSMC transfected with ati-miR133a. Together, these data indicate that IL-19-mediated reduction in oxLDL uptake is likely mediated by miR133a-mediated reduction in LDLRAP1 protein.
2.7 miR133a and LDLRAP1 regulate proliferation of human VSMC
Effects of miR133a on human VSMC proliferation have not been reported. We first determined that miR133a could reduce proliferation of human VSMC. For these experiments, VSMC were transduced with AdmiR133a or empty vector control virus. Figures 8A and 8B show that infection of human VSMC with Ad133a results in significantly less proliferation of those cells, compared with scrambled control virus (98.6 ×103+/−9.5 × 103 vs 50.6 × 103+/−3.2 × 103, P<0.01). Limited literature suggests LDLRAP1 may have cell-specific functions unrelated to LDLR internalization [40]. To determine if LDLRAP1 played a role in VSMC proliferation, human VSMC were transfected with LDLRAP1-specific siRNA, and VSMC proliferation quantitated. Figures 8C and 8D show that LDLRAP1 siRNA effectively reduced LDLRAP1 protein expression and that reduction of LDLRAP1 significantly reduced VSMC proliferation compared with scrambled controls (79.8 × 103+/−9.4 × 103 vs 3.17 × 103+/−10.0 × 103, P<0.1). These are the first data to show a role for LDLRAP1 in VSMC proliferation. This suggests an important role in VSMC pathophysiology and vascular proliferative diseases such as allograft vasculopathy and vascular restenosis.
Figure 8.

miR133a and LDLRAP1 regulate human VSMC proliferation. A. miR133a reduces VSMC proliferation. Human VSMC were transduced with 4 MOI of Adscrambled control vector or AdmiR133a, equal numbers seeded, and VSMC counted at 3 and 6 days post-seeding. B. Relative expression of miR133a in adenoviral transduced VSMC. VSMC were infected with AdmiR133a or Adcontrol vector, RNA isolated and miR133a abundance detected by quantitative RT-PCR. C. LDLRAP1 knockdown reduces VSMC proliferation. VSMC were transfected with LDLRAP1 or scrambled siRNA, equal numbers seeded, and VSMC counted at 3 and 6 days postseeding with a BioRad TC20 automated cell counter, n=3. D. western blot verifying that LDLRAP1 siRNA reduces LDLRAP1 protein expression in human VSMC. Asterisks indicate significant reduction in the treated versus control VSMC. E. miR133a levels are significantly increased in plasma from hyperlipidemic patients. Plasma was isolated from normal, or patients with plasma cholesterol >200mg/dl. RNA was isolated, and miR133a levels quantitated by quantitative RT-PCR. N=9 each. ** indicate P<0.01, *** P<0.001.
2.8 Plasma miR133a levels are increased in hyperlipidemia patients
MicroRNA’s are present in many bio fluids, and several studies have attempted to correlate circulating miRNA levels with a specific disease state, particularly vascular diseases [41–43]. Correlation between circulating miR133a and hyperlipidemia in human subjects has not been reported. To draw an association between miR133a and lipid homeostasis, the abundance of miR133a in plasma from normal and hyperlipidemic patients was quantitated by RT-PCR. Figure 8E shows a significant increase in miR133a in plasma from hyperlipidemic (cholesterol >200mg/dl) compared with normolipidemic patients. While the functional ramifications of this increase are not yet clear, miR133a might be a useful diagnostic since its expression is muscle specific, and it does suggest that miR133a abundance in plasma might be considered a biomarker that correlates with increased cholesterol.
3.0 Discussion
We have shown that IL-19 is atheroprotective, and the present study indicates that one mode of protection may be a decrease in lipid uptake by VSMC. Characterization of this mechanism has led to several novel findings. The first novel aspect of this study is that IL-19 induces miR133a expression in VSMC. The miR133 family consists of three mi RNA transcripts; miR133a-1, 133a-2, and 133b, all expressed exclusively in muscle. miR133a-1 and 133a-2 are identical in sequence but are transcribed from different regions [44,45]. They differ from miR133b by only 2nt at their 3′ end. IL-19 did not upregulate expression of miR133b, which is consistent with miR133b expression restricted to skeletal muscle [37,38]. Most studies on miR133a focus on cardiac myocytes, and miR133a participates in heart development, myoblast proliferation and differentiation [22,44–46]. Our observation that IL-19 induced miR133a expression is novel in that cytokine induction of miR133a in VSMC, and IL-19 induction of any miRNA has not been reported.
The TargetScan prediction algorithm predicts that four regions within LDLRAP1 mRNA are a potential target for miR133a, and a second novel finding of this study is that transfection of miR133a mimic reduces LDLRAP1 mRNA and protein abundance in VSMC. We also show that miR133a reduces LDLRAP1 mRNA stability, and targets LDLRAP1 3′UTR LDLRAP1 is a cytosolic adaptor protein which interacts with the cytoplasmic tail of the LDL receptor, presumably to internalize the LDL receptor when it engages with LDL [31,32]. Mutations inLDLRAP1 lead to LDL receptor malfunction and cause the Autosomal Recessive Hypercholesterolemia (ARH) disorder in humans [33,34]. Since IL-19 induced miR133a expression, we reasoned that IL-19 might also reduce LDLRAP1, and several experiments tightly link IL-19 with LDLRAP1 expression. First, IL-19 treatment of VSMC induces a reduction in LDLRAP1 mRNA and protein abundance. Second, LDLRAP1 mRNA and protein are increased in VSMC from IL-19 knock out mice, suggesting that anti-inflammatory stimuli such as IL-19 can reduce LDLRAP1 expression in a miR133a-dependent mechanism in VSMC. Third, IL-19 cannot reduce LDLRAP1 protein abundance in VSMC transfected with anti-miR to miR133a.
Little to no characterization of LDLRAP1 expression in VSMC or vascular tissue has been reported. A third novel finding of this study is the demonstration that LDLRAP1 expression is upregulated in TNFa and oxLDL-stimulated cultured, and in atherosclerotic plaque VSMC, but is barely detectible in medial VSMC which were not in the vicinity of plaque. The LDLRAP1 promoter contains an NF-κB consensus site, further suggesting that in VSMC, LDLRAP1 expression is inflammation-responsive.
Most studies on LDLRAP1 expression and function are performed in hepatocytes. ARH patients have much lower clearance of LDL from the circulation compared to normal subjects and subsequently, increased atherosclerosis [33]. This hypercholesterolemia is assumed to be a result of impaired LDL internalization by hepatocytes, and disruption of LDLRAP1 results in 80% reduction in LDL internalization in cultured hepatocytes [31,32,35]. Importantly, LDLRAP1 function has been posited to be tissue specific [40], as hepatocytes and lymphoblasts from ARH patients display impaired LDL internalization, but internalization is normal in fibroblasts from the same patients. The transformation of VSMC into foam cells is a key step in atherogenesis [2]. Surprisingly, no investigation into a function for LDLRAP1 in lipid internalization in VSMC has been reported. We observed that addition of IL-19 decreases cholesterol uptake in mouse and human VSMC, and uptake was increased in IL-19−/− VSMC. miR133a transfection also decreased lipid uptake in VSMC. Reduction of LDLRAP1 by transfection of specific siRNA also reduced uptake in VSMC. Addition of IL-19 to either of miR133a or LDLRAP1 siRNA transfected VSMC did not further decrease lipid uptake. Importantly, IL-19 was unable to decrease lipid uptake in VSMC transfected with miR133a anti-miR, implying that miR133a is the major mechanism whereby IL-19 reduces lipid uptake in VSMC. This is the first study to implicate LDLRAP1 as regulating lipid homeostasis in VSMC. Because LDLRAP1 is preferentially expressed in activated VSMC, it may contribute to VSMC foam cell formation indicative of advanced atherosclerosis. IL-19 reduction of VSMC oxLDL uptake is in contrast to our previous report showing that IL-19 increases oxLDL uptake in macrophages [14], which is also an atheroprotective mechanism [47]. We speculate that IL-19 reduces oxLDL uptake in VSMC but not macrophages because miR133a is muscle specific. VSMC expression of miR133a may be part of IL-19′s novel counter-regulatory protective response to reduce local oxLDL uptake and VSMC foam cell formation. While these findings implicate miR133a as a molecular link between anti-inflammatory processes and lipid homeostasis in VSMC, it does not define if reduction of LDLRAP1 is the sole mechanism for IL-19 reduction in lipid uptake in VSMC. Future studies should investigate if VSMC from ARH patients have altered LDL internalization.
Previous studies link miR133a with cardiomyocyte proliferation [44]. Mice lacking miR133a demonstrated increased proliferation of cardiomyocytes, but not cardiac fibroblasts. The same study showed that over expression of miR133a resulted in diminished proliferation of cardiac myocytes. In a second study, it was shown that miR133a participates in mouse VSMC phenotype modulation and intimal hyperplasia which the authors speculated was through targeting of SRF [28]. Our study using only 4 MOI of AdmiR133a confirms a role for miR133a in regulation of human VSMC proliferation. Over expression of miR133a could potentially decrease VSMC proliferation by forcing the cell to maintain the differentiated, contractile phenotype. However, our current study could represent an alternative mechanism that miR133a reduces proliferation because it reduces LDLRAP1 abundance as we demonstrated reduction of LDLRAP1 with specific siRNA resulted in a dramatic decrease of VSMC proliferation. Indeed, limited literature suggests LDLRAP1 has functions outside of LDLR internalization. For example, fibroblasts from some ARH patients grow more slowly, and one report suggests LDLRAP1 may participate in mitosis [40]. VSMC in which LDLRAP1 is knocked down are comparable to ARH patients which have a non-functional LDLRAP1 protein, suggesting a function for LDLRAP1 in VSMC proliferation. Also, LDLRAP1 is expressed in squid, which lack LDL receptors, suggesting a function not related to LDLR internalization [48]. Together with data showing LDLRAP1 expression in neointimal VSMC which are primarily proliferative, this may link LDLRAP1 expression with VSMC proliferation, suggesting a role in vascular proliferative diseases such as restenosis and allograft vasculopathy.
MicroRNA’s are present in many biological fluids and they can be released by multiple mechanisms ranging from tissue injury and cellular breakdown to active secretion in exosomes [49]. There are several examples where abundance of miRNA’s have been correlated with vascular diseases in rodents and humans [16,50]. For example, circulating levels miR-33a and miR-33b, which have been found to play a role in cholesterol transport and regression of atherosclerosis, were found to be increased in plasma of hypercholesteremic children [17,41]. Compared with other miRNA’s, miR133a is a particularly attractive surrogate in that its expression is muscle specific, and one study suggested that miR133a may represent a biomarker of acute muscle toxicity [51]. Although our study examined a small sample size, miR133a abundance was significantly greater in plasma from hyperlipidemic patients compared with normolipidemic patients and, future studies may implicate miR133a as a useful diagnostic for muscleopathies as well as vascular disease.
3.1 Conclusions
IL-19 is atheroprotective [13], and we have recently shown that IL-19 increases oxLDL uptake and efflux in macrophage [14]. The present study suggests that one mode of IL-19 athero-protection may be a decrease in lipid uptake and foam cell formation by VSMC. Because miRNA133a expression is muscle-specific, it may explain how IL-19 can decrease oxLDL uptake in VSMC, but not in macrophages. We propose that while LDLRAP1 function in liver and macrophage is essential for cholesterol uptake and maintenance of normal cholesterol levels, reduction of LDLRAP1 in VSMC is atheroprotective. Because miRNA133a can decrease oxLDL uptake in VSMC but not in macrophages, it may represent a novel target to reduce plaque size and vulnerability to rupture.
4.0 Materials and Methods
4.1 Cells and culture
Primary human coronary artery vascular endothelial cells, and human coronary artery vascular smooth muscle cells were obtained as cryopreserved secondary culture from Lonza Corporation [Allendale, NJ] and maintained as we described [11–13]. Cells were used from passage 3–5. For mouse VSMC, abdominal aorta from wild-type and IL-19 knockout mice were excised, endothelial layer removed, and VSMC isolated as described [37]. VSMC were cultured in DMEM supplemented with 15% fetal calf serum (FCS). Greater than 95% of isolated cells were SMC actin positive, and VSMC from passage 3 to 5 were used. Bone marrow-derived macrophages (BMDM) were generated as we described [13,14]. Briefly, femurs and tibiae were flushed with sterile DMEM, collected cells washed, resuspended in DMEM+5% fetal bovine serum (FBS), and cultured overnight to remove adherent cells. Non-adherent cells were cultured for 6 days in DMEM+10% FBS in the presence of 100ng/ml M-CSF (Peprotech). Adherent cells were then detached by incubation with Versene solution (GIBCO). For proliferation, VSMC infected by adenovirus or transfected with siRNA were seeded at 103 VSMC/ml in 12 well trays as we described [52]. VSMC were trypsinized and counted on a BioRad TC20 automated cell counter and expressed as VSMC/ml. All animal procedures followed Temple University IACUC approved protocols.
4.2 DiI LDL uptake and Luciferase assay
To measure DiI LDL uptake, 1×106 VSMC were plated in 12 well plates in DMEM +1%BSA, with or without 100ng/ml IL-19. After overnight culture VSMC were incubated in the dark with DiI-oxLDL (5ug/ml) (Kalen Biomedical) for 4 hours on 37°C. After the incubation cells were washed 3X with PBS, detached with Versene, harvested with 1% BSA in HBSS and subjected to flow cytometry analysis (FACS Calibur, Becton Dickinson) as we described [14]. The mean of DiI-oxLDL fluorescence intensity was obtained from 10,000 cells. Data were calculated and expressed as mean fluorescence intensity (MFI). Targeting of LDLRAP1 3′UTR by miR133a was assayed by luciferase assay. NIH 3T3 cells were transfected with luciferase preceded by the LDLRAP1 3′UTR (SwitchGear Genomics, Inc), and luciferase assay performed as we described [14]. NIH 3T3 cells were obtained from the American Type Culture Collection and cultured in DMEM containing 10% FBS.
4.3 Transfection and siRNA knockdown
Gene silencing was performed using ON-TARGET plus SMARTpool LDLRAP1 siRNA, which contains a mixture of four siRNAs which target human LDLRAP1 (10 nM) purchased from Dharmacon, Inc. as we have described [14]. miR133a and miR133a anti-mer was purchased from Dharmacon, Inc. Transfection of VSMC was performed using the AMAXA Nucleofector™ Kit (Amaxa, Inc.) following the manufacturer’s instructions as we described [14]. The miR133a adenovirus was constructed using the Invitrogen Blocki-PolII miR RNAi expression system using oligonucleotides representing the miR133a sequence: top 5′ TGCTACAATGCTTTGCTAGAGCTGGTAAAATGGAACCAAATCGCCTCTTCAATGGATTTGGTCC CCTTCAACCAGCTGTAGCTATGCATTGA; bottom 5′-CCTGTCAATGCATAGCTACAGCTGGTTGAAGGGGACCAAATCCATTGAAGAGGCGATTTGGTT CCATTTTACCAGCTCTAGCAAAGCATTGT. Scrambled control adenovirus is a control provided by Invitrogen. Both were used at 4MOI.
4.4 Immunohistochemistry
Ligated mouse carotid arteries and human coronary arteries were collected as part of studies described previously [12,13,39,53]. Tissue sections were fixed in 10% buffered formalin, embedded in paraffin, sectioned at 5μm, deparaffinized in xylene and rehydrated through graded alcohols. Endogenous peroxidase activity was blocked with 1.5% hydrogen peroxide in methanol for 15 min. Tissue sections were blocked in 10% goat serum, incubated with primary antibody at 0.5μg/ml in 1%BSA/PBS and were applied for 1 hour, followed by incubation with biotinylated secondary antibody (1:200), followed by avidin-biotin peroxidase complex each for 30 minutes. Non-specific identical IgG isotype control (Neomarkers # NC-100-P, and Biolegend #400601) antibodies were used as negative controls. The reaction product was visualized using DAB (Vector Labs) as the chromogenic substrate, which produces a reddish-brown stain. The sections were counterstained with hematoxylin.
4.5 RNA extraction and quantitative RT-PCR
RNA from cultured cells, aorta, or spleen was isolated and reverse transcribed into cDNA as we have described, and target genes amplified using an Eppendorf Realplex4 Mastercycler [12,13,39]. RNA from mouse aorta was isolated as part of a previously published study [13] and adhered to IACUC approved protocols. Multiple mRNAs (Ct values) were quantitated simultaneously by the Eppendorf software. Primer pairs were purchased from Integrated DNA Technologies, (Coralville, IA), SYBR green used for detection. The following primer pairs were used: Human LDLRAP1: F TCTTTTCCATCCTGAGGTGC, R: TAAGGTGTCCGGTTACTCCA Mouse LDLRAP1: FTGTTGGGTGATGCTGTAGTC, RGCTCACACTCAGAAAGGGAA. miR133a was detected using a kit from Qiagen, Inc. (catalog numbers MS00031423, MS00032305, and MS00033740). Human plasma samples were obtained in compliance with the Declaration of Helsinki and approved by the Internal Institutional Review Board (IRB) of Temple University School of Medicine. Written informed consent (English/Spanish) was obtained from every individual involved in this study. All study participants (human subjects) providing plasma samples were recruited from the Department of Ophthalmology at Temple University Hospital and for the Control Group, from the Sol Sherry Thrombosis Research Center, Temple University School of Medicine. Twelve milliliters of whole blood was obtained from either patients or control subjects. Blood was processed within 30 minutes of phlebotomy by high speed centrifugation [3,000 rpm] to obtain plasma poor in platelets and subsequently aliquoted in Eppendorf tubes and stored at −80°C for further analysis. miR133a abundance in human plasma was performed by Exiqon, Inc. Total RNA was extracted from 200μl plasma using the miRCURY™ biofluids RNA isolation kit. Each RNA sample was reverse transcribed [RT] into cDNA and run on the miRCURY LNATM Universal RT microRNA PCR PicknMix and amplified in a Roche Lightcycler 480. Raw Cp values and melting points are quantitated by the Lightcycler software. Normalization is performed based on the average of the assays detected in all samples as this is shown to be the best normalization for qPCR studies involving numerous assays Normalized Cq = average Cq (n=30) - assay Cq (sample) [53].
4.6 Western blotting
Protein extracts VSMC were made as described [11–14] and separated by SDS-PAGE, transferred to nitrocellulose membrane, incubated with a 1:5000 dilution of LDLRAP1, or 1:10000 dilution of GAPDH primary antibody (Biogenesis, Inc.), and a 1:8000 dilution of secondary antibody. Equal loading of protein extracts on gels was verified by Ponceau S staining of the membrane, and blotting with the housekeeping protein anti-GAPDH [1:7000 dilution, and reactive proteins were visualized using enhanced chemiluminescence. The intensity of each band was quantitated using image analysis software (NIH Image, Frederick, MD).
4.7 Statistical analysis
Results are expressed as mean ± SEM. Differences between groups were evaluated with the use of Student’s t test, or ANOVA, where appropriate. Differences were considered significant when p<0.05. Analysis of statistical significance of miR133a abundance in normal versus hypercholesteremic patients was determined by the Benjamini-Hochberg analysis correction with a significance of 0.05 and performed by Exiqon, Inc.
Highlights.
IL-19 induces miR133a expression in VSMC
miR133a targets LDLRAP1 which regulates LDLR internalization
miR133a and LDLRAP1 siRNA reduce VSMC foam cell formation
IL-19 driven miR133a expression links lipid metabolism and anti-inflammation
Acknowledgments
Funding:
This work was supported by grants HL115575 and HL117724 from the National Heart Lung, and Blood Institute of the National Institutes of Health, and Grant 13GRNT1685003 from the American Heart Association to MVA. K.G was supported by American Heart Association post-doctoral fellowship 11POST7530001. MR was supported by American Heart Association pre-doctoral fellowship 16PRE31220005.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
The authors declare no actual or potential conflict of interest including any financial, personal or other relationships with other people or organizations.
References
- 1.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 2.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;5:812–819. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pei H, Wang Y, Miyoshi T, Zhang Z, Matsumoto AH, Helm GA, Tellides G, Shi W. Direct evidence for a crucial role of the arterial wall in control of atherosclerosis susceptibility. Circulation. 2006;114:2382–2389. doi: 10.1161/CIRCULATIONAHA.106.640185. [DOI] [PubMed] [Google Scholar]
- 5.Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. 2010;47:168–80. doi: 10.1159/000250095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raines E, Ferri N. Cytokines affecting endothelial and smooth muscle cells in vascular disease. J Lipid Res. 2005;46:1081–109. doi: 10.1194/jlr.R500004-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Geng YJ, Libby P. Progression of atheroma: a struggle between death and procreation. Arterioscler Thromb Vasc Biol. 2002;22:1370–1380. doi: 10.1161/01.atv.0000031341.84618.a4. [DOI] [PubMed] [Google Scholar]
- 8.Gallagher G, Dickensheets H, Eskdale J, Izotova LS, Mirochnitchenko OV, Peat JD, Vazquez N, Pestka S, Donnelly RP, Kotenko SV. Cloning, expression and initial characterization of interleukin-19 IL-19, a novel homologue of human interleukin-10 IL-10. Genes Immun. 2000;1:442–450. doi: 10.1038/sj.gene.6363714. [DOI] [PubMed] [Google Scholar]
- 9.Gallagher G, Eskdale J, Jordan W, Peat J, Campbell J, Boniotto M, Lennon GP, Dickensheets H, Donnelly RP. Human interleukin-19 and its receptor: a potential role in the induction of Th2 responses. International Immunopharmacology. 2004;4:615–626. doi: 10.1016/j.intimp.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Oral H, Kotenko S, Yilmaz M, Mani O, Zumkehr J, Blaser K, Akdis C, Akdis M. Regulation of T cells and cytokines by the interleukin-10 IL-10-family cytokines IL-19, IL-20, IL-22, IL-24 and IL-26. Eur J Immunol. 2006;36:380–8. doi: 10.1002/eji.200425523. [DOI] [PubMed] [Google Scholar]
- 11.Tian Y, Sommerville LJ, Cuneo A, Kelemen SE, Autieri MV. Expression and Suppressive Effects of Interleukin-19 on Vascular Smooth Muscle Cell Proliferation, Signaling, and Development of Intimal Hyperplasia. American Journal of Pathology. 2008;173:901–909. doi: 10.2353/ajpath.2008.080163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain S, Gabunia K, Kelemen SE, Panetti TS, Autieri MV. The anti-inflammatory cytokine interleukin 19 is expressed by and angiogenic for human endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:167–175. doi: 10.1161/ATVBAHA.110.214916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellison S, Gabunia K, Kelemen SE, England RN, Scalia R, Richards JM, Orr AW, Traylor JG, Jr, Rogers T, Cornwell W, Berglund LM, Goncalves I, Gomez MF, Autieri MV. Attenuation of experimental atherosclerosis by interleukin-19. Arterioscler Thromb Vasc Biol. 2013;33:2316–2324. doi: 10.1161/ATVBAHA.113.301521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabunia K, Ellison S, Kelemen S, Kako F, Cornwell WD, Rogers TJ, Datta PK, Ouimet M, Moore KJ, Autieri MV. IL-19 Halts Progression of Atherosclerotic Plaque, Polarizes, and Increases Cholesterol Uptake and Efflux in Macrophages. Am J Pathol. 2016;186:1361–1374. doi: 10.1016/j.ajpath.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rayner KJ, Moore KJ. MicroRNA control of high-density lipoprotein metabolism and function. Circ Res. 2014;114:183–92. doi: 10.1161/CIRCRESAHA.114.300645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun X, He S, Wara AK, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, Croce K, Feinberg MW. Systemic delivery of microRNA-181b inhibits nuclear factor-κB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ Res. 2014;114:32–40. doi: 10.1161/CIRCRESAHA.113.302089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;2:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Virtue A, Mai J, Yn Y, Meng S, Tran T, Jiang X, Wang H, Yang XF. Structural evidence of antiatherogenic microRNAs. Front Biosci Landmark Ed. 2011;16:3133–3145. doi: 10.2741/3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Näär AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramirez CM, Dávalos A, Goedeke L, Salerno AG, Warrier N, Cirera-Salinas D, Suárez Y, Fernández-Hernando C. MicroRNA-758 regulates cholesterol efflux through posttranscriptional repression of ATP-binding cassette transporter A1. Arterioscler Thromb Vasc Biol. 2011;31:2707–2714. doi: 10.1161/ATVBAHA.111.232066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Aguiar Vallim TQ, Tarling EJ, Kim T, Civelek M, Baldán Á, Esau C, Edwards PA. MicroRNA-144 regulates hepatic ATP binding cassette transporter A1 and plasma high-density lipoprotein after activation of the nuclear receptor farnesoid X receptor. Circ Res. 2013;112:1602–1612. doi: 10.1161/CIRCRESAHA.112.300648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grange RW, Richardson JA, Bassel-Duby R, Olson EN. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J Clin Invest. 2011;121:3258–3268. doi: 10.1172/JCI46267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meder B, Katus HA, Rottbauer W. Right into the heart of microRNA-133a. Genes Dev. 2008;22:3227–3231. doi: 10.1101/gad.1753508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proceedings of the National Academy of Sciences. 2006;103:8721–8726. doi: 10.1073/pnas.0602831103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 26.van Rooij E, Liu N, Olson EN. MicroRNAs flex their muscles. Trends Genet. 2008;24:159–166. doi: 10.1016/j.tig.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Williams AH, Liu N, van Rooij E, Olson EN. MicroRNA control of muscle development and disease. Curr Opin Cell Biol. 2009;21:461–469. doi: 10.1016/j.ceb.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011;109:880–893. doi: 10.1161/CIRCRESAHA.111.240150. [DOI] [PubMed] [Google Scholar]
- 29.Miano JM, Small EM. MicroRNA133a: a new variable in vascular smooth muscle cell phenotypic switching. Circ Res. 2011;109:825–837. doi: 10.1161/CIRCRESAHA.111.254656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agarwal V, Bell GW, Nam J, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sirinian MI, Belleudi F, Campagna F, Ceridono M, Garofalo T, Quagliarini F, Verna R, Calandra S, Bertolini S, Sorice M, Torrisi MR, Arca M. Adaptor protein ARH is recruited to the plasma membrane by low density lipoprotein LDL binding and modulates endocytosis of the LDL/LDL receptor complex in hepatocytes. J Biol Chem. 2005;280:38416–38423. doi: 10.1074/jbc.M504343200. [DOI] [PubMed] [Google Scholar]
- 32.Garuti R, Jones C, Li WP, Michaely P, Herz J, Gerard RD, Cohen JC, Hobbs HH. The modular adaptor protein autosomal recessive hypercholesterolemia ARH promotes low density lipoprotein receptor clustering into clathrin-coated pits. J Biol Chem. 2005;280:40996–41004. doi: 10.1074/jbc.M509394200. [DOI] [PubMed] [Google Scholar]
- 33.Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, Calandra S, Bertolini S, Cossu F, Grishin N, Barnes R, Cohen JC, Hobbs HH. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292:1394–1398. doi: 10.1126/science.1060458. [DOI] [PubMed] [Google Scholar]
- 34.Zuliani G, Arca M, Signore A, Bader G, Fazio S, Chianelli M, Bellosta S, Campagna F, Montali A, Maioli M, Pacifico A, Ricci G, Fellin R. Characterization of a new form of inherited hypercholesterolemia: familial recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1999;19:802–809. doi: 10.1161/01.atv.19.3.802. [DOI] [PubMed] [Google Scholar]
- 35.Jones C, Hammer RE, Li WP, Cohen JC, Hobbs HH, Herz J. Normal sorting but defective endocytosis of the low density lipoprotein receptor in mice with autosomal recessive hypercholesterolemia. J Biol Chem. 2003;278:29024–29030. doi: 10.1074/jbc.M304855200. [DOI] [PubMed] [Google Scholar]
- 36.Sirinian MI, Belleudi F, Campagna F, Ceridono M, Garofalo T, Quagliarini F, Verna R, Calandra S, Bertolini S, Sorice M, Torrisi MR, Arca M. Adaptor protein ARH is recruited to the plasma membrane by low density lipoprotein LDL binding and modulates endocytosis of the LDL/LDL receptor complex in hepatocytes. J Biol Chem. 2005;280:38416–38423. doi: 10.1074/jbc.M504343200. [DOI] [PubMed] [Google Scholar]
- 37.van Rooij E, Liu N, Olson EN. MicroRNAs flex their muscles. Trends Genet. 2008;24:59–66. doi: 10.1016/j.tig.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 38.McCarthy JJ. MicroRNA-206: the skeletal muscle-specific myomiR. Biochim Biophys Acta. 2008;1779:682–691. doi: 10.1016/j.bbagrm.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellison S, Gabunia K, Richards JM, Kelemen SE, England RN, Rudic D, Azuma YT, Monroy MA, Eguchi S, Autieri MV. IL-19 reduces ligation-mediated neointimal hyperplasia by reducing vascular smooth muscle cell activation. Am J Pathol 1. 2014;84:2134–2143. doi: 10.1016/j.ajpath.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun XM, Patel DD, Acosta JC, Gil J, Soutar AK. Premature senescence in cells from patients with autosomal recessive hypercholesterolemia ARH: evidence for a role for ARH in mitosis. Arterioscler Thromb Vasc Biol. 2011;31:2270–2277. doi: 10.1161/ATVBAHA.111.232223. [DOI] [PubMed] [Google Scholar]
- 41.Martino F, Carlomosti F, Avitabile D, Persico L, Picozza M, Barillà F, Arca M, Montali A, Martino E, Zanoni C, Parrotto S, Magenta A. Circulating miR-33a and miR-33b are up-regulated in familial hypercholesterolaemia in paediatric age. Clin Sci Lond. 2015;129:963–972. doi: 10.1042/CS20150235. [DOI] [PubMed] [Google Scholar]
- 42.Wang F, Long G, Zhao C, Li H, Chaugai S, Wang Y, Chen C, Wang DW. Atherosclerosis-related circulating miRNAs as novel and sensitive predictors for acute myocardial infarction. PLoS One. 2014;9:e105734. doi: 10.1371/journal.pone.0105734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu HQ, Liang C, He ZQ, Fan M, Wu ZG. Circulating miR-214 is associated with the severity of coronary artery disease. J Geriatr Cardiol. 2013;10:34–38. doi: 10.3969/j.issn.1671-5411.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell. 2010;18:510–525. doi: 10.1016/j.devcel.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meder B, Katus HA, Rottbauer W. Right into the heart of microRNA-133a. Genes Dev. 2008;22:3227–3231. doi: 10.1101/gad.1753508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Eck M, Bos IS, Kaminski WE, Orsó E, Rothe G, Twisk J, Böttcher A, Van Amersfoort ES, Christiansen-Weber TA, Fung-Leung WP, Van Berkel TJ, Schmitz G. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc Natl Acad Sci U S A. 2002;99:6298–6303. doi: 10.1073/pnas.092327399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mameza MG, Lockard JM, Zamora E, Hillefors M, Lavina ZS, Kaplan BB. Characterization of the adaptor protein ARH expression in the brain and ARH molecular interactions. J Neurochem. 2007;103:927–94. doi: 10.1111/j.1471-4159.2007.04854.x. [DOI] [PubMed] [Google Scholar]
- 49.Zorio E, Medina P, Rueda J, Millán JM, Arnau MA, Beneyto M, Marín F, Gimeno JR, Osca J, Salvador A, España F, Estellés A. Insights into the role of microRNAs in cardiac diseases: from biological signalling to therapeutic targets. Cardiovasc Hematol Agents Med Chem. 2009;7:82–90. doi: 10.2174/187152509787047676. [DOI] [PubMed] [Google Scholar]
- 50.Deng L, Blanco FJ, Stevens H, Lu R, Caudrillier A, McBride M, McClure JD, Grant J, Thomas M, Frid M, Stenmark K, White K, Seto AG, Morrell NW, Bradshaw AC, MacLean MR, Baker AH. MicroRNA-143 Activation Regulates Smooth Muscle and Endothelial Cell Crosstalk in Pulmonary Arterial Hypertension. Circ Res. 2015;117:870–883. doi: 10.1161/CIRCRESAHA.115.306806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calvano J, Achanzar W, Murphy B, DiPiero J, Hixson C, Parrula C, Burr H, Mangipudy R, Tirmenstein M. Evaluation of microRNAs-208 and 133a/b as differential biomarkers of acute cardiac and skeletal muscle toxicity in rats. Toxicol Appl Pharmacol. 2015;S0041-008X:30142–30153. doi: 10.1016/j.taap.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 52.Sommerville LJ, Xing C, Kelemen SE, Eguchi S, Autieri MV. Inhibition of allograft inflammatory factor-1 expression reduces development of neointimal hyperplasia and p38 kinase activity. Cardiovasc Res. 2009;81:206–215. doi: 10.1093/cvr/cvn242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F. Vandesompele A novel and universal method for microRNA RT-qPCR data normalization. J Genome Biol. 2009;10:R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]