

Abstract
Localized brain tissue damage activates surrounding astrocytes, which significantly influences subsequent long-term pathological processes. Most existing focal brain injury models in rodents employ craniotomy to localize mechanical insults. However, the craniotomy procedure itself induces gliosis. To investigate perilesional astrocyte activation under conditions where the skull is intact, we created focal brain injuries using light exposure through a cranial window made by thinning the skull that does not induce gliosis. The lesion size is maximal at ~12 h and shows substantial recovery over the subsequent 30 days. Two distinct types of perilesional reactive astrocyte, identified by GFAP upregulation and hypertrophy, were found. In proximal regions, the reactive astrocytes proliferated and expressed nestin, whereas in regions distal to the injury core, astrocytes showed increased GFAP expression but did not proliferate, lacked nestin expression, and displayed different morphology. Simply making the window did not induce any of these changes. There were also significant numbers of neurons in the recovering cortical tissue. In the recovery region, reactive astrocytes radially extended processes, which appeared to influence the shapes of neuronal nuclei. The proximal reactive astrocytes also formed a cell layer, which appeared to serve as a protective barrier, blocking the spread of IgG deposition and migration of microglia from the lesion core to surrounding tissue. The recovery was preceded by perilesional accumulation of leukocytes expressing vascular endothelial growth factor. These results suggest that under intact skull conditions, focal brain injury is followed by perilesional reactive astrocyte activities that foster cortical tissue protection and recovery.
Keywords: cranial window, gliosis, hemorrhage, nestin, microglia
Introduction
Reactive astrocytes, characterized by hypertrophic morphology and upregulation of glial fibrillary acidic protein (GFAP), are common manifestations following focal brain injury (Norenberg, 1994). The robust growth and propagation of reactive astrocytes suggest that they have important roles in either secondary injury or wound healing (McGraw et al., 2001); however the roles indicated in previous studies are not necessarily consistent. For example, opposing influences of reactive astrocytes has been reported on neuronal network regeneration (Fitch & Silver, 1997; Oberheim et al., 2008) and inflammation (Merrill & Benveniste, 1996; Bush et al., 1999). These diverse actions are probably due to the heterogeneity of reactive astrocytes, which has been proposed based on studies of their structure, proliferation, and gene expression (Ridet et al., 1997). The classification of reactive astrocyte subtypes generated in clinically relevant animal models and the assignment of each subtype to a set of functions represent crucial information for research establishing effective treatments for brain injury.
Existing rodent focal brain injury models, such as fluid-percussion injury (FPI) (McIntosh et al., 1989) and controlled cortical impact (CCI) (Cherian et al., 1996), are not necessarily appropriate for studying gliosis due to the influence and artifacts of craniotomy used for localizing mechanical impact. Impairment of synaptic plasticity in sham-treated animals in FPI (D’Ambrosio et al., 1998; Sanders et al., 2000) has been attributed to the upregulation of inducible nitric oxide synthase (NOS) expression, which presumably reflects cytokine production gliosis (Min et al., 2003). Furthermore, a recent study demonstrated craniotomy-related artifactual gliosis and synaptic instability (Xu et al., 2007). Thus, the origin and roles of reactive astrocytes in existing focal brain injury models are complicated by craniotomy-related gliosis, and may reflect experimental artifacts rather than clinically relevant pathological processes.
In this study, we investigated astrocyte activation in response to focal brain injury without resorting to craniotomy. For this purpose, we made a thinned skull cranial window in mice and exposed the underlying small cortical region to intense light. The thinned-skull cranial window has already been shown not to cause artifactual gliosis, and to preserve normal glial phenotypes (Xu et al., 2007). This novel method, which we have designated “photo injury,” has been used successfully to generate reproducible cortical injuries that resemble human closed-head contusions. We histologically characterized tissue degeneration and the impressive recovery that follows, as well as the reactive astrocyte subtypes found in photo injury. Long-term spread of tissue degeneration was not observed, and a number of structural aspects of perilesional reactive astrocytes suggested their contribution to the recovery process. We also examined the expression of vascular endothelial growth factor, VEGF, which has been shown to be associated with cortical tissue recovery (Ruiz de Almodovar et al., 2009), and found a unique distribution prior to recovery, which is considered to characterize the pathological processes of photo injury. As astrocytes were solely activated by the cortical lesion without craniotomy-related artifacts, we propose that the heterogeneity and functional activities of reactive astrocytes seen here reflect the innate responses of cortical astrocytes to localized tissue damage.
Materials and Methods
Animal experiments
All experiments were approved by the Institutional Animal Care and Use Committee of the University of New Mexico (IACUC). Photo injury was generated in 6 – 10-week-old male C57BL6 mice (Harlan, Indianapolis, IN). A thinned-skull cranial window (approx. 0.5 mm in diameter and approx. 20 μm in thickness) was created over the right primary somatosensory cortex (1 mm posterior to bregma and 3 mm lateral from the midline) by scraping the skull using micro-drill burrs (19007-07M; FST, Foster City, CA) and a microsurgical blade (Nordland Blade #6900; Salivan, Charlotte, NC). For light exposure, the mouse was mounted on a microscope stage and the head was fixed with the cranial window centered under a 20× water immersion microscope objective (XLUMPLFLN 20×W NA1.0; Olympus, Center Valley, PA). NADH autofluorescence of cerebral parenchyma was imaged by two-photon microscopy (Ultima Multiphoton Microscopy System; Prairie Technologies, Middleton, WI), as shown in Fig. 1B. This information was used to set the focus of light exposure to the same depth between mice. Based on the results of preliminary investigations regarding light exposure conditions, injury light was delivered from a 90 W halogen bulb, with the housing mounted directly on a microscope camera port and focused 200 μm below the cortical surface by the objective. In preliminary experiments, it was determined that a 2.5-min exposure was optimal to cause total neuronal cell loss down to a depth of 80% – 90% of cortical thickness by 12 h after exposure (Supporting Information 1). The animals were anesthetized with 1.0% – 1.5% isoflurane in N2O:O2 (70:30). To avoid the neuroprotective effects of isoflurane (Zhou et al., 2010), the mouse was disconnected from anesthesia within 5 min after completion of light exposure, and the scalp was sutured within this 5-min period. To avoid unpredictable pain in a novel brain injury model, the sedative buprenorphine (0.1 mg/kg, s.c.) was administered every 12 h for the first 2 days after surgery. For bromodeoxyuridine (BrdU) labeling, BrdU (50 mg/kg, i.p.) was administered on days 1 though 6. All mice were housed under a 12 h light: 12 h dark cycle with food and water available ad libitum.
Fig. 1.
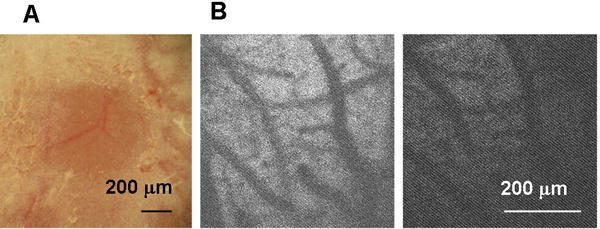
Thinned-skull cranial window (TSCW) and cortical light exposure. (A) Low-magnification bright field image of TSCW. (B) NADH autofluorescence through TSCW. Two photon microscopy images (Ex 740 nm/Em 450 m) before (left) and after (right) light exposure through a 20× microscope objective (90 W halogen bulb, 2.5 min).
Histological analysis
Mice were sacrificed at given time points, and tissue was subjected to histological analysis. After sodium pentobarbital overdose (150 mg/kg, i.p), mice were transcardially perfused with ice-cold 0.1 M PBS, followed by 4% (w/v) paraformaldehyde in 0.1 M PBS. Brains were post-fixed overnight, and sliced at a thickness of 50 μm in PBS using a Vibratome (Leica Microsystems, Buffalo Grove, IL). Slices of the maximum horizontal distance of lesion, presumably including the central region of the lesion, were selected for mounting onto slide glasses and air-dried. For immunofluorescence staining, slices were permeabilized with 1% (w/v) Triton X-100, blocked with 5% goat normal serum, and incubated with primary antibodies as follows: rabbit polyclonal anti-GFAP (1:200; DAKO, Carpinteria, CA); mouse monoclonal anti-NeuN (1:200; BD Pharmingen, San Diego, CA); mouse monoclonal anti-nestin (1:200; Millipore, Billerica, MA); rat monoclonal anti-BrdU (1:100; Accurate Chemical, Westbury, NY); rat monoclonal anti-CD68 (1:100; AbD Serotec, Raleigh, NC) and rabbit polyclonal anti-VEGF (1:100; Abcam, Cambridge, MA). Immunofluorescence was visualized using FITC- or Cy3-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA), and coverslipped with ProlongGold™ antifade mounting media (Invitrogen, Eugene, OR). For BrdU double staining, samples were treated with 2 N hydrochloric acid (37°C, 30 min), and then neutralized with 0.1 M sodium borate (pH 8.5, 10 min). For nestin and VEGF immunostaining, antigen retrieval was performed using 0.01 M citric acid (pH 6.0) at 100°C for 15 min, followed by 10 min in PBS at room temperature. Fluorescence images were obtained using an epifluorescence microscope (IX70; Olympus) equipped with a cooled-CCD camera (QuantiFire; Optronics, Goleta, CA) (Fig. 3A, 6, 9, 10A) or two-photon excitation microscopy and Z-stack reconstruction (Fig. 3B, 5, 7, 8, 10B).
Fig. 3.
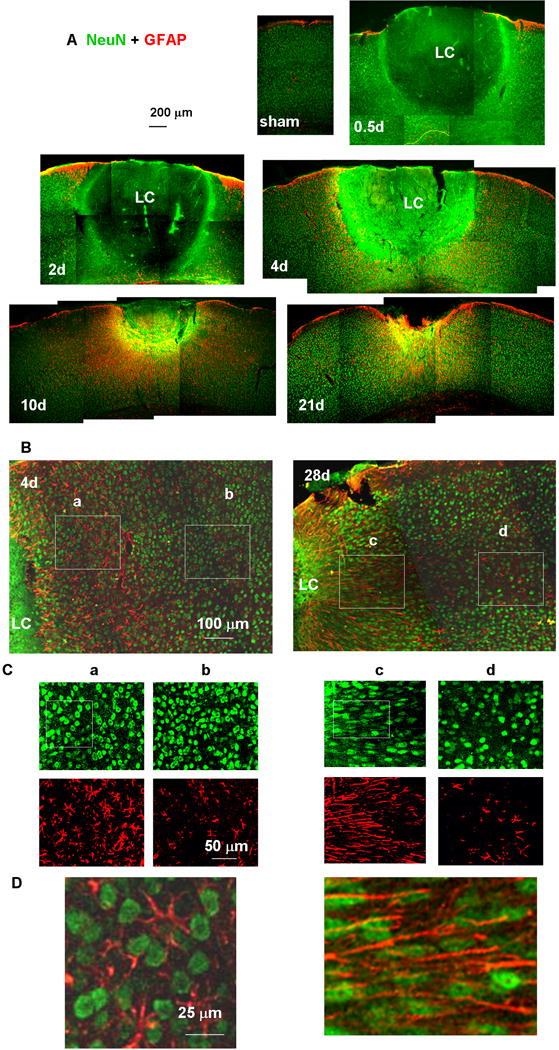
Tissue sections showing the lesion core (LC) and perilesional reactive astrocytes. Neuronal nuclei and astrocytes were stained with anti-NeuN (green) and anti-GFAP (red) antibodies, respectively. (A) Low-magnification images of a sham-treated (no light injury, fixed 4 days after skull thinning) mouse cortex underlying TSCW and injured cortex at 0.5, 2, 4, 10, and 21 days after photo injury. (B) Higher magnification images of the perilesional region on days 4 and 28. The NeuN (green)- and GFAP (red)-positive structures in Boxes (a) – (d) are further magnified in (C). On day 4, the morphology of neuronal nuclei was similar between the regions (a) proximal and (b) distant to LC. On day 28, neuronal nuclei showed pronounced elongation running along the direction of GFAP fibers in (c) the proximal region, but there were no significant differences from day 4 in (d) the distal region. In (D), the boxed regions shown in (C) are further magnified and merged to emphasize the elongation of NeuN staining nuclei along the GFAP-staining fibers. In this figure only, GFAP labeling is shown in red, reserving green for NeuN, which gave a clearer definition of nuclear shape.
Fig. 6.
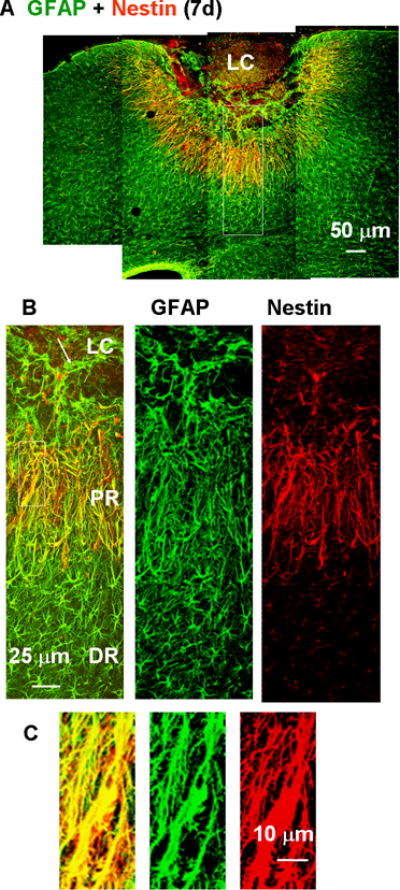
Nestin expression in proximal reactive astrocytes 7 days after the photo injury demonstrated by double immunofluorescence staining for nestin (red) and GFAP (green). (A) Low-magnification image of lesioned cortex. (B) Higher magnification images of the box in (A). GFAP-positive structures in the proximal region (PR) were positive for nestin, but those in the lesion core (LC) and distal region (DR) were negative for nestin. The arrow in the left-hand panel indicates extension of a GFAP-positive structure into the LC. (C) Higher magnification images of the boxed region in (B). The left panel shows the high degree of overlap of GFAP and nestin staining.
Fig. 9.
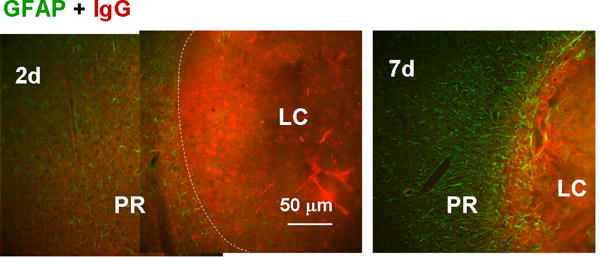
Blockade of blood component diffusion by the proximal reactive astrocyte layer. Reactive astrocytes and blood components were stained with anti-GFAP and anti-IgG antibodies, respectively. On day 2, IgG immunoreactivity was diffuse outside of the LC (approximate boundary indicated by the dashed line); whereas on day 7, it was limited to within the LC, which was surrounded by reactive astrocytes.
Fig. 10.
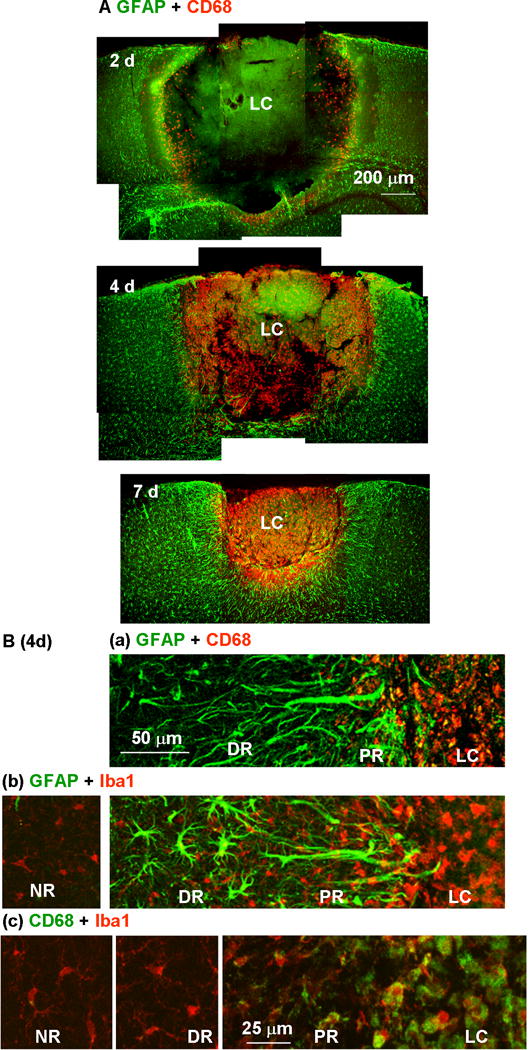
Restriction of microglial distribution by the proximal reactive astrocyte cell layer. (A) Reactive astrocytes and activated microglia in the injured cortex were stained with anti-GFAP (green) and anti-CD68 (red) antibodies, respectively, 2, 4, and 7 days after photo injury. (B) Higher magnification images on day 4 illustrated the distribution of microglia at different activation states in the lesion core (LC), proximal region (PR), distal region (DR), and normal region (NR). Microglia were stained with anti-CD68 (a) or the pan-microglial marker, Iba1 (b). The label colors are indicated above each set of panels. In the panels in (c), microglia stained for CD68 (green) and Iba1 (red) were further magnified for comparison of structures in different regions.
Fig. 5.
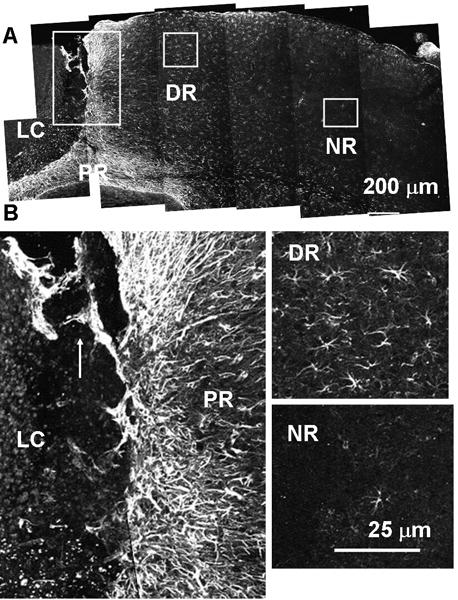
Two distinct types of reactive astrocytes found after photo injury. Anti-GFAP immunostaining on post-injury day 12. Lesion core (LC), and proximal (PR), distal (DR), and normal (NR) region. (A) Low-magnification image. Each box is magnified in (B). In the PR, GFAP-positive fibers extended radially from the LC (isomorphic), and also showed migration into the LC (arrow). In DR, GFAP-positive fibers extended randomly from the cell body (anisomorphic). In the NR, GFAP-positive cells were rarely found.
Fig. 7.
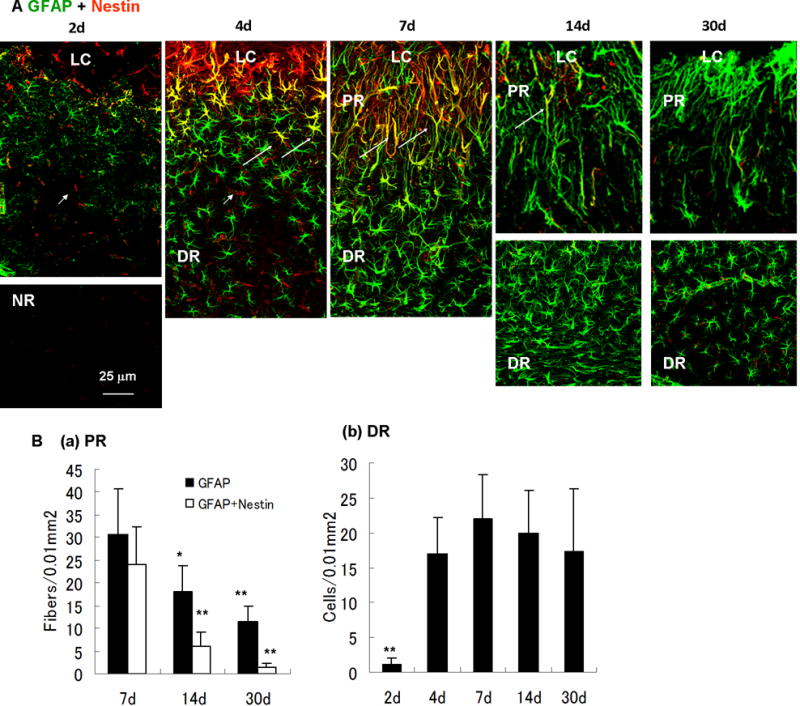
Time-dependent changes of reactive astrocytes after photo injury. (A) Time course of nestin (red) and GFAP (green) immunoreactivity in reactive astrocytes (days 2, 4, 7, 14, and 30). Perilesional GFAP expression was upregulated more than the normal region (NR) by day 2. Structural difference of reactive astrocytes between the lesion core (LC), the proximal region (PR) and the distal region (DR) was established by day 7. Nestin-expressing reactive astrocytes were clearly identified by day 4, reached the maximum level on day 7, and decreased to below the limit of detection by day 30. On day 4, the morphology of reactive astrocytes was uniform, whereas nestin-expressing reactive astrocytes indicated by arrows were localized in the proximity of the LC. On days 14 and 30, the DR is shown in separate pictures because the region of high-density reactive astrocytes was remote from the LC, reflecting the enlargement of the PR. Arrowheads in day 2 and 4 indicate nestin-positive and capillary-like structures without GFAP immunoreactivity, presumably reflecting proliferating endothelial cells in damaged vessels. (B) Cell densities of reactive astrocytes. Density of radial fibers in PR (GFAP-positive or GFAP/nestin-positive) or cell bodies of GFAP-positive reactive astrocytes in DR were counted in areas of 100 μm × 100 μm. Means ± SD (n = 3 images at each time point), ** P < 0.01, * P < 0.05 ANOVA and Dunnett’s test for multiple comparisons with 7 days.
Fig. 8.
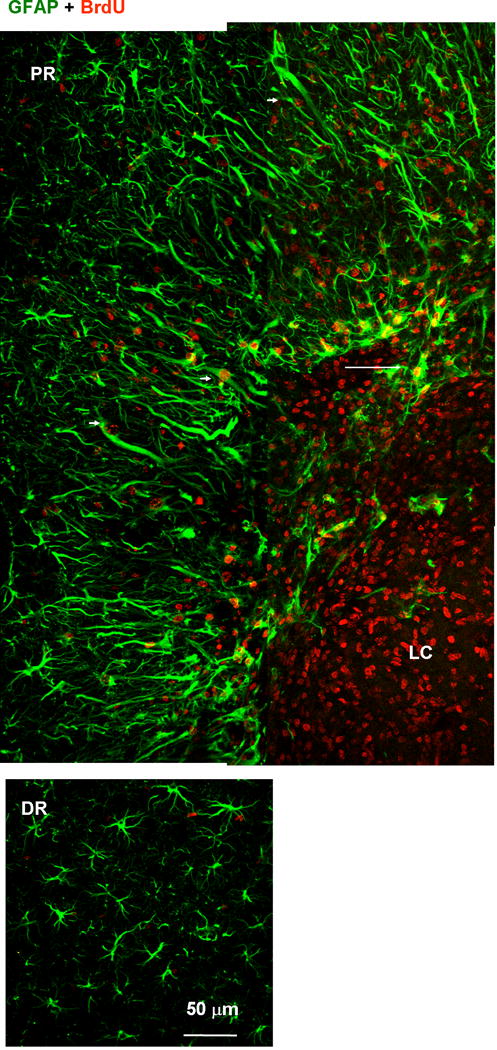
Proliferative nature of proximal reactive astrocytes. Tissue was labeled with anti-GFAP (green) and anti-BrdU (red) antibodies. BrdU was administrated between 1 and 6 days after photo injury and animals were sacrificed on day 7. Fluorescence images of the lesion core (LC) and proximal region (PR) (upper panels), and distal region (DR) (lower). The BrdU-positive but GFAP-negative nuclei in the LC likely correspond to microglia (see also Fig. 10). In the PR, the majority of GFAP-positive structures were co-localized with BrdU-positive nuclei. Arrowheads indicate BrdU-positive reactive astrocytes extending processes radially. In the DR, no significant co-localization of BrdU and GFAP was found. The arrow indicates the extension of GFAP-positive structure into LC.
Each histological data represents more than three images each from an independent mouse subjected to the same experimental condition. Numbers of images described in statistical analyses also refer to independent mice.
Results
Macroscopic aspects of photo injury
The thinned-skull cranial window (TSCW) was sufficiently transparent to allow visualization of cortical vessels under low-magnification bright field observation (Fig. 1A). Two-photon microscope observations taken through the TSCW also allowed visualization of cortical vessels by NADH autofluorescence (Fig. 1B, left). The injury-inducing light exposure caused no immediate structural changes, but reduced NADH autofluorescence was seen, presumably due to photobleaching (Fig. 1B, right). No structural changes or bleeding were evident on bright field observation immediately after light exposure (data not shown). These results indicated that the light exposure did not cause rapid macroscopic structural changes, including hemorrhage.
The time-dependent changes in appearance of injured cortex are shown in Fig. 2. At 1 h post-injury (Fig. 2, 1 h), the ipsilateral hemisphere (right) was indistinguishable from the contralateral side. In contrast, a cortical region much larger than the TSCW (approximately 0.5 mm in diameter as mentioned in Materials and Methods) showed hemorrhage by day 2 (Fig. 2, 2d). Red blood cell deposition, which did not appear as a hematoma, was visible macroscopically even after fixation. The deposition disappeared by day 7, and the corresponding region became discolored, presumably reflecting tissue degeneration (Fig. 2, 7d). The lesion formed a cavity that was significantly smaller than the initial hemorrhagic region by day 30 (Fig. 2, 30d-1), and the periphery of the cavity showed normal macroscopic appearance even in unfixed brain samples (Fig. 2, 30d-2). These results indicated that photo injury caused delayed and spreading hemorrhage and subsequent tissue degeneration, both of which are representative pathological changes observed following contusion (Finnie & Blumbergs, 2002). In addition, the gross appearance suggested significant tissue recovery within the initial lesion, because the final cavity was much smaller than the initial hemorrhagic region (compare Fig. 2, 2d with 30d-1 and 30d-2; see also below).
Fig. 2.
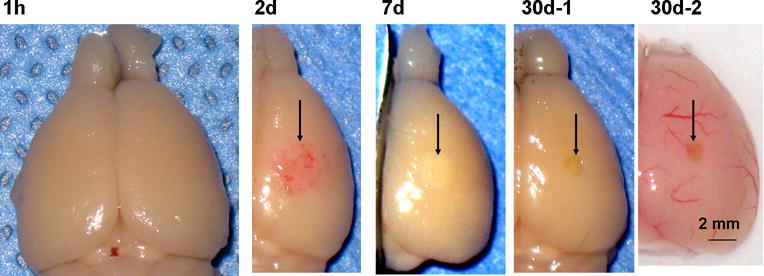
Low-magnification photographs of lesioned brains after photo injury 1 h – 30 days after injury. As mentioned in Materials and Methods, photo injury was induced in the right somatosensory cortex. Brains were dissected after paraformaldehyde fixation by transcardiac perfusion (1 h, 2 d, 7 d, and 30d-1) or without fixation (30d-2). Post-injury tissue changes are indicated by arrows in subsequent frames.
To clarify the mechanisms underlying photo injury, the change in temperature by light exposure was measured as detailed in Supplemental Information 2. The results indicated that the tissue temperature at the light focus increased by approximately 15°C. Thus, tissue degeneration by photo injury was likely attributable to moderate heating. As heat stroke is a major symptom following such a brain temperature increase, changes in cerebral blood flow after light exposure were measured as detailed in Supplemental Information 3. The results indicated that blood flow was normal, at least within 1 h after light exposure. Thus, immediate ischemia, which is a consequence of heat stroke, is unlikely the initial step in photo injury. At present, the mechanisms underlying the tissue degeneration associated with photo injury remain to be determined.
Cortical tissue degeneration and gliosis after photo injury
Neurodegeneration and astrocyte activation after photo injury were assessed by immunostaining for the neuronal marker NeuN and the astrocytic marker GFAP. The sham-treated mouse cortex 4 days after surgical treatment to create the TSCW (Fig. 3A, sham) showed no upregulation of GFAP and a normal distribution of NeuN-positive nuclei, in agreement with previous results (Xu et al., 2007). By 12 hours after photo injury (Fig. 3A, 0.5d), a large cortical region centered around the focus of light exposure showed total loss of NeuN-positive nuclei, which is designated hereafter as the lesion core (LC). The diffuse green color of the LC at various intensities (Fig. 3A, 0.5d, 2d, 4d, 10d) was due to the use of mouse antibody in a mouse sample containing mouse IgG due to hemorrhage, consistent with the IgG immunoreactivity shown in Fig. 9. The specificities of immunostaining using mouse primary antibodies are also mentioned on Fig. 9. The LC was surrounded by an area of increased GFAP immunoreactivity. By day 10 (Fig. 3A, 10d), the size of the LC had decreased, whereas the perilesional GFAP-positive region had broadened. This change is referred to below as perilesional broadening. By day 21 (Fig. 3A, 21d), the LC consisted of a cavity that was significantly smaller than that on day 4. This reduction in lesion size was consistent with the macroscopic observations shown in Fig. 2, in which the cavity was significantly smaller than the initial hemorrhagic region.
The proximal perilesional region included a large number of NeuN-positive nuclei, comparable to the density seen in the distal normal region both before (Fig. 3B, day 4) and after (Fig. 3B, day 28) perilesional broadening. These findings indicated that the broadened perilesional region was more complex than simple glial scar tissue, which normally consists only of glial cells and extracellular matrix proteins (Silver & Miller, 2004). It is also noteworthy that the broadened region showed alterations in the morphology of NeuN-positive neuronal nuclei (Fig. 3C and D). Before perilesional broadening (day 4), the morphology of NeuN-positive nuclei was round in both the perilesional region (Fig. 3Ca and D left) and the distal region (Fig. 3Cb). After perilesional broadening (day 28), however, the NeuN-positive nuclei showed pronounced elongation running along the direction of adjacent GFAP-positive fibers in the perilesional region (Fig. 3Cc and D right), whereas the NeuN-positive nuclei in the distal region showed the same round morphology (Fig. 3Cd) as seen on day 4 (Fig. 3Ca and Cb). These results suggested that the perilesional tissue recovered as a result of broadening of neuron-containing tissue with neuronal remodeling. In addition, the region of recovery and remodeling showed a high density of radially extending GFAP fibers.
The reduction of LC size was assessed quantitatively by measuring the maximum horizontal extent of the region lacking NeuN-positive nuclei. As shown in Fig. 4, the extent of the LC was constant between 12 h and 4 days after photo injury. This metric then began to decrease by day 7 and reached approximately one third of the initial extent by day 30. These results indicated that photo injury was followed by a reproducible LC size reduction, with the intervening space being occupied by neuron-containing tissue.
Fig. 4.
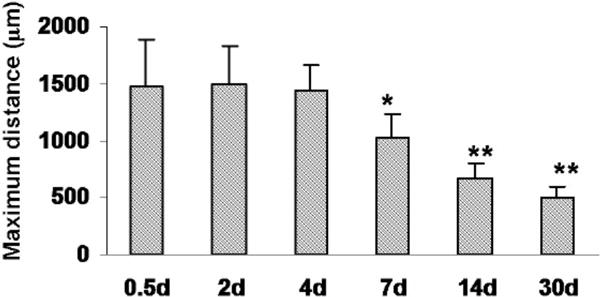
Progression of LC size changes following injury. Maximum horizontal distances of the area lacking NeuN staining were plotted at 0.5, 2, 4, 6, 14, and 30 days after photo injury. Means ± SD (n = 3 at each time point), ** P < 0.01, * P < 0.05 ANOVA and Dunnett’s test for multiple comparisons with 0.5 days.
Reactive astrocyte subtypes following photo injury
The structures of reactive astrocytes following photo injury were characterized by GFAP immunostaining, focusing on three regions showing marked differences, i.e., the proximal region (PR), the distal region (DR), and the normal region (NR) on day 12 (Fig. 5A). As shown at higher magnification in Fig. 5B (left), the PR included a dense layer of GFAP-positive fibers radially extending from the LC, and these were morphologically identical to the GFAP fibers found in the remodeling perilesional region on day 28 (Fig. 3Cc). In addition, there were generally distinct GFAP-positive structures extending from the PR into the LC, as indicated by the arrow in the figure. Similar extensions of GFAP-positive structures can be seen in the micrograph in Fig. 3A (GFAP-positive structure in LC on day 10), and also in Figs. 7B (arrow) and 8B (arrow). These extensions presumably reflect invasion of the LC by PR reactive astrocytes and may be one of the mechanisms of perilesional broadening. A similar migration of reactive astrocytes into the lesion site was demonstrated in a spinal cord injury model and proposed as an important mechanism for scar formation (Hsu et al., 2008). Meanwhile, the DR contained a far lower density of GFAP-positive cells than the PR. In this region, the astrocytes extended fibers in all directions (Fig. 5B right, upper panel). The NR remote from the LC included very few GFAP-positive cells, as seen in the normal mouse cortex (Fig. 5B right, lower panel). Taken together, these results indicated that photo injury is followed by production of two distinct types of reactive astrocytes, i.e., PR and DR reactive astrocytes.
The PR- and DR reactive astrocytes were further characterized with respect to expression of the neural stem cell marker nestin (Lendahl et al., 1990). As shown in Fig. 6A & B, photo injury resulted in nestin expression in the PR reactive astrocytes on day 7. Meanwhile, the DR reactive astrocytes did not express nestin. The overlap of GFAP- and nestin-positive structures is shown at higher magnification in Fig. 6C.
We further characterized the genesis of these two reactive astrocyte subtypes by longitudinal GFAP and nestin immunostaining (Fig. 7). GFAP immunoreactivity became detectable by day 2 and DR reactive astrocytes were found by day 4. Radially extended GFAP fibers of the PR reactive astrocytes became common by day 7, and these fibrous structures remained until at least day 30. Thus, the PR reactive astrocytes were spatially and temporally correlated to the perilesional neuronal tissue broadening, which started by day 7 (see Fig. 4). The time course of changes in nestin expression indicated that nestin appeared by day 4, peaked around day 7, and decreased to a level below the limit of detection by day 30. Interestingly, the nestin-expressing PR reactive astrocytes on day 4 were structurally indistinguishable from nestin-negative DR reactive astrocytes. Thus, it was suggested that the PR- and DR reactive astrocytes are distinct, not only structurally but also immunologically, and the immunological difference precedes the structural difference. Perilesional vessel-like structures also appeared nestin-positive on day 2 and 4 (Fig. 7A, arrowheads). As mentioned in Fig. 11, these structures presumably reflected VEGF increase and revascularization during the early stage of injury.
Fig. 11.
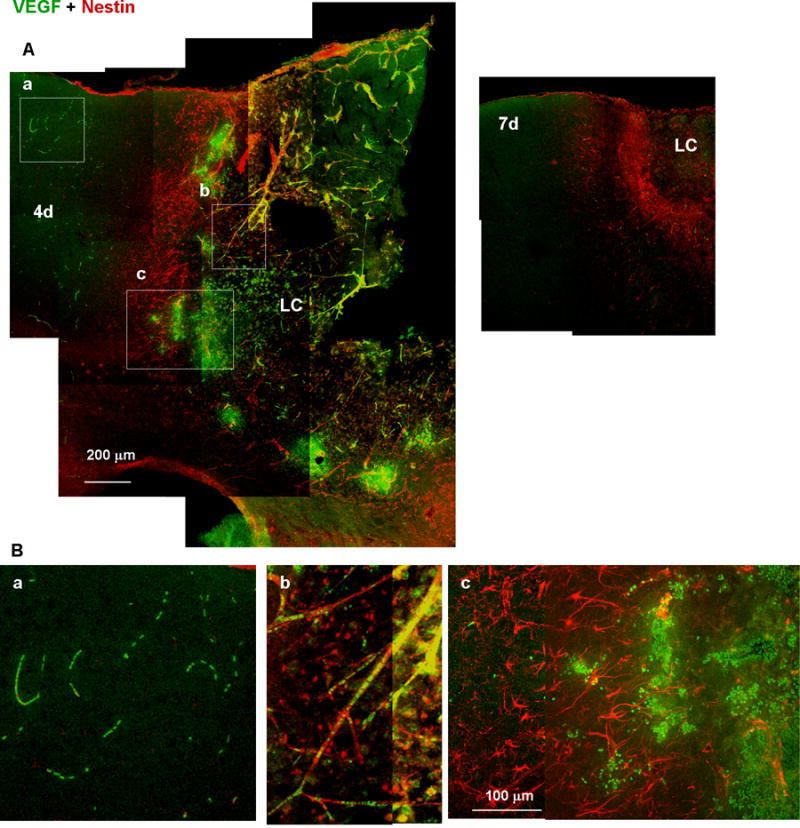
Accumulation of VEGF-expressing leukocytes at the early stages of wound healing. (A) Low-magnification images of tissue labeled with anti-VEGF (green) and anti-nestin (red) antibodies. VEGF immunoreactivity was distributed in both the distal region (a), which is outside of the nestin-positive proximal reactive astrocyte layer, and in the lesion core (LC; b and c), which is inside of the reactive astrocyte layer, 4 days after injury (4d), but undetectable on day 7 (7d). (B) High-magnification images of the boxes in (A). VEGF immunoreactivity was localized in leukocytes in the presumed distal region capillaries (a) or LC vessels (b), or accumulated at the border between the reactive astrocyte layer and the LC.
Images obtained by GFAP and nestin double staining were statistically analyzed to determine time-dependent changes in reactive astrocyte densities (Fig. 7B). As the PR reactive astrocytes were difficult to count because GFAP rarely visualizes cell bodies, the number of radially extended GFAP-positive and GFAP/nestin double-positive fibers were quantified in the PR. As shown in Fig. 7B(a), the density of reactive astrocyte fiber reached the maximum level on day 7 (GFAP; 30.6 ± 10.1 in an area of 100 μm × 100 μm, means ± SD, n = 3 images, GFAP/nestin; 24.2 ± 8.1), and was significantly reduced on day 14 (GFAP; 18.0 ± 5.7, GFAP/nestin; 6.0 ± 3.1) and day 30 (GFAP; 11.6 ± 3.2, GFAP/nestin; 1.4 ± 0.9). The density of DR reactive astrocytes increased significantly from day 2 (1.2 ± 0.8 in an area of 100 μm × 100 μm, means ± SD, n = 3 images) to day 4 (17.1 ± 5.2), and remained constant on day 7 (22.2 ± 6.3), day 14 (20.0 ± 6.1), and day 30 (17.4 ± 8.9). These results were consistent with the observations described above.
We also compared proliferation of these two reactive astrocyte populations by BrdU labeling as shown in Fig. 8. Briefly, 78 ± 33% (n = 3 images) of BrdU-labeled nuclei were co-localized with GFAP-positive fibers in the PR (arrowheads in Fig. 8, upper panel), whereas the co-localization in the DR was 7 ± 5%, which was significantly lower than in the PR (P < 0.01, t test), indicating that the PR and DR reactive astrocytes were distinct with respect to proliferation. The BrdU-positive nuclei lacking GFAP immunoreactivity in the LC (Fig. 8, upper panel) likely belonged to the microglia, because this cell population increased in the LC (see Fig. 10, below). The GFAP-positive structures extending from the PR to the LC, as indicated by arrows in Fig. 6B and Fig. 8, were largely negative for nestin (Fig. 6B), but showed marked labeling by BrdU (Fig. 8). We hypothesized that the proliferated PR reactive astrocytes migrated to the LC after the level of nestin expression had decreased.
Potential neuroprotective functions of PR reactive astrocytes
As previous reports suggests that perilesional proliferative reactive astrocytes encapsulate the lesion (Bush et al., 1999; Sofroniew, 2009), we examined the influence of PR reactive astrocytes on physical diffusion from the LC to the perilesional region. For this purpose, the IgG immunoreactivity in the perilesional region representing diffusion of blood components from hemorrhagic LC was evaluated. Figure 9 shows double labeling for GFAP and IgG immunoreactivity in and around the LC at 2 and 7 days after injury. On day 2 (left), the density of reactive astrocytes was low and IgG immunoreactivity was clearly detectable in the PR and beyond. On day 7 (right) there were many more reactive astrocytes, especially lined up along the LC-PR boundary, and IgG staining in the PR was virtually undetectable despite heavy labeling in the LC. The mean fluorescence intensity of IgG immunostaining in the region between 25 and 50 μm outside of the LC-PR border was 49% ± 14% (mean ± SD, n = 3 images) of that in the region between 25 and 50 μm inside the border on day 2, which was significantly higher than the value of 20% ± 11% on day 7 (n = 3, P < 0.01, t test). This pattern where IgG was limited to the LC persisted until day 21, when the LC consisted of a cavity. These results suggested that diffusion of blood components leaked from the damaged vessels in the LC to the perilesional region was being blocked by the PR reactive astrocytes. As blood components such as thrombin and hemoglobin are neurotoxic (Vanderveldt & Regan, 2004; Xue et al., 2006), and blood-brain barrier disruption is neurodegenerative (Kim et al., 2003), this diffusion barrier function is likely neuroprotective. The diffuse pattern of IgG staining, i.e., direct staining with the second antibody used for mouse primary antibodies, excludes the possibility that NeuN-positive nuclei (Fig. 3) and nestin-positive fibers (Fig. 7) reflect nonspecific staining in damaged tissue of the host species.
The influence of the PR reactive astrocytes on the cellular diffusion was also examined by immunohistological localization of activated microglis, which has been reported to be associated with neurotoxicity (Kaushal & Schlichter, 2008). Figure 10A shows co-immunostaining for the activated microglial marker CD68 and GFAP. On day 2 (top), activated microglia were diffusely present in the inner portion of the LC and had propagated throughout the LC by day 4. The LC was approximately the same size on days 2 and 4, but the marked increase in microglial number was restricted to this region without detectable invasion into the PR. The segregation persisted long after the LC had undergone progressive shrinkage (Fig. 10A, day 7). It is likely that tissue clearance by activated microglia is at least partially responsible for the size reduction of the LC, but they were prevented from attacking surrounding tissue. Examination of the LC-PR boundary region on day 4 at higher magnification indicated the distinct distributions of PR reactive astrocytes and activated microglia (Fig. 10Ba). CD68 immunoreactivity was seen only within the layer of GFAP immunoreactivity, reinforcing the above suggestion that the PR reactive astrocytes inhibited microglial migration into the perilesional region. Thus, this reactive astrocyte subtype prevented cellular diffusion at the PR-LC boundary. The morphology and density of microglia stained for the pan-microglial marker Iba1 (Imai et al., 1996) were also distinct in the LC (Fig. 10Bb and Bc). The LC included packed amoeboid microglia, whereas the DR and NR included relatively few process-bearing microglia. Neither the structure nor density of microglia showed a significant difference between the DR and NR. These data indicated that the microglia outside the PR reactive astrocyte layer were largely normal.
Perilesional accumulation of VEGF-expressing leukocytes
The expression of VEGF was examined as a potential mechanism underlying cortical tissue recovery, which is a characteristic of the wound healing process following photo injury. As shown in Fig. 11A, VEGF immunoreactivity was distributed in both the LC and DR 4 days after injury, but was undetectable on day 7. VEGF immunoreactivity appeared in small particles as demonstrated in high-magnification images on day 4 (Fig. 11B). These particles were presumably leukocytes, because they were arrayed as in capillaries in the DR (Fig. 11Ba), or colocalized with nestin-positive fibers, which were presumably damaged vessels including proliferating nestin-positive endothelial cells (Suzuki et al., 2010), in the LC (Fig. 11Bb). Since such nestin-positive vessel-like structures were more prominent during the early stage as shown in day 2 and day 4 in Fig. 7A, the accumulation of the VEGF-expressing leukocyte was temporally correlated with the appearance of proliferating endothelial cell, suggesting that VEGF upregulated revascularization prior to tissue regeneration. As the VEGF-expressing leukocytes accumulated at the border between nestin-positive proximal reactive astrocytes and LC as in Fig. 11Bc, they were likely spread from damaged vessels in the LC and infiltrated to the border. These results indicated that VEGF-expressing leukocytes accumulated in the perilesional region of photo injury, and likely contributed to the revascularization in the early stages of wound healing.
Discussion
Here, we discuss the actions of reactive astrocytes following focal brain injury in intact-skull mice using a novel method, termed photo injury. In this method, the precipitating insult is delivered through the unopened skull thereby avoiding craniotomy-related artifacts, such as gliosis, which is common in existing rodent focal brain injury models. As reported elsewhere (Xu et al., 2007) and confirmed here, preparation of the thinned-skull cranial window did not induce gliosis. The injury-generating light exposure did not change the exposed tissue structure immediately, unlike mechanical insults, but caused delayed hemorrhage and the development of a degenerative lesion, which spread from the initial insult site, similar to mechanical or chemical insults. As the hemorrhage showed robust red blood cell deposition, but not hematoma, it was suggested that vascular damage leaking blood cells into parenchyma spread from the light exposure site of injury. If the vascular damage was limited to the light exposure site, such robust blood leakage should have formed a hematoma rather than the broad deposit actually observed. The lesion reached maximum size by 12 h after photo injury, and its linear extent at the cortical surface was approximately threefold larger than the cranial window. Beginning around 5 – 7 days post-insult, evidence of tissue recovery and remodeling was seen that persisted for the 30-day observation period, and resulted in a radical reduction in size of the LC (see Fig. 4). The beginning of the recovery period was marked by the appearance of two distinct reactive astrocyte subtypes. In the PR, reactive astrocytes proliferated, expressed nestin, and extended processes, many of which had a radial orientation out from the LC. In the DR, the reactive astrocytes lacked both BrdU and nestin labeling, and processes were extended in all directions from the cell body. The perilesional tissue expansion may also reflect the contraction of lesion core by microglial phagocytotic activity, as the LC was filled with microglia prior to tissue recovery (Fig. 10A).
Our results implied that reactive astrocytes in the PR have a positive effect on tissue recovery, including neuronal cells, and confirmed the previously proposed beneficial functions of reactive astrocytes (Sofroniew, 2009). Taken together, our findings suggest that these astrocytes played a key role in “walling off” the extremely damaged LC volume, i.e., preventing diffusion of toxic blood components into relatively undamaged regions (Fig. 9) and constraining the range of activated microglia (Fig. 10). These protective functions agree with the results of previous studies showing that the selective ablation of proliferating reactive astrocytes, presumably corresponding to the PR reactive astrocytes, in stab injury, CCI and spinal cord injury models (Bush et al., 1999; Faulkner et al., 2004; Myer et al., 2006; Okada et al., 2006), caused lesional expansion due to the increased presence of toxic substances, such as glutamate and leukocytes. Moreover, the processes of these astrocytes appeared to predominate in the perilesional volume that expanded over days 7 – 30 to fill in the former LC (Figs. 5 & 6). This expanding volume contained not only post-lesionally generated astrocytes, but surprisingly also NeuN-positive cells, presumably neurons, which at later stages in our observation period showed unexplained nuclear elongation parallel to the astrocyte processes (Fig. 3C & D). At present, it is not clear whether these were newly differentiated (putative) neurons or whether they had migrated in from nearby regions. To date, we have succeeded in detecting neither BrdU-NeuN co-labeling nor immunoreactivity for the neuroblast marker, doublecortin, in any of the populations studied (data not shown), suggesting that the migration of preexisting mature NeuN-positive neuron is the more important factor. However, activated astrocytes obtained after in vivo stab wound injury have recently been shown to be capable of generating a neuronal phenotype when transferred to tissue culture conditions (Buffo et al., 2008). Therefore, it is possible that some of the NeuN-positive cells in the expanded PR volume were newly generated neurons, and that our BrdU labeling protocols were inadequate. Further studies are planned to investigate this interesting possibility.
Previous reviews suggested the existence of two structurally different post-lesional reactive astrocyte subtypes in areas proximal and distal to wounds, as seen here (Ridet et al., 1997; Sofroniew, 2009). It was suggested that the proximal reactive astrocytes develop scar tissue, which consists of gliotic tissue at the interface between lesion and normal tissue, whereas our findings indicating both the proximal astrocytes and neurons in the tissue expanding into the injury void (Fig. 3) suggested a novel tissue regenerative function of this reactive astrocyte subtype prior to scar formation. Other studies have demonstrated a number of degenerative functions of reactive astrocytes following different types of insult (Merrill, 1992; Rosenberg et al., 2001; Seiffert et al., 2004). The outcomes of injury-related changes are obviously complex and dependent on the model used.
The DR reactive astrocytes may support the survival of neurons in ways different from the above. For example, non-proliferative reactive astrocytes, similar to the DR reactive astrocytes, are neuroprotective due to increased glutamate uptake (Beurrier et al., 2010), as well as producing a greater nutrient supply to neurons (Escartin et al., 2007). Thus, this reactive astrocyte subtype would be beneficial for neuronal survival, especially under excitotoxic conditions, such as would exist in the DR after injury, due to spillover of glutamate and K+ from the LC. Indeed, similar non-proliferating reactive astrocytes have been shown to be widespread in excitotoxicity models, such as kainite lesions (Mitchell et al., 1993). Thus, one of the anticipated protective functions of the DR reactive astrocytes is to attenuate the excitotoxic environment for neuronal survival.
The degenerative mechanism underlying photo injury remains to be determined; however, the experiment detailed in Supplemental Information 2 suggested that light exposure likely caused tissue damage by moderate heating. As the heat stress associated with raising the core body temperature to 40°C – 43°C causes serious brain damage in mice (Sminia et al., 1994), this moderate focal heating by light exposure is thought to be sufficient to create localized brain injury. Heat stroke, which involves impaired cerebral blood flow by coagulation and/or inflammation following exposure of the brain tissue to high temperatures (Bouchama & Knochel, 2002), may also have contributed to the brain damage seen here. Indeed, the appearance and time course of lesions following acute focal ischemia by photo thrombosis share many aspects with those following photo injury (Schroeter et al., 2002; Carmichael, 2005).
Nevertheless, another experiment detailed in Supplemental Information 3 excluded the immediate contributions of blood flow impairment. However, delayed loss of blood flow (vasospasm), which is observed in a large proportion of brain injury patients (Werner & Engelhard, 2007), is still a possible mechanism that may have contributed to degeneration following photo injury. We assumed that light exposure generated delayed and spreading hemorrhage and tissue degeneration in the LC within 12 h due to protein denaturation and/or metabolic stress. Thus, we hypothesized that the LC was the result of secondary injury spread during the initial 12 h, which persisted for at least the following 3.5 days. Secondary injury reflects progressive cellular and biochemical degenerative processes that occur within minutes to days after primary injury from a variety of mechanical insults, and is a very significant cause of brain tissue deterioration in hospitalized brain injury patients (Moppett, 2007). The processes following photo injury, hemorrhage, and the subsequent tissue loss, are proposed to be equivalent in some respects to those of contusion, as mentioned in regard to Fig. 2. Therefore, photo injury provides a novel opportunity for studying contusion, which is a major clinical issue in traumatic brain injury, without artifacts caused by craniotomy.
VEGF-expressing leukocytes accumulated in the vessels of both the proximal and distal perilesional region, as well as migrating into the proximal perilesional parenchyma on post-injury day 4, but disappeared on day 7. This unique VEGF distribution prior to perilesional enlargement likely underlies the prominent tissue recovery in the photo injury model, as improvements of the outcome of brain injury by VEGF have been demonstrated in previous studies using treatments with VEGF or its neutralizing antibody (Krum & Khaibullina, 2003; Shen et al., 2006). VEGF is beneficial by promoting revascularization, astrocyte activation, neuronal survival, and neurite growth after injury, but detrimental by increasing blood-brain barrier permeability (Ruiz de Almodovar et al., 2009). Since VEGF-expressing leukocytes distributed both LC and capillaries in DR, whereas activated microglia was limited within LC, VEGF is supposed to play a key role in activating DR-reactive astrocytes, which is remote from the site of microglial cytokine production. Further studies using the photo injury model will address the contribution of VEGF to the activation of two different types of reactive astrocytes and perilesional tissue recovery. Previous studies have demonstrated VEGF expression in neurons, reactive astrocytes, vascular endothelial cells, and leukocyte during the early stages of brain injury (Nag et al., 1997; Chodobski et al., 2003), especially neutrophils after traumatic brain injury (Chodobski et al., 2003) and macrophages after stroke (Kovacs et al., 1996). Future studies will address the types of perilesional VEGF-expressing leukocytes in photo injury, and the contribution of leukocytes to perilesional tissue recovery. If leukocytes are involved in cortical tissue recovery, local and/or systemic inflammation, which affects differentiation of leukocytes and may alter their VEGF expression, is an important factor determining the extent of recovery. Furthermore, avoiding craniotomy, which limits cortical inflammation, likely explained the reproducible recovery seen in photo injury.
The three most remarkable aspects of focal brain injury in intact-skull mice were as follows: (1) severe and spreading secondary injury; (2) reproducible size of the LC (approx. 1.5 mm in maximum horizontal dimension as shown in Fig. 4); and (3) substantial cortical tissue recovery, likely involving the PR reactive astrocytes. To our knowledge, there have been no previous reports of longitudinal changes in lesion size, both progressive tissue degeneration and recovery, following FPI. The lesions following CCI have been shown to spread without tissue recovery for up to 1 month (Chen et al., 2003). In clinical situations, the long-term consequences of brain injury also vary widely between patients, and are largely unpredictable. However, a significant number of patients show brain tissue recovery (Fiehler et al., 2002; Mitsias et al., 2004), which has not been confirmed in conventional animal models. The wide variation between animals and the lack of tissue recovery in conventional animal models may be attributed to the craniotomy, which likely causes inflammation due to the leakage of inflammatory blood cells from damaged vessels (Soares et al., 1995) and microglial activation following mechanical stimulation (Davalos et al., 2005). If this is the case, the post-lesional pathological processes in the conventional models are complicated by various artifacts of inflammation. The pathological processes after closed-head injury are expected to be characterized by a lack of inflammation following skull fracture. The influence of inflammation on the growth of the PR reactive astrocytes and their functions for cortical recovery is an important question to be addressed in future studies. Finally, the remarkable tissue recovery and quantitative reproducibility of the long-term pathological processes make photo injury a desirable tool, which can be used to elucidate the mechanisms underlying cortical recovery.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH/NCRR Centers of Biomedical Research Excellence (PR15636), the Department of Neurosurgery, University of New Mexico. Grant-in-Aid for Scientific Research on Innovative area “Cellular and Molecular Basis for Neuro-vascular wiring” (#23122513, Japan), Grant-in-Aid for Scientific Research (B) (#23300133, Japan), the Smoking Research Foundation (Japan) and Takeda Science Foundation (Japan).
Abbreviations
- CCI
controlled cortical impact
- DR
distal region
- FPI
fluid-percussion injury
- GFAP
glial fibrillary acidic protein
- LC
lesion core
- NOS
nitric oxide synthase (NOS)
- NR
normal region
- PR
proximal region
- TSCW
thinned-skull cranial window
References
- Beurrier C, Faideau M, Bennouar KE, Escartin C, Kerkerian-Le Goff L, Bonvento G, Gubellini P. Ciliary neurotrophic factor protects striatal neurons against excitotoxicity by enhancing glial glutamate uptake. PLoS One. 2010;5:e8550. doi: 10.1371/journal.pone.0008550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchama A, Knochel JP. Heat stroke. N Engl J Med. 2002;346:1978–1988. doi: 10.1056/NEJMra011089. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Gotz M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Exp Neurol. 2003;182:87–102. doi: 10.1016/s0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- Cherian L, Robertson CS, Goodman JC. Secondary insults increase injury after controlled cortical impact in rats. J Neurotrauma. 1996;13:371–383. doi: 10.1089/neu.1996.13.371. [DOI] [PubMed] [Google Scholar]
- Chodobski A, Chung I, Kozniewska E, Ivanenko T, Chang W, Harrington JF, Duncan JA, Szmydynger-Chodobska J. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience. 2003;122:853–867. doi: 10.1016/j.neuroscience.2003.08.055. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Selective loss of hippocampal long-term potentiation, but not depression, following fluid percussion injury. Brain Res. 1998;786:64–79. doi: 10.1016/s0006-8993(97)01412-1. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Escartin C, Pierre K, Colin A, Brouillet E, Delzescaux T, Guillermier M, Dhenain M, Deglon N, Hantraye P, Pellerin L, Bonvento G. Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J Neurosci. 2007;27:7094–7104. doi: 10.1523/JNEUROSCI.0174-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehler J, Foth M, Kucinski T, Knab R, von Bezold M, Weiller C, Zeumer H, Rother J. Severe ADC decreases do not predict irreversible tissue damage in humans. Stroke. 2002;33:79–86. doi: 10.1161/hs0102.100884. [DOI] [PubMed] [Google Scholar]
- Finnie JW, Blumbergs PC. Traumatic brain injury. Vet Pathol. 2002;39:679–689. doi: 10.1354/vp.39-6-679. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Silver J. Glial cell extracellular matrix: boundaries for axon growth in development and regeneration. Cell Tissue Res. 1997;290:379–384. doi: 10.1007/s004410050944. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Bourguignon LY, Adams CM, Peyrollier K, Zhang H, Fandel T, Cun CL, Werb Z, Noble-Haeusslein LJ. Matrix metalloproteinase-9 facilitates glial scar formation in the injured spinal cord. J Neurosci. 2008;28:13467–13477. doi: 10.1523/JNEUROSCI.2287-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GW, Gasche Y, Grzeschik S, Copin JC, Maier CM, Chan PH. Neurodegeneration in striatum induced by the mitochondrial toxin 3-nitropropionic acid: role of matrix metalloproteinase-9 in early blood-brain barrier disruption? J Neurosci. 2003;23:8733–8742. doi: 10.1523/JNEUROSCI.23-25-08733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs Z, Ikezaki K, Samoto K, Inamura T, Fukui M. VEGF and flt. Expression time kinetics in rat brain infarct. Stroke. 1996;27:1865–1872. doi: 10.1161/01.str.27.10.1865. discussion 1872–1863. [DOI] [PubMed] [Google Scholar]
- Krum JM, Khaibullina A. Inhibition of endogenous VEGF impedes revascularization and astroglial proliferation: roles for VEGF in brain repair. Exp Neurol. 2003;181:241–257. doi: 10.1016/s0014-4886(03)00039-6. [DOI] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- McGraw J, Hiebert GW, Steeves JD. Modulating astrogliosis after neurotrauma. J Neurosci Res. 2001;63:109–115. doi: 10.1002/1097-4547(20010115)63:2<109::AID-JNR1002>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- Merrill JE. Tumor necrosis factor alpha, interleukin 1 and related cytokines in brain development: normal and pathological. Dev Neurosci. 1992;14:1–10. doi: 10.1159/000111642. [DOI] [PubMed] [Google Scholar]
- Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- Min KJ, Jou I, Joe E. Plasminogen-induced IL-1beta and TNF-alpha production in microglia is regulated by reactive oxygen species. Biochem Biophys Res Commun. 2003;312:969–974. doi: 10.1016/j.bbrc.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Mitchell J, Sundstrom LE, Wheal HV. Microglial and astrocytic cell responses in the rat hippocampus after an intracerebroventricular kainic acid injection. Exp Neurol. 1993;121:224–230. doi: 10.1006/exnr.1993.1089. [DOI] [PubMed] [Google Scholar]
- Mitsias PD, Ewing JR, Lu M, Khalighi MM, Pasnoor M, Ebadian HB, Zhao Q, Santhakumar S, Jacobs MA, Papamitsakis N, Soltanian-Zadeh H, Hearshen D, Patel SC, Chopp M. Multiparametric iterative self-organizing MR imaging data analysis technique for assessment of tissue viability in acute cerebral ischemia. AJNR Am J Neuroradiol. 2004;25:1499–1508. [PMC free article] [PubMed] [Google Scholar]
- Moppett IK. Traumatic brain injury: assessment, resuscitation and early management. Br J Anaesth. 2007;99:18–31. doi: 10.1093/bja/aem128. [DOI] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Nag S, Takahashi JL, Kilty DW. Role of vascular endothelial growth factor in blood-brain barrier breakdown and angiogenesis in brain trauma. J Neuropathol Exp Neurol. 1997;56:912–921. doi: 10.1097/00005072-199708000-00009. [DOI] [PubMed] [Google Scholar]
- Norenberg MD. Astrocyte responses to CNS injury. J Neuropathol Exp Neurol. 1994;53:213–220. doi: 10.1097/00005072-199405000-00001. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M. Loss of astrocytic domain organization in the epileptic brain. J Neurosci. 2008;28:3264–3276. doi: 10.1523/JNEUROSCI.4980-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, Razhagi A, Miller K, Gearing A. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893:104–112. doi: 10.1016/s0006-8993(00)03294-7. [DOI] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, Lambrechts D, Mazzone M, Carmeliet P. Role and therapeutic potential of VEGF in the nervous system. Physiol Rev. 2009;89:607–648. doi: 10.1152/physrev.00031.2008. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Sick TJ, Perez-Pinzon MA, Dietrich WD, Green EJ. Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 2000;861:69–76. doi: 10.1016/s0006-8993(00)01986-7. [DOI] [PubMed] [Google Scholar]
- Schroeter M, Jander S, Stoll G. Non-invasive induction of focal cerebral ischemia in mice by photothrombosis of cortical microvessels: characterization of inflammatory responses. J Neurosci Methods. 2002;117:43–49. doi: 10.1016/s0165-0270(02)00072-9. [DOI] [PubMed] [Google Scholar]
- Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen F, Su H, Fan Y, Chen Y, Zhu Y, Liu W, Young WL, Yang GY. Adeno-associated viral-vector-mediated hypoxia-inducible vascular endothelial growth factor gene expression attenuates ischemic brain injury after focal cerebral ischemia in mice. Stroke. 2006;37:2601–2606. doi: 10.1161/01.STR.0000240407.14765.e8. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Sminia P, van der Zee J, Wondergem J, Haveman J. Effect of hyperthermia on the central nervous system: a review. Int J Hyperthermia. 1994;10:1–30. doi: 10.3109/02656739409009328. [DOI] [PubMed] [Google Scholar]
- Soares HD, Hicks RR, Smith D, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci. 1995;15:8223–8233. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Namiki J, Shibata S, Mastuzaki Y, Okano H. The Neural Stem/Progenitor Cell Marker Nestin Is Expressed in Proliferative Endothelial Cells, but Not in Mature Vasculature. Journal of Histochemistry & Cytochemistry. 2010;58:721–730. doi: 10.1369/jhc.2010.955609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderveldt GM, Regan RF. The neurotoxic effect of sickle cell hemoglobin. Free Radic Res. 2004;38:431–437. doi: 10.1080/10715760310001638010. [DOI] [PubMed] [Google Scholar]
- Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci. 2007;10:549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- Xue M, Hollenberg MD, Yong VW. Combination of thrombin and matrix metalloproteinase-9 exacerbates neurotoxicity in cell culture and intracerebral hemorrhage in mice. J Neurosci. 2006;26:10281–10291. doi: 10.1523/JNEUROSCI.2806-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Lekic T, Fathali N, Ostrowski RP, Martin RD, Tang J, Zhang JH. Isoflurane posttreatment reduces neonatal hypoxic-ischemic brain injury in rats by the sphingosine-1-phosphate/phosphatidylinositol-3-kinase/Akt pathway. Stroke. 2010;41:1521–1527. doi: 10.1161/STROKEAHA.110.583757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.