

Abstract
Primary sclerosing cholangitis (PSC) is a disease of the biliary tract, which has been documented in the literature since 1867. This disease has a strong predilection for affecting men and can be seen in individuals as young as 2 years of age. PSC has a strong associated with inflammatory bowel disease, more commonly with ulcerative colitis, and is also part of the clinical spectrum of IgG4-related diseases. Small-duct PSC, a variant of PSC, also has an association with inflammatory bowel disease. The exact pathogenesis of PSC is not well understood at present, however, is likely a combination of a genetic predisposition with alteration of the molecular structure of the gut. Abnormal serum liver chemistry and presence of certain autoimmune markers are usually the first indicators leading to a diagnosis of PCS, however, these may often be normal in early stages of this disease. The diagnosis is made by cholangiography, which is now considered the gold standard. PSC is a known pre-malignant condition. Such patients have an increased risk of developing cholangiocarcinoma, gallbladder neoplasia, and colon cancer. Many new treatment modalities have emerged in the recent past, including anti-tumor necrosis factor- α and anti-integrins; however, liver transplantation is the only known cure for PSC. Despite past and present research, PSC remains an enigmatic biliary disease with few viable treatment options.
Keywords: Primary sclerosing cholangitis, Cholestasis, Inflammatory bowel disease, Autoimmune, Gallbladder neoplasia, Cholangiocarcinoma, IgG4 related disease, Colon cancer, Liver transplant
Core tip: Primary sclerosing cholangitis (PSC) is a fascinating disease with numerous and overlapping theorized pathogenetic models. An autoimmune etiology is in part due to its association with inflammatory bowel disease and autoimmune hepatitis, and inclusion within the IgG4 spectrum of diseases. Though PSC has been documented in the literature for more than a century, only sparse details exist regarding its true pathogenetics and even less about successful medical therapy. More rigorous research is needed to truly understand and treat this disease entity.
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a cholestatic liver and biliary tract disease associated with chronic inflammation of the biliary epithelium that cannot be attributed to another cause. This inflammatory process results in multifocal intra- and/or extrahepatic biliary strictures and fibrosis eventually leading to biliary cirrhosis and malignancy[1,2]. PSC often goes undiagnosed since approximately 40%-50% of patients with this disease are asymptomatic[3-5]. Fatigue, fever, jaundice, pruritus, and vague upper abdominal discomfort are the most commonly described symptoms at the time of diagnosis[3,6].
PSC has a strong association with inflammatory bowel disease (IBD)[7-9] with approximately 60%-80% of patients with PSC having coexisting ulcerative colitis (UC)[10,11].
PSC is a challenging condition whose pathogenesis continues to remain elusive despite extensive research of this disease in the 21st century. The only know cure for PSC is liver transplantation (LT) and symptomatic management with ursodeoxycholic acid and immunosuppressive agents.
Here we present a comprehensive review of the pathogenesis and clinical spectrum of PSC.
EPIDEMIOLOGY
PSC was first described in 1867 by Hoffman[12]. The incidence of PSC greatly varies from country to country but is increasing over time as evidenced by population-based studies from the United States, United Kingdom, and Northern Europe[13]. This variability may be related to human leukocyte antigen (HLA)-susceptibility among different ethnic groups and also varying frequency of IBD among populations across the world[14]. The incidence rate of PSC in the United States ranges from 0 to 0.92 per 100000 inhabitants per year, with no PSC patients being identified in Alaska between 1984 and 2000[4,15,16]. The prevalence of PSC in the United States is reported to be 13.6 per 100000 inhabitants[4]. Norway has the highest incidence rate at 1.31 per 100000 inhabitants and a prevalence of 8.5 per 100000[17]. The true prevalence of PSC may be higher than the aforementioned estimates both nationally and internationally because cholangiography may not be widely available in many parts of the world and patients with PSC may have normal levels of serum alkaline phosphatase (ALP)[18].
PSC can present itself at any age. The youngest individual documented to have PSC was under 2 years old[19]. UC is a major risk factor for the development of PSC, with approximately 60%-80% of patients having the PSC-UC phenotype[10,11]. However, only 4% of patients with UC have concomitant PSC[10,11].
PATHOGENESIS
The pathogenic mechanisms of PSC remain incompletely understood. Much like other autoimmune diseases, it has been theorized that the development of PSC is more likely to occur in a genetically susceptible individual after exposure to a trigger. PSC is the result of a complex immune-mediated response rather than a true autoimmune disease as it does not present with classic autoimmune features: female predominance, pathogenic autoantibodies, and response to immunosuppressive medications[20].
There is a 100-fold increased risk of developing PSC among siblings, however, the specific pattern of inheritance is much more complex[21,22].
Numerous studies have attempted to identify specific genes, which either predispose or protect an individual from the development of PSC. Many loci within the major histocompatibility complex have been linked to increased risk of PSC[23-25]. These are several class II HLA haplotypes including DRB1*0301- DRB3*0101-DQA1*0501-DQB1*0201, DRB1*1301-DRB3*0101-DQA1*0103-DQB1*0603, DRB1*1501-DRB5*0101-DQA1*0102- DQB1*0602, DRB1*0101-DQA1*0101, and B*0801[26-32].
HLA haplotypes associated with decreased risk of disease are DRB1*0401-DRB4*0103-DQA1*03-DQB1*0302, DRB1*0701-DQA1*0201-DQB1*0303, DRB4*0202-DRB1*1101-DQA1*0501-DQB1*0301, and MICA*002[29,31,32].
Due to the association of IBD with PSC, a “leaky gut” hypothesis has also been postulated[33]. Translocation of gastrointestinal (GI) flora from an inflamed GI tract to the portal venous system causes a systemic inflammatory response, which may disrupt the tight junctions in biliary epithelial cells[34,35]. This alteration exposes cholangiocytes to bile acids that could promote injury and inflammation[36].
Immune activation to an antigen (or a cross-reactive autoantigen or an enteric microbiome) also leads to inflammation of the gut and of the biliary tree[37]. Toll-like receptor and nucleotide oligomerization domain-like receptors assist in the detection of pathogens, which results in the secretion of pro-inflammatory cytokines[38]. Tumor necrosis factor (TNF)-α, transforming growth factor β1, interleukin (IL)-1β, and IL-6, along with involvement of CD8+ and CD4+ T cells have been proposed to cause myofibroblast activation and fibrosis[38,39]. IL-2 has also been proposed as a key player in the regulation and programming of the immune system. IL-2 receptor α gene deficiency in mice causes biliary inflammation resembling PSC[40]. Integrin ligands, intracellular adhesions molecule 1, and vascular cell adhesion molecule 1 are also expressed by the biliary epithelium and contribute to the recruitment of inflammatory leukocytes that play a role in development of biliary inflammation seen in PSC[41]. Gut-specific T and B cells can be programmed to perpetuate biliary inflammation after encountering an enteric pathogen, providing an important rationale of how liver and gut inflammation may be linked[42].
A critical driver of disease development may be an altered biliary mucosal milieu, giving rise to biliary colonization of non-commensal bacteria[43]. Reduced biliary Proteobacteria and increased firmicutes due to a non-functional galactoside 2-α-L-fucosyltransferase 2 (FUT-2) enzyme appears to alter the commensal bacteria in the biliary tree[44].
The pathogenesis of PSC is likely the result of an amalgamation of a heightened immune response to a pathogen in a host with both an altered biliary mucosal milieu and a genetic predisposition to PSC.
DIAGNOSIS OF PSC
Signs and symptoms
Approximately 40%-50% of patients with PSC have no symptoms at initial presentation[3-5]. Among the symptomatic patients, fatigue, fever, jaundice, pruritus, and vague upper abdominal discomfort are most commonly described[3,6]. A sudden onset of jaundice, however, should prompt the clinician to inquire about an obstructive biliary process. As approximately 60%-70% of patients with PSC have coexisting UC, GI bleeding may also be seen in these patients.
Serologic markers
The hallmark of PSC is an elevation of alkaline phosphatase (ALP). ALP may vary throughout the course of disease and may also be normal in patients with PSC[45]. Improvement of serum ALP during the disease is a predictor of better outcomes and prolonged transplant-free survival[46]. Serum alanine (ALT) and aspartate (AST) aminotransferase levels may also be elevated to 2- to 3-fold above the upper limit of normal[10,47]. Serum bilirubin is usually normal at time of diagnosis of PSC, however, may be elevated in patients with advanced disease, malignancy, or superimposed choledocholithiasis[47,48].
Detectable autoantibodies are found in as many as 97% of patients with PSC[49]. The most commonly noted autoantibodies are anti-smooth muscle antibodies (ASMA) and antinuclear antibodies (ANA), which can be seen in up to 75% of patients[50]. Perinuclear antineutrophil cytoplasmic antibody and anti P-40 autoantibody can also be detected in approximately 30%-80% of patients with PSC and UC[51,52].
Proteinase-3 antineutrophil cytoplasmic antibody (PR3-ANCA) has been studied extensively for disease severity in patients with UC[53,54]. More recently, when measured using chemiluminescence immunoassay, PR3-ANCA was seen in 38.5% of patients with PSC compared to only 10.6% of patients with liver disease suggesting it is a better biomarker for the diagnosis of PSC[55]. Numerous other autoantibodies have been detected in patients with PSC, including anticardiolipin antibodies, thyroperoxidase, and rheumatoid factor[49]. These autoantibodies, however, are not routinely assessed for the diagnosis of PSC, as they may not be present in patients with PSC, and furthermore do not correlate with disease severity or disease prognosis.
Imaging
Cholangiography is considered the gold standard for the diagnosis of PSC. Historically, endoscopic retrograde cholangiopancreatography (ERCP) was the initial diagnostic procedure of choice, however, magnetic resonance cholangiopancreatography (MRCP) has become the preferred method of diagnosis of PSC in the past decade due to comparable specificity (> 90%) and sensitivity (80%-90%), associated lower cost, less invasive testing, and fewer complications[56-58]. The characteristic features include multifocal annular stricturing within intrahepatic and/or extrahepatic bile ducts, with alternating normal or slightly dilated segments of bile ducts (Figure 1), giving rise to the typical beads-on-a-string appearance[59]. Diffuse involvement of the hepatobiliary system may be seen, including stricturing of the gallbladder, cystic duct and pancreatic duct, however, approximately 25% of patients have isolated intrahepatic involvement[59]. Although MRCP is recommended as the initial imaging modality for the diagnosis of PSC, ERCP may be necessary in patients with a non-diagnostic MRCP or those who require therapeutic intervention for bile duct strictures.
Figure 1.
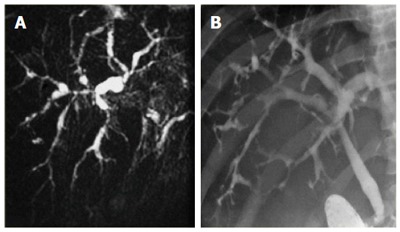
Imaging and endoscopy demonstrating primary sclerosing cholangitis. A: MRCP of a patient with PSC demonstrating intrahepatic stricturing with alternating normal and dilated segments of bile ducts; B: ERCP of a patient with PSC with similar findings. (Reproduced from Radiology Assistant. Levy AD, Chief of Gastrointestinal Radiology, Department of Radiologic Pathology, Armed Forces Institute of Pathology, Washington, D.C, United States). PSC: Primary sclerosing cholangitis; MRCP: Magnetic resonance cholangiopancreatography; ERCP: Endoscopic retrograde cholangiopancreatography.
Histologic features
A liver biopsy is rarely needed to confirm a diagnosis of PSC if characteristic cholangiographic findings are seen. Additional reasons why liver biopsies are not routinely obtained is the pathognomonic periductal fibrosis or “onion skinning” is not a common histologic finding in PSC[60]. Patients may have non-specific histologic findings and findings may be patchy and include more than one histological stage in at a given time indicating high sample variability[61].
When determining the stage of fibrosis, newer surrogate markers of cirrhosis including FibroSure (LabCorp) and transient ultrasound elastography (Echosens) have limited the need for a liver biopsy.
VARIANT PSC SYNDROME
Small-duct PSC
Individuals with biochemical markers and histologic features suggestive of PSC with normal cholangiography are considered to have small-duct PSC[62]. Small-duct PSC represents a small proportion of patients with PSC and may even be an earlier stage of PSC[63]. This subgroup of PSC has been less studied than the classic large-duct PSC. Several studies suggest better prognosis in individuals with small-duct PSC, however, available data on this population is limited due to lack of long-term follow up[50,63,64]. A recent study with one of the longest follow-up of patients with small-duct PSC suggested that approximately a fourth of patients’ progress to classic PSC in an average of 8 years[65]. Furthermore, individuals with small-duct PSC may progress to end-stage liver disease even without developing large duct disease and cholangiocarcinoma does not seem to occur in patients with small-duct disease in the absence of progression to large-duct PSC[65].
Lastly, the association of small-duct PSC appears to be stronger with Crohn’s colitis than with UC[8,66].
Overlap syndrome (autoimmune hepatitis-PSC)
An overlap syndrome of PSC with autoimmune hepatitis (AIH) is more often diagnosed in younger adults, adolescents and in children[67,68]. Patients who exhibit features of both hepatocellular and cholestatic disease with the presence of ANA antibodies, the presence or absence of ASMA antibodies, and/or histological changes in the absence of AMA antibodies are considered to have autoimmune sclerosing cholangitis[69-71].
The diagnosis of AIH is based on the presence of characteristic clinical, laboratory and histologic findings, abnormal levels of serum globulins, and the presence of typical autoantibodies which ultimately provides the clinician with either a “definite” or a “probable” diagnosis of AIH based on the modified scoring system of AIH[72-74]. ANA and ASMA are typically seen in type 1 AIH whereas liver/ kidney microsomal type 1 antibody and liver cytosol type 1 antibody are observed in type 2 AIH[75]. Histologic lesions typically present in AIH are periportal lymphocytic or lymphoplasmacytic infiltration (interface hepatitis) with hepatocyte swelling[75]. In case of fulminant presentation, massive necrosis may also be present[75]. Treatment is largely based on immunosuppressive therapy with corticosteroids and azathioprine.
Many similarities exist between PSC and AIH, including the autoimmune serology, histologic findings, and the disease response to treatment with immunosuppressive agents. The International Autoimmune Hepatitis Group scoring system can help in the making a diagnosis of AIH, however, it is not recommended in making a diagnosis of AIH-PSC overlap syndrome[72]. In 2001, a cohort of 55 patients was evaluated to assess this overlap syndrome[67]. Approximately 50% of these patients had bile duct changes diagnostic of sclerosing cholangitis (SC) at the time of presentation and all but one of these patients would have been diagnosed with AIH type 1. Several other studies have also assessed this overall phenomenon in similar populations[19,76]. It is possible that the juvenile form of SC may represent an early stage of PSC in patients with concomitant AIH and progression to PSC is delayed with early use of immunosuppressive therapy.
IgG4-related Sclerosing cholangitis
A common manifestation of IgG4-related diseases (IgG4-RD) is IgG4-sclerosing cholangitis (ISC). Type 1 autoimmune pancreatitis (AIP) is the leading manifestation of IgG4-RD, affecting approximately 60% of patients with IgG4-RD, followed by sialadenitis affecting 34% of patients, followed by tubulointerstitial nephritis (23%), dacryoadenitis (23%), and periaortitis (20%)[77]. ISC is seen in approximately 20%-88% of patients with IgG4-RD[77-79]. Individuals with ISC are commonly diagnosed with concomitant AIP[80]. Similar to PSC and unlike classic autoimmune disease, ISC is more commonly seen in males with a male-to-female ratio of 4:1[77].
Analogous to PSC, patients typically present with vague abdominal pain. Individuals may also present with obstructive jaundice with concurrent AIP[79,81].
An elevated serum level of IgG4 is the most sensitive and specific method of diagnosing ISC, however, elevated levels of IgG4 may be seen in approximately 10% of patients with PSC, in approximately 15% of those with cholangiocarcinoma, and also in approximately 7% of patients who may have other ailments[82-84]. Several additional serologic abnormalities may be seen in patients with ISC including elevated levels of IgG (approximately 60%), ANA positivity (approximately 40%), rheumatoid factor (approximately 20%), and IgE elevation (approximately 30%)[85-87].
Cholangiography may reveal multifocal biliary strictures, thickened bile duct wall and gallbladder wall thickening without vascular invasion[88].
Histologically, ISC demonstrates transmural fibro-inflammation with both fibrosis and inflammation evenly distributed from the mucosal surface to subserosa[89]. Immunostaining of the biopsy sample for IgG4 demonstrates diffuse infiltration of IgG4-positive plasma cells.
An approach for the diagnosis of ISC is the HISTORt criteria, which includes features on histology, imaging, serology, other organ involvement, and response to treatment with corticosteroids, and was initially utilized for the diagnosis of AIP and has been extended to include additional IgG4-related biliary diseases[78,90].
As the diagnostic algorithm suggests, rapid disease remission is achieved with immunosuppression using high-dose steroids[91]. Relapse of ISC can be seen in 30%-50% of patients and affected individuals may require re-induction of remission with additional high-dose steroids[91,92].
Long-term outcomes of patients with ISC are inconclusive and it remains unclear whether patients progress to end-stage liver disease or go on to develop cholangiocarcinoma.
PSC AND ASSOCIATED CONDITIONS
IBD and colorectal neoplasia
As previously mentioned, PSC has a strong association with IBD[7-9] with approximately 60%-70% of patients with PSC having coexisting UC, which often precedes the diagnosis of PSC or is diagnosed concomitantly[10,11]. Thus patients with PSC should undergo colonoscopic evaluation with biopsies despite the absence of typical symptoms[10]. If an initial evaluation does not reveal IBD, a repeat colonoscopy should be performed every 5 years to either confirm or exclude IBD[93].
Patients with IBD in the setting of PSC are considered to have a different phenotype (PSC-UC), which predicts a milder clinical course of the disease[9,94-96]. There also appears to be a right-sided predominance of diseased colonic mucosa, inflammation observed in the ileum and milder histologic inflammation in patients with PSC-IBD[97,98]. Due to the association of multiple malignancies with PSC, this disease entity should be considered a premalignant condition.
Patients with PSC-IBD have a significantly increased risk of developing colorectal malignancy compared to those with UC alone[99]. The cumulative CRC risk after 10 years of disease is 9% and 2%, which increases to 21%-30% and 5% in patients with PSC-IBD and isolated UC, respectively[99]. Moreover, colorectal cancer (CRC) and dysplasia are most often located in the right colon[95,96,98], which are associated with a worse prognosis when compared with left-sided colon cancer[100]. Annual or biennial surveillance colonoscopy is recommended in patients with PSC-IBD from the time of PSC diagnosis[101].
Cholangiocarcinoma
The most important risk is that of cholangiocarcinoma, which is several hundred times higher in patients with PSC than in patients without this disease[102]. Cholangiocarcinoma occurs in 1%-2% of patients annually following a diagnosis of PSC and is frequently detected within the first 1-3 years after the initial diagnosis[103,104].
Diagnosing cholangiocarcinoma in patients with PSC poses a tremendous challenge, as distinguishing between a benign dominant stricture from ductal cholangiocarcinoma requires the use of serologic, imaging, and ERCP over time. There are no designated risk stratification criteria, however, a commonly used approach involves annual MRCP or ultrasound examinations in conjunction with serum carbohydrate antigen 19-9 (CA19-9)[10]. If an individual is noted to have increasing serum levels of CA19-9, dominant strictures on imaging, and/or deterioration in either clinical status or liver test results, further assessment is made with an ERCP[105]. It is important to note that some individuals may not produce CA19-9 due to genetic reasons thus disease surveillance with this serologic test will not prove beneficial[106].
Routine brush cytology evaluation detects cholangiocarcinoma with low sensitivity (40%)[10] and near 100% specificity. Fluorescent in situ hybridization is used in conjunction with brush cytology specimens to apply a probe to subpopulations of cells with chromosome amplifications to assess for aneusomy. Individuals with the presence of a dominant stricture and polysomy [5 or more cells which have gained 2 or more chromosomes (3, 7, 17, and band 9p21)] are eventually diagnosed with cholangiocarcinoma with 88% specificity[107]. Cholangioscopy allows direct biliary visualization and directed biopsies of the dominant stricture and has been reported to have increased sensitivity and specificity to > 90%[108]. It is also being used to interrogate indeterminate strictures in an effort to enhance detection of cholangiocarcinoma. Confocal laser microscopy has not been studied in dominant strictures in PSC despite a high reported sensitivity and moderate specificity for indeterminate strictures in general[109]. Intraductal ultrasound may also offer improved diagnostic yield but is not widely adopted since its initial report for diagnosis of indeterminate strictures[110].
Gallbladder neoplasia
Concurrent abnormalities such as gallstone disease and PSC involving the gallbladder or cystic duct are seen in approximately 41% of patients with PSC[111]. This population is also at an increased risk of developing gallbladder neoplasia, although the exact prevalence is unknown[112]. Although the malignant potential of gallbladder polyps smaller than 8 mm is small[113], the 2010 American Association for the Study of Liver Diseases (AASLD) guidelines recommend annual ultrasound and cholecystectomy if lesions are detected, regardless of the size[10]. Despite annual surveillance for gallbladder polyps, there is a lack of consensus regarding the malignancy potential of small polyps and surgical intervention in patients with advanced liver disease poses its own set of risks. It is important for the clinician to weigh the risks vs benefits of routine surveillance and surgical intervention.
TREATMENT OF PSC
Medical management
Historically, ursodeoxycholic acid (UDCA) has been used for the symptomatic improvement in cholestatic pruritus, which can be a debilitating consequence of PSC, and to improve abnormal liver chemistries. High dose UDCA (28-30 mg/kg per day) has been shown to increase the risk of colonic neoplasia in patients with PSC-IBD[114]. Additionally, there is a lack of definitive evidence that the use of moderate dose UDCA (15-20 mg/kg per day) improves survival in PSC patients or is efficacious in the prevention of colorectal cancer in those with PSC-IBD or biliary neoplasia[115,116]. Although the current AASLD guidelines recommend against the routine use of UDCA in patients with PSC, clinical practice varies between centers[10].
Azathioprine and steroids are recommended for use in patients with AIH as well as those with AIH-PSC overlap syndrome[10]. However, the use of immunosuppressive therapy (azathioprine, cyclosporine, tacrolimus, and methotrexate) and anti-TNF agent, infliximab, failed to demonstrate sustained improvement in abnormal liver chemistries and prevention of progression to end-stage liver disease in patients with PSC[117-120].
As newer investigations shine light on the link between biliary inflammation, bile acid homeostasis, and the gut microbiota, there is increasing interest in pharmacologic treatment of PSC with antimicrobial agents. The use of non-absorbable antibiotics, such as vancomycin, demonstrated improvement in liver chemistries in a small subset of patients[121]. An improvement in liver biochemistries was also observed with use of absorbable antimicrobials including metronidazole, azithromycin, and minocycline[122-124]. More data points are necessary to provide the clinician with definitive and accurate treatment options for PSC.
Novel treatment strategies, including the use of biologic therapy against lymphocytic trafficking in the pathogenesis of PSC, are being investigated. Vedolizumab (Millennium Pharmaceuticals, Takeda) is a gut-specific monoclonal antibody that selectively targets against α4β7 heterodimer resulting in improvement in gut histology and mucosal T-cell infiltration[125]. It was approved by the Food and Drug Administration for induction and maintenance therapy for moderate to severe UC and Crohn’s disease in 2014[126,127]. Its use in patients with PSC-IBD is theorized to take effect by the presence of gut adhesion molecules and the entero-hepatic expression in PSC, however, the clinical utility of vedolizumab in PSC-IBD patients remains under investigation[37,128].
Management of biliary strictures
Patients with worsening symptoms over the disease course require investigation to exclude the presence of an extrahepatic dominant biliary stricture. A dominant stricture is defined as an area of stenosis ≤ 1.5 mm in the common bile duct or ≤ 1 mm in the common hepatic duct and is present in approximately 50% of PSC patients[10,129].
Dominant strictures are treated by either dilation alone or with dilation and placement of temporary plastic biliary stents during the ERCP[130]. It is important to note that PSC patients undergoing ERCP should be provided with prophylactic antibiotics to prevent possible cholangitis[131]. The duration of endoscopic therapy is variable and can range from 6 weeks to 12 mo before strictures resolve. ERCP with repeated stenting may be required in some patients who are refractory to dilation[130]. Due to the risk of cholangiocarcinoma masquerading as a dominant stricture, brush cytology and/or biopsy samples should be obtained during the endoscopic procedure[132]. The utility of ERCP is solely for the exclusion of cholangiocarcinoma and to provide therapy for dominant biliary strictures and does not modify the progression of the disease[130].
Liver transplantation
Due to the lack of durable pharmacologic and endoscopic therapy, liver transplantation (LT) remains the sole curative option in patients with progressive disease. PSC is the fifth most frequent indication for LT in the United States[133]. Intractable pruritus, recurrent bacterial cholangitis, and perihilar cholangiocarcinoma are additional indications for LT in PSC patients.
Post-transplant acute rejection can be seen within the first 30 d of transplantation, but usually resolves with systemic corticosteroids and does not appear to alter graft survival[134].
In patients with cholangiocarcinoma, LT in conjunction with neoadjuvant chemotherapy and radiation should be considered[135,136].
Patients with PSC-IBD may develop worsening disease post LT and approximately 14%-30% of patients with PSC may go on to develop de novo IBD up to 10 years post LT[10]. Patients should be monitored with serial serum ALP measurements post LT as increasing levels indicate recurrence of disease. Recurrent PSC, despite LT, is seen in 30%-50% of patients within 10 years of transplantation[137]. To date, no medical therapy has been identified to halt the progression or recurrence of disease.
CONCLUSION
PSC is a fascinating and largely elusive entity within the realm of hepatobiliary diseases. This is a chronic cholestatic liver disease, which has a tremendous impact on survival of those who are affected. Over time, many treatment modalities have been evaluated, however only LT is a known therapeutic option in these patients. Although newer drugs continue to be investigated for the treatment of PSC, effective treatment options remain limited. Future research in genomic-based therapies will hopefully allow for alteration of the natural course of this disease.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Peer-review started: October 28, 2016
First decision: December 19, 2016
Article in press: March 20, 2017
P- Reviewer: Vento S S- Editor: Gong ZM L- Editor: A E- Editor: Wang CH
References
- 1.Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet. 2013;382:1587–1599. doi: 10.1016/S0140-6736(13)60096-3. [DOI] [PubMed] [Google Scholar]
- 2.Ponsioen CY, Lam K, van Milligen de Wit AW, Huibregtse K, Tytgat GN. Four years experience with short term stenting in primary sclerosing cholangitis. Am J Gastroenterol. 1999;94:2403–2407. doi: 10.1111/j.1572-0241.1999.01364.x. [DOI] [PubMed] [Google Scholar]
- 3.Broomé U, Olsson R, Lööf L, Bodemar G, Hultcrantz R, Danielsson A, Prytz H, Sandberg-Gertzén H, Wallerstedt S, Lindberg G. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996;38:610–615. doi: 10.1136/gut.38.4.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bambha K, Kim WR, Talwalkar J, Torgerson H, Benson JT, Therneau TM, Loftus EV, Yawn BP, Dickson ER, Melton LJ. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology. 2003;125:1364–1369. doi: 10.1016/j.gastro.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 5.Tischendorf JJ, Hecker H, Krüger M, Manns MP, Meier PN. Characterization, outcome, and prognosis in 273 patients with primary sclerosing cholangitis: A single center study. Am J Gastroenterol. 2007;102:107–114. doi: 10.1111/j.1572-0241.2006.00872.x. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan MM. Medical approaches to primary sclerosing cholangitis. Semin Liver Dis. 1991;11:56–63. doi: 10.1055/s-2008-1040423. [DOI] [PubMed] [Google Scholar]
- 7.Boonstra K, van Erpecum KJ, van Nieuwkerk KM, Drenth JP, Poen AC, Witteman BJ, Tuynman HA, Beuers U, Ponsioen CY. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:2270–2276. doi: 10.1002/ibd.22938. [DOI] [PubMed] [Google Scholar]
- 8.Halliday JS, Djordjevic J, Lust M, Culver EL, Braden B, Travis SP, Chapman RW. A unique clinical phenotype of primary sclerosing cholangitis associated with Crohn’s disease. J Crohns Colitis. 2012;6:174–181. doi: 10.1016/j.crohns.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 9.O’Toole A, Alakkari A, Keegan D, Doherty G, Mulcahy H, O’Donoghue D. Primary sclerosing cholangitis and disease distribution in inflammatory bowel disease. Clin Gastroenterol Hepatol. 2012;10:439–441. doi: 10.1016/j.cgh.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, Gores GJ. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51:660–678. doi: 10.1002/hep.23294. [DOI] [PubMed] [Google Scholar]
- 11.Olsson R, Danielsson A, Järnerot G, Lindström E, Lööf L, Rolny P, Rydén BO, Tysk C, Wallerstedt S. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology. 1991;100:1319–1323. [PubMed] [Google Scholar]
- 12.Hoffman C. Verschluss der gallenwege durch verdickung der wandungen. Archives of Pathology, Anatomy and Psysiology. 1867;39:206–215. [Google Scholar]
- 13.Molodecky NA, Kareemi H, Parab R, Barkema HW, Quan H, Myers RP, Kaplan GG. Incidence of primary sclerosing cholangitis: a systematic review and meta-analysis. Hepatology. 2011;53:1590–1599. doi: 10.1002/hep.24247. [DOI] [PubMed] [Google Scholar]
- 14.Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol. 2012;56:1181–1188. doi: 10.1016/j.jhep.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 15.Hurlburt KJ, McMahon BJ, Deubner H, Hsu-Trawinski B, Williams JL, Kowdley KV. Prevalence of autoimmune liver disease in Alaska Natives. Am J Gastroenterol. 2002;97:2402–2407. doi: 10.1111/j.1572-0241.2002.06019.x. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan GG, Laupland KB, Butzner D, Urbanski SJ, Lee SS. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis. Am J Gastroenterol. 2007;102:1042–1049. doi: 10.1111/j.1572-0241.2007.01103.x. [DOI] [PubMed] [Google Scholar]
- 17.Boberg KM, Aadland E, Jahnsen J, Raknerud N, Stiris M, Bell H. Incidence and prevalence of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis in a Norwegian population. Scand J Gastroenterol. 1998;33:99–103. doi: 10.1080/00365529850166284. [DOI] [PubMed] [Google Scholar]
- 18.Lindor KD, Kowdley KV, Harrison ME. ACG Clinical Guideline: Primary Sclerosing Cholangitis. Am J Gastroenterol. 2015;110:646–659; quiz 660. doi: 10.1038/ajg.2015.112. [DOI] [PubMed] [Google Scholar]
- 19.Wilschanski M, Chait P, Wade JA, Davis L, Corey M, St Louis P, Griffiths AM, Blendis LM, Moroz SP, Scully L. Primary sclerosing cholangitis in 32 children: clinical, laboratory, and radiographic features, with survival analysis. Hepatology. 1995;22:1415–1422. [PubMed] [Google Scholar]
- 20.Pollheimer MJ, Halilbasic E, Fickert P, Trauner M. Pathogenesis of primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2011;25:727–739. doi: 10.1016/j.bpg.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergquist A, Lindberg G, Saarinen S, Broomé U. Increased prevalence of primary sclerosing cholangitis among first-degree relatives. J Hepatol. 2005;42:252–256. doi: 10.1016/j.jhep.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Donaldson PT. Genetics of liver disease: immunogenetics and disease pathogenesis. Gut. 2004;53:599–608. doi: 10.1136/gut.2003.031732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlsen TH, Franke A, Melum E, Kaser A, Hov JR, Balschun T, Lie BA, Bergquist A, Schramm C, Weismüller TJ, et al. Genome-wide association analysis in primary sclerosing cholangitis. Gastroenterology. 2010;138:1102–1111. doi: 10.1053/j.gastro.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 24.Melum E, Franke A, Schramm C, Weismüller TJ, Gotthardt DN, Offner FA, Juran BD, Laerdahl JK, Labi V, Björnsson E, et al. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nat Genet. 2011;43:17–19. doi: 10.1038/ng.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Folseraas T, Melum E, Franke A, Karlsen TH. Genetics in primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2011;25:713–726. doi: 10.1016/j.bpg.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Farrant JM, Doherty DG, Donaldson PT, Vaughan RW, Hayllar KM, Welsh KI, Eddleston AL, Williams R. Amino acid substitutions at position 38 of the DR beta polypeptide confer susceptibility to and protection from primary sclerosing cholangitis. Hepatology. 1992;16:390–395. doi: 10.1002/hep.1840160217. [DOI] [PubMed] [Google Scholar]
- 27.Mehal WZ, Lo YM, Wordsworth BP, Neuberger JM, Hubscher SC, Fleming KA, Chapman RW. HLA DR4 is a marker for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1994;106:160–167. doi: 10.1016/s0016-5085(94)95085-7. [DOI] [PubMed] [Google Scholar]
- 28.Olerup O, Olsson R, Hultcrantz R, Broome U. HLA-DR and HLA-DQ are not markers for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1995;108:870–878. doi: 10.1016/0016-5085(95)90463-8. [DOI] [PubMed] [Google Scholar]
- 29.Spurkland A, Saarinen S, Boberg KM, Mitchell S, Broome U, Caballeria L, Ciusani E, Chapman R, Ercilla G, Fausa O, et al. HLA class II haplotypes in primary sclerosing cholangitis patients from five European populations. Tissue Antigens. 1999;53:459–469. doi: 10.1034/j.1399-0039.1999.530502.x. [DOI] [PubMed] [Google Scholar]
- 30.Underhill JA, Donaldson PT, Doherty DG, Manabe K, Williams R. HLA DPB polymorphism in primary sclerosing cholangitis and primary biliary cirrhosis. Hepatology. 1995;21:959–962. [PubMed] [Google Scholar]
- 31.Mells GF, Kaser A, Karlsen TH. Novel insights into autoimmune liver diseases provided by genome-wide association studies. J Autoimmun. 2013;46:41–54. doi: 10.1016/j.jaut.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Eksteen B. Advances and controversies in the pathogenesis and management of primary sclerosing cholangitis. Br Med Bull. 2014;110:89–98. doi: 10.1093/bmb/ldu008. [DOI] [PubMed] [Google Scholar]
- 33.O’Mahony CA, Vierling JM. Etiopathogenesis of primary sclerosing cholangitis. Semin Liver Dis. 2006;26:3–21. doi: 10.1055/s-2006-933559. [DOI] [PubMed] [Google Scholar]
- 34.Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. 2013;182:375–387. doi: 10.1016/j.ajpath.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheth P, Samak G, Shull JA, Seth A, Rao R. Protein phosphatase 2A plays a role in hydrogen peroxide-induced disruption of tight junctions in Caco-2 cell monolayers. Biochem J. 2009;421:59–70. doi: 10.1042/BJ20081951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakisaka S, Kawaguchi T, Taniguchi E, Hanada S, Sasatomi K, Koga H, Harada M, Kimura R, Sata M, Sawada N, et al. Alterations in tight junctions differ between primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology. 2001;33:1460–1468. doi: 10.1053/jhep.2001.25086. [DOI] [PubMed] [Google Scholar]
- 37.Das KM. Relationship of extraintestinal involvements in inflammatory bowel disease: new insights into autoimmune pathogenesis. Dig Dis Sci. 1999;44:1–13. doi: 10.1023/a:1026629528233. [DOI] [PubMed] [Google Scholar]
- 38.Matsushita H, Miyake Y, Takaki A, Yasunaka T, Koike K, Ikeda F, Shiraha H, Nouso K, Yamamoto K. TLR4, TLR9, and NLRP3 in biliary epithelial cells of primary sclerosing cholangitis: relationship with clinical characteristics. J Gastroenterol Hepatol. 2015;30:600–608. doi: 10.1111/jgh.12711. [DOI] [PubMed] [Google Scholar]
- 39.Liaskou E, Jeffery LE, Trivedi PJ, Reynolds GM, Suresh S, Bruns T, Adams DH, Sansom DM, Hirschfield GM. Loss of CD28 expression by liver-infiltrating T cells contributes to pathogenesis of primary sclerosing cholangitis. Gastroenterology. 2014;147:221–232.e7. doi: 10.1053/j.gastro.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169:4850–4860. doi: 10.4049/jimmunol.169.9.4850. [DOI] [PubMed] [Google Scholar]
- 41.Afford SC, Humphreys EH, Reid DT, Russell CL, Banz VM, Oo Y, Vo T, Jenne C, Adams DH, Eksteen B. Vascular cell adhesion molecule 1 expression by biliary epithelium promotes persistence of inflammation by inhibiting effector T-cell apoptosis. Hepatology. 2014;59:1932–1943. doi: 10.1002/hep.26965. [DOI] [PubMed] [Google Scholar]
- 42.Lalor PF, Tuncer C, Weston C, Martin-Santos A, Smith DJ, Adams DH. Vascular adhesion protein-1 as a potential therapeutic target in liver disease. Ann N Y Acad Sci. 2007;1110:485–496. doi: 10.1196/annals.1423.051. [DOI] [PubMed] [Google Scholar]
- 43.Folseraas T, Melum E, Rausch P, Juran BD, Ellinghaus E, Shiryaev A, Laerdahl JK, Ellinghaus D, Schramm C, Weismüller TJ, et al. Extended analysis of a genome-wide association study in primary sclerosing cholangitis detects multiple novel risk loci. J Hepatol. 2012;57:366–375. doi: 10.1016/j.jhep.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wannhoff A, Hov JR, Folseraas T, Rupp C, Friedrich K, Anmarkrud JA, Weiss KH, Sauer P, Schirmacher P, Boberg KM, et al. FUT2 and FUT3 genotype determines CA19-9 cut-off values for detection of cholangiocarcinoma in patients with primary sclerosing cholangitis. J Hepatol. 2013;59:1278–1284. doi: 10.1016/j.jhep.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Stanich PP, Björnsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis. 2011;43:309–313. doi: 10.1016/j.dld.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to & lt; 1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2013;58:329–334. doi: 10.1016/j.jhep.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 47.Talwalkar JA, Lindor KD. Primary sclerosing cholangitis. Inflamm Bowel Dis. 2005;11:62–72. doi: 10.1097/00054725-200501000-00009. [DOI] [PubMed] [Google Scholar]
- 48.Steele IL, Levy C, Lindor KD. Primary sclerosing cholangitis--approach to diagnosis. MedGenMed. 2007;9:20. [PMC free article] [PubMed] [Google Scholar]
- 49.Angulo P, Peter JB, Gershwin ME, DeSotel CK, Shoenfeld Y, Ahmed AE, Lindor KD. Serum autoantibodies in patients with primary sclerosing cholangitis. J Hepatol. 2000;32:182–187. doi: 10.1016/s0168-8278(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 50.Björnsson E, Boberg KM, Cullen S, Fleming K, Clausen OP, Fausa O, Schrumpf E, Chapman RW. Patients with small duct primary sclerosing cholangitis have a favourable long term prognosis. Gut. 2002;51:731–735. doi: 10.1136/gut.51.5.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bansi DS, Fleming KA, Chapman RW. Importance of antineutrophil cytoplasmic antibodies in primary sclerosing cholangitis and ulcerative colitis: prevalence, titre, and IgG subclass. Gut. 1996;38:384–389. doi: 10.1136/gut.38.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mandal A, Dasgupta A, Jeffers L, Squillante L, Hyder S, Reddy R, Schiff E, Das KM. Autoantibodies in sclerosing cholangitis against a shared peptide in biliary and colon epithelium. Gastroenterology. 1994;106:185–192. doi: 10.1016/s0016-5085(94)95271-x. [DOI] [PubMed] [Google Scholar]
- 53.Arias-Loste MT, Bonilla G, Moraleja I, Mahler M, Mieses MA, Castro B, Rivero M, Crespo J, López-Hoyos M. Presence of anti-proteinase 3 antineutrophil cytoplasmic antibodies (anti-PR3 ANCA) as serologic markers in inflammatory bowel disease. Clin Rev Allergy Immunol. 2013;45:109–116. doi: 10.1007/s12016-012-8349-4. [DOI] [PubMed] [Google Scholar]
- 54.Mahler M, Bogdanos DP, Pavlidis P, Fritzler MJ, Csernok E, Damoiseaux J, Bentow C, Shums Z, Forbes A, Norman GL. PR3-ANCA: a promising biomarker for ulcerative colitis with extensive disease. Clin Chim Acta. 2013;424:267–273. doi: 10.1016/j.cca.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Stinton LM, Bentow C, Mahler M, Norman GL, Eksteen B, Mason AL, Kaplan GG, Lindkvist B, Hirschfield GM, Milkiewicz P, et al. PR3-ANCA: a promising biomarker in primary sclerosing cholangitis (PSC) PLoS One. 2014;9:e112877. doi: 10.1371/journal.pone.0112877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Angulo P, Pearce DH, Johnson CD, Henry JJ, LaRusso NF, Petersen BT, Lindor KD. Magnetic resonance cholangiography in patients with biliary disease: its role in primary sclerosing cholangitis. J Hepatol. 2000;33:520–527. doi: 10.1034/j.1600-0641.2000.033004520.x. [DOI] [PubMed] [Google Scholar]
- 57.Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology. 2004;40:39–45. doi: 10.1002/hep.20287. [DOI] [PubMed] [Google Scholar]
- 58.Berstad AE, Aabakken L, Smith HJ, Aasen S, Boberg KM, Schrumpf E. Diagnostic accuracy of magnetic resonance and endoscopic retrograde cholangiography in primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2006;4:514–520. doi: 10.1016/j.cgh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 59.MacCarty RL, LaRusso NF, Wiesner RH, Ludwig J. Primary sclerosing cholangitis: findings on cholangiography and pancreatography. Radiology. 1983;149:39–44. doi: 10.1148/radiology.149.1.6412283. [DOI] [PubMed] [Google Scholar]
- 60.Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med. 1995;332:924–933. doi: 10.1056/NEJM199504063321406. [DOI] [PubMed] [Google Scholar]
- 61.Angulo P, Larson DR, Therneau TM, LaRusso NF, Batts KP, Lindor KD. Time course of histological progression in primary sclerosing cholangitis. Am J Gastroenterol. 1999;94:3310–3313. doi: 10.1111/j.1572-0241.1999.01543.x. [DOI] [PubMed] [Google Scholar]
- 62.Wee A, Ludwig J, Coffey RJ, LaRusso NF, Wiesner RH. Hepatobiliary carcinoma associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hum Pathol. 1985;16:719–726. doi: 10.1016/s0046-8177(85)80158-1. [DOI] [PubMed] [Google Scholar]
- 63.Angulo P, Maor-Kendler Y, Lindor KD. Small-duct primary sclerosing cholangitis: a long-term follow-up study. Hepatology. 2002;35:1494–1500. doi: 10.1053/jhep.2002.33202. [DOI] [PubMed] [Google Scholar]
- 64.Broomé U, Glaumann H, Lindstöm E, Lööf L, Almer S, Prytz H, Sandberg-Gertzén H, Lindgren S, Fork FT, Järnerot G, et al. Natural history and outcome in 32 Swedish patients with small duct primary sclerosing cholangitis (PSC) J Hepatol. 2002;36:586–589. doi: 10.1016/s0168-8278(02)00036-3. [DOI] [PubMed] [Google Scholar]
- 65.Björnsson E, Olsson R, Bergquist A, Lindgren S, Braden B, Chapman RW, Boberg KM, Angulo P. The natural history of small-duct primary sclerosing cholangitis. Gastroenterology. 2008;134:975–980. doi: 10.1053/j.gastro.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 66.Björnsson E. Small-duct primary sclerosing cholangitis. Curr Gastroenterol Rep. 2009;11:37–41. doi: 10.1007/s11894-009-0006-6. [DOI] [PubMed] [Google Scholar]
- 67.Gregorio GV, Portmann B, Karani J, Harrison P, Donaldson PT, Vergani D, Mieli-Vergani G. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology. 2001;33:544–553. doi: 10.1053/jhep.2001.22131. [DOI] [PubMed] [Google Scholar]
- 68.McNair AN, Moloney M, Portmann BC, Williams R, McFarlane IG. Autoimmune hepatitis overlapping with primary sclerosing cholangitis in five cases. Am J Gastroenterol. 1998;93:777–784. doi: 10.1111/j.1572-0241.1998.224_a.x. [DOI] [PubMed] [Google Scholar]
- 69.Ben-Ari Z, Dhillon AP, Sherlock S. Autoimmune cholangiopathy: part of the spectrum of autoimmune chronic active hepatitis. Hepatology. 1993;18:10–15. [PubMed] [Google Scholar]
- 70.Czaja AJ. The variant forms of autoimmune hepatitis. Ann Intern Med. 1996;125:588–598. doi: 10.7326/0003-4819-125-7-199610010-00009. [DOI] [PubMed] [Google Scholar]
- 71.Czaja AJ, Carpenter HA, Santrach PJ, Moore SB. Autoimmune cholangitis within the spectrum of autoimmune liver disease. Hepatology. 2000;31:1231–1238. doi: 10.1053/jhep.2000.7878. [DOI] [PubMed] [Google Scholar]
- 72.Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, Chapman RW, Cooksley WG, Czaja AJ, Desmet VJ, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929–938. doi: 10.1016/s0168-8278(99)80297-9. [DOI] [PubMed] [Google Scholar]
- 73.Krawitt EL. Autoimmune hepatitis. N Engl J Med. 2006;354:54–66. doi: 10.1056/NEJMra050408. [DOI] [PubMed] [Google Scholar]
- 74.Vergani D, Alvarez F, Bianchi FB, Cançado EL, Mackay IR, Manns MP, Nishioka M, Penner E. Liver autoimmune serology: a consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J Hepatol. 2004;41:677–683. doi: 10.1016/j.jhep.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 75.Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli-Vergani G, Vergani D, Vierling JM. Diagnosis and management of autoimmune hepatitis. Hepatology. 2010;51:2193–2213. doi: 10.1002/hep.23584. [DOI] [PubMed] [Google Scholar]
- 76.Debray D, Pariente D, Urvoas E, Hadchouel M, Bernard O. Sclerosing cholangitis in children. J Pediatr. 1994;124:49–56. doi: 10.1016/s0022-3476(94)70253-5. [DOI] [PubMed] [Google Scholar]
- 77.Inoue D, Yoshida K, Yoneda N, Ozaki K, Matsubara T, Nagai K, Okumura K, Toshima F, Toyama J, Minami T, et al. IgG4-related disease: dataset of 235 consecutive patients. Medicine (Baltimore) 2015;94:e680. doi: 10.1097/MD.0000000000000680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, Clain JE, Pearson RK, Petersen BT, Vege SS, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006;4:1010–106; quiz 934. doi: 10.1016/j.cgh.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 79.Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, Topazian MD, Clain JE, Pearson RK, Petersen BT, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–715. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 80.Zen Y, Kawakami H, Kim JH. IgG4-related sclerosing cholangitis: all we need to know. J Gastroenterol. 2016;51:295–312. doi: 10.1007/s00535-016-1163-7. [DOI] [PubMed] [Google Scholar]
- 81.Björnsson E, Chari ST, Smyrk TC, Lindor K. Immunoglobulin G4 associated cholangitis: description of an emerging clinical entity based on review of the literature. Hepatology. 2007;45:1547–1554. doi: 10.1002/hep.21685. [DOI] [PubMed] [Google Scholar]
- 82.Mendes FD, Jorgensen R, Keach J, Katzmann JA, Smyrk T, Donlinger J, Chari S, Lindor KD. Elevated serum IgG4 concentration in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2006;101:2070–2075. doi: 10.1111/j.1572-0241.2006.00772.x. [DOI] [PubMed] [Google Scholar]
- 83.Ngwa TN, Law R, Murray D, Chari ST. Serum immunoglobulin G4 level is a poor predictor of immunoglobulin G4-related disease. Pancreas. 2014;43:704–707. doi: 10.1097/MPA.0000000000000118. [DOI] [PubMed] [Google Scholar]
- 84.Oseini AM, Chaiteerakij R, Shire AM, Ghazale A, Kaiya J, Moser CD, Aderca I, Mettler TA, Therneau TM, Zhang L, et al. Utility of serum immunoglobulin G4 in distinguishing immunoglobulin G4-associated cholangitis from cholangiocarcinoma. Hepatology. 2011;54:940–948. doi: 10.1002/hep.24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kamisawa T, Anjiki H, Egawa N, Kubota N. Allergic manifestations in autoimmune pancreatitis. Eur J Gastroenterol Hepatol. 2009;21:1136–1139. doi: 10.1097/meg.0b013e3283297417. [DOI] [PubMed] [Google Scholar]
- 86.Okazaki K, Uchida K, Fukui T. Recent advances in autoimmune pancreatitis: concept, diagnosis, and pathogenesis. J Gastroenterol. 2008;43:409–418. doi: 10.1007/s00535-008-2190-9. [DOI] [PubMed] [Google Scholar]
- 87.Sah RP, Pannala R, Zhang L, Graham RP, Sugumar A, Chari ST. Eosinophilia and allergic disorders in autoimmune pancreatitis. Am J Gastroenterol. 2010;105:2485–2491. doi: 10.1038/ajg.2010.236. [DOI] [PubMed] [Google Scholar]
- 88.Kojima E, Kimura K, Noda Y, Kobayashi G, Itoh K, Fujita N. Autoimmune pancreatitis and multiple bile duct strictures treated effectively with steroid. J Gastroenterol. 2003;38:603–607. [PubMed] [Google Scholar]
- 89.Zen Y, Harada K, Sasaki M, Sato Y, Tsuneyama K, Haratake J, Kurumaya H, Katayanagi K, Masuda S, Niwa H, et al. IgG4-related sclerosing cholangitis with and without hepatic inflammatory pseudotumor, and sclerosing pancreatitis-associated sclerosing cholangitis: do they belong to a spectrum of sclerosing pancreatitis? Am J Surg Pathol. 2004;28:1193–1203. doi: 10.1097/01.pas.0000136449.37936.6c. [DOI] [PubMed] [Google Scholar]
- 90.Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing the Mayo Clinic’s HISORt criteria. J Gastroenterol. 2007;42 Suppl 18:39–41. doi: 10.1007/s00535-007-2046-8. [DOI] [PubMed] [Google Scholar]
- 91.Hart PA, Zen Y, Chari ST. Recent Advances in Autoimmune Pancreatitis. Gastroenterology. 2015;149:39–51. doi: 10.1053/j.gastro.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 92.Hart PA, Topazian MD, Witzig TE, Clain JE, Gleeson FC, Klebig RR, Levy MJ, Pearson RK, Petersen BT, Smyrk TC, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut. 2013;62:1607–1615. doi: 10.1136/gutjnl-2012-302886. [DOI] [PubMed] [Google Scholar]
- 93.Fevery J, Henckaerts L, Van Oirbeek R, Vermeire S, Rutgeerts P, Nevens F, Van Steenbergen W. Malignancies and mortality in 200 patients with primary sclerosering cholangitis: a long-term single-centre study. Liver Int. 2012;32:214–222. doi: 10.1111/j.1478-3231.2011.02575.x. [DOI] [PubMed] [Google Scholar]
- 94.Joo M, Abreu-e-Lima P, Farraye F, Smith T, Swaroop P, Gardner L, Lauwers GY, Odze RD. Pathologic features of ulcerative colitis in patients with primary sclerosing cholangitis: a case-control study. Am J Surg Pathol. 2009;33:854–862. doi: 10.1097/PAS.0b013e318196d018. [DOI] [PubMed] [Google Scholar]
- 95.Loftus EV, Harewood GC, Loftus CG, Tremaine WJ, Harmsen WS, Zinsmeister AR, Jewell DA, Sandborn WJ. PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut. 2005;54:91–96. doi: 10.1136/gut.2004.046615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sokol H, Cosnes J, Chazouilleres O, Beaugerie L, Tiret E, Poupon R, Seksik P. Disease activity and cancer risk in inflammatory bowel disease associated with primary sclerosing cholangitis. World J Gastroenterol. 2008;14:3497–3503. doi: 10.3748/wjg.14.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Broomé U, Bergquist A. Primary sclerosing cholangitis, inflammatory bowel disease, and colon cancer. Semin Liver Dis. 2006;26:31–41. doi: 10.1055/s-2006-933561. [DOI] [PubMed] [Google Scholar]
- 98.Claessen MM, Lutgens MW, van Buuren HR, Oldenburg B, Stokkers PC, van der Woude CJ, Hommes DW, de Jong DJ, Dijkstra G, van Bodegraven AA, et al. More right-sided IBD-associated colorectal cancer in patients with primary sclerosing cholangitis. Inflamm Bowel Dis. 2009;15:1331–1336. doi: 10.1002/ibd.20886. [DOI] [PubMed] [Google Scholar]
- 99.Soetikno RM, Lin OS, Heidenreich PA, Young HS, Blackstone MO. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: a meta-analysis. Gastrointest Endosc. 2002;56:48–54. doi: 10.1067/mge.2002.125367. [DOI] [PubMed] [Google Scholar]
- 100.Yahagi M, Okabayashi K, Hasegawa H, Tsuruta M, Kitagawa Y. The Worse Prognosis of Right-Sided Compared with Left-Sided Colon Cancers: a Systematic Review and Meta-analysis. J Gastrointest Surg. 2016;20:648–655. doi: 10.1007/s11605-015-3026-6. [DOI] [PubMed] [Google Scholar]
- 101.Laine L, Kaltenbach T, Barkun A, McQuaid KR, Subramanian V, Soetikno R. SCENIC international consensus statement on surveillance and management of dysplasia in inflammatory bowel disease. Gastrointest Endosc. 2015;81:489–501.e26. doi: 10.1016/j.gie.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 102.Farges O, Malassagne B, Sebagh M, Bismuth H. Primary sclerosing cholangitis: liver transplantation or biliary surgery. Surgery. 1995;117:146–155. doi: 10.1016/s0039-6060(05)80078-9. [DOI] [PubMed] [Google Scholar]
- 103.Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Lööf L, Danielsson A, Hultcrantz R, Lindgren S, Prytz H, Sandberg-Gertzén H, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002;36:321–327. doi: 10.1016/s0168-8278(01)00288-4. [DOI] [PubMed] [Google Scholar]
- 104.Fevery J, Verslype C, Lai G, Aerts R, Van Steenbergen W. Incidence, diagnosis, and therapy of cholangiocarcinoma in patients with primary sclerosing cholangitis. Dig Dis Sci. 2007;52:3123–3135. doi: 10.1007/s10620-006-9681-4. [DOI] [PubMed] [Google Scholar]
- 105.Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology. 2011;54:1842–1852. doi: 10.1002/hep.24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Narimatsu H, Iwasaki H, Nakayama F, Ikehara Y, Kudo T, Nishihara S, Sugano K, Okura H, Fujita S, Hirohashi S. Lewis and secretor gene dosages affect CA19-9 and DU-PAN-2 serum levels in normal individuals and colorectal cancer patients. Cancer Res. 1998;58:512–518. [PubMed] [Google Scholar]
- 107.Bangarulingam SY, Bjornsson E, Enders F, Barr Fritcher EG, Gores G, Halling KC, Lindor KD. Long-term outcomes of positive fluorescence in situ hybridization tests in primary sclerosing cholangitis. Hepatology. 2010;51:174–180. doi: 10.1002/hep.23277. [DOI] [PubMed] [Google Scholar]
- 108.Tischendorf JJ, Krüger M, Trautwein C, Duckstein N, Schneider A, Manns MP, Meier PN. Cholangioscopic characterization of dominant bile duct stenoses in patients with primary sclerosing cholangitis. Endoscopy. 2006;38:665–669. doi: 10.1055/s-2006-925257. [DOI] [PubMed] [Google Scholar]
- 109.Meining A, Chen YK, Pleskow D, Stevens P, Shah RJ, Chuttani R, Michalek J, Slivka A. Direct visualization of indeterminate pancreaticobiliary strictures with probe-based confocal laser endomicroscopy: a multicenter experience. Gastrointest Endosc. 2011;74:961–968. doi: 10.1016/j.gie.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 110.Farrell RJ, Agarwal B, Brandwein SL, Underhill J, Chuttani R, Pleskow DK. Intraductal US is a useful adjunct to ERCP for distinguishing malignant from benign biliary strictures. Gastrointest Endosc. 2002;56:681–687. doi: 10.1067/mge.2002.128918. [DOI] [PubMed] [Google Scholar]
- 111.Mendes F, Lindor KD. Primary sclerosing cholangitis: overview and update. Nat Rev Gastroenterol Hepatol. 2010;7:611–619. doi: 10.1038/nrgastro.2010.155. [DOI] [PubMed] [Google Scholar]
- 112.Karlsen TH, Schrumpf E, Boberg KM. Gallbladder polyps in primary sclerosing cholangitis: not so benign. Curr Opin Gastroenterol. 2008;24:395–399. doi: 10.1097/MOG.0b013e3282f5727a. [DOI] [PubMed] [Google Scholar]
- 113.Eaton JE, Thackeray EW, Lindor KD. Likelihood of malignancy in gallbladder polyps and outcomes following cholecystectomy in primary sclerosing cholangitis. Am J Gastroenterol. 2012;107:431–439. doi: 10.1038/ajg.2011.361. [DOI] [PubMed] [Google Scholar]
- 114.Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, Harnois D, et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am J Gastroenterol. 2011;106:1638–1645. doi: 10.1038/ajg.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lindström L, Boberg KM, Wikman O, Friis-Liby I, Hultcrantz R, Prytz H, Sandberg-Gertzén H, Sangfelt P, Rydning A, Folvik G, et al. High dose ursodeoxycholic acid in primary sclerosing cholangitis does not prevent colorectal neoplasia. Aliment Pharmacol Ther. 2012;35:451–457. doi: 10.1111/j.1365-2036.2011.04966.x. [DOI] [PubMed] [Google Scholar]
- 116.Wolf JM, Rybicki LA, Lashner BA. The impact of ursodeoxycholic acid on cancer, dysplasia and mortality in ulcerative colitis patients with primary sclerosing cholangitis. Aliment Pharmacol Ther. 2005;22:783–788. doi: 10.1111/j.1365-2036.2005.02650.x. [DOI] [PubMed] [Google Scholar]
- 117.Talwalkar JA, Gossard AA, Keach JC, Jorgensen RA, Petz JL, Lindor RN. Tacrolimus for the treatment of primary sclerosing cholangitis. Liver Int. 2007;27:451–453. doi: 10.1111/j.1478-3231.2007.01441.x. [DOI] [PubMed] [Google Scholar]
- 118.Cullen SN, Chapman RW. Review article: current management of primary sclerosing cholangitis. Aliment Pharmacol Ther. 2005;21:933–948. doi: 10.1111/j.1365-2036.2005.02407.x. [DOI] [PubMed] [Google Scholar]
- 119.Hommes DW, Erkelens W, Ponsioen C, Stokkers P, Rauws E, van der Spek M, ten Kate F, van Deventer SJ. A double-blind, placebo-controlled, randomized study of infliximab in primary sclerosing cholangitis. J Clin Gastroenterol. 2008;42:522–526. doi: 10.1097/MCG.0b013e3181662426. [DOI] [PubMed] [Google Scholar]
- 120.Epstein MP, Kaplan MM. A pilot study of etanercept in the treatment of primary sclerosing cholangitis. Dig Dis Sci. 2004;49:1–4. doi: 10.1023/b:ddas.0000011827.87103.2e. [DOI] [PubMed] [Google Scholar]
- 121.Abarbanel DN, Seki SM, Davies Y, Marlen N, Benavides JA, Cox K, Nadeau KC, Cox KL. Immunomodulatory effect of vancomycin on Treg in pediatric inflammatory bowel disease and primary sclerosing cholangitis. J Clin Immunol. 2013;33:397–406. doi: 10.1007/s10875-012-9801-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boner AL, Peroni D, Bodini A, Delaini G, Piacentini G. Azithromycin may reduce cholestasis in primary sclerosing cholangitis: a case report and serendipitous observation. Int J Immunopathol Pharmacol. 2007;20:847–849. doi: 10.1177/039463200702000423. [DOI] [PubMed] [Google Scholar]
- 123.Färkkilä M, Karvonen AL, Nurmi H, Nuutinen H, Taavitsainen M, Pikkarainen P, Kärkkäinen P. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: a randomized placebo-controlled trial. Hepatology. 2004;40:1379–1386. doi: 10.1002/hep.20457. [DOI] [PubMed] [Google Scholar]
- 124.Silveira MG, Torok NJ, Gossard AA, Keach JC, Jorgensen RA, Petz JL, Lindor KD. Minocycline in the treatment of patients with primary sclerosing cholangitis: results of a pilot study. Am J Gastroenterol. 2009;104:83–88. doi: 10.1038/ajg.2008.14. [DOI] [PubMed] [Google Scholar]
- 125.Podolsky DK, Lobb R, King N, Benjamin CD, Pepinsky B, Sehgal P, deBeaumont M. Attenuation of colitis in the cotton-top tamarin by anti-alpha 4 integrin monoclonal antibody. J Clin Invest. 1993;92:372–380. doi: 10.1172/JCI116575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, Van Assche G, Axler J, Kim HJ, Danese S, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369:699–710. doi: 10.1056/NEJMoa1215734. [DOI] [PubMed] [Google Scholar]
- 127.Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, Lukas M, Fedorak RN, Lee S, Bressler B, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369:711–721. doi: 10.1056/NEJMoa1215739. [DOI] [PubMed] [Google Scholar]
- 128.Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, Sands BE, Hanauer SB, Targan S, Rutgeerts P, et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med. 2012;367:1519–1528. doi: 10.1056/NEJMoa1203572. [DOI] [PubMed] [Google Scholar]
- 129.Stiehl A, Rudolph G, Klöters-Plachky P, Sauer P, Walker S. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. J Hepatol. 2002;36:151–156. doi: 10.1016/s0168-8278(01)00251-3. [DOI] [PubMed] [Google Scholar]
- 130.ASGE Standards of Practice Committee, Chathadi KV, Chandrasekhara V, Acosta RD, Decker GA, Early DS, Eloubeidi MA, Evans JA, Faulx AL, Fanelli RD, Fisher DA, Foley K, Fonkalsrud L, Hwang JH, Jue TL, Khashab MA, Lightdale JR, Muthusamy VR, Pasha SF, Saltzman JR, Sharaf R, Shaukat A, Shergill AK, Wang A, Cash BD, DeWitt JM. The role of ERCP in benign diseases of the biliary tract. Gastrointest Endosc. 2015;81:795–803. doi: 10.1016/j.gie.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 131.ASGE Standards of Practice Committee, Banerjee S, Shen B, Baron TH, Nelson DB, Anderson MA, Cash BD, Dominitz JA, Gan SI, Harrison ME, Ikenberry SO, Jagannath SB, Lichtenstein D, Fanelli RD, Lee K, van Guilder T, Stewart LE. Antibiotic prophylaxis for GI endoscopy. Gastrointest Endosc. 2008;67:791–798. doi: 10.1016/j.gie.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 132.Baluyut AR, Sherman S, Lehman GA, Hoen H, Chalasani N. Impact of endoscopic therapy on the survival of patients with primary sclerosing cholangitis. Gastrointest Endosc. 2001;53:308–312. doi: 10.1016/s0016-5107(01)70403-8. [DOI] [PubMed] [Google Scholar]
- 133.Bjøro K, Brandsaeter B, Foss A, Schrumpf E. Liver transplantation in primary sclerosing cholangitis. Semin Liver Dis. 2006;26:69–79. doi: 10.1055/s-2006-933565. [DOI] [PubMed] [Google Scholar]
- 134.Graziadei IW, Wiesner RH, Marotta PJ, Porayko MK, Hay JE, Charlton MR, Poterucha JJ, Rosen CB, Gores GJ, LaRusso NF, et al. Long-term results of patients undergoing liver transplantation for primary sclerosing cholangitis. Hepatology. 1999;30:1121–1127. doi: 10.1002/hep.510300501. [DOI] [PubMed] [Google Scholar]
- 135.Darwish Murad S, Kim WR, Harnois DM, Douglas DD, Burton J, Kulik LM, Botha JF, Mezrich JD, Chapman WC, Schwartz JJ, et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology. 2012;143:88–98.e3; quiz e14. doi: 10.1053/j.gastro.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rosen CB, Darwish Murad S, Heimbach JK, Nyberg SL, Nagorney DM, Gores GJ. Neoadjuvant therapy and liver transplantation for hilar cholangiocarcinoma: is pretreatment pathological confirmation of diagnosis necessary? J Am Coll Surg. 2012;215:31–8; discussion 38-40. doi: 10.1016/j.jamcollsurg.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 137.Fosby B, Karlsen TH, Melum E. Recurrence and rejection in liver transplantation for primary sclerosing cholangitis. World J Gastroenterol. 2012;18:1–15. doi: 10.3748/wjg.v18.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]