

Abstract
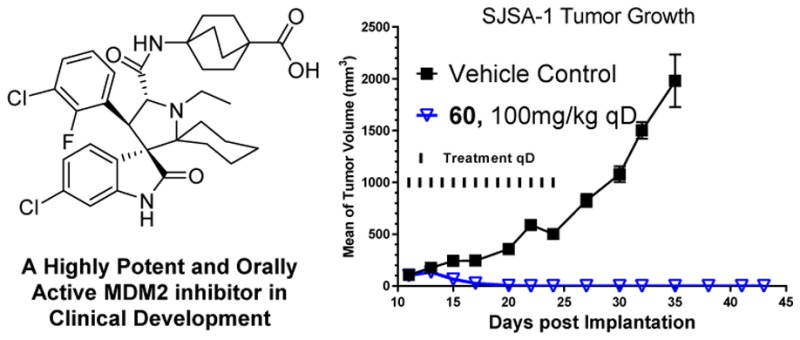
We previously reported the design of spirooxindoles with two identical substituents at the carbon-2 of the pyrrolidine core as potent MDM2 inhibitors. In this paper we describe an extensive structure–activity relationship study of this class of MDM2 inhibitors, which led to the discovery of 60 (AA-115/APG-115). Compound 60 has a very high affinity to MDM2 (Ki < 1 nM), potent cellular activity, and an excellent oral pharmacokinetic profile. Compound 60 is capable of achieving complete and long-lasting tumor regression in vivo and is currently in phase I clinical trials for cancer treatment.
Introduction
The tumor suppressor function of p53 is compromised in essentially all human cancers. In about half of human cancers, TP53, the gene encoding p53 protein, is mutated or deleted, and this inactivates the tumor suppressor function of p53.1 In the remaining 50%, p53 retains its wild-type status but its function is effectively inhibited by the murine double minute 2 (MDM2) protein through a direct protein–protein interaction.2−8 Through direct binding, the MDM2 protein blocks the transactivation domain of p53, transports p53 from the nucleus to the cytoplasm, and ubiquitinates p53 for proteasomal degradation.6 Small-molecule inhibitors designed to block the MDM2–p53 interaction (hereafter called MDM2 inhibitors) can liberate the tumor suppressor function of p539−13 and may have a promising therapeutic potential for cancer treatment. Intense research efforts have resulted in advancement of several potent, selective, non-peptide MDM2 inhibitors (1–5),14−20 shown in Figure 1, into clinical development.
Figure 1.

Chemical structures of representative MDM2 inhibitors in clinical trials.
Our laboratory has designed and optimized spirooxindoles as a new class of MDM2 inhibitors and has advanced compound 2 (MI-77301) into clinical development.18 One shortcoming of 2 and its earlier analogues that we have found is that they slowly isomerize in solution.21,22 To overcome this stability issue, we recently reported the design of new spirooxindoles containing two identical substituents at C-2 of the pyrrolidine ring, which can undergo a rapid and irreversible conversion to a single diastereoisomer.21,22 In the present study, we report an extensive structure–activity relationship (SAR) study for this class of MDM2 inhibitors. This study has led to the discovery of 60 (AA-115/APG-115), a highly potent, chemically stable and efficacious MDM2 inhibitor, which has entered clinical development for cancer treatment (ClinicalTrials.gov identifier for 60: NCT02935907).
Results and Discussion
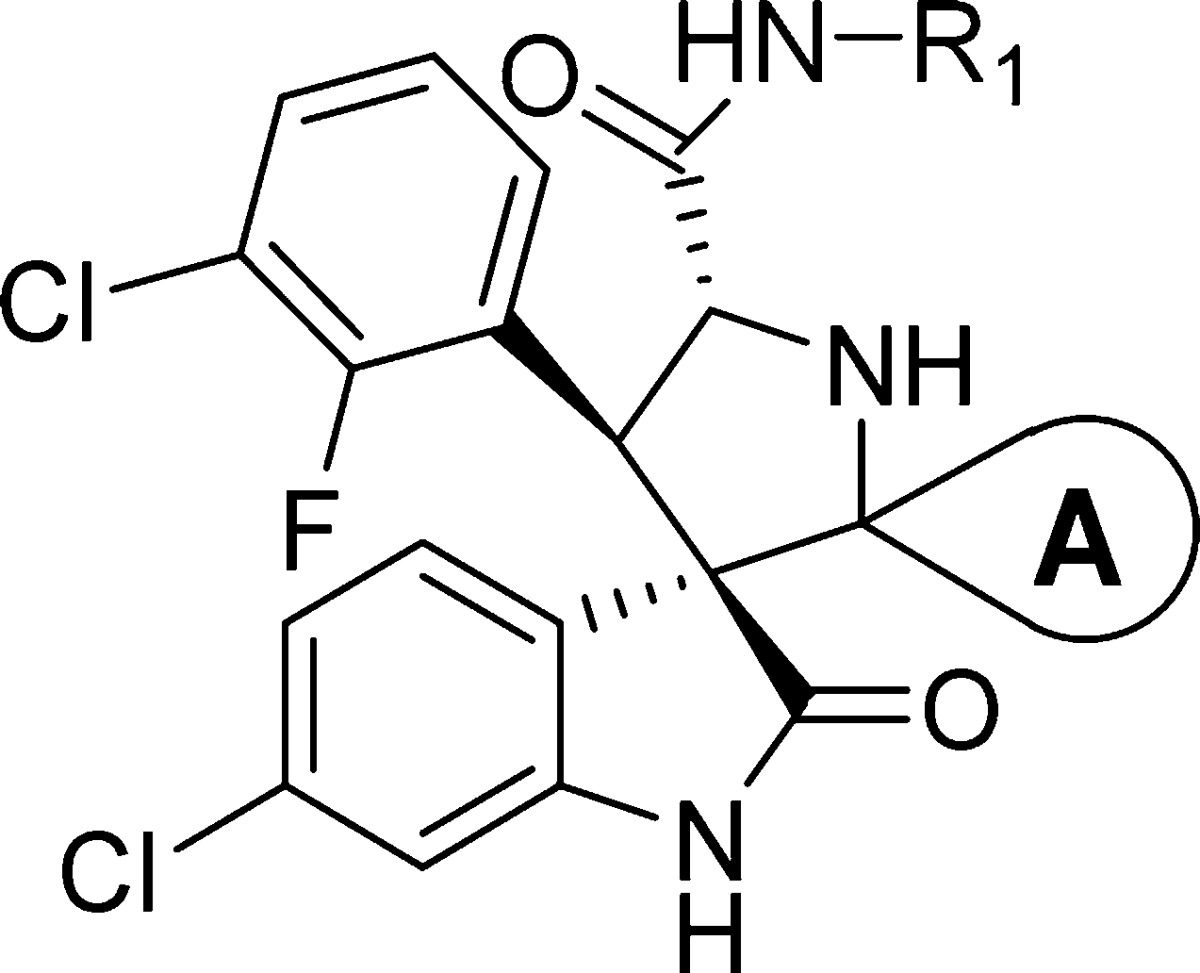
Our exploration started with lead molecule 6(21) (Figure 2A) that binds to MDM2 with Ki = 2.9 nM, inhibits the growth of the SJSA-1 cancer cell line with IC50 = 190 nM, and achieves strong tumor growth inhibition but not complete tumor regression in an SJSA-1 osteosarcoma xenograft model in mice.21 In a search for superior compounds, we have performed extensive modifications of 6.
Figure 2.

(A) Chemical structure of compound 6 and five regions (A–E) where chemical modifications were made. (B) Cocrystal structure of 2 (light blue spheres, PDB code 5TRF) and model of the binding mode of 6 (green sticks) in complex with MDM2.
To guide our chemical modifications, we modeled the structure of compound 6 in a complex with MDM2 based upon the cocrystal structure of compound 2 complexed with human MDM2 (Figure 2B).18 Our model shows that the oxindolephenyl, 3-chloro-2-fluorophenyl, and cyclohexyl groups of 6 project into the Trp23, Leu26, and Phe19 p53 binding pockets of the MDM2 protein and the 4-hydroxyl of the N-cyclohexylcarboxamide group forms a hydrogen bond with Lys94 of MDM2 (Figure 2B). Guided by this model, we performed extensive modifications on five regions of the molecule (labeled as sites A–E, Figure 2A).
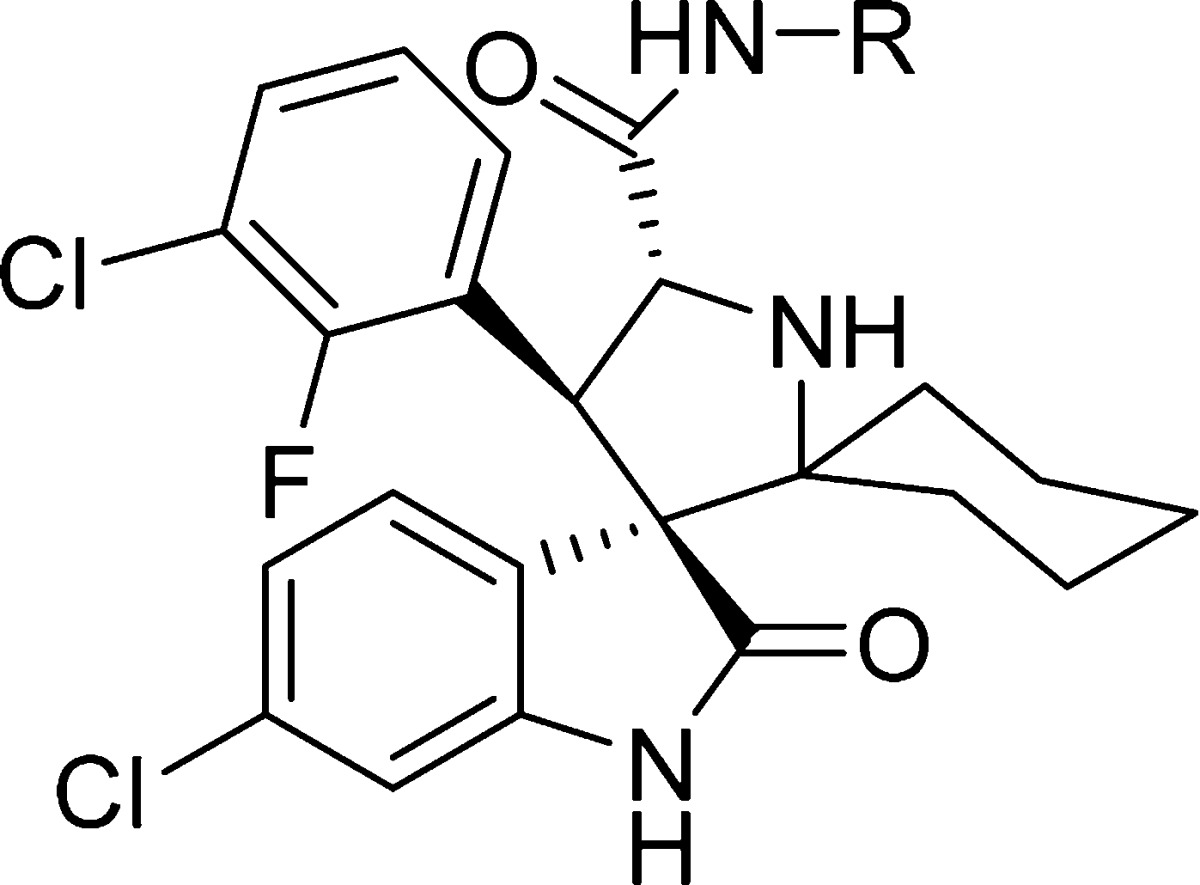
We began by replacing the 4-hydroxycyclohexyl group at site D in compound 6 with a methyl group, giving compound 7. Since compounds 6 and 7 have similar binding affinities to MDM2, we have used either compound 6 or 7 as the template for modifications of site A (Table 1). Replacement of the cyclohexyl group at site A with 4-piperidinyl group led to compound 8, which has no appreciable binding up to 2 μM to MDM2. Methylation or acetylation of the amine group in 8 resulted in compound 9 or 10, respectively, and these also have very weak affinities for MDM2. Replacement of the cyclohexyl group at site A in 6 with 4-tetrahydro-2H-pyran generated compound 11, which has a moderate binding affinity to MDM2 (Ki = 75.9 nM). Introduction of two fluorine substituents at the 4-position of the cyclohexyl group in compound 6 resulted in compound 12, which has a good affinity to MDM2 (Ki = 9.8 nM) but is 4 times less potent than 6. However, introduction of two methyl groups at the same 4-position in 6 yielded compound 15, which has a Ki value of <1 nM to MDM2 and is several times more potent than 6. Replacement of the cyclohexyl group with a cyclobutyl group generated compound 13,21 which is 6 times less potent than 6. Installation of methyl groups at the 3-position of the cyclobutyl ring led to compound 14, which is equally potent as compound 6 and 7 times more potent than compound 13. Hence, our SAR data for site A clearly show that a hydrophobic group is highly desirable for achieving a strong binding affinity to MDM2, and cyclohexyl, 4,4′-dimethylcyclohexyl, and 3,3′-cyclobutyl groups are the most preferred.
Table 1. Structure–Activity Relationship of Pyrrolidine-2-spirocyclohexyl Substituentc.

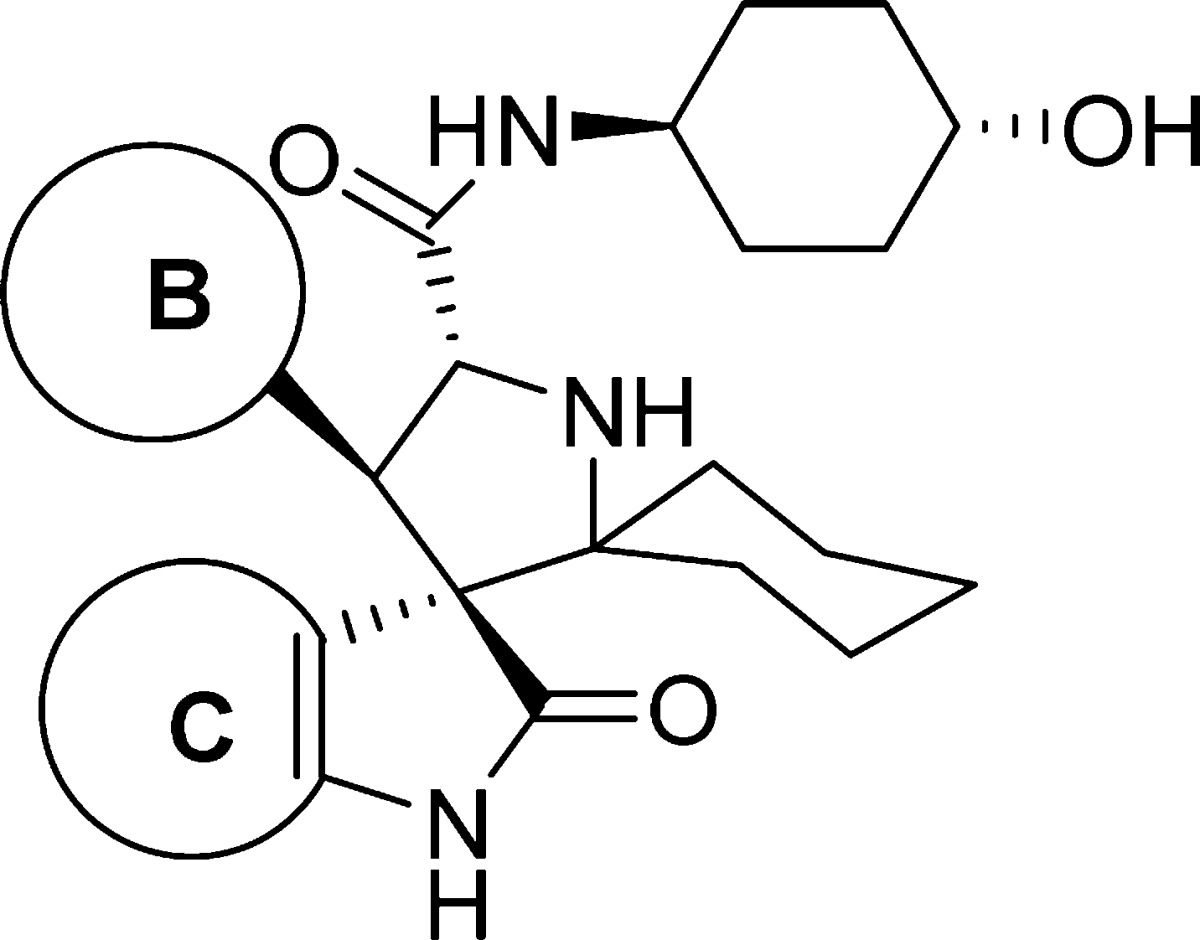
Next, we explored modifications of the 3-chloro-2-fluorophenyl and the oxindolephenyl substituents (sites B and C in Figure 2A, respectively). To improve solubility, we inserted one or more nitrogen atoms into the aryl rings, producing compounds 16, 17, 18, and 19 (Table 2). Insertion of a nitrogen into the 3-chloro-2-fluorophenyl substituent (ring B) gave 16, which has a Ki of 77 nM, 26 times weaker than 6. Interestingly, although the Trp23 pocket is hydrophobic in nature, compounds 17, 18, and 19 containing a pyridinyl or a pyrimidinyl oxindole group (ring C) are all quite potent with Ki = 13–14 nM. Our data, however, are consistent with a previous study, which showed that a compound containing a chloropyridyl oxindole group is a potent MDM2 inhibitor.23 This previous study also showed that replacing the oxindole with a thienopyrrolone led to potent MDM2 inhibitors.23 We therefore synthesized compound 20 containing a thienopyrrolone, which has a Ki of 6.5 nM and is 2-fold less potent than 6. We found that the thiophene group in 20 can be oxidized during the oxidative removal of the chiral auxiliary, preventing further investigation of this compound.
Table 2. SAR of the B and C Aryl Ringsa.

Values are average of at least two tests.
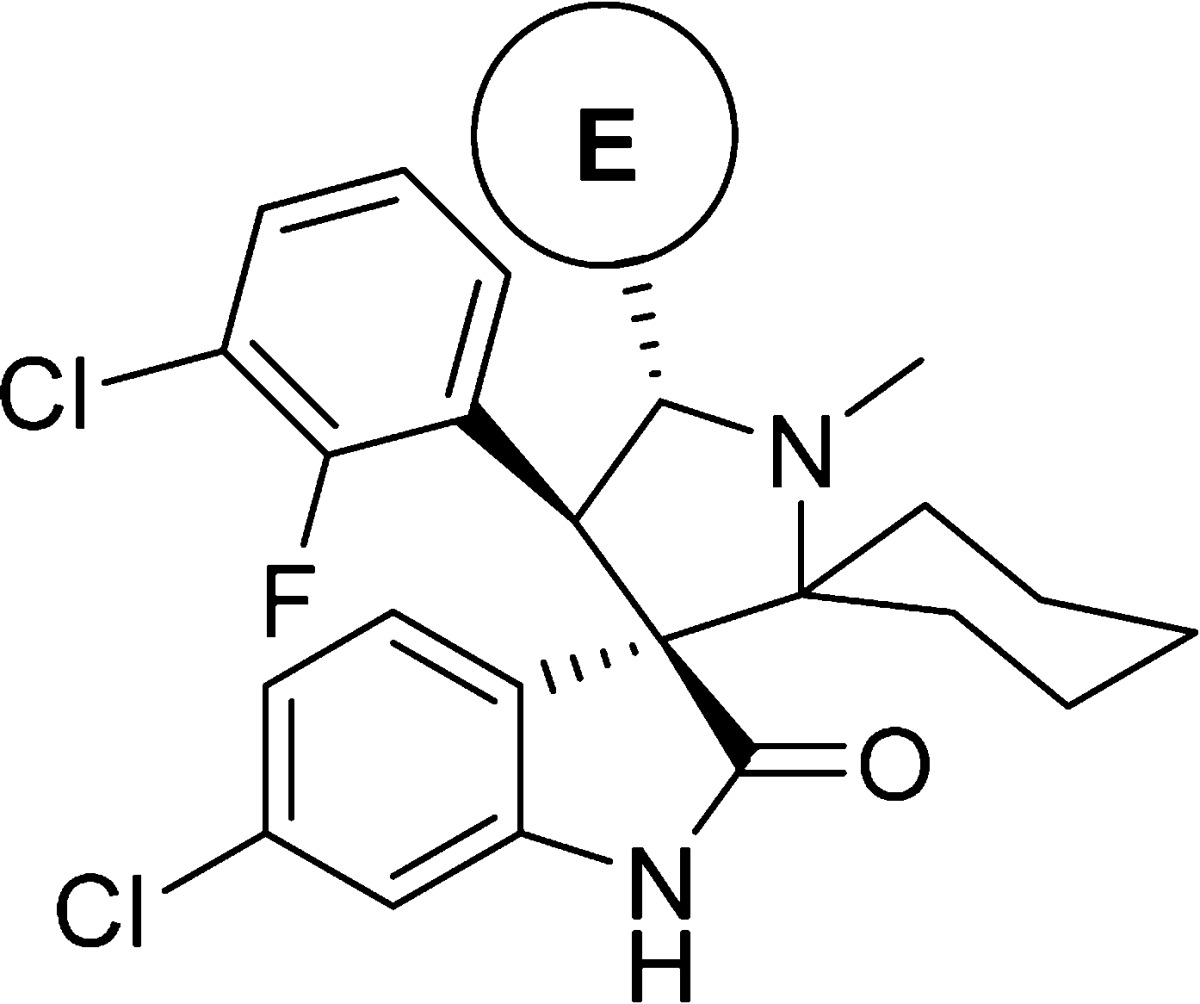
Our modeling of 6 and the cocrystal structure of 2 (Figure 2B) showed that the carbonyl of the amide (site E) forms a hydrogen bond with His96 of the MDM2 protein but the amino group does not interact with the MDM2 protein. We evaluated a series of five-membered heteroaromatic rings as the amide bioisoteres (Table 3). Among these replacements, we found that heteroaromatic rings with carboxylic acid substituents, e.g., 21, 22, 26, and 27 have the best binding affinities with Ki values between 1 and 3 nM. Unfortunately, these compounds show weak cell growth inhibitory activity (IC50 > 2 μM, Table 3) in the SJSA-1 cancer cells, probably because the negatively charged acid group in these compounds decreases their cell permeability. We therefore converted the acid group in compound 21 to an amide but found that the resulting compound 28 is 20 times less potent than 21.
Table 3. Carboxamide (E) Bioisostere Replacementc.

Value of only one experimnent.
NT = not tested.
Values are average of at least two experiments.
Our modeling of 6 and the cocrystal structure of 2 also showed that the 4-hydroxyl on the cyclohexylcarboxamide substituent of 6 forms a hydrogen bond with Lys94 of the MDM2 protein (Figure 2B). We synthesized a series of compounds containing either a terminal carboxylic acid or a sulfone to investigate the effect on binding (compounds 29–38) (Table 4). These modifications resulted in a series of compounds with high affinities. In particular, compounds 31, 32, and 33 bind to MDM2 with Ki < 1 nM and are more than 3 times more potent than 6.
Table 4. SAR of Carboxamide Substituent (D)d.

Reference (21).
NT = not tested.
Value of one time testing.
Values are average of at least two tests.
We tested the cell growth inhibitory activity of compounds 29–38 in the SJSA-1 cell line, and the data are summarized in Table 4. Among this series of compounds, 32, 33, and 38 have the best antiproliferative activity with IC50 values of 0.22, 0.15, and 0.24 μM, respectively.
We further evaluated compound 33 for its pharmacodynamic (PD) effect in the SJSA-1 xenograft tissue. Our PD data showed that a single, oral administration of 33 at 100 mg/kg is very effective in inducing upregulation of MDM2, p53, and p21 proteins at both 3 and 6 h time-points in the SJSA-1 tumor tissue, indicating strong activation of p53 (Figure 3A). Upon the basis of its promising PD data, we evaluated compound 33 for its antitumor efficacy in the SJSA-1 xenograft model (Figure 3B,C). While compound 33 can effectively retard tumor growth, it failed to achieve tumor regression. During the course of our research, we also observed that compound 33 can decompose in solution, particularly in cell culture media (panels A and B in Scheme 1). To analyze the potential structural features affecting the stability of 33, we determined the stability of compound 31 with a flexible butanoic acid group, compound 6 with a 4-hydroxylcyclohexyl group, and 33. Compound 6 had negligible (<1%) decomposition (panel C in Scheme 1). On the other hand, compound 31 showed 18% decomposition after 2 days in cell culture media, which is greater than that observed for compound 33 (panel D in Scheme 1). On the basis of these results, we proposed that the carboxylic group assists in the decomposition of 33.
Figure 3.

(A) Pharmacodynamics (PD) effect of 33 in SJSA-1 tumors in mice. Mice were given a single, oral dose of the vehicle, or 33 at 100 mg/kg and sacrificed at indicated time points. West blotting was performed on harvested tumor tissue to probe MDM2, p53, p21, PAPR, cleaved PARP (cl-PAPR) proteins. (B) Antitumor activity of 33 in the SJSA-1 osteosarcoma tumor xenograft model in mice. Compound 33 was administered via oral gavage at 100 mg/kg daily for 14 days. (C) Body weight change of mice during administration of 33.
Scheme 1. Proposed Mechanism of Decomposition for 33.

(A) Stability of compound 33 in various solutions. Compound 33 was incubated in CH3CN/H2O, MeOH/H2O, or cell culture media, and the % composition of 33 in the solutions was determined daily for 2 days by UPLC (ultraperformance liquid chromatography) and plotted. (B, C, D) UPLC/MS spectrum of 33, 6, and 31, respectively, after incubation for 2 days in cell culture media. Peaks are labeled with retention time followed by molecular weight.
We next performed further chemical modifications of 33 with the goal to improve its chemical stability, cellular potency, and in vivo efficacy.
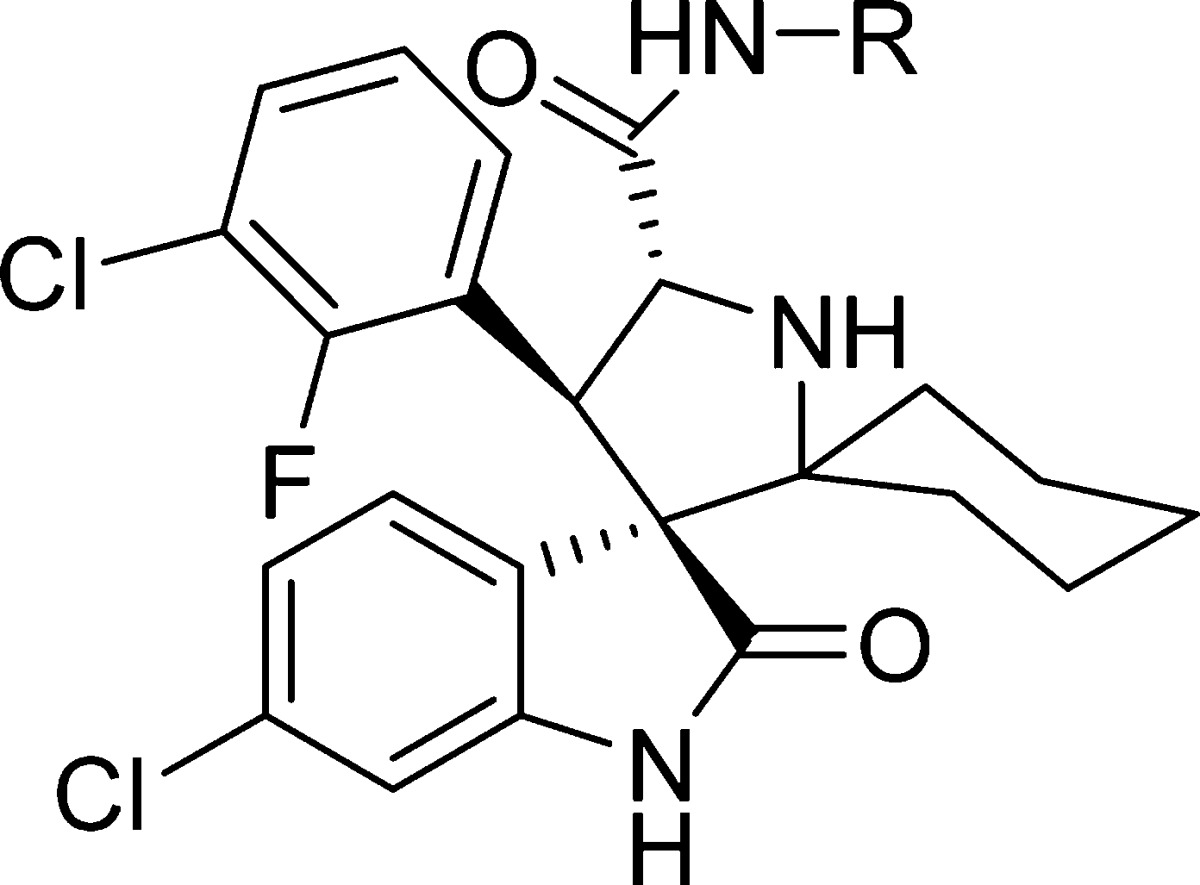
We investigated if replacement of the cyclohexyl group with an even more rigid aryl group can prevent the decomposition. A series of compounds containing an N-arylcarboxamide substituent were synthesized and tested (Table 5). These compounds all display very high binding affinities to MDM2, superior to that of 6 and equivalent to that of 33. The cell growth inhibition of SJSA-1 cells by these compounds was determined (Table 5). Compounds 39 (MI-1061)21 and 44 displayed the best cellular activity among the compounds in this series and are 3 times more potent than compound 33 in inhibition of cell growth in the SJSA-1 cell line. Compound 39 was found in our previous study21 to be very stable in solution. Furthermore, 39 was capable of achieving partial tumor regression in the SJSA-1 xenograft model as shown in our previous report.21
Table 5. SAR of (Aryl acid)carboxamidesd.

Value of one time testing.
NT = not tested.
Reference (21).
Values are average of at least three tests.
We next combined all the favorable modifications on sites A and D and designed and synthesized compound 54 (Scheme 2). Compound 54 has a very high binding affinity to MDM2 (IC50 = 2.4 nM, Ki < 1 nM, Scheme 2) and potently inhibits cell growth of SJSA-1 cells with IC50 = 13 nM (Scheme 2).
Scheme 2. Design of Compound 54 by Combining the Optimal Substituents of Sites A and D.

Reference (21).
We assessed the antitumor activity of compound 54 in the SJSA-1 xenograft model in mice, with compound 2 included as the control (Figure 4). Compound 54 effectively inhibits tumor growth in a dose-dependent manner compared to the vehicle control. While compound 54 at 50 mg/kg daily dosing effectively retards tumor growth, at 100 mg/kg it achieves a maximum of 87% tumor regression during the treatment. However, while compound 2 at 100 mg/kg achieves complete tumor regression, compound 54 fails to do so.
Figure 4.

(A) Antitumor activity of compound 54, in comparison to compound 2 in the SJSA-1 osteosarcoma tumor xenograft model in mice. Compound 54 was administered via oral gavage at 50 or 100 mg/kg daily for 14 days and compound 2 was administered via oral gavage at 100 mg/kg daily for 14 days. (B) Changes in body weight of mice during animal dosing.
Pharmacokinetic (PK) studies in rats (Table 6) showed that with oral administration, compounds 39 and 54 achieve considerably lower Cmax and AUC values than compound 2, suggesting that further improvement of the oral PK for compounds 39 and 54 is needed in order to achieve stronger in vivo antitumor activity. Since 39 and 54 achieve comparable tumor regression in the SJSA-1 xenograft model and 39 is less hydrophobic than 54, we have selected compound 39 for further optimization of its PK properties and in vivo efficacy.
Table 6. Pharmacokinetics Table for Comparison of 39, 54, 56, 59, 60, and 2 in Rats.
| compound |
||||||
|---|---|---|---|---|---|---|
| 39 | 54 | 56 | 59 | 60 | 2 | |
| po dose (mg/kg) | 25 | 25 | 25 | 25 | 25 | 25 |
| po Cmax (ng/mL) | 1553 | 968 | 8234 | 4391 | 5453 | 4547 |
| po AUC (h·ng/mL) | 6799 | 10188 | 73603 | 35205 | 39083 | 17230 |
| po T1/2 (h) | 4.0 | 4.5 | 4.3 | 3.9 | 4.6 | 1.6 |
| iv dose (mg/kg) | 10 | 10 | 10 | 10 | 10 | 5 |
| iv AUC (h·ng/mL) | 8633 | 14767 | 82241 | 28825 | 38265 | 4630 |
| iv CL (L h–1 kg–1) | 1.16 | 0.690 | 0.119 | 0.352 | 0.257 | 1.03 |
| iv Vss(L/kg) | 2.14 | 2.45 | 0.734 | 1.10 | 1.25 | 2.98 |
| F (%) | 31.5 | 27.6 | 35.0 | 48.6 | 40.3 | 74.9 |
| antitumor efficacy | 86% regression | 87% regression | no regression | 100% regression | 100% regression | 100% regression |
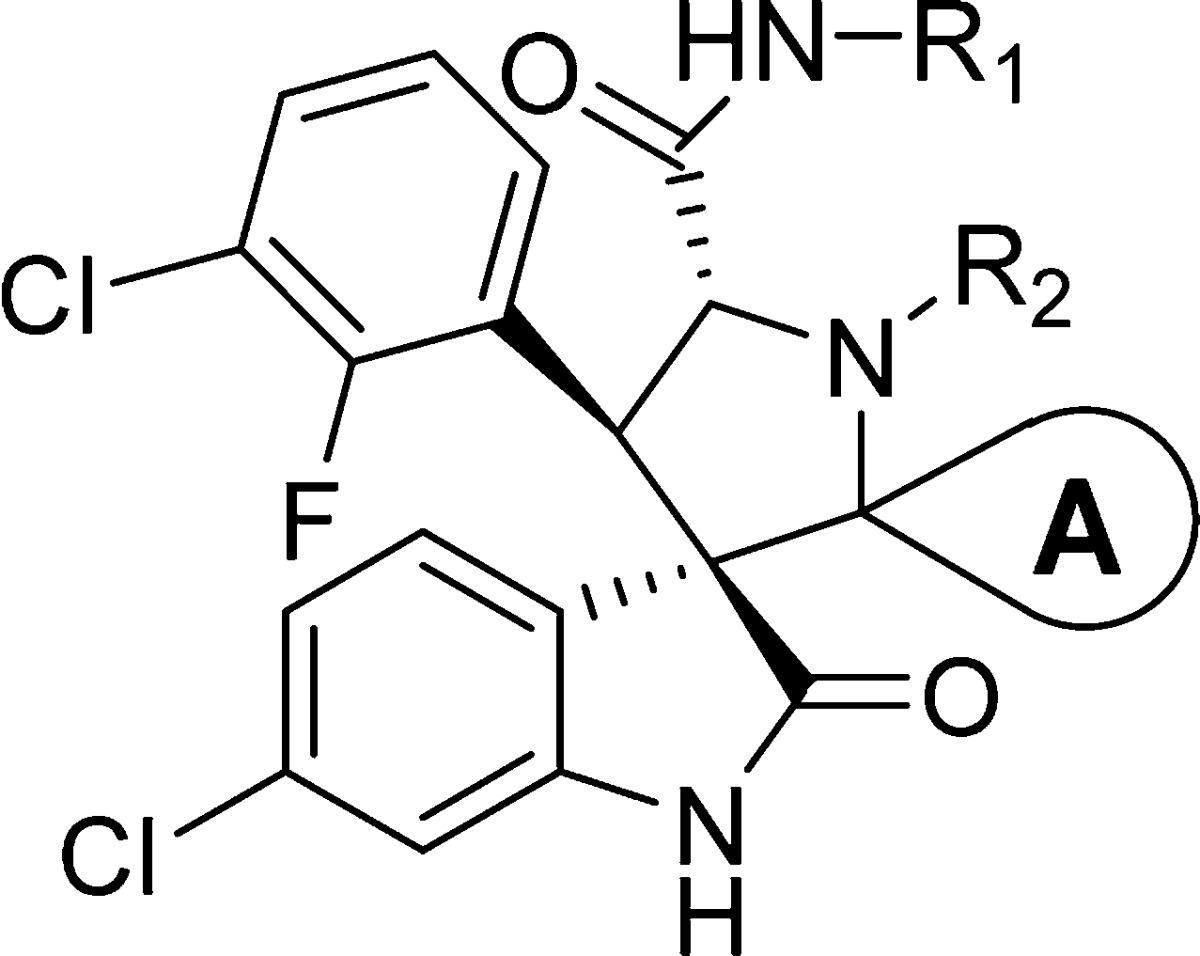
In our attempt to further improve the oral PK properties of compound 39 while retaining its high binding affinity to MDM2 and cellular potency, we investigated the replacement of the benzoic acid with nonclassical benzoic acid mimetics such as a bicyclo[1.1.1]pentane-1-carboxylic acid24 or a bicyclo[2.2.2]octane-1-carboxylic acid. These efforts led to the design and synthesis of compounds 55 and 56 (Scheme 3). Compound 55 binds to MDM2 with a high affinity (IC50 = 6.4 nM, Ki < 1 nM) but is 5 times less potent than compound 39 in inhibition of cell growth in the SJSA-1 cell line. In comparison, compound 56 has a high binding affinity to MDM2 (IC50 = 3.7 nM, Ki < 1 nM) and is as potent as compound 39 in inhibition of cell growth in the SJSA-1 cell line (Scheme 3 and Table 7).
Scheme 3. Replacement of the Benzoic Acid in Compound 39 with Nonclassical Benzoic Acid Bioisosteres.

Table 7. SAR of Benzoic Acid Nonclassical Bioisosteres and Alkylation of the Pyrrolidine Nitrogenc.

Value from one experiment.
Reference (21).
Values are average of at least two independent experiments.
A PK study in rats showed that 56 has a higher plasma exposure than compound 2 based upon both the Cmax (8234 vs 4547 μg/L) and AUC values (73603 vs 17230 h·μg/L). However, our PD experiment showed that oral administration of compound 56 at 100 mg/kg induces only modest p53 activation and no cleavage of PARP and caspase-3 in the SJSA-1 xenograft tumor tissue in mice (Figure 6A). Consistent with the PD data, compound 56 at 100 mg/kg administered daily for 14 days demonstrates only a very moderate tumor growth inhibition in the SJSA-1 xenograft model in mice (Figure 5A). Hence, the antitumor activity of 56 is much inferior to that achieved by 2, 39, and 54. Because of its excellent in vitro cellular potency and excellent plasma exposure, the modest p53 activation and antitumor activity achieved by compound 56 suggests that this compound has a poor penetration into the xenograft tumor tissue. Accordingly, we investigated strategies with which to improve the tissue penetration while retaining high binding affinity to MDM2, potent cellular activity, and good oral bioavailability of compound 56.
Figure 6.

Pharmacodynamic (PD) analysis of the effect of MDM2 inhibitors in SJSA-1 tumors. Mice bearing SJSA-1 tumors were dosed with a single oral dose of 56, 57, 58, 59, 60 and 2 at 100 mg/kg and the tumors were harvested at 6 and 24 hours for western blot analysis. (A, B) PD of 56, 57 and 58 show modest activation of p53 as seen with the low levels of p53, MDM2, and p21; (C) Compounds 59, 60 and 2 show robust activation of p53 as seen with increased levels of p53, MDM2, ubiquitinated MDM2 (ub-MMD2) and p21, and induce strong apoptosis as seen with increased levels of cleaved PARP (cl-PAPR) and cleaved caspase-3 (cl-Casp3).
Figure 5.

(A) Antitumor activity of compounds 39 and 56 in the SJSA-1 osteosarcoma xenograft model in mice. (B) Body weight change of mice during administration period.
We investigated three strategies to improve the tissue penetration: (1) decreasing the lipophilicity of the molecule; (2) reducing the acidity of carboxylic acids; and (3) increasing the basicity of the nitrogen atom in the pyrrolidine core.25
Fluorine substitution on aliphatic groups is known to decrease lipophilicity.26−28 We therefore synthesized compound 57 with a 4,4-difluorocyclohexyl group at carbon-2 of the pyrrolidine core (Table 7). For the second strategy, we replaced the carboxylic acid group with an acyl sulfonamide bioisostere, which resulted in 58 (Table 7). For the third strategy, we installed a methyl or ethyl group on the nitrogen of the pyrrolidine core, which led to 59 and 60, respectively (Table 7).
Compound 57 binds to MDM2 with a high affinity but is less potent than 56 and is 2 times less potent than 56 in inhibition of cell growth in the SJSA-1 cell line (Table 7). Compound 58 is 2 times less potent than 56 in binding to MDM2 and is 4 times less potent than 56 in inhibition of cell growth in the SJSA-1 cell line (Table 7). The PD study showed that oral administration of 57 or 58 at 100 mg/kg only has a modest effect on activation of p53 and induction of cleavage of PARP and caspase-3 (Figure 6B).
In comparison, compounds 59 and 60 bind to MDM2 with a high binding affinity (Ki < 1 nM, Table 7). Both compounds potently inhibit cell growth in the SJSA-1 cell line with IC50 values of 70 and 60 nM, respectively, and are thus as potent as compound 56 (Table 7). PD experiments showed that a single, oral dose of 59 and 60 at 100 mg/kg achieves strong p53 activation, with the effect persisting for 24 h in the SJSA-1 xenograft tumor tissue (Figure 6C). Compounds 59 and 60 also achieve strong apoptosis induction in the SJSA-1 xenograft tumor, based upon cleavage of PARP and caspase-3 (Figure 6C).
We next tested 59 and 60 for the antitumor activity in the SJSA-1 xenograft model (Figure 7). Compound 59 or 60 orally administered at 30 mg/kg daily for 14 days effectively inhibits tumor growth. Compound 59 or 60 orally administered at 100 mg/kg daily for 14 days effectively achieves complete and long-lasting tumor regression. In comparison, compound 56 at 100 mg/kg inhibits tumor growth only moderately in the same experiment (Figure 7A). Compounds 56, 59, and 60 cause no or minimal weight loss during the entire experiment (Figure 7B).
Figure 7.

(A) Antitumor activity of compound 59, 60 in comparison to 56 in the SJSA-1 osteosarcoma xenograft model in mice. Compound 59 and 60 were administered at 30 mg/kg or 100 mg/kg daily for 14 days and 56 at 100 mg/kg daily for 14 days. (B) Animal weight changes of mice during the experiment.
Our PK studies in rats with oral administration showed that 59 and 60 achieve a Cmax value similar to that of 2 but have a 2 times higher AUC than compound 2 (Table 6).
We evaluated compounds 56, 59, and 60 for their activity and selectivity in a number of human cancer cell lines of different tumor types (Table 8). Consistent with their mechanism of action, these three compounds potently inhibit cell growth in cancer cell lines with wild-type p53 and display outstanding selectivity in cancer cell lines with deleted p53. Compound 60 is the most potent compound, achieving IC50 values of 38, 18, and 104 nM in the RS4;11 acute leukemia, LNCaP prostate cancer, and HCT116 colon cancer cell lines, respectively.
Table 8. Cell Growth Inhibition Activity of 56, 59, and 60 in Different Human Cancer Cell Linesa.
| cell growth inhibition (IC50) |
|||||
|---|---|---|---|---|---|
| cell line | tumor type | p53 status | 56 | 59 | 60 |
| SJSA-1 | osteosarcoma | wild-type | 89 ± 33 (nM) | 70 ± 21(nM) | 60 ± 22 (nM) |
| Saos2 | osteosarcoma | null | 26.7 ± 5.1 (μM) | 25 ± 6 (μM) | 22.7 ± 4.7 (μM) |
| RS4;11 | leukemia | wild-type | 62 ± 26 (nM) | 56 ± 18 (nM) | 38 ± 5 (nM) |
| LNCaP | prostate cancer | wild-type | 36 ± 19 (nM) | 30 ± 15 (nM) | 18 ± 13 (nM) |
| PC3 | prostate cancer | null | 12.3 ± 2.5 (μM) | 24 ± 5 (μM) | 22 ± 7.2 (μM) |
| HCT116 | colon cancer | wild-type | 137 ± 31 (nM) | 117 ± 33 (nM) | 104 ± 36 (nM) |
| HCT116 p53–/– | colon cancer | knockout | 14 ± 2 (μM) | 18 ± 8 (μM) | 8 ± 1 (μM) |
IC50 values are average of at least three independent experiments.
We next evaluated compound 60 for its antitumor activity in the RS4;11 acute leukemia xenograft model in mice. Compound 60 effectively inhibits tumor growth in a dose-dependent manner and achieves partial tumor regression at 100 mg/kg, daily, oral administration for 14 days, or at 200 mg/kg weekly dosing for 3 weeks (Figure 8). Compound 60 is also well tolerated at all doses and schedules in this experiment.
Figure 8.

(A) Antitumor activity of compound 60 in the RS4; 11 acute lymphoblastic leukemia xenograft model in mice. Compound 60 was administered at 50 mg/kg, 75 mg/kg, or 100 mg/kg daily for 14 days and 200 mg/kg weekly for 3 weeks. (B) Animal body weight of mice during administration.
We evaluated the chemical stability of compound 60 in three different solutions and found that this compound is very stable in solutions for a period of 7 days (Figure 9). In comparison, compound 2 slowly isomerizes in solutions and retains only 93–96% purity after 7 days (Figure 9).
Figure 9.

Stability comparison of compounds 2 and 60 in (A) 1:1 CH3CN to H2O; (B) 1:1 MeOH to H2O; (C) cell culture media.
Chemistry
To explore achiral substituents in carbon-2 of the pyrrolidine core, the compounds in Table 1 were synthesized using the method21 outlined in Scheme 4. The azomethine ylide generated in situ by reaction of the chiral amine (62) with the cyclic ketone (63) was allowed to react with 61 in a 1,3-dipolar cycloaddition reaction, generating the chiral spirooxindoles (6a–15a). Reaction of this morpholine (6a–15a) with an aliphatic amine led to the open-ring compound (6b–15b). Upon oxidation with ceric ammonium nitrate (CAN), 6c–15c were formed as a mixture of two diastereoisomers. “Aging” in acetonitrile/water resulted in formation of the single diastereoisomers 6–15.
Scheme 4. Synthesis of Compounds 6–15.

Reagents and conditions: (a) toluene, reflux, 2 h; (b) R1NH2, THF, reflux overnight; (c) CH3CN/H2O (1:1), CAN, 15 min; (d) CH3CN/H2O, overnight.
Scheme 5 shows the method by which the B and C aryl rings were replaced. First the intermediates 16b–20b were synthesized by condensation of the pyrrolidones (16a–20a) with an arylaldehyde. Subsequently, the compounds (16–20) in Table 2 were obtained by using the intermediates (16b–20b), which are equivalent to 61, in the synthetic method outlined in Scheme 4.
Scheme 5. Synthesis of Compounds 16–20.

Reagents and conditions: (a) MeOH, piperidine, reflux.
Compounds containing various five-membered heteroaromatic rings as bioisostere replacements for the carboxamide group E were synthesized as outlined in Schemes 6 and 7. The N-methylpyrrolidine (65) was generated by reductive amination of 64 with paraformaldehyde and NaBH(OAc)3. Hydrolysis of the methyl ester (65) was followed by conversion of the acid (66) to the corresponding carboxamide (67). Refluxing of 67 with Lawesson’s reagent produced the thioamide (68) which was reacted with an α-bromoketone in a Hantzsch thiazole synthesis reaction, generating the thiazoles 69 and 70.29 Basic hydrolysis of the esters produced the acid analogues 21 and 27. Compound 21 was further elaborated by amide coupling, generating the carboxamide (28).
Scheme 6. Synthesis of Compounds 21, 27, and 28.

Reagents and conditions: (a) DCM/AcOH (1:1), paraformaldehyde, NaBH(OAc)3; (b) THF/MeOH/H2O, LiOH·H2O; (c) DCE, CDI, DIEA, 50 °C 30 min, then NH4OH; (d) DCM, Lawesson’s reagent, reflux overnight; (e) (1) THF, BrCH2COR, Et3N, reflux overnight, (2) DME, TFAA, pyridine, −20 °C to rt, 30 min; (f) THF/MeOH/H2O (1:1:1), NaOH, rt, prep-HPLC.
Scheme 7. Synthesis of Compounds 22–26.

Reagents and conditions: (a) DCE, CDI, DIEA, DMAP, 50 °C for 30 min, then 71, 74, 76, 78, and reflux; (b) (1) THF, Deoxo-Fluor, −20 °C for 30 min, (2) BrCCl3, DBU at rt overnight; (c) THF/H2O (2:1), LiOH·H2O, rt for 30 min, then preparative HPLC; (d) pyridine, 100 °C for 4 h; (e) DCM, PPh3, I2, Et3N, rt; (f) DCM, Lawesson’s reagent, reflux overnight.
Several five-membered heteroaromatic rings were formed from the carboxylic acid (66). The carboxylic acid (66) was activated with CDI (1,1′-carbonyldiimidazole), and this was followed by addition of commercially available compounds 71, 74, 76, 78, resulting in the formation of intermediates 72, 75, 77, 79, respectively. Cyclization to the respective five-membered heteroaromatic compounds (73,3023,3124,3225, 80) was accomplished under the conditions outlined in Scheme 7. The carboxylic acid compounds 22 and 26 were produced by hydrolysis of the esters in compounds 73 and 80, respectively.
The compounds in Tables 4 and 5 were obtained by the synthesis outlined in Scheme 8. Activation of the carboxylic acid (29) by HATU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate) followed by addition of an alkylamine gave the N-alkylcarboxamide intermediates 30, 36, 38, 31–35, 37, 55, and 56. When N-arylamines were used, HATU failed to accomplish the coupling, but the coupling was successful when the acid was activated with Ph2POCl 17 and produced the N-arylcarboxamides 39–48, 50, and 51. Basic hydrolysis of the esters produced the carboxylic acid compounds listed in Tables 4 and 5. Further elaboration of the acids (56 to give 58; 39 to give 49 and 52; and 44 to give 53) was accomplished by CDI activation of the carboxylic acid followed by reaction with the respective amine.
Scheme 8. Synthesis of Compounds in Tables 4 and 5.

Reagents and conditions: (a) DCM, HATU, DIEA, alkylamine; (b) THF/MeOH/H2O (1:1:1), LiOH·H2O for alkyl esters or NaOH for aryl esters; (c) DCM, Ph2POCl, DIEA 30 min, then arylamine and DMAP; (d) DCE, CDI, DIEA, DMAP, 50 °C for 30 min, then R2NH2 and reflux.
Reductive amination of 56 with paraformaldehyde or acetaldehyde produced 59 and 60 as shown in Scheme 9.
Scheme 9. Synthesis of Compound 59 and 60.

Reagents and conditions: (a) DCM/AcOH (1:1), paraformaldehyde or acetaldehyde, NaBH(OAc)3, rt overnight.
Conclusion
In this study, we have performed an extensive SAR study on spirooxindoles containing two substituents at the carbon-2 of the pyrrolidine core as MDM2 inhibitors. Extensive modifications in five different regions of the molecule have led to discovery of a number of potent and highly efficacious MDM2 inhibitors. In particular, compound 60 has a very high binding affinity to MDM2 (Ki < 1 nM), potently activates wild-type p53, inhibits cell growth with low nanomolar IC50 values in human cancer cell lines carrying wild-type p53, and demonstrates an outstanding cellular selectivity over human cancer cell lines with deleted p53. Compound 60 is very stable in solutions, has excellent oral pharmacokinetics, and effectively activates p53 in the SJSA-1 xenograft tumor tissue in mice following a single oral administration. Significantly, 60 achieves complete and long-lasting regression of the SJSA-1 xenograft tumors in mice and demonstrates strong antitumor activity in the RS4;11 acute leukemia model at well tolerated dose schedules. Compound 60 has been advanced into phase I clinical development for the treatment of human cancer.
Experimental Section
General Information
Unless otherwise stated, all commercial reagents were used as supplied without further purification and all reactions were performed under a nitrogen atmosphere in dry solvents under anhydrous conditions. NMR spectra were obtained on either a Bruker 300 UltraShield spectrometer at a 1H frequency of 300 MHz and 13C frequency of 75 MHz or Bruker 400 Ascend spectrometer at a 1H frequency of 400 MHz and 13C frequency of 100 MHz. Chemical shifts (δ) are reported in parts per million (ppm) relative to an internal standard. The final products were purified on a preparative HPLC (Waters 2545, quaternary gradient module) with a SunFire Prep C18 OBD 5 μm, 50 mm × 100 mm reverse phase column. The mobile phase was a gradient of solvent A (H2O with 0.1% TFA) and solvent B (MeOH with 0.1% TFA or MeCN with 0.1% TFA) at a flow rate of 40 mL/min and 1%/2 min increase of solvent B or a flow rate of 60 mL/min and 1%/min increase of solvent B. All final compounds have purity of ≥95% as determined by Waters ACQUITY UPLC using reverse phase column (SunFire, C18-5 μm, 4.6 mm × 150 mm) and a solvent gradient of A (H2O with 0.1% of TFA) and solvent D (MeOH with 0.1% of TFA) or A (H2O with 0.1% of TFA) and solvent B (CH3CN with 0.1% of TFA). ESI mass spectrum analysis was performed on a Thermo-Scientific LCQ Fleet mass spectrometer. No PAINS liability was found in any of the compounds presented as determined by analysis in the online filter SmartsFilter (http://pasilla.health.unm.edu/tomcat/biocomp/smartsfilter).
Compounds 6, 13, 29, 39 and intermediate 64 were reported previously.21 Compounds 16b–20b were synthesized as reported previously.23
Compounds 7–20 were synthesized according to our reported method21 which is described below for the synthesis of compound 6.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (6)21
In a round-bottom flask, (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)indolin-2-one (500 mg, 1.62 mmol), (5R,6S)-5,6-diphenyl-2-morpholinone (492 mg, 1.94 mmol), and cyclohexanone (4.86 mmol) were suspended in toluene (10 mL) and heated at reflux for 2 h at 140 °C. Then the reaction was allowed to cool to room temperature and the solvent was removed by rotoevaporation. The crude product was purified by column chromatography (the compound was eluted with 100% DCM) to give 665 mg (64% yield) of 6a as a pale yellow solid. trans-4-Aminocyclohexanol (475 mg, 4.13 mmol) was added to a solution of 6a (530 mg, 0.826 mmol) in THF (20 mL) and heated at reflux overnight. Then the reaction was cooled to room temperature and the solvent was removed by rotoevaporation. The crude 6b was purified by column chromatography to produce 224 mg of 6b. Cerium ammonium nitrate (324 mg, 0.592 mmol) was added to a solution of the resulting 6b (224 mg, 0.296 mmol) in MeCN (6 mL) and stirred for 5 min at room temperature, then H2O (6 mL) was added. After stirring the reaction for an additional 10 min, it was quenched with saturated sodium bicarbonate, brine was added, and the solution was extracted with EtOAc. The EtOAc solution was dried over sodium sulfate, filtered through Celite and the solvent was removed by rotoevaporation to produce crude 6c as a mixture of diastereisomers. The crude material was dissolved in a 3:1 mixture of MeOH/H2O that was acidified with TFA and aged. After 2 days this solution was purified by preparative HPLC. The combined fractions of the pure compound were concentrated and then redissolved in a minimum amount of MeCN, H2O was added, and the solution was frozen and lyophilized to produce the TFA salt of 6 (116 mg, 70% yield) as a white powder. Chemical data were reported previously.21
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-methyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (7)
7 was obtained using the synthetic method described for the preparation of 6. 1H NMR (300 MHz, CD3OD) δ ppm 8.29 (br s, 1H), 7.63 (t, 1H, J = 7.2 Hz), 7.51 (dd, 1H, J = 2.4, 8.2 Hz), 7.39 (t, 1H, J = 7.6 Hz), 7.21–7.07 (m, 2H), 6.77 (d, 1H, J = 1.5 Hz), 5.13 (d, 1H, J = 10.9 Hz), 4.83 (d, 1H, J = 11.0 Hz), 2.83 (d, 1H, J = 8.3 Hz), 2.73 (s, 3H), 2.17 (d, 1H, J = 15.3 Hz), 2.04–1.68 (m, 5H), 1.52 (q, 1H, J = 14.6 Hz), 1.31–1.09 (m, 2H); ESI-MS m/z 476.25 (M + 1)+.
(3R,4′S,5′R)-6-Chloro-4′-(3-chloro-2-fluorophenyl)-N-methyl-2-oxodispiro[indoline-3,3′-pyrrolidine-2′,4″-piperidine]-5′-carboxamide (8)
Starting with tert-butyl 4-oxopiperidine-1-carboxylate in place of cyclohexanone, compound 8-Boc (compound 8 with N-Boc piperidine) was prepared according to the synthetic method described for the preparation of 6. Compound 8-Boc was dissolved in trifluoroacetic acid and stirred. After 30 min the trifluoroacetic acid was removed and the crude was purified by HPLC to give 8 (TFA salt). 1H NMR (300 MHz, CD3OD) δ ppm 8.24 (br s, 1H), 7.64 (t, 1H, J = 7.2 Hz), 7.49 (dd, 1H, J = 2.2, 8.1 Hz), 7.27 (t, 1H, J = 7.3 Hz), 7.13–7.03 (m, 2H), 6.68 (s, 1H), 4.79 (d, 1H, J = 9.6 Hz), 4.64 (d, 1H, J = 9.6 Hz), 3.70 (t, 1H, J = 13.1 Hz), 3.44–3.18 (m, 3H), 2.77 (d, 3H, J = 4.3 Hz), 2.39 (d, 1H, J = 14.5 Hz), 2.10–1.88 (m, 2H), 1.50–1.26 (m, 1H). ESI-MS m/z 477.17 (M + 1)+.
(3R,4′S,5′R)-6-Chloro-4′-(3-chloro-2-fluorophenyl)-N,1″-dimethyl-2-oxodispiro[indoline-3,3′-pyrrolidine-2′,4″-piperidine]-5′-carboxamide (9)
Starting with 1-methylpiperidin-4-one in place of cyclohexanone, compound 9 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (300 MHz, CD3OD) δ ppm 8.25 (br s, 1H), 7.62 (t, 1H, J = 7.3 Hz), 7.47 (dd, 1H, J = 2.1, 8.2 Hz), 7.25 (t, 1H, J = 7.5 Hz), 7.12–7.01 (m, 2H), 6.77 (d, 1H, J = 1.6 Hz), 4.76 (d, 1H, J = 9.5 Hz), 4.62 (d, 1H, J = 9.5 Hz), 3.75 (t, 1H, J = 12.5 Hz), 3.52–3.40 (m, 2H), 3.24–3.12 (m, 1H), 2.86 (s, 3H), 2.76 (d, 3H, J = 4.0 Hz), 2.40 (d, 1H, J = 14.4 Hz), 2.12–1.86 (m, 2H), 1.54–1.34 (m, 1H); ESI-MS m/z 491.08 (M + 1)+.
(3R,4′S,5′R)-1″-Acetyl-6-chloro-4′-(3-chloro-2-fluorophenyl)-N-methyl-2-oxodispiro[indoline-3,3′-pyrrolidine-2′,4″-piperidine]-5′-carboxamide (10)
Starting with 1-acetylpiperidin-4-one in place of cyclohexanone, compound 10 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (300 MHz, CD3OD) δ ppm 7.63 (t, 1H, J = 7.8 Hz), 7.47 (dd, 1H, J = 2.6, 8.2 Hz), 7.31 (t, 1H, J = 8.3 Hz), 7.15–7.04 (m, 2H), 6.75 (d, 1H, J = 1.7 Hz), 4.77 (d, 1H, J = 10.1 Hz), 4.61 (d, 0.5H, J = 16.4 Hz, rotamer), 4.43 (d, 0.5H, J = 11.9 Hz, rotamer), 4.00 (d, 0.5H, J = 13.1 Hz, rotamer), 3.85 (d, 0.5H, J = 12.6 Hz, rotamer), 3.81–3.68 (m, 1H), 2.76 (s, 3H), 2.56–2.40 (m, 1H), 2.16–1.76 (m, 5H), 1.45–1.11 (m, 2H); ESI-MS m/z 519.17 (M + 1)+.
(3R,4′S,5′R)-6-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-2-oxo-2″,3″,5″,6″-tetrahydrodispiro[indoline-3,3′-pyrrolidine-2′,4″-pyran]-5′-carboxamide (11)
Starting with tetrahydro-4H-pyran-4-one in place of cyclohexanone, compound 11 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (300 MHz, CD3OD) δ ppm 8.19 (d, J = 7.9 Hz 1H), 7.63 (ddd, 1H, J = 1.5, 6.5, 7.9 Hz), 7.51 (dd, 1H, J = 2.3, 8.2 Hz), 7.37 (t, 1H, J = 8.3 Hz), 7.19–7.07 (m, 2H), 6.80 (d, 1H, J = 1.9 Hz), 5.02 (d, 1H, J = 10.8 Hz), 4.74 (d, 1H, J = 10.8 Hz), 4.11–3.93 (m, 2H), 3.87 (dd, 1H, J = 3.9, 12.4 Hz), 3.69–3.55 (m, 2H), 3.50–3.38 (m, 1H), 2.62 (d, 1H, J = 13.2 Hz), 2.26–2.12 (m, 1H), 2.04–1.73 (m, 4H), 1.70–1.17 (m, 5H), 1.08 (ddd, 1H, J = 3.5, 12.7, 24.0 Hz); ESI-MS m/z 562.67 (M + 1)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-4,4-difluoro-N-((1r,4R)-4-hydroxycyclohexyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (12)
Starting with 4,4-difluorocyclohexan-1-one in place of cyclohexanone, compound 12 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (300 MHz, CD3OD) δ ppm 8.12 (d, 1H, J = 8.1 Hz), 7.62 (t, 1H, J = 7.2 Hz), 7.49 (dd, 1H, J = 2.3, 8.2 Hz), 7.33 (t, 1H, J = 8.3 Hz), 7.16–7.05 (m, 2H), 6.78 (d, 1H, J = 1.9 Hz), 4.77 (d, 1H, J = 10.3 Hz), 3.70–3.41 (m, 2H), 2.74–1.64 (m, 11H), 1.48–1.21 (m, 4H), 1.18–1.02 (m, 1H); ESI-MS m/z 596.75 (M + 1)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-3,3-dimethyl-2″-oxodispiro[cyclobutane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (14)
Starting with 3,3-dimethylcyclobutan-1-one in place of cyclohexanone, compound 14 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (400 MHz, CD3OD) δ ppm 7.61 (dd, 1H, J = 1.9, 8.2 Hz), 7.52 (ddd, 1H, J = 1.5, 6.4, 7.9 Hz), 7.39 (ddd, 1H, J = 1.5, 7.3, 8.6 Hz), 7.18–7–11 (m, 2H), 6.89 (d, 1H, J = 1.9 Hz), 4.92 (d, 1H, J = 10.9 Hz), 4.46 (d, 1H, J = 10.9 Hz), 3.68–3.58 (m, 1H), 3.50–3.39 (m, 1H), 2.78 (dd, 2H, J = 14.5, 39.1 Hz), 2.37 (d, 1H, J = 14.2 Hz), 1.95–1.76 (m, 3H), 1.69–1.59 (m, 1H), 1.38–1.17 (m, 7H), 0.98 (ddd, 1H, J = 3.6, 12.9, J = 24.3 Hz), 0.54 (s, 3H); ESI-MS m/z 560.25 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-4,4-dimethyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (15)
Starting with 4,4-dimethylcyclohexan-1-one in place of cyclohexanone, compound 15 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (400 MHz, CD3OD) δ ppm 7.62 (t, 1H, J = 7.9 Hz), 7.48 (dd, 1H, J = 1.4, 7.8 Hz), 7.35–7.25 (m, 1H), 7.15–7.04 (m, 2H), 6.78 (d, 1H, J = 1.7 Hz), 4.73 (d, 1H, J = 9.9 Hz), 3.67–3.57 (m, 1H), 3.52–3.43 (m, 1H), 2.08–1.64 (m, 8H), 1.58–1.42 (m, 2H), 1.41–1.20 (m, 6H), 0.98 (s, 3H), 0.73 (s, 3H); ESI-MS m/z 588.25 (M + H)+.
(3′R,4′R,5′R)-6″-Chloro-4′-(5-chloropyridin-3-yl)-N-((1r,4R)-4-hydroxycyclohexyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (16)
Starting with (E)-6-chloro-3-((5-chloropyridin-3-yl)methylene)indolin-2-one (16b) in place of (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)indolin-2-one (61), compound 16 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (300 MHz, CD3OD) δ ppm 8.45 (d, 1H, J = 2.1 Hz), 8.24 (d, 1H, J = 1.7 Hz), 7.89 (s, 1H), 7.60 (d, 1H, J = 8.2 Hz), 7.14 (dd, 1H, J = 1.8, 8.2 Hz), 6.78 (d, 1H, J = 1.8 Hz), 5.10 (d, 1H, J = 10.9 Hz), 4.47 (d, 1H, J = 10.9 Hz), 3.73–3.57 (m, 1H), 3.50–3.36 (m, 1H), 2.83 (d, 1H, J = 12.5 Hz), 2.17 (d, 1H, J = 14.3 Hz), 2.03–1.70 (m, 8H), 1.70–1.13 (m, 7H), 1.08–0.88 (m, 1H); ESI-MS m/z 543.75 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-2″-oxo-1″,2″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-pyrrolo[3,2-c]pyridine]-5′-carboxamide (17)
Starting with (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)-1,3-dihydro-2H-pyrrolo[3,2-c]pyridin-2-one (17b) in place of (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)indolin-2-one (61), compound 17 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (400 MHz, CD3OD) δ ppm 8.32 (s, 1H), 7.58 (t, 1H, J = 6.7 Hz), 7.36–7.27 (m, 1H), 7.11 (t, 1H, J = 8.6 Hz), 6.81 (s, 1H) 3.67–3.57 (m, 1H), 3.55–3.45 (m, 1H), 2.01–1.51 (m, 10H), 1.42–1.00 (m, 8H); ESI-MS m/z 561.25 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-2″-oxo-1″,2″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-pyrrolo[2,3-b]pyridine]-5′-carboxamide (18)
Starting with (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)-1,3-dihydro-2H-pyrrolo[2,3-b]pyridin-2-one (18b) in place of (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)indolin-2-one (61), compound 18 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (300 MHz, CD3OD) δ ppm 7.89 (dd, 1H, J = 2.14, 7.84 Hz), 7.61 (t, 1H, J = 7.43 Hz), 7.40 (t, 1H, J = 7.14 Hz), 7.17 (t, 1H, J = 8.40 Hz), 7.12 (d, 1H, J = 7.96 Hz), 5.07–4.94 (m, 1H), 4.78 (d, 1H, J = 10.91 Hz), 3.71–3.57 (m, 1H), 3.51–3.39 (m, 1H), 2.89–2.66 (m, 1H), 2.17–2.02 (m, 1H), 1.99–1.43 (m, 10H), 1.43–1.11 (m, 5H), 1.09–0.93 (m, 1H); ESI-MS m/z 561.58 (M + H)+.
(3′R,4′S,5′R)-2″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-6″-oxo-6″,7″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,5″-pyrrolo[2,3-d]pyrimidine]-5′-carboxamide (19)
Starting with (E)-2-chloro-5-(3-chloro-2-fluorobenzylidene)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (19b) in place of (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)indolin-2-one (61), compound 19 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (400 MHz, CD3OD) δ ppm 8.55 (d, 1H, J = 2.2 Hz), 7.55 (t, 1H, J = 7.8 Hz), 7.39 (t, 1H, J = 7.0 Hz), 7.16 (t, 1H, J = 7.9 Hz), 3.67–3.56 (m, 1H), 3.53–3.43 (m, 1H), 1.99–1.48 (m, 10H), 1.42–1.04 (m, 8H); ESI-MS m/z 562.33 (M + H)+.
(3′R,4′R,5′R)-2″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-5″-oxo-4″,5″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,6″-thieno[3,2-b]pyrrole]-5′-carboxamide (20)
Starting with (Z)-2-chloro-6-(3-chloro-2-fluorobenzylidene)-4,6-dihydro-5H-thieno[3,2-b]pyrrol-5-one (20b) in place of (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)indolin-2-one (61), compound 20 was prepared according to the synthetic method described for the preparation of 6. 1H NMR (400 MHz, CD3OD) δ ppm 7.40 (td, 2H, J = 6.8, 15.1 Hz), 7.15 (t, 1H, J = 8.0 Hz), 6.71 (s, 1H), 4.58 (d, 1H, J = 10.3 Hz), 4.45 (d, 1H, J = 10.4 Hz), 3.68–3.57 (m, 1H), 3.53–3.43 (m, 1H), 2.16 (d, 1H, J = 11.4 Hz), 2.00–1.79 (m, 4H), 1.77–1.46 (m, 7H), 1.40–1.07 (m, 6H); ESI-MS m/z 566.25 (M + H)+.
2-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indolin]-5′-yl)thiazole-4-carboxylic Acid (21)
Ethyl 3-bromo-2-oxopropanoate (48 mg, 0.244 mmol) and triethylamine (0.033 mL, 0.244 mmol), were added to a solution of 68 (30 mg, 0.061 mmol) in THF (3 mL) and refluxed overnight. Then the reaction was cooled, and the solvent was removed. The crude product was redissolved in DME (3 mL), and to this solution, at −20 °C, was added trifluoracetic anhydride (0.025 mL, 0.183 mmol) and pyridine (0.025 mL, 0.305 mmol), and then the reaction was allowed to warm to rt. After 30 min, saturated sodium bicarbonate solution was added and the reaction was extracted with EtOAc. The EtOAc solution was dried over sodium sulfate, filtered, and purified by column chromatography to produce 18 mg of compound 69. Lithium hydroxide monohydrate (50 mg, 1.19 mmol) was added to a solution of 69 (18 mg, 0.031 mmol) in THF/MeOH/H2O (1:1:1, 3 mL). After 2 h the reaction was quenched with ammonium chloride, and brine was added and extracted with EtOAc. The EtOAc solution was dried over sodium sulfate, filtered, concentrated and the crude was purified by preparative HPLC to produce compound 21 (TFA salt). 1H NMR (300 MHz, CD3OD) δ ppm 8.34 (s, 1H), 7.70 (t, 1H, J = 6.8 Hz), 7.53 (d, 1H, J = 7.1 Hz), 7.27 (t, 1H, J = 7.5 Hz), 7.13–7.03 (m, 2H), 6.73 (d, 1H, J = 1.8 Hz), 5.78–5.59 (m, 1H), 4.81–4.68 (m, 1H), 3.10 (s, 3H), 2.51–2.38 (m, 1H), 2.25 (d, 1H, J = 15.1 Hz), 2.13–1.94 (m, 1H), 1.87–1.10 (m, 7H); ESI-MS m/z 560.33 (M + H)+.
General Procedure A: Amide Coupling to Compound 66
CDI (3 equiv), DIEA (5 equiv), DMAP (1 equiv) were added to a suspension of compound 66 (1 equiv) in 1,2-dichloroethane and heated at 50 °C. After 30 min, the respective amine (5 equiv) was added and the reaction was refluxed overnight. Then the solvent was removed and the crude product was purified to produce the carboxamide.
2-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indolin]-5′-yl)oxazole-4-carboxylic Acid (22)
Compound 72 was produced according to general procedure A. At −20 °C, Deoxo-Fluor (50 μL) was added to a solution of 72 (51 mg) in THF (3 mL). After 30 min bromotrichloromethane (500 μL) and DBU (400 μL) were added and the reaction was allowed to warm to room temperature. After 5 h the reaction was quenched with saturated sodium bicarbonate (stirred for 5 min) and then extracted with EtOAc. The EtOAc solution was dried over sodium sulfate, filtered, and purified by column chromatography to produce 34 mg of compound 73. Lithium hydroxide monohydrate (60 mg, 1.43 mmol) was added to a solution of 73 (34 mg, 0.061 mmol) in THF/H2O (1:1, 4 mL). After 2 h the reaction was quenched with TFA, concentrated and the crude was redissolved in 3:1 MeOH/H2O and purified by HPLC to produce 22 (TFA salt) as a white powder. 1H NMR (400 MHz, CD3OD) δ ppm 8.50 (s, 1H), 7.62–7.53 (m, 2H), 7.24 (t, 1H, J = 7.3 Hz), 7.07 (dd, 1H, J = 1.7, 8.2 Hz), 7.03 (t, 1H, J = 8.1 Hz), 6.73 (d, 1H, J = 1.8 Hz), 5.34 (d, 1H, J = 10.3 Hz), 5.02 (d, 1H, J = 11.6 Hz), 2.97 (s, 3H), 2.33 (d, 1H, J = 13.1 Hz), 2.15 (d, 1H, J = 14.1 Hz), 1.95–1.85 (m, 1H), 1.74–1.44 (m, 5H), 1.38–1.26 (m, 1H), 1.19–1.05 (m, 1H); ESI-MS m/z 544.42 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-5′-(3-methyl-1,2,4-oxadiazol-5-yl)dispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indolin]-2″-one (23)
Starting with 50 mg of compound 66, compound 75 was obtained using general procedure A. Crude 75 was dissolved in pyridine (5 mL) and heated to 100 °C. After 5 h the solution was cooled and the solvent was removed by rotoevaporation and then the crude product was purified by HPLC to produce 23 (TFA salt) as a white powder. 1H NMR (400 MHz, CD3OD) δ ppm 7.60 (ddd, 1H, J = 1.5, 6.4, 7.9 Hz), 7.50 (dd, 1H, J = 3.0, 8.2 Hz), 7.25 (ddd, 1H, J = 1.6, 7.2, J = 8.6 Hz), 7.07–7.01 (m, 2H), 6.72 (d, 1H, J = 1.9 Hz), 5.30 (d, 1H, J = 11.0 Hz), 4.98 (d, 1H, J = 11.1 Hz), 2.99 (s, 3H), 2.32 (s, 3H), 2.24 (d, 1H, J = 13.2 Hz), 2.19–2.10 (m, 1H), 1.86–1.47 (m, 6H), 1.21 (ddd, 1H, J = 5.0, 12.6 Hz, 13.9 Hz), 1.15–1.02 (m, 1H); ESI-MS m/z 514.92 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-5′-(5-methyl-1,3,4-oxadiazol-2-yl)dispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indolin]-2″-one (24)
Starting with 77 mg of compound 66, 63 mg of 77 was obtained using general procedure A. A solution of 77 (30 mg, 0.056 mmol) in DCM (3 mL) was slowly added to a solution of PPh3 (30 mg, 0.112 mmol), Et3N (31 μL), and iodine (28 mg, 0.112 mmol) in DCM (1 mL). After 3 h, H2O and brine were added and extracted with EtOAc. The EtOAc solution was dried over sodium sulfate, filtered, concentrated and the crude product was purified by HPLC to produce compound 24 (TFA salt) as a white powder. 1H NMR (400 MHz, CD3OD) δ ppm 7.60 (t, 1H, J = 7.9 Hz), 7.51 (dd, 1H, J = 2.9, 8.2 Hz), 7.25 (t, 1H, J = 8.3 Hz), 7.08–7.00 (m, 2H), 6.72 (d, 1H, J = 1.9 Hz), 5.29 (d, 1H, J = 11.1 Hz), 4.95 (d, 1H, J = 10.8 Hz), 2.95 (s, 3H), 2.52 (s, 3H), 2.24 (d, 1H, J = 13.8 Hz), 2.15 (dd, 1H, J = 2.3, 14.1 Hz), 1.86–1.02 (m, 8H); ESI-MS m/z 515.25 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-5′-(5-methyl-1,3,4-thiadiazol-2-yl)dispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indolin]-2″-one (25)
Lawesson’s reagent (45 mg, 0.112 mmol) was added to a solution of 77 (30 mg, 0.056 mmol) in DCM and refluxed. After standing overnight the solvent was removed and the crude was purified by HPLC to produce compound 25 (TFA salt) as a white powder. 1H NMR (400 MHz, CD3OD) δ ppm 7.69 (t, 1H, J = 7.9 Hz), 7.46 (dd, 1H, J = 2.8, 8.2 Hz), 7.25 (t, 1H, J = 8.4 Hz), 7.08 (dt, 1H, J = 1.1, 8.1 Hz), 7.03 (dd, 1H, J = 2.0, 8.2 Hz), 6.72 (d, 1H, J = 1.9 Hz), 5.50 (d, 1H, J = 10.2 Hz), 4.63 (d, 1H, J = 11.0 Hz), 3.00 (s, 3H), 2.71 (s, 3H), 2.27 (d, 1H, J = 13.0 Hz), 2.23–2.15 (m, 1H), 1.88 (t, 1H, J = 11.6 Hz), 1.83–1.60 (m, 4H), 1.60–1.49 (m, 1H), 1.31–1.07 (m, 2H); ESI-MS m/z 531.17 (M + H)+.
2-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indolin]-5′-yl)-5-methyloxazole-4-carboxylic Acid (26)
Starting with 44 mg of compound 66, 25 mg of 79 was obtained using general procedure A. Cyclization to generate 80 (13 mg) was accomplished according to the same procedure used to obtain 24. Lithium hydroxide monohydrate (30 mg, 0.715 mmol) was added to a solution of 80 (13 mg, 0.023 mmol) in THF/H2O (1:1, 2 mL). After 2 h the reaction was quenched with TFA, concentrated and the crude was redissolved in 3:1 MeOH/H2O and purified by HPLC to produce 26 (TFA salt) as a white powder. 1H NMR (400 MHz, CD3OD) δ ppm 7.60 (t, 1H, J = 7.2 Hz), 7.52 (dd, 1H, J = 2.7, 8.1 Hz), 7.21 (t, 1H, J = 8.3 Hz), 7.07–6.98 (m, 2H), 6.70 (d, 1H, J = 1.9 Hz), 5.07 (d, 1H, J = 10.8 Hz), 4.91 (d, 1H, J = 11.7 Hz), 2.83 (s, 3H), 2.59 (s, 3H), 2.31–2.23 (m, 1H), 2.12–2.04 (m, 1H), 1.85–1.02 (m, 8H); ESI-MS m/z 558.75 (M + H)+.
2-(2-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indolin]-5′-yl)thiazol-4-yl)acetic Acid (27)
Compound 27 was produced using the same synthetic strategy used for 21. 1H NMR (400 MHz, CD3OD) δ ppm 7.65 (t, 1H, J = 6.7 Hz), 7.57 (d, 1H, J = 7.1 Hz), 7.42 (s, 1H), 7.27 (t, 1H, J = 8.2 Hz), 7.13–7.04 (m, 2H), 6.75 (d, 1H, J = 1.9 Hz), 5.92–5.70 (m, 1H), 3.78 (s, 2H), 3.20 (s, 3H), 2.58–2.44 (m, 1H), 2.26 (d, 1H, J = 13.8 Hz), 2.22–2.08 (m, 1H), 1.83–1.43 (m, 5H), 1.38–1.27 (m, 1H), 1.25–1.13 (m, 1H); ESI-MS m/z 574.08 (M + H)+.
2-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indolin]-5′-yl)thiazole-4-carboxamide (28)
Starting with acid 21 in place of 66, compound 28 was produced using general procedure A. Crude 28 was purified by HPLC to produce 28 as its TFA salt. 1H NMR (400 MHz, CD3OD) δ ppm 8.11 (s, 1H), 7.75 (t, 1H, J = 7.2 Hz), 7.42 (d, 1H, J = 8.1 Hz), 7.26 (t, 1H, J = 8.46 Hz), 7.09 (t, 1H, J = 7.83), 7.01 (dd, 1H, J = 1.7, 8.1 Hz), 6.71 (d, 1H, J = 1.8 Hz), 3.00 (s, 3H), 2.36–2.16 (m, 2H), 2.09–2.00 (m, 1H), 1.91–1.52 (m, 5H), 1.38–1.24 (m, 1H), 1.21–1.06 (m, 1H); ESI-MS m/z 559.42 (M + H)+.
General Procedure B: Amide Coupling of Alkylamines
HATU (2 equiv) and DIEA (4 equiv) were added to a suspension of acid compound 29 (1 equiv) in DCM. After 10 min, alkylamine (4 equiv) was added. After 12 h, the reaction was concentrated and purified by column chromatography to produce alkyl amide compounds.
General Procedure C: Hydrolysis of Alkyl Esters
LiOH·H2O (3 equiv) was added to a solution of ester (1 equiv) in THF/MeOH/H2O (1:1:1). After 2 h, TFA was added and the solution was concentrated, redissolved in MeOH/H2O (3:1), and purified by reverse phase preparative HPLC. The pure fractions were combined and lyophilized to give the carboxylic acid products (TFA salt) as a white powder.
General Procedure D: Amide Coupling of Arylamines
Ph2POCl (3 equiv) and DIEA (5 equiv) were added to a suspension of compound 29 (1 equiv) in DCM. After 30 min, arylamine (4 equiv) and DMAP (1 equiv) were added. After 12 h, the reaction was concentrated and purified by column chromatography to produce arylamide compounds.
General Procedure E: Hydrolysis of Aryl Esters
NaOH (3 equiv) was added to a solution of ester (1 equiv) in THF/MeOH/H2O (1:1:1). After 2 h, TFA was added and the reaction was concentrated, redissolved in MeOH/H2O (3:1), and purified by reverse phase preparative HPLC. The pure fractions were combined and lyophilized to give the carboxylic acid products as the TFA salt as a white powder.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-(methylsulfonyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (30)
Starting with compound 29 in place of 66, compound 30 was obtained according to general procedure A. 1H NMR (300 MHz, CD3OD) δ ppm 7.61 (t, 1H, J = 7.5 Hz), 7.53 (dd, 1H, J = 2.1, 8.2 Hz), 7.33 (t, 1H, J = 8.0 Hz), 7.17–7.06 (m, 2H), 6.76 (d, 1H, J = 1.7 Hz), 4.98 (d, 1H, J = 10.4 Hz), 3.09 (s, 3H), 2.60 (d, 1H, J = 13.7 Hz), 2.09 (d, 1H, J = 16.1 Hz), 2.01–1.43 (m, 6H), 1.34–1.06 (m, 2H); ESI-MS m/z 540.08 (M + 1)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)butanoic Acid (31)
31 was obtained according to general procedure B followed by general procedure C. 1H NMR (300 MHz, CD3OD) δ ppm 7.64 (t, 1H, J = 7.1 Hz), 7.51 (dd, 1H, J = 2.2, 8.3 Hz), 7.39 (t, 1H, J = 7.5 Hz), 7.17 (t, 1H, J = 8.1 Hz), 7.11 (dd, 1H, J = 1.6, 8.2 Hz), 6.78 (d, 1H, J = 1.6 Hz), 5.12 (d, 1H, J = 11.0 Hz), 4.80 (d, 1H, J = 11.1 Hz), 3.21–3.04 (m, 1H), 2.81 (d, 1H, J = 7.4 Hz), 2.17 (d, 1H, J = 12.0 Hz), 2.06 (t, 2H, J = 7.4 Hz), 2.01–1.84 (m, 3H), 1.84–1.40 (m, 5H), 1.32–1.12 (m, 2H); ESI-MS m/z 548.42 (M + 1)+.
(1R,3r)-3-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)cyclobutane-1-carboxylic Acid (32)
32 was obtained by general procedure B followed by general procedure C. 1H NMR (300 MHz, CD3OD) δ ppm 7.65 (t, 1H, J = 7.0 Hz), 7.49 (dd, 1H, J = 1.7, 8.0 Hz), 7.40 (t, 1H, J = 7.3 Hz), 7.18 (t, 1H, J = 8.0 Hz), 7.11 (d, 1H, J = 8.3 Hz), 6.79 (s, 1H), 5.06 (d, J = 10.8, 1H), 4.79 (d, 1H, J = 10.8 Hz), 4.47 (p, 1H, J = 8.1 Hz), 2.96–2.71 (m, 2H), 2.63–2.38 (m, 2H), 2.29–2.08 (m, 2H), 2.05–1.83 (m, 4H), 1.76 (d, 2H, J = 16.2 Hz), 1.62–1.39 (m, 1H), 1.33–1.11 (m, 2H) ESI-MS m/z 560.50 (M + 1)+.
(1R,4r)-4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)cyclohexane-1-carboxylic Acid (33)
33 was obtained by general procedure B followed by general procedure C. 1H NMR (300 MHz, CD3OD) δ ppm 7.64 (t, 1H, J = 7.1 Hz), 7.49 (d, 1H, J = 8.1 Hz), 7.39 (t, 1H, J = 7.5 Hz), 7.17 (t, 1H, J = 8.1 Hz), 7.11 (d, 1H, J = 8.1 Hz), 6.79 (s, 1H), 5.08 (d, 1H, J = 11.1 Hz), 4.79 (d, 1H, J = 11.1 Hz), 3.72–3.54 (m, 1H), 2.84 (d, 1H, J = 8.4 Hz), 2.25–2.07 (m, 2H), 2.04–1.84 (m, 6H), 1.77 (d, 2H, J = 12.0 Hz), 1.63 (d, 1H, J = 13.0 Hz), 1.56–1.34 (m, 3H), 1.32–1.11 (m, 3H), 0.99–0.82 (m, 1H); 13C NMR (75 MHz, CD3OD) δ ppm 179.01, 178.21, 167.66, 157.76 (d, JC–F = 248.87 Hz), 145.27, 137.07, 132.35, 129.50, 128.91, 126.48 (d, JC–F = 4.74 Hz), 123.39, 122.45, 122.31, 122.05, 121.87, 111.77, 73.24, 67.76, 62.25, 46.72, 46.68, 43.15, 32.33, 32.09, 32.07, 31.37, 28.79, 28.72, 25.35, 23.11, 21.70; ESI-MS m/z 588.33 (M + 1)+.
(1S,4s)-4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)cyclohexane-1-carboxylic Acid (34)
34 was obtained according to general procedure B followed by general procedure C. 1H NMR (300 MHz, CD3OD) δ ppm 7.67 (t, 1H, J = 6.61 Hz), 7.49 (dd, 1H, J = 2.0, 8.1 Hz), 7.37 (t, 1H, J = 7.5 Hz), 7.16 (t, 1H, J = 7.9 Hz), 7.10 (dd, 1H, J = 1.7, 8.3 Hz), 6.79 (d, 1H, J = 1.7 Hz), 5.20 (d, 1H, J = 11.3 Hz), 4.76 (d, 1H, J = 11.2 Hz), 3.89–3.75 (m, 1H), 2.84 (d, 1H, J = 9.1 Hz), 2.41–2.29 (m, 1H), 2.20 (d, 1H, J = 15.3 Hz), 2.03–1.12 (m, 16H); ESI-MS m/z 588.50 (M + H)+.
(1R,4r)-4-(((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)methyl)cyclohexane-1-carboxylic Acid (35)
35 was obtained according to general procedure B followed by general procedure C. 1H NMR (300 MHz, CD3OD) δ ppm 8.36–8.26 (m, 1H), 7.67 (t, 1H, J = 6.8 Hz), 7.52 (d, 1H, J = 8.2 Hz), 7.42 (t, 1H, J = 7.5 Hz), 7.20 (t, 1H, d J = 8.0 Hz), 7.12 (dd, 1H, J = 1.4, 8.2 Hz), 6.78 (d, 1H, J = 1.5 Hz), 5.14 (d, 1H, J = 11.1 Hz), 4.78 (d, 1H, J = 11.3 Hz), 3.48–3.34 (m, 1H), 2.90–2.64 (m, 2H), 2.19 (d, 1H, J = 11.3 Hz), 2.09–1.70 (m, 8H), 1.61–1.11 (m, 8H), 0.79–0.59 (m, 2H); ESI-MS m/z 602.58 (M + 1)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-(1,1-dioxidotetrahydro-2H-thiopyran-4-yl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (36)
36 was obtained according to general procedure B. 1H NMR (300 MHz, CD3OD) δ ppm 7.64 (t, 1H, J = 7.1 Hz), 7.49 (dd, 1H, J = 2.1, 8.1 Hz), 7.41 (t, 1H, J = 7.5 Hz), 7.18 (t, 1H, J = 9.9 Hz), 7.11 (dd, 1H, J = 1.6, 8.2 Hz), 6.79 (d, 1H, J = 1.6 Hz), 5.10 (d, 1H, J = 11.1 Hz), 4.80 (d, 1H, J = 11.2 Hz), 4.10–3.94 (m, 1H), 3.27–2.91 (m, 3H), 2.88–2.74 (m, 2H), 2.35–1.64 (m, 10H), 1.52 (q, 1H, J = 13.9 Hz), 1.31–1.11 (m, 2H); ESI-MS m/z 594.50 (M + 1)+.
2-(4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)piperidin-1-yl)acetic Acid (37)
37 was obtained according to general procedure B followed by general procedure C. 1H NMR (300 MHz, CD3OD) δ ppm 7.66 (t, 1H, J = 7.1 Hz), 7.48 (dd, 1H, J = 2.1, 8.2 Hz), 7.39 (t, 1H, J = 7.3 Hz), 7.17 (t, 1H, J = 8.0 Hz), 7.10 (dd, 1H, J = 1.6, 8.1 Hz), 6.79 (d, 1H, J = 1.7 Hz), 5.12 (d, 1H, J = 11.0 Hz), 4.80 (d, 1H, J = 10.9 Hz), 4.05–3.89 (m, 2H), 3.79–3.39 (m, 2H), 3.29–3.04 (m, 2H), 2.79 (d, 1H, J = 9.4 Hz), 2.17 (d, 2H, J = 9.4 Hz), 2.03–1.42 (m, 8H), 1.42–1.33 (m, 2H), 1.31–1.10 (m, 2H); ESI-MS m/z 603.67 (M + 1)+.
(3′R,4′S,5′R)-N-(1-Acetylazetidin-3-yl)-6″-chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (38)
38 was obtained according to general procedure B. 1H NMR (300 MHz, CD3OD) δ ppm 7.64 (t, 1H, J = 7.2 Hz), 7.50 (dd, 1H, J = 1.8, 8.2 Hz), 7.40 (t, 1H, J = 7.7 Hz), 7.17 (t, 1H, J = 8.1 Hz), 7.11 (dd, 1H, J = 1.7, 8.2 Hz), 6.79 (s, 1H), 5.10 (d, 1H, J = 10.6 Hz), 4.64–4.36 (m, 2H), 4.29–4.11 (m, 1H), 4.09–3.56 (m, 2H), 2.79 (d, 1H, J = 9.8 Hz), 2.16 (d, 1H, J = 14.9 Hz), 2.01–1.68 (m, 8H), 1.64–1.37 (m, 1H), 1.34–1.11 (m, 2H); ESI-MS m/z 560.08 (M + 1)+.
3-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)benzoic Acid (40)
40 was obtained according to general procedure D followed by general procedure E. 1H NMR (300 MHz, CD3OD) δ ppm 8.14 (s, 1H), 7.84–7.67 (m, 3H), 7.55 (dd, 1H, J = 1.9, 8.2 Hz), 7.48–7.34 (m, 2H), 7.19 (t, 1H, J = 8.0 Hz), 7.12 (dd, 1H, J = 1.6, 8.2 Hz), 6.79 (d, 1H, J = 1.6 Hz), 5.29 (d, 1H, J = 11.0 Hz), 4.97 (d, 1H, J = 10.7 Hz), 2.96–2.84 (m, 1H), 2.18 (d, J = 14.0 Hz 1H), 2.08–1.85 (m, 3H), 1.78 (d, 2H, J = 12.2 Hz), 1.63–1.42 (m, 1H), 1.35–1.13 (m, 2H); ESI-MS m/z 582.58 (M + H)+.
4-(((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)methyl)benzoic Acid (41)
41 was obtained according to general procedure B followed by general procedure E. 1H NMR (300 MHz, CD3OD) δ ppm 7.87 (d, 2H, J = 8.2 Hz), 7.66 (t, 1H, J = 7.0 Hz), 7.50 (dd, 1H, J = 2.3, 8.3 Hz), 7.40 (t, 1H, J = 7.6 Hz), 7.22–7.08 (m, 2H), 7.04 (d, 2H, J = 8.2 Hz), 6.78 (d, 1H, J = 1.6 Hz), 5.20 (d, 1H, J = 11.2 Hz), 4.80 (d, 1H, J = 11.1 Hz), 4.66 (d, 1H, J = 15.3 Hz), 4.20 (d, 1H, J = 15.3 Hz), 2.83 (d, 1H, J = 10.0 Hz), 2.20 (d, 1H, J = 15.8 Hz), 2.04–1.85 (m, 3H), 1.77 (d, 2H, J = 11.9 Hz), 1.52 (q, 1H, J = 13.7 Hz), 1.33–1.10 (m, 2H); ESI-MS m/z 596.33 (M + 1)+.
2-(4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)phenyl)acetic Acid (42)
42 was obtained according to general procedure D followed by general procedure E. 1H NMR (400 MHz, CD3OD) δ ppm 7.70 (t, 1H, J = 7.8 Hz), 7.52 (dd, 1H, J = 2.5, 8.2 Hz), 7.47 (d, 2H, J = 8.6 Hz), 7.35 (t, 1H, J = 8.1 Hz), 7.24 (d, 2H, J = 8.6 Hz), 7.16 (t, 1H, J = 8.0 Hz), 7.10 (dd, 1H, J = 1.9, 8.2 Hz), 6.78 (d, 1H, J = 1.9 Hz), 5.20 (d, 1H, J = 9.8 Hz), 4.93 (d, 1H, J = 10.8 Hz), 3.57 (s, 2H), 2.87–2.69 (m, 1H), 2.13 (d, 1H, J = 13.3 Hz), 1.99–1.70 (m, 5H), 1.64–1.48 (m, 1H), 1.27–1.12 (m, 2H); ESI-MS m/z 596.42 (M + H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)-2-methoxybenzoic Acid (43)
43 was obtained according to general procedure D followed by general procedure E. 1H NMR (300 MHz, CD3OD) δ ppm 7.83 (d, 1H, J = 8.51 Hz), 7.70 (t, 1H, J = 6.92 Hz), 7.62 (s, 1H), 7.52 (dd, 1H, J = 2.17, 8.08 Hz), 7.35 (t, 1H, J = 7.08 Hz), 7.20–7.02 (m, 3H), 6.78 (d, 1H, J = 1.71 Hz), 5.24–5.09 (m, 1H), 4.94 (d, 1H, J = 10.49 Hz), 3.89 (s, 3H), 2.80–2.58 (m, 1H), 2.09 (d, 1H, J = 14.12 Hz), 2.01–1.47 (m, 6H), 1.26–1.09 (m, 2H); ESI-MS m/z 612.42 (M + H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)-3-methoxybenzoic Acid (44)
44 was obtained according to general procedure D followed by general procedure E. 1H NMR (300 MHz, CD3OD) δ ppm 8.26 (d, 1H, J = 8.6 Hz), 7.79 (t, 1H, J = 7.1 Hz), 7.66 (dd, 1H, J = 1.4, 8.4 Hz), 7.60 (s, 1H), 7.51 (dd, 1H, J = 2.2, 8.3 Hz), 7.40 (t, 1H, J = 8.2 Hz), 7.21 (t, 1H, J = 8.6 Hz), 7.10 (dd, 1H, J = 1.8, 8.1 Hz), 6.79 (d, 1H, J = 1.7 Hz), 5.49–5.23 (m, 1H), 3.80 (s, 3H), 2.68–2.47 (m, 1H), 2.21–1.52 (m, 7H), 1.35–1.07 (m, 2H); ESI-MS m/z 612.17 (M + 1)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)-2-fluorobenzoic Acid (45)
45 was obtained according to general procedure D followed by general procedure E. 1H NMR (300 MHz, CD3OD) δ ppm 7.90 (t, 1H, J = 8.4 Hz), 7.74–7.61 (m, 2H), 7.55 (dd, 1H, J = 2.5, 8.2 Hz), 7.38 (t, 1H, J = 7.2 Hz), 7.26 (dd, 1H, J = 1.7, 8.6 Hz), 7.19 (t, 1H, J = 8.2 Hz), 7.12 (dd, 1H, J = 1.8, 8.2 Hz), 6.80 (d, 1H, J = 1.7 Hz), 5.31 (d, 1H, J = 10.8 Hz), 4.97 (d, 1H, J = 10.8 Hz), 2.94–2.84 (m, 1H), 2.20 (d, 1H, J = 15.7 Hz), 2.06–1.85 (m, 3H), 1.79 (d, 2H, J = 10.9 Hz), 1.65–1.43 (m, 1H), 1.34–1.11 (m, 2H); ESI-MS m/z 600.42 (M + H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)-3-fluorobenzoic Acid (46)
46 was obtained according to general procedure D followed by general procedure E. 1H NMR (300 MHz, CD3OD) δ ppm 8.21 (t, 1H, J = 8.0 Hz), 7.91–7.79 (m, 1H), 7.79–7.67 (m, 2H), 7.52 (dd, 1H, J = 2.4, 8.2 Hz), 7.37 (t, 1H, J = 7.3 Hz), 7.25–7.06 (m, 2H), 6.79 (d, 1H, J = 1.7 Hz), 5.41 (d, 1H, J = 9.4 Hz), 2.81–2.67 (m, 1H), 2.14 (d, 1H, J = 14.7 Hz), 2.00–1.84 (m, 3H), 1.84–1.70 (m, 2H), 1.68–1.47 (m, 1H), 1.41–1.09 (m, 2H); ESI-MS m/z 600.83 (M + H)+.
5-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)picolinic Acid (47)
47 was obtained according to general procedure D followed by general procedure E. 1H NMR (300 MHz, CD3OD) δ ppm 8.80 (s, 1H), 8.27 (d, 1H, J = 8.8 Hz), 8.15 (d, 1H, J = 8.7 Hz), 7.71 (t, 1H, J = 7.2 Hz), 7.55 (d, 1H, J = 8.7 Hz), 7.37 (t, 1H, J = 7.1 Hz), 7.18 (t, 1H, J = 7.9 Hz), 7.12 (d, 1H, J = 8.1 Hz), 6.80 (s, 1H), 5.30 (d, 1H, J = 11.1 Hz), 5.00 (d, 1H, J = 10.8 Hz), 2.90–2.75 (m, 1H), 2.16 (d, 1H, J = 16.9 Hz), 2.06–1.84 (m, 3H), 1.78 (d, 2H, J = 13.2 Hz), 1.66–1.42 (m, 1H), 1.32–1.11 (m, 2H); ESI-MS m/z 583.96 (M + 1)+.
5-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)furan-2-carboxylic Acid (48)
48 was obtained according to general procedure D followed by general procedure E. 1H NMR (300 MHz, CD3OD) δ ppm 7.66 (t, 1H, J = 7.23 Hz), 7.49 (dd, 1H, J = 2.46, 8.24 Hz), 7.31 (t, 1H, J = 7.44 Hz), 7.22 (d, 1H, J = 3.60 Hz), 7.16–7.04 (m, 2H), 6.76 (d, 1H, J = 1.68 Hz), 6.51 (d, 1H, J = 3.53 Hz), 5.06–4.90 (m, 2H), 2.53–2.37 (m, 1H), 2.06–1.50 (m, 7H), 1.25–1.00 (m, 2H); ESI-MS m/z 572.42 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-(4-((methylsulfonyl)carbamoyl)phenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (49)
Starting with compound 39 in place of 66, compound 49 was obtained according to general procedure A. 1H NMR (300 MHz, CD3OD) δ ppm 7.89 (d, 2H, J = 8.8 Hz), 7.78–7.66 (m, 3H), 7.52 (dd, 1H, J = 2.5, 8.4 Hz), 7.36 (t, 1H, J = 7.8 Hz), 7.17 (t, 1H, J = 7.6 Hz), 7.11 (dd, 1H, J = 1.7, 8.2 Hz), 6.79 (d, 1H, J = 1.7 Hz), 5.31–5.15 (m, 1H), 3.35 (s, 3H), 2.89–2.70 (m, 1H), 2.13 (d, 1H, J = 13.6 Hz), 2.03–1.70 (m, 5H), 1.68–1.46 (m, 1H), 1.35–1.12 (m, 2H); ESI-MS m/z 659.67 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxo-N-(4-sulfamoylphenyl)dispiro- [cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (50)
50 was obtained according to general procedure D. 1H NMR (300 MHz, CD3OD) δ ppm 7.60–7.50 (m, 3H), 7.39 (dd, 1H, J = 2.24, 8.22 Hz), 7.30 (t, 1H, J = 6.96 Hz), 7.12–7.02 (m, 2H), 6.73 (d, 1H, J = 1.59 Hz), 6.64 (d, 2H, J = 8.68 Hz), 4.80 (d, 1H, J = 10.19 Hz), 4.62 (d, 1H, J = 10.28 Hz), 2.24 (d, 1H, J = 13.09 Hz), 1.94 (d, 1H, J = 13.01 Hz), 1.88–1.41 (m, 6H), 1.20–0.93 (m, 2H); ESI-MS m/z 617.17 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-(4-(methylsulfonyl)phenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (51)
51 was obtained according to general procedure D. 1H NMR (300 MHz, CD3OD) δ ppm 7.91 (d, 2H, J = 8.80 Hz), 7.82 (d, 2H, J = 8.85 Hz), 7.70 (t, 1H, J = 6.71 Hz), 7.52 (dd, 1H, J = 2.42, 8.17 Hz), 7.35 (t, 1H, J = 7.58 Hz), 7.16 (t, 1H, J = 8.05 Hz), 7.10 (dd, 1H, J = 1.84, 8.21 Hz), 6.78 (d, 1H, J = 1.80 Hz), 5.20 (d, 1H, J = 14.03 Hz), 4.94 (d, 1H, J = 10.48 Hz), 3.09 (s, 3H), 2.80–2.63 (m, 1H), 2.11 (d, 1H, J = 13.18 Hz), 2.01–1.46 (m, 6H), 1.31–1.08 (m, 2H); ESI-MS m/z 616.58 (M + H)+.
(3′R,4′S,5′R)-N-(4-Carbamoylphenyl)-6″-chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (52)
Starting with compound 39 in place of 66, compound 52 was obtained according to general procedure A. 1H NMR (400 MHz, CD3OD) δ ppm 7.85 (d, 2H, J = 8.9,Hz), 7.70 (t, 1H, J = 7.9 Hz), 7.65 (d, 2H, J = 8.8 Hz), 7.51 (dd, 1H, J = 2.4, 8.2 Hz), 7.34 (t, 1H, J = 7.1 Hz), 7.15 (t, 1H, J = 8.0 Hz), 7.09 (dd, 1H, J = 1.9, 8.2 Hz), 6.78 (d, 1H, J = 1.9 Hz), 5.22–5.10 (m, 1H), 4.93 (d, 1H, J = 9.6 Hz), 2.81–2.58 (m, 1H), 2.09 (d, 1H, J = 12.4 Hz), 1.99–1.67 (m, 5H), 1.67–1.50 (m, 1H), 1.28–1.10 (m, 2H); ESI-MS m/z 581.33 (M + H)+.
(3′R,4′S,5′R)-N-(4-Carbamoyl-2-methoxyphenyl)-6″-chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (53)
Starting with compound 44 in place of 66, compound 53 was obtained according to general procedure A. 1H NMR (400 MHz, CD3OD) δ ppm 8.24 (d, 1H, J = 8.4 Hz), 7.75 (t, 1H, J = 7.0 Hz), 7.55–7.45 (m, 3H), 7.35 (t, 1H, J = 7.0 Hz), 7.15 (t, 1H, J = 7.9 Hz), 7.06 (dd, 1H, J = 1.9, 8.2 Hz), 6.76 (d, 1H, J = 1.9 Hz), 5.34–5.10 (m, 1H), 3.85 (s, 3H), 2.57–2.31 (m, 1H), 2.18–1.96 (m, 2H), 1.91–1.57 (m, 5H), 1.25–1.05 (m, 2H); ESI-MS m/z 611.25 (M + H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-4,4-dimethyl-2″-oxodispiro[cyclo- hexane-1,2'-pyrrolidine-3′,3″-indoline]-5'-carboxamido)benzoic Acid (54)
54 was obtained according to general procedure D followed by general procedure E. 1H NMR (400 MHz, CD3OD) δ ppm 7.98 (d, 2H, J = 8.7 Hz), 7.70 (t, 1H, J = 7.7 Hz), 7.64 (d, 2H, J = 8.7 Hz), 7.57 (dd, 1H, J = 2.4, 8.2 Hz), 7.38 (t, 1H, J = 8.2 Hz), 7.19 (t, 1H, J = 8.1 Hz), 7.13 (dd, 1H, J = 1.9, 8.2 Hz), 6.81 (d, 1H, J = 1.9 Hz), 5.29 (d, 1H, J = 10.5 Hz), 4.97 (d, 1H, J = 10.8 Hz), 2.75 (d, 1H, J = 13.6 Hz), 2.13 (dt, 1H, J = 4.2, 14.0 Hz), 2.04 (d, 1H, J = 13.4 Hz), 1.87 (dt, 1H, J = 3.4, 14.2 Hz), 1.64 (d, 1H, J = 14.7 Hz), 1.53–1.34 (m, 3H), 1.02 (s, 3H), 0.77 (s, 3H); 13C NMR (100 MHz, CD3OD) δ ppm 178.48, 169.09, 168.51, 157.71 (d, JC–F = 249.3 Hz), 145.30, 142.77, 136.94, 132.18, 131.88(2C), 129.50, 128.91, 128.25, 126.32 (d, JC–F = 4.6 Hz), 123.29, 122.77, 122.28 (d, JC–F = 19.1 Hz), 120.51(2C), 111.75, 72.59, 68.33, 62.99, 46.84, 35.75, 34.45, 32.21, 30.00, 28.07, 27.96, 23.63; ESI-MS m/z 610.25 (M + H)+.
3-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[1.1.1]pentane-1-carboxylic Acid (55)
55 was obtained using the same synthetic strategy described for 56. 1H NMR (300 MHz, CD3OD) δ ppm 7.61 (t, 1H, J = 6.55 Hz), 7.49 (dd, 1H, J = 2.34, 8.20 Hz), 7.39 (t, 1H, J = 6.90 Hz), 7.15 (t, 1H, J = 8.53 Hz), 7.10 (dd, 1H, J = 1.94, 8.22 Hz), 6.78 (d, 1H, J = 1.88 Hz), 4.98 (d, 1H, J = 10.87 Hz), 4.78 (d, 1H, J = 10.92 Hz), 2.84–2.71 (m, 1H), 2.26 (s, 6H), 2.14 (d, 1H, J = 13.90 Hz), 2.02–1.67 (m, 5H), 1.60–1.38 (m, 1H), 1.31–1.10 (m, 2H); ESI-MS m/z 572.25 (M + H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (56)
HATU (616 mg, 1.62 mmol) and DIEA (0.550 mL, 3.24 mmol) were added to a suspension of acid 29 (500 mg, 1.08 mmol) in DCM (15 mL) and stirred. After 10 min, methyl 4-aminobicyclo[2.2.2]octane-1-carboxylate (396 mg, 2.16 mmol) and DMAP (132 mg, 1.08 mmol) were added to the reaction. After overnight, the solvent was removed in vacuo and the crude was purified by column chromatography to give 549 mg of interemediate 56-ester. LiOH·H2O (110 mg, 2.62 mmol) and sodium hydroxide (105 mg, 2.62 mmol) were added to a solution of interemidiate 56-ester (549 mg, 0.873 mmol) dissolved in a mixture of THF (3 mL), H2O (3 mL), and MeOH (3 mL). After the hydrolysis was complete, as determined by TLC, the reaction was quenched with TFA (3 mL) and stirred. After 5 min, the solution was concentrated in vacuo (not to dryness) and the resulting oil was redissolved in CH3CN and H2O (1:1) and the solution was purified by preparative HPLC. The purified fractions were combined, concentrated in vacuo, redissolved in H2O, frozen, and lyophilized to give 56 (TFA salt) as a white powder. 1H NMR (300 MHz, CD3OD) δ ppm 7.63 (t, 1H, J = 6.84 Hz), 7.45 (d, 1H, J = 6.76 Hz), 7.35 (t, 1H, J = 7.21 Hz), 7.18–7.04 (m, 2H), 6.77 (dd, 1H, J = 1.26 Hz), 4.68 (d, 1H, J = 10.61 Hz), 2.73–2.48 (m, 1H), 2.16–1.98 (m, 1H), 1.98–1.43 (m, 18H), 1.27–1.02 (m, 2H); 13C NMR (100 MHz, CD3OD) δ ppm 180.79, 178.22, 167.86, 157.78 (d, JC–F = 248.9 Hz), 145.23, 137.07, 132.32, 129.39, 128.99, 126.45 (d, JC–F = 4.9 Hz), 123.39, 122.50, 122.17, 122.15 (d, JC–F = 19.3 Hz), 111.77, 73.22, 67.72, 62.58, 52.82, 46.96 (d, JC–F = 3.5 Hz), 38.96, 32.17, 31.36, 30.41(3C), 29.45(3C), 25.35, 23.08, 21.74; ESI-MS m/z 614.92 (M + H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-4,4-difluoro-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (57)
57 was obtained using the same synthetic strategy described for 56. 1H NMR (300 MHz, CD3OD) δ ppm 7.71 (s, 1H), 7.63 (t, 1H, J = 6.61 Hz), 7.50 (dd, 1H, J = 2.08, 8.18 Hz), 7.36 (t, 1H, J = 7.54 Hz), 7.18–7.05 (m, 2H), 6.79 (d, 1H, J = 1.83 Hz) 4.96 (d, 1H, J = 10.48 Hz), 4.71 (d, 1H, J = 10.51 Hz), 2.78 (d, 1H, J = 14.25 Hz), 2.59–1.91 (m, 6H), 1.91–1.70 (m, 12H), 1.53–1.33 (m, 1H); ESI-MS m/z 650.92 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-(4-((methylsulfonyl)carbamoyl)-bicyclo[2.2.2]octan-1-yl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (58)
CDI (49 mg, 0.303 mmol), DIEA (88 μL, 0.505 mmol), and DMAP (cat.) were added to a solution of 56 (62 mg, 0.101 mmol) in 1,2-dichloroethane (10 mL), and the reaction was heated to 40 °C. After 20 min, methanesulfonamide (96 mg, 1.01 mmol) was added and the reaction was refluxed. After overnight, the sovent was removed in vacuo and the crude was purified by prepartive HPLC to give 58 (TFA salt) as a white solid. 1H NMR (300 MHz, CD3OD) δ ppm 7.64 (t, 1H, J = 7.23 Hz), 7.45 (dd, 1H, J = 1.93, 8.22 Hz), 7.36 (t, 1H, J = 7.23 Hz), 7.18–7.04 (m, 2H), 6.77 (d, 1H, J = 1.66 Hz), 4.69 (d, 1H, J = 10.70 Hz), 3.19 (s, 3H), 2.75–2.52 (m, 1H), 2.21–1.99 (m, 1H), 1.99–1.44 (m, 17H), 1.41–1.27 (m, 1H), 1.27–1.03 (m, 2H); ESI-MS m/z 691.42 (M + H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (59)
Paraformaldehyde (15 mg, 0.506 mmol) was added to a solution of compound 56 (20 mg, 0.028 mmol) dissolved in AcOH (1 mL). After 15 min sodium triacetoxyborohydride (59 mg, 0.28 mmol) was added, and after reacting overnight, the reaction was quenched with saturated ammonium chloride solution and extracted with EtOAc. The EtOAc solvent was evaporated in vacuo, and the resulting oil was redissolved in a solution of MeCN and H2O (1:1 with 0.1% TFA) and purified by preparative HPLC. The pure 59 fractions were combined, concentrate in vacuo, redissolved in H2O (with minimum amount of MeCN), frozen, and lyophilized to give 59 as the TFA salt as a white powder. 1H NMR (300 MHz, CD3OD) δ ppm 7.94 (s, 1H), 7.61–7.52 (m, 2H), 7.40 (t, 1H, J = 7.32 Hz), 7.19–7.08 (m, 2H), 6.78 (d, 1H, J = 1.56 Hz), 4.99 (d, 1H, J = 11.86 Hz), 4.63 (d, 1H, J = 12.06 Hz), 3.27 (s, 3H), 2.61–2.48 (m, 1H), 2.32–2.14 (m, 2H), 1.88–1.40 (m, 18H), 1.37–1.12 (m, 1H); 13C NMR (100 MHz, CD3OD) δ ppm 180.74, 178.21, 166.55, 157.74 (d, JC–F = 249.6 Hz), 144.90, 137.11, 132.39, 129.80, 129.09, 126.30 (d, JC–F = 4.7 Hz), 124.00, 123.43, 122.31 (d, JC–F = 19.2 Hz), 121.63 (d, JC–F = 13.6 Hz), 111.77, 77.98, 73.06, 67.67, 52.98, 46.56, 40.34, 38.97, 34.28, 30.38(3C), 29.44(3C), 28.97, 24.95, 23.57, 22.07; ESI-MS m/z 628.83 (M + H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (60)
60 was obtained using the same synthetic strategy described for 59. 1H NMR (300 MHz, CD3OD) δ ppm 7.63 (t, 1H, J = 7.04 Hz), 7.56–7.48 (m, 2H), 7.42 (t, 1H, J = 7.39 Hz), 7.18 (t, 1H, J = 7.96 Hz), 7.10 (d, 1H, J = 8.06 Hz), 6.79 (s, 1H), 5.08–4.96 (m, 1H), 4.57 (d, 1H, J = 11.85 Hz), 4.18–3.99 (m, 1H), 3.87–3.69 (m, 1H), 2.70–2.54 (m, 1H), 2.36–2.13 (m, 2H), 1.94–1.45 (m, 18H), 1.39 (t, 3H, J = 6.65 Hz), 1.32–1.14 (m, 1H); 13C NMR (100 MHz, CD3OD) δ ppm 180.95, 180.06, 177.01, 157.71 (d, JC–F = 247.1 Hz), 144.91, 135.39, 130.37, 129.74 (d, JC–F = 1.6 Hz), 129.34 (d, JC–F = 2.5 Hz), 126.86 (d, JC–F = 12.5 Hz), 126.47, 125.46 (d, JC–F = 4.6 Hz), 122.27, 121.50 (d, JC–F = 19.7 Hz), 110.69, 74.84, 72.58, 71.05, 51.51, 48.09, 47.36, 39.17, 37.56, 30.84(3C), 29.61(3C), 29.25, 26.37, 24.25, 23.14, 17.04; ESI-MS m/z 642.50 (M + H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-methyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carbothioamide (68)
Intermediate 64(21) (1.0 g, 2.09 mmol) was dissolved in a mixture of DCM (10 mL) and AcOH (10 mL). Paraformaldehyde (1.13 g, 37.62 mmol) was added, and after 10 min NaBH(OAc)3 (4.43 g, 20.9 mmol) was added in portions. After 4 h, as determined by TLC, the reaction was complete. Saturated NH4Cl (aq) was added to the reaction which was then extracted with EtOAc. The EtOAc solution was washed with NaOH (1 M) and brine, then dried over Na2SO4, filtered, concentrated, and purified by column chromatography to give 764 mg of intermediate 65. Sodium hydroxide (320 mg, 8.0 mmol) was added to a solution of intermediate 65 (764 mg, 1.6 mmol) dissolved in a mixture of THF (3 mL), H2O (3 mL), and MeOH (3 mL), and the solution was heated to 45–50 °C. After hydrolysis was complete, as determined by TLC, the reaction was cooled, neutralized with 2 M HCl, and extracted with EtOAc. The EtOAc solution was dried over Na2SO4, filtered, and concentrated to give the acid intermediate 66 as a solid that was used without further purification. CDI (315 mg, 1.94 mmol) and DIEA (0.450 mL, 2.59 mmol) were added to a solution of 66 (300 mg, 0.648 mmol) in THF (15 mL) and heated to 50 °C. After 30 min ammonium hydroxide was was added. After standing overnight, H2O was added and the reaction was extracted with EtOAc. The EtOAc solution was dried over sodium sulfate, filtered, concentrated, and purified by column chromatography to give 240 mg of compound 67. Lawesson’s reagent (102 mg, 0.252 mmol) was added to a solution of compound 67 (60 mg, 0.126 mmol) in DCM (3 mL) and refluxed overnight. Then the reaction was cooled, the solvent was removed, and the crude was column chromatographed to produce 60 mg of 68. 1H NMR (400 MHz, CD3OD) δ ppm 7.66 (ddd, 1H, J = 1.4, 6.4, 7.9 Hz), 7.53 (dd, 1H, J = 2.5, 8.3 Hz), 7.38 (t, 1H, J = 7.3 Hz), 7.15 (t, 1H, J = 8.0 Hz), 7.10 (dd, 1H, J = 1.9, 8.2 Hz), 6.77 (d, 1H, J = 1.9 Hz), 5.48 (br s, 1H), 4.71 (d, 1H, J = 11.2 Hz), 3.37 (s, 3H), 2.38 (d, 1H, J = 14.2 Hz), 2.19–2.02 (m, 1H), 1.99–1.85 (m, 1H), 1.83–1.61 (m, 4H), 1.50–1.36 (m, 1H), 1.35–1.16 (m, 2H); ESI-MS m/z 492.67 (M + H)+.
Fluorescence Polarization-Based (FP-Based) Protein Binding Assay, Cell Growth Inhibition Assay, Pharmacodynamics (PD) Study in SJSA-1 Xenograft Model, and in Vivo Efficacy Study
These assays were performed according to the previously reported procedures.21 All the in vivo studies were performed under an animal protocol (PRO00005315) approved by the University Committee on Use and Care of Animals of the University of Michigan, in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Computational Methods
Docking studies were performed using our previously reported structure of MDM2 in complex with 2 (PDB code 5TRF)18 with the GOLD33,34 program (version 5.2). For each genetic algorithm (GA) run, a maximum number of 200 000 operations were performed on a population of five islands of 100 individuals. Operator weights for crossover, mutation, and migration were set to 95, 95, and 10, respectively, and the Chemscore35 fitness function was used to evaluate the docked conformations. Docking was terminated after 10 runs for each ligand, and the top 10 conformations were saved for analysis of the predicted binding modes.
Acknowledgments
This study is supported in part by grants from the National Institutes of Health (Grant R01CA121279 to S.W.), the National Cancer Institute Cancer Center Core grant (P30CA046592) to the University of Michigan Comprehensive Cancer Center, a grant from Ascentage Pharma Group to the University of Michigan (to S.W.), the Key New Drug Creation project of the major science and technology program (Grant 2012ZX09401005, China), National “863” Grant (2012AA020305, China), and Science & Technology Innovation Team Grant of Guangdong Province (Grant 2013Y114) to Jiangsu Ascentage Biomed Development Inc.
Glossary
Abbreviations Used
- THF
tetrahydrofuran
- CAN
ceric ammonium nitrate
- DCM
dichloromethane
- MeOH
methanol
- DCE
1,2-dichloroethane
- CDI
1,1′-carbonyldiimidazole
- AcOH
acetic acid
- DIEA
N,N′-diisopropylethylamine
- DME
1,2-dimethoxyethane
- TFA
trifluoroacetic acid
- TFAA
trifluoracetic anhydride
- HPLC
high performance liquid chromatography
- UPLC
ultraperformance liquid chromatography
- DMAP
4-(dimethylamino)pyridine
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- rt
room temperature
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01665.
The authors declare the following competing financial interest(s): The University of Michigan has filed a patent on AA-115 and its analogues, and Shaomeng Wang, Angelo Aguilar, Jianfeng Lu, Liu Liu, and Donna McEachern are co-inventors of the patent. The patent application covering AA-115 and its analogues has been licensed by Ascentage Pharma Group for clinical development in which Shaomeng Wang is a co-founder, owns stock, and serves as a paid consultant. Shaomeng Wang and the University of Michigan also receive a research grant from Ascentage Pharma Group.
Supplementary Material
References
- Hainaut P.; Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv. Cancer Res. 1999, 77, 81–137. 10.1016/S0065-230X(08)60785-X. [DOI] [PubMed] [Google Scholar]
- Vousden K. H.; Lane D. P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Momand J.; Zambetti G. P.; Olson D. C.; George D.; Levine A. J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. 10.1016/0092-8674(92)90644-R. [DOI] [PubMed] [Google Scholar]
- Freedman D. A.; Wu L.; Levine A. J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. 10.1007/s000180050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond G. L.; Hu W. W.; Levine A. J. MDM2 is a central node in the p53 pathway: 12 years and counting. Curr. Cancer Drug Targets 2005, 5, 3–8. 10.2174/1568009053332627. [DOI] [PubMed] [Google Scholar]
- Wu X.; Bayle J. H.; Olson D.; Levine A. J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Juven-Gershon T.; Oren M. Mdm2: the ups and downs. Mol. Med. 1999, 5, 71–83. [PMC free article] [PubMed] [Google Scholar]
- Momand J.; Wu H. H.; Dasgupta G. MDM2--master regulator of the p53 tumor suppressor protein. Gene 2000, 242, 15–29. 10.1016/S0378-1119(99)00487-4. [DOI] [PubMed] [Google Scholar]
- Vassilev L. T.; Vu B. T.; Graves B.; Carvajal D.; Podlaski F.; Filipovic Z.; Kong N.; Kammlott U.; Lukacs C.; Klein C.; Fotouhi N.; Liu E. A. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Vassilev L. T. p53 Activation by small molecules: application in oncology. J. Med. Chem. 2005, 48, 4491–4499. 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]
- Shangary S.; Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008, 14, 5318–5324. 10.1158/1078-0432.CCR-07-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. M.; Zhao Y. J.; Bernard D.; Aguilar A.; Kumar S. Targeting the MDM2-p53 protein-protein interaction for new cancer therapeutics. Top. Med. Chem. 2012, 8, 57–79. 10.1007/978-3-642-28965-1_2. [DOI] [Google Scholar]
- Carry J. C.; Garcia-Echeverria C. Inhibitors of the p53/hdm2 protein-protein interaction-path to the clinic. Bioorg. Med. Chem. Lett. 2013, 23, 2480–2485. 10.1016/j.bmcl.2013.03.034. [DOI] [PubMed] [Google Scholar]
- Ray-Coquard I.; Blay J. Y.; Italiano A.; Le Cesne A.; Penel N.; Zhi J.; Heil F.; Rueger R.; Graves B.; Ding M.; Geho D.; Middleton S. A.; Vassilev L. T.; Nichols G. L.; Bui B. N. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Aguilar A.; Bernard D.; Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu B.; Wovkulich P.; Pizzolato G.; Lovey A.; Ding Q.; Jiang N.; Liu J. J.; Zhao C.; Glenn K.; Wen Y.; Tovar C.; Packman K.; Vassilev L.; Graves B. Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q.; Zhang Z.; Liu J. J.; Jiang N.; Zhang J.; Ross T. M.; Chu X. J.; Bartkovitz D.; Podlaski F.; Janson C.; Tovar C.; Filipovic Z. M.; Higgins B.; Glenn K.; Packman K.; Vassilev L. T.; Graves B. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- Wang S.; Sun W.; Zhao Y.; McEachern D.; Meaux I.; Barriere C.; Stuckey J. A.; Meagher J. L.; Bai L.; Liu L.; Hoffman-Luca C. G.; Lu J.; Shangary S.; Yu S.; Bernard D.; Aguilar A.; Dos-Santos O.; Besret L.; Guerif S.; Pannier P.; Gorge-Bernat D.; Debussche L. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. 10.1158/0008-5472.CAN-14-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D.; Li Z.; Rew Y.; Gribble M.; Bartberger M. D.; Beck H. P.; Canon J.; Chen A.; Chen X.; Chow D.; Deignan J.; Duquette J.; Eksterowicz J.; Fisher B.; Fox B. M.; Fu J.; Gonzalez A. Z.; Gonzalez-Lopez De Turiso F.; Houze J. B.; Huang X.; Jiang M.; Jin L.; Kayser F.; Liu J. J.; Lo M. C.; Long A. M.; Lucas B.; McGee L. R.; McIntosh J.; Mihalic J.; Oliner J. D.; Osgood T.; Peterson M. L.; Roveto P.; Saiki A. Y.; Shaffer P.; Toteva M.; Wang Y.; Wang Y. C.; Wortman S.; Yakowec P.; Yan X.; Ye Q.; Yu D.; Yu M.; Zhao X.; Zhou J.; Zhu J.; Olson S. H.; Medina J. C. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- Holzer P.; Masuya K.; Furet P.; Kallen J.; Valat-Stachyra T.; Ferretti S.; Berghausen J.; Bouisset-Leonard M.; Buschmann N.; Pissot-Soldermann C.; Rynn C.; Ruetz S.; Stutz S.; Chene P.; Jeay S.; Gessier F. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J. Med. Chem. 2015, 58, 6348–6358. 10.1021/acs.jmedchem.5b00810. [DOI] [PubMed] [Google Scholar]
- Aguilar A.; Sun W.; Liu L.; Lu J.; McEachern D.; Bernard D.; Deschamps J. R.; Wang S. Design of chemically stable, potent, and efficacious MDM2 inhibitors that exploit the retro-mannich ring-opening-cyclization reaction mechanism in spiro-oxindoles. J. Med. Chem. 2014, 57, 10486–10498. 10.1021/jm501541j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Liu L.; Sun W.; Lu J.; McEachern D.; Li X.; Yu S.; Bernard D.; Ochsenbein P.; Ferey V.; Carry J. C.; Deschamps J. R.; Sun D.; Wang S. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J. Am. Chem. Soc. 2013, 135, 7223–7234. 10.1021/ja3125417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Chu X. J.; Liu J. J.; Ding Q.; Zhang J.; Bartkovitz D.; Jiang N.; Karnachi P.; So S. S.; Tovar C.; Filipovic Z. M.; Higgins B.; Glenn K.; Packman K.; Vassilev L.; Graves B. Discovery of potent and orally active p53-MDM2 inhibitors RO5353 and RO2468 for potential clinical development. ACS Med. Chem. Lett. 2014, 5, 124–127. 10.1021/ml400359z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepan A. F.; Subramanyam C.; Efremov I. V.; Dutra J. K.; O’Sullivan T. J.; DiRico K. J.; McDonald W. S.; Won A.; Dorff P. H.; Nolan C. E.; Becker S. L.; Pustilnik L. R.; Riddell D. R.; Kauffman G. W.; Kormos B. L.; Zhang L.; Lu Y.; Capetta S. H.; Green M. E.; Karki K.; Sibley E.; Atchison K. P.; Hallgren A. J.; Oborski C. E.; Robshaw A. E.; Sneed B.; O’Donnell C. J. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active gamma-secretase inhibitor. J. Med. Chem. 2012, 55, 3414–3424. 10.1021/jm300094u. [DOI] [PubMed] [Google Scholar]
- Kerns E. H.; Di L.. Plasma Protein Binding. In Drug-like Properties: Concepts, Structure Design and Methods; Academic Press: San Diego, CA, 2008; Chapter 14, pp 187–196. [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Smart B. E. Fluorine substituent effects (on bioactivity). J. Fluorine Chem. 2001, 109, 3–11. 10.1016/S0022-1139(01)00375-X. [DOI] [Google Scholar]
- Huchet Q. A.; Kuhn B.; Wagner B.; Fischer H.; Kansy M.; Zimmerli D.; Carreira E. M.; Muller K. On the polarity of partially fluorinated methyl groups. J. Fluorine Chem. 2013, 152, 119–128. 10.1016/j.jfluchem.2013.02.023. [DOI] [Google Scholar]
- Moody C. J.; Bagley M. C. Total synthesis of (+)-nostocyclamide. J. Chem. Soc., Perkin Trans. 1 1998, 601–607. 10.1039/a704094f. [DOI] [Google Scholar]
- Phillips A. J.; Uto Y.; Wipf P.; Reno M. J.; Williams D. R. Synthesis of functionalized oxazolines and oxazoles with DAST and Deoxo-Fluor. Org. Lett. 2000, 2, 1165–1168. 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- Borg S.; Estennebouhtou G.; Luthman K.; Csoregh I.; Hesselink W.; Hacksell U. Synthesis of 1,2,4-oxadiazole-derived, 1,3,4-oxadiazole-derived, and 1,2,4-triazole-derived dipeptidomimetics. J. Org. Chem. 1995, 60, 3112–3120. 10.1021/jo00115a029. [DOI] [Google Scholar]
- Wipf P.; Venkatraman S. Total synthesis of (−)-muscoride A. J. Org. Chem. 1996, 61, 6517–6522. 10.1021/jo960891m. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved protein-ligand docking using GOLD. Proteins: Struct., Funct., Genet. 2003, 52, 609–623. 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- Eldridge M. D.; Murray C. W.; Auton T. R.; Paolini G. V.; Mee R. P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425–445. 10.1023/A:1007996124545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.