

Abstract
Genetic analyses can provide information about human evolutionary history that cannot always be gleaned from other sources. We evaluated evidence of selective pressure due to introduced infectious diseases in the genomes of two indigenous southern African San groups—the ‡Khomani who had abundant contact with other people migrating into the region and the more isolated Ju|’hoansi. We used a dual approach to test for increased selection on immune genes compared with the rest of the genome in these groups. First, we calculated summary values of statistics that measure genomic signatures of adaptation to contrast selection signatures in immune genes and all genes. Second, we located regions of the genome with extreme values of three selection statistics and examined these regions for enrichment of immune genes. We found stronger and more abundant signals of selection in immune genes in the ‡Khomani than in the Ju|’hoansi. We confirm this finding within each population to avoid effects of different demographic histories of the two populations. We identified eight immune genes that have potentially been targets of strong selection in the ‡Khomani, whereas in the Ju|’hoansi, no immune genes were found in the genomic regions with the strongest signals of selection. We suggest that the more abundant signatures of selection at immune genes in the ‡Khomani could be explained by their more frequent contact with immigrant groups, which likely led to increased exposure and adaptation to introduced infectious diseases.
Keywords: population genetics, human migrations, introduced diseases, immune genes, adaptation
1. Background
Infectious diseases have impacted human populations throughout history. While studies of contemporary diseases benefit from modern methods which allow rapid collection and dissemination of information about the diseases' effects on populations, studying diseases in the past is more challenging. Some historical disease events, such as the plague that struck Europe in the fourteenth century, are relatively well understood, but in other cases little or conflicting information is available about the diseases and their impacts. One such event is the series of epidemics caused by colonization of the Americas by Europeans. By some accounts, disease killed more than 90% of the native population and caused widespread social disruption, but other reports suggest smaller impacts (see varying estimates of population mortality rates in Dobyns [1] and Crosby [2]).
Despite uncertainty about the disease-related effects of European colonization of the Americas, that interaction has been studied much more extensively than have the migrations into Africa, in particular southern Africa. In fact, Europeans were not the first immigrant group to settle in southern Africa. There is evidence for at least two earlier within-Africa population movements. One may be associated with the introduction of pastoralism to southern Africa around 2 000 years ago [3–7]. Admixture studies find a small fraction of east African pastoralist ancestry in southern African pastoralist Khoekhoe populations, indicating that the cultural practice of pastoralism may have been transported to southern Africa by a relatively small number of east African individuals who assimilated into the local populations [3,8]. Despite the low number of immigrants participating in this migration, the introduction of pastoralism likely resulted in a large increase in disease burden, particularly zoonotic disease. A later migration of Bantu-speaking farmers from west and central Africa (e.g. [9]) arrived in the south around 1 200 years ago [10]. This migration was a larger-scale movement of people and resulted in the many Bantu-speaking groups found in southern Africa today (e.g. [8,9,11]). This later migration that involved many individuals and the new cultural practice of farming is likely to be a better candidate for a large-scale effect on disease burden. While many of the aboriginal southern African San populations remained mobile hunter–gatherers, they may have been exposed to diseases associated with sedentary or herding lifestyles through interactions with immigrant groups and local groups that adopted those modes of subsistence.
More recently, European colonists began arriving in southern Africa around 1650. They first settled close to the southern coast where they primarily came into contact with indigenous groups living close to the African south coast (Khoekhoe herders and Tuu-speaking San groups, most probably ancestral to, e.g. ‡Khomani and Karretjie groups). This interaction resulted in disease epidemics, including several documented smallpox epidemics in the 1700s that killed up to 90% of the Cape Khoekhoe groups [12]. However, while some effects of diseases introduced during European colonization are better understood than those due to earlier within-Africa movements, questions remain even about that period.
Studies of both the within-Africa and European migrations are hindered by the lack of pre-arrival indigenous population records, which makes it impossible to estimate the impacts of introduced diseases using traditional measures of mortality and morbidity. Genetic analyses, however, offer another account of population history and enable us to find signatures of past events that we could not otherwise measure. Episodes of natural selection, as would occur during epidemics of introduced infectious diseases, are expected to leave signatures in the genome [13] such as extended lengths of haplotype homozygosity (measured with long-range haplotype scores such as iHS [14] and XP-EHH [15]), differentiation between populations (e.g. FST), and change along a specific lineage in a three-way population differentiation comparison (the population branch statistic, PBS [16]). These different statistics would capture signals of natural selection at different time points, from very recent (perhaps a couple of hundred years) to far back in time (beyond human emergence) [13].
We compared genome-wide population-genetic data for signals of disease-related selection in the ‡Khomani and the Ju|’hoansi peoples of southern Africa. The indigenous peoples of southern Africa are the San people (hunter–gatherers) and the closely related Khoekhoe people (pastoralists) who belong to a common branch of the human lineage that diverged more than 100 000 years ago from all other modern humans, thus representing the earliest diversification event among modern humans [8,17,18]. The ‡Khomani is a San group that historically resided in the southern Kalahari region of southern Africa while the Ju|’hoansi, another San group, historically resided in the northwestern part of southern Africa (figure 1). These two populations are estimated to share common ancestry 35 000 years ago [8]. Their different geographical locations have resulted in disparate levels of contact with outside groups entering southern Africa within the last 2 000 years. The Ju|’hoansi population has been isolated throughout its history and has had low levels of contact and gene flow with outside groups, whereas the ‡Khomani population has experienced much more contact and gene flow with both immigrants practising farming and the local indigenous groups that adopted pastoralism [3,8,19,20].
Figure 1.
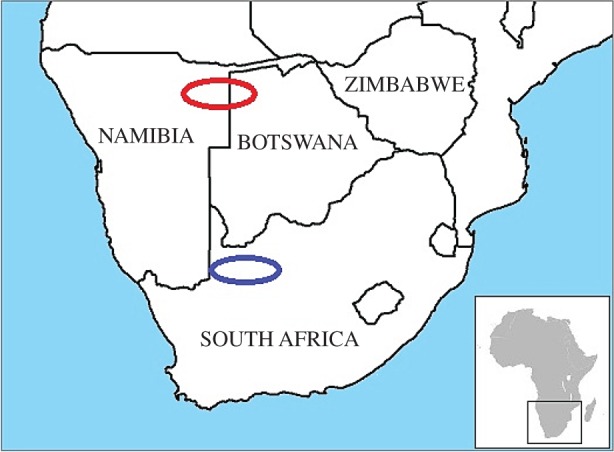
Sampling locations of the Ju|’hoansi (red), a population with a history of isolation, and the ‡Khomani (blue), a population with abundant contact with Khoekhoe pastoralists, Bantu-speaking farmers, and European colonists. (Online version in colour.)
We used two methods to compare signals of selection in these populations. Since any statistic designed to detect selection will also—at least to some extent—be affected by demography (such as bottlenecks, admixture, and expansions), we first explicitly contrasted summary statistic values of genetic tests for selection in immune genes versus all genes in the two populations. As demography affects genetic variation at all (autosomal) genes in a population equally, this approach allowed us to control for possible biases due to demography. Next, we examined regions of the genome that were in the top fraction for three selection statistics (iHS, PBS, and FST) for enrichment of immune genes. We combined three summary statistics to reduce the number of false positives [21]. This dual approach allowed us to examine signals of selection on immune genes throughout the genome as well as in regions with the strongest indicators of selection, resulting in a fuller understanding of infectious disease-related selection than using a single method.
2. Results
(a). Signals of selection in immune genes versus all genes
We first tested whether there was a difference in selection signals in the full set of immune SNPs compared to the full set of (non-immune) genic SNPs (using a Student's t-test and/or a Mann–Whitney U-test), and whether such a difference varied between the Ju|’hoansi and the ‡Khomani. Using a weighted block jackknife approach, we examined to what extent a signal among SNPs was due to a few genomic regions with tightly linked SNPs (see below). The full list of genes (80 922 genes) overlapped with 642 560 SNPs in our dataset. The immune gene list (855 genes) contained 33 578 SNPs (5.2% of the full list). In both the Ju|’hoansi and the ‡Khomani, iHS values were significantly greater for immune genes compared with all genes (figure 2, Ju|’hoansi p = 0.026; ‡Khomani p =5.1 × 10−6, Mann–Whitney U-test).
Figure 2.

iHS results (a) quantile-quantile (qq) plot for SNPs in immune genes (x-axis) versus SNPs in all genes (y-axis) in the Ju|'hoansi (mean |iHS| = 0.791 among 13 126 SNPs in immune genes and mean |iHS| = 0.778 among 415 266 SNPs in other genes, p = 0.0263 based on Mann–Whitney U-test), (b) distribution of |iHS| at SNPs in immune genes (red) and at SNPs in all genes (black) in the Ju|'hoansi, (c) qq plot for SNPs in immune genes (x-axis) versus SNPs in all genes (y-axis) in the ‡Khomani (mean |iHS| = 0.805 among 14 422 SNPs in immune genes and mean |iHS| = 0.778 among 458 852 SNPs in other genes, p = 5.08 × 10−6 based on Mann–Whitney U-test), (d) distribution of |iHS| at SNPs in immune genes (red) and at SNPs in all genes (black) in the ‡Khomani. (Online version in colour.)
To test whether frequency changes were different between SNPs in immune genes and SNPs in other genes in the two populations, we used the PBS statistic that produces a three-way population topology proportional to differentiation among groups (we used the Herero, a Bantu-speaking group, as an outgroup). The length of the Ju|’hoansi branch based on SNPs in immune genes is slightly shorter (mean = 0.029) than the branch based on SNPs in all genes (mean = 0.030, Mann–Whitney U-test p-value = 0.010). By contrast, the ‡Khomani branch based on SNPs in immune genes is much longer (mean = 0.015) than the branch length based on SNPs in all genes (mean = 0.00017, Mann–Whitney U-test p-value = 0.0023) indicating stronger selection in immune genes than all genes in the ‡Khomani (figure 3a,b,e,f).
Figure 3.

PBS and BLD analysis for Ju|'hoansi versus ‡Khomani (p-values based on Mann–Whitney U-test) (a) qq plot of PBS values for SNPs in immune genes (n = 19 737, x-axis) and SNPs in all genes (n = 627 769, y-axis) in Ju|'hoansi (mean PBS = 0.0289 among SNPs in immune genes and mean PBS = 0.0300 among SNPs in other genes, p = 0.00997), (b) qq plot of PBS values for SNPs in immune genes (x-axis) and SNPs in all genes (y-axis) in ‡Khomani (mean PBS = 0.00147 among SNPs in immune genes and mean PBS = 0.00017 among SNPs in other genes, p = 0.0023), (c) qq-plot for BLD values of SNPs in immune genes (x-axis) and SNPs in all genes (y-axis) in Ju|'hoansi versus ‡Khomani (mean BLD = 0.0274 among SNPs in immune genes and mean BLD = 0.0298 among SNPs in other genes, p = 0.00527), (d) distribution of BLD at SNPs in immune genes (red) and at SNPs in all genes (black), (e) tree representation of PBS values for SNPs in immune genes, and (f) tree representation of PBS values for SNPs in other genes. (Online version in colour.)
In addition to testing whether values are different for SNPs in immune genes and SNPs in other genes for each population separately, we tested whether the distribution of the difference between Ju|’hoansi and ‡Khomani was different for SNPs in immune genes and for SNPs in other genes. By contrasting FST-based population branch lengths for different categories of SNPs, we computed the difference between PBS(Ju|’hoansi) and PBS(‡Khomani) (i.e. Branch Length Difference, BLD) for SNPs in immune genes and SNPs in other genes. These two distributions were then compared. BLD was significantly smaller (Mann–Whitney U-test p = 0.0053) among immune SNPs (mean = 0.027) than among genic SNPs (mean = 0.030) (figure 3c,d). BLD being smaller among immune SNPs is consistent with a relatively longer ‡Khomani branch at immune SNPs than at other genic SNPs. That both means are positive indicates a longer Ju|’hoansi branch, probably an effect of a larger proportion of Bantu-speaking admixture (Bantu-speaking Herero, used as an outgroup) in ‡Khomani than in Ju|’hoansi, consistent with previous observations [8]. Using the East African Maasai as an outgroup instead of the Herero in the PBS analysis led to very similar results (data not shown).
The XP-EHH statistic captures differences between pairs of populations in extended haplotype homozygosity that signals local adaptation. XP-EHH values were significantly (t-test p = 4.7 × 10−9) more negative at SNPs located within immune genes (mean = −0.052) compared with SNPs in all genes (mean = −0.012; figure 4). Negative XP-EHH values for the Ju|’hoansi and ‡Khomani comparison corresponds to longer haplotypes in ‡Khomani relative to haplotypes in Ju|’hoansi, consistent with relatively stronger selection on immune genes in ‡Khomani than in Ju|’hoansi. To verify that this result is not due to bias caused by comparing a small set of genes to a much larger set of genes, we replaced immune SNPs with a random set of SNPs from the full set of genic SNPs and then performed the same XP-EHH analysis contrasting Ju|’hoansi and ‡Khomani. We repeated these 100 times and only four of these were significant at p < 0.05 (close to the expected five out of 100 under the null model of no difference) with a minimum p-value of 0.0029 (compared to the observed p-value of 4.7 × 10−9).
Figure 4.

XP-EHH analysis for Ju|'hoansi versus ‡Khomani (a) qq-plot for SNPs in immune genes (x-axis) versus SNPs in all genes (y-axis); (mean XP-EHH = −0.0523 among 20 662 SNPs in immune genes and mean XP-EHH = −0.0122 among 648 000 SNPs in other genes, p = 4.7 × 10−9 based on Student's t-test) and (b) distribution at SNPs in immune genes (red) and at SNPs in all genes (black). (Online version in colour.)
To test the effect of populations used for our comparisons, we also performed the iHS, XP-EHH, PBS, and BLD analyses using the Karretjie [8], another San population exposed to migrant populations, in place of the ‡Khomani. Results from this comparison corroborate the results presented here: the Ju|’hoansi appear to be less affected by selection on immune genes than the Karretjie (electronic supplementary material, figures S4–S6).
It is important to point out that we assess significance using statistical tests that assume independence among SNPs. A weighted block jackknife analysis to study the effect of linkage shows that the only statistic that has non-overlapping 95% CIs between immune and genic SNPs is |iHS| in ‡Khomani (see the electronic supplementary material, figure S1), which points to a very distinct difference between signals of recent selection on immune and genic SNPs. It also suggests that there are specific regions driving the signal. To investigate this closer, we identified blocks (of 5 Mbp) that were driving the difference between the immune and genic SNPs for each statistic (see Material and methods). The results are shown in the electronic supplementary material, table S1.
(b). Genomic regions with strong indications of selection according to multiple tests
In addition to examining differences in summary values of selection statistics at immune and all genes, we also identified genomic regions with the strongest signals of selection, as indicated by high values of three separate selection statistics, and evaluated these regions for enrichment of immune genes. We first selected the 10 most significant iHS windows in each population and compared these windows with the same genomic location in the other population. We visually examined the 20 pairs of windows to determine whether there was evidence of selection also in the other population. Of the top 10 iHS windows in the Ju|’hoansi, six showed evidence of selection in the ‡Khomani as well, indicating that most of the genome-regions with the strong selection affect both groups. Of these four windows with Ju|’hoansi-specific evidence of selection, two contained no genes and the other two contained no immune genes. Because we were searching for regions with high values for all three summary statistics that contained immune genes, and no iHS windows in the Ju|’hoansi passed the iHS selection step, none were considered further in our analysis (table 1). By contrast, nine of the top 10 iHS windows in the ‡Khomani showed evidence of selection unique to that population. Of these nine windows, seven contained genes with immune function and were considered for FST analysis.
Table 1.
Results of analyses of genomic regions with strong signals of selection. Values are the number of windows (SNPs) remaining significant after each step of the filtering process.
| Ju|’hoansi | ‡Khomani | |
|---|---|---|
| iHS windows with unique selection and immune gene(s) | 0 | 7 |
| windows with high-FST SNPs | — | 5 (21) |
| windows with high-PBS SNPs | — | 4 (8) |
We used a 99th percentile FST cut-off of 0.238 (mean FST = 0.0184) to determine extremely differentiated Ju|’hoansi–‡Khomani SNPs. Of the seven regions in the ‡Khomani selected during the iHS step, five contained SNPs with FST values above this cut-off. The number of significant SNPs ranged from one to 11 per window (21 in total).
We then calculated PBS values for the 21 SNPs selected via the combined iHS and FST steps. Because all of the regions being examined were chosen due to selection in the ‡Khomani, we focused on SNPs for which the ‡Khomani (not the Ju|’hoansi or Herero) had the long branch (a cut-off of 2.5 times the second-longest branch). This resulted in four windows containing eight SNPs that showed strong evidence of selection and differentiation due to adaptation along the ‡Khomani lineage. Varying the iHS, FST, and PBS cut-offs did not qualitatively change our results (table 2).
Table 2.
Analyses of genomic regions with strong signals of selection. The numbers in the tables indicate the number of all (left) and immune (right) genes within 100 kb of SNPs that were in the top fractions of all three summary statistics for the given combination of top fraction cut-off values.
| total genes, 10 iHS windows |
immune genes, 10 iHS windows |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
FST |
FST |
|||||||||
| Ju|’hoansi | 1% | 0.50% | 0.10% | 1% | 0.50% | 0.10% | ||||
| 2.5 | 5 | 5 | 0 | 2.5 | 1 | 1 | 0 | |||
| PBS | 5 | 5 | 5 | 0 | PBS | 5 | 1 | 1 | 0 | |
| 10 | 5 | 5 | 0 | 10 | 1 | 1 | 0 | |||
| ‡Khomani | 1% | 0.50% | 0.10% | 1% | 0.50% | 0.10% | ||||
| 2.5 | 48 | 36 | 1 | 2.5 | 13 | 7 | 0 | |||
| PBS | 5 | 39 | 33 | 1 | PBS | 5 | 8 | 6 | 0 | |
| 10 | 34 | 32 | 1 | 10 | 7 | 5 | 0 | |||
Three of the four regions selected by this three-test process contain SNPs with distinct signs of selection along the ‡Khomani branch in close proximity to immune genes (table 3). SNPs in the fourth region were 600 kb away from the nearest immune gene (HSPD1). Owing to the distance, this region was excluded from further analyses. The eight immune genes in the three retained regions were two members of the Fc-receptor-like cluster (FCRL4 and FCRL5), located on chromosome 1 around 157.5 Mb (electronic supplementary material, figure S7a); the Butyrophilin family (BTN2A1, 2A2, 3A1, 3A2, and 3A3, located in the extended MHC on chromosome 6, around 26.4 Mb (electronic supplementary material, figure S7b); and PRSS16, also in the extended MHC region on chromosome 6 around 27.5 Mb (electronic supplementary material, figure S7c).
Table 3.
SNPs in the top fraction of all three summary statistics (at standard cut-offs) and nearby immune genes.
| SNP name | SNP location | immune gene(s) within 100 kb | |
|---|---|---|---|
| ‡Khomani | kgp15319500 | Chr1:157436510 | FCRL4 |
| ‡Khomani | kgp9250245 | Chr1:157460150 | FCRL5, FCRL4 |
| ‡Khomani | kgp1160934 | Chr1:157485720 | FCRL5, FCRL4 |
| ‡Khomani | rs1412676 | Chr1:157539317 | FCRL5, FCRL4 |
| ‡Khomani | kgp9844954 | Chr6:26380608 | BTN3A2, BTN3A3, BTN3A1, BTN2A2, BTN2A1 |
| ‡Khomani | rs13194491 | Chr6:27037080 | PRSS16 |
| ‡Khomani | kgp1961233 | Chr6:27172761 | PRSS16 |
3. Discussion
(a). Different levels of contact and exposure to diseases may explain deviant selection at immune genes
The indigenous populations of southern Africa experienced different levels of interactions and exposure to groups migrating into the region based on their historical locations. Their past demographic histories coupled with varying degrees of interactions with external groups and concomitant exposure to their unfamiliar diseases, may explain signatures of selection at immune genes. Here, we have used the Ju|’hoansi as a representative group for a population with minimal exposure to incoming farmer/herder cultures in the past 2 000 years and the ‡Khomani as a representative of an exposed population in the same time period. We find several lines of genetic evidence in accord with our hypothesis that the ‡Khomani underwent stronger selection on immune function than did the Ju|’hoansi.
(b). Evidence for stronger selection on immune genes in the ‡Khomani
Using the framework based on extended lengths of haplotype homozygosity [14,15], we find that iHS values are significantly higher for immune genes than for all genes in both the Ju|’hoansi and ‡Khomani (figure 2). This result indicates that the immune system may have been a target of selection in both populations over a long period of their history in southern Africa. However, XP-EHH values for immune gene regions and genic regions show smaller values at SNPs in immune genes in the Ju|’hoansi and ‡Khomani (figure 4), indicating that while selection on the immune system may have occurred in both populations, it has likely to have had a stronger effect in the ‡Khomani.
The divergent selection pressure between the populations is further demonstrated by PBS and BLD analyses. Branch lengths as estimated by PBS are significantly longer for immune genes than for all genes in the ‡Khomani, with the converse true in the Ju|’hoansi (figure 3). BLD values are significantly smaller at immune genes than at all genes suggesting historically stronger directional selection at immune genes in the ‡Khomani than in the Ju|’hoansi. While the direction of selection is not indicated by XP-EHH and BLD for the comparison of immune genes versus all genes (and it could in principle be explained by stronger directional selection at non-immune genes in Ju|’hoansi than in ‡Khomani), the iHS and PBS analyses show that it is immune genes that adapted faster.
The large difference in power between the framework based on extended lengths of haplotype homozygosity (which had much smaller p-values) and the FST-based framework suggests that linkage disequilibrium patterns, rather than frequency differences, contain most of the information, perhaps indicating more recent selection (e.g. [15]). A more careful inspection of the XP-EHH and BLD distributions (figures 3 and 4) suggests that while the significant result for BLD is based on a few outliers, the XP-EHH result is due to a general left-skew of the XP-EHH values. That the selection tests give different results may also be due to the fact that they capture different aspects of selection. iHS is best at detecting recent selective sweeps that have not gone to fixation [14], while PBS and XP-EHH measure more ancient events [15,22]. To precisely determine the age of the selective events that gave rise to the genomic signals is difficult. The Ju|’hoansi and the ‡Khomani diverged around 35 000 years ago [8], which provides an upper limit for the time of selection. The signals could be a result of selection during any or all of the three major population movements we are aware of, or could be a result of earlier unknown migration events. Alternatively, the signals could be due to adaptation to disease exposure in general as a result of repeated exposure to immigrants and unfamiliar diseases.
We note that although we contrast the statistics on a genome-wide scale, the qq-plots suggest that the signals are driven by relatively few regions. In fact, a more conservative weighted block jackknife analysis to identify the genomic regions driving the difference between immune and genic SNPs (electronic supplementary material, table S1) shows that the test statistics are generally driven by distinct regions of the genome. The MHC region, for example, drives |iHS| signals, but not XP-EHH, PBS, and BLD (electronic supplementary material, table S1). These regions are also distinct from those identified using stringent cut-offs for the three-test statistics. This effect may be caused by the fact that the test statistics are sensitive to selection events of different time frames. This observation also suggests that the difference between the Ju|’hoansi and ‡Khomani is not due to a single event, rather it is the result of a combination of events (such as the greater exposure of the ‡Khomani to both Bantu-speaking and European migrants or to other factors that may have introduced differences prior to their exposure to more recent immigrant populations).
One potential concern is that not only are the ‡Khomani more likely to have suffered from a higher disease burden than the Ju|’hoansi, they also have a larger proportion of genomic material of Bantu-speaker ancestry. Since the farming Bantu-speaker populations likely also experienced an increased disease exposure as a consequence of their change in subsistence mode, it is possible that the difference in selection signals between San populations at immune genes compared to all genes merely reflects different levels of admixture. However, this scenario predicts a stronger difference between immune genes and all genes in the Bantu-speaking population compared to the San populations. We do not find that effect with either iHS (see the electronic supplementary material, figure S2) or PBS (electronic supplementary material, figure S3) for the Bantu-speaking population suggesting that such a scenario does not explain the difference in disease adaptation between the ‡Khomani and the Ju|’hoansi.
Finally, pathogen load has been shown to be correlated to climate, specifically to precipitation and temperature [23]. At least for recent climate data, we could not detect any difference with respect to precipitation and temperature between the geographical areas of the Ju|’hoansi and the ‡Khomani (electronic supplementary material, table S2). However, to fully investigate this possibility, climatic data over long timescales is required, and such a test would also require a strong assumption of geographically stable populations.
(c). Immune genes in regions of the genome with strongest signals of selection
Our second approach, using extreme values of the selection statistics to locate regions of the genome with strong indications of selection unique to each population, also indicates that selection on immune genes has been a stronger force in the ‡Khomani than the Ju|’hoansi. While there were seven potentially immune SNPs in the ‡Khomani that were highly significant for all three tests, no SNPs in the Ju|’hoansi met these criteria.
The eight genes located near significant SNPs have a range of roles in the immune system, some more well defined than others. PRSS16 on chromosome 6 encodes a thymus-specific serine protease involved in MHC class II antigen presentation to T cells during positive selection [24], and it shows a dramatic signal of selection in ‡Khomani (electronic supplementary material, figure S7c). Another selected region on chromosome 6 contains the butyrophilin (BTN) genes, including BTN2A1, 2A2, 2A3, 3A1, and 3A2 (electronic supplementary material, figure S7b). BTN family members are structurally similar to B7 co-stimulators and those whose function has been investigated are inhibitory co-stimulators with immunosuppressive function [25]. A third region with extreme signals of selection in the ‡Khomani contains two members of a family of Fc receptor-like genes, FCRL4 and FCRL5 (electronic supplementary material, figure S7a) on chromosome 1. These are B-cell membrane receptor proteins with both inhibitory and stimulatory signalling subunits [26].
Several of the genes identified as putative targets of selection have inhibitory functions, but further investigation will be required to know whether the variants selected in the ‡Khomani lead to up- or downregulation of these genes, i.e. whether selection favoured increased or decreased immune response. The type of immune response that would be beneficial depends on the diseases to which a population is exposed. While certain diseases are more efficiently fought with an increased immune response, others, including some influenza pandemics [27,28] and SARS [29], cause damage via over-activation of the immune system.
The roles of these genes in response to specific infectious diseases are still unknown. One disease known to have affected indigenous southern African populations is the repeated epidemics of smallpox during European colonization [12], which had severe impact on the affected populations. Studies of the vaccinia virus, the closely related poxvirus from which the smallpox vaccine was derived [30], have shown that one way poxviruses evade the immune response is by blocking signalling pathways, particularly those activating the Toll-like receptor [31,32] and complement pathways [33,34], two important components of the innate immune system. Several of the immune system genes identified in this study are involved in signalling in the adaptive immune system, which could potentially compensate for the downregulation of the innate system caused by smallpox. A recent study identified two recently emerged alleles in the ‡Khomani that also affect signalling, indicating this may have been a common target of selection [35]. This study also implicates a very high diversity at the KIR2DL1 gene (a gene that interacts HLA-C in the major histocompatibility complex) in ‡Khomani with some variants originating in this population and subsequently transmitted to neighbouring populations. None of the statistics that we employ suggests selection (or population-specific selection) at this locus however (data not shown). As functional analyses of more immune system genes become available, it may be possible to make more definitive links between genes apparently under selection and their roles in various diseases, both those known to have affected the indigenous populations, such as smallpox and influenza during European colonization, as well as unknown diseases that may have affected the populations during the earlier migrations.
It is possible that the genetic variants under selection in the immune genes in ‡Khomani were introduced via admixture (i.e. adaptive introgression) from Bantu-speaking populations, such as the Herero. There is clear evidence of genetic material from Bantu-speaking populations in the ‡Khomani (e.g. [8]). Although we cannot rule out adaptive introgression for single immune regions, we note that (i) the differentiation (FST) between the ‡Khomani and the Herero is very similar for immune genes and other genes (0.0538 versus 0.0528, p > 0.05, t-test) and (ii) contrasting the frequency of the immune SNPs likely to be under selection (table 3) across worldwide populations (electronic supplementary material, figure S8), suggest that the frequency changes have occurred specifically in ‡Khomani (six of seven SNPs showed substantial change, in contrast to one for the Ju|’hoansi and none for the Herero). We hypothesize that the majority adaptive immune gene-variants in ‡Khomani are endogenous. However, regardless of the origin (introgressed or not) of these variants, they are under stronger selection in the ‡Khomani than in the Ju|’hoansi.
It is important to note that many genes not associated with immune response are likely to have experienced different selective pressures in the ‡Khomani and the Ju|’hoansi. This would decrease our power to detect a difference between immune and all genes even if there are population-specific differences in selection pressure on the immune system. Additionally, the simultaneous use of three stringent criteria for candidate regions of selection in our second approach potentially omits true signals of selection and differentiation (but is important for avoiding false-positive results). That we find signals of differential selection at immune genes in the ‡Khomani compared to the Ju|’hoansi using two conservative approaches increases our confidence in the results.
4. Conclusion
Our results indicate that selective pressure on immune genes has been strong for indigenous southern African populations, but also that it was a considerably stronger force in the ‡Khomani than the more isolated Ju|’hoansi. The regions with the strongest signals of selection in the Ju|’hoansi contained no immune genes while there were at least eight immune genes in the regions with the strongest signals of selection in the ‡Khomani, supporting the theory of less selective pressure on Ju|’hoansi immune system genes. Our findings suggest that rapid adaptation of immune function can result from contact with external groups and their unfamiliar diseases.
5. Material and methods
(a). Study populations and SNP data preparation
We used 2 286 795 high-quality filtered SNPs typed by the Illumina Omni 2.5M SNP array ([8], data available at [36]). Related and exceptionally admixed individuals were removed from the analysis, resulting in sample sizes of 17 Ju|’hoansi individuals and 17 ‡Khomani individuals (for further details of sampling and processing of the data, see [8]). Sampling locations for the Ju|’hoansi and ‡Khomani are indicated in figure 1.
(b). Summary statistics for immune genes and all genes
We first compared summary statistics for SNPs in immune system genes and SNPs in all genes to examine differences in selection between the two in each population. We created a list of SNPs in all genes using the hg19 gene list (http://genome.ucsc.edu/cgi-bin/hgTables?command=start). We assembled the start and end positions of each gene and combined any overlapping intervals. SNPs in the resulting intervals were selected from the full SNP dataset, yielding the list of all genic SNPs. Defining a set of immune genes is not straightforward as the immune system is complicated and involved in many interactions. We examined several lists of immune genes and chose the Immunome Database [37,38], because its clear inclusion/exclusion criteria define a core immune gene set. Genes included in the Immunome must have a specifically immune function, or if a part of another system, pathway, or interaction, the gene must have a clear role in immune processes [37]. This allowed us to detect selection on immune function while avoiding potential confounding effects of selection on non-immune roles of genes with broader functions. Of the 893 genes in the Immunome, 38 were either on sex chromosomes or only on certain haplotypes of autosomes and so were excluded from the analysis, resulting in a list of 855 immune genes. SNPs in these genes were selected from the full SNP dataset, yielding the list of immune SNPs.
We calculated four summary statistics for these two sets of SNPs. We computed the integrated haplotype score (iHS [14], calculated following Pickrell et al. [19]) and the population-specific branch length (PBS [16]) for the Ju|’hoansi and ‡Khomani separately. The relative iHS (XP-EHH [15]) and the BLD were calculated between the Ju|’hoansi and ‡Khomani. Both PBS and BLD rely on a rescaling of pairwise FST values according to the relationship T = − ln(1 − FST). We used Weir & Cockerham [39] to calculate the pairwise FST values between Ju|’hoansi, ‡Khomani, and Herero (a Bantu-speaking population used as the outgroup, n = 8). No conditioning on SNPs being polymorphic in any of the populations was performed. The BLD between Ju|’hoansi and ‡Khomani was calculated as T(Ju|’hoansi,Herero) – T(‡Khomani,Herero). Note that BLD between population ‘pop1’ and population ‘pop2’ using ‘pop0’ as the outgroup, by construction, equals PBS(pop1,pop2,pop0)-PBS(pop2,pop1,pop0). For |iHS|, PBS, and BLD, that are not normally distributed, we used the Mann–Whitney U-test to assess whether the distribution of the statistics were different for SNPs in immune genes and SNPs in all genes. For XP-EHH (which closely follows a normal distribution), we assessed statistical significance using the Student's t-test.
To determine what genomic regions were driving the difference between immune SNPs and genic SNPs, we followed Busing et al. [40] in performing a weighted block jackknife analysis. We divided the genome into 5 Mbp blocks, removed each individually, and re-calculated the p-value of the difference between immune and genic SNPs. We ordered the blocks according to how much their removal increased the p-value (lowered the significance). The dataset was then decimated by removing first the top block on this list, then the second block, and so on. For each additional block that was removed, the p-value for the difference between immune and genic SNPs was calculated. This was repeated until the difference between the two categories of SNPs was no longer significant (p > 0.05). In this way, we identified the 5 Mb blocks driving the difference between immune and genic SNPs.
(c). Extracting genomic regions with strong indications of selection
To select genes with strong signatures of selection, we combined three summary statistics, examining only genes within 100 kb of SNPs that had high values for all three. Selected SNPs belonged to the top 10 iHS windows, top 1% FST values, and had a PBS branch length ratio above 2.5 (the branch length for a given SNP was 2.5 times as long in the population of interest as in the second-longest population). The top iHS windows were assessed by calculating p-values for non-overlapping 200 kb windows [19]. Adjacent windows with p-values below 0.01 were merged and assigned the lowest p-value among the merged windows. The cut-offs for each test were applied separately to the full dataset. More stringent cut-offs were also investigated for each summary statistic to examine the impact of stringency on the results (table 2). A high value for all three summary statistics indicated that selection had acted on the corresponding genomic region in only one of the populations due to the differentiation required to generate significant FSTand PBS values.
SNPs with high values for all three tests were checked for proximity (±100 kb) to immune genes. Gene functions were investigated using GeneCards [41,42], the UCSC Genome Browser [43,44], and literature searches. Genes were considered immune-related if there was strong evidence for a functional role in immune processes. As we were interested in selection due to infectious disease, genes involved strictly in autoimmune or tumour-related disease were not included.
Supplementary Material
Acknowledgements
We are grateful to all subjects who participated in our research and would like to thank members of the Working Group of Indigenous Minorities in Southern Africa (WIMSA) and the South African San Council for facilitating research among the San people. Computations were performed at the Swedish National Infrastructure for Computing (SNIC-UPPMAX).
Ethics
This study investigates published data, see Schlebusch et al. [8] for ethical description.
Data accessibility
This study investigates published data [8] for a data description and access. The data can be accessed at: http://jakobssonlab.iob.uu.se/data/.
Authors' contributions
C.M.S. and M.J. conceived the project; K.A.O., P.Sj., and P.Sk. analysed data; K.A.O., P.Sj., C.M.S., and M.J. wrote the paper; C.M.S. and H.S. conducted community engagement and fieldwork; H.S. contributed the samples. All authors read and approved the manuscript.
Competing interests
The authors declare no competing interests.
Funding
This project was supported by grants from the Erasmus Mundus Programme (to K.O.), the Wenner-Gren Foundations (to C.M.S.), the South African Medical Research Council (to H.S.), Göran Gustafsson foundation, Knut and Alice Wallenberg Foundation and the Swedish Research Council (to M.J.).
References
- 1.Dobyns HF. 1993. Disease transfer at contact. Annu. Rev. Anthropol. 22, 273–291. ( 10.1146/annurev.an.22.100193.001421) [DOI] [Google Scholar]
- 2.Crosby AW. 1976. Virgin soil epidemics as a factor in the aboriginal depopulation in America. William Mary Q. 33, 289–299. ( 10.2307/1922166) [DOI] [PubMed] [Google Scholar]
- 3.Breton G, Schlebusch CM, Lombard M, Sjödin P, Soodyall H, Jakobsson M. 2014. Lactase persistence alleles reveal partial east African ancestry of southern African Khoe pastoralists. Curr. Biol. 24, 852–858. ( 10.1016/j.cub.2014.02.041) [DOI] [PubMed] [Google Scholar]
- 4.Macholdt E, Lede V, Barbieri C, Mpoloka SW, Chen H, Slatkin M, Pakendorf B, Stoneking M. 2014. Tracing pastoralist migrations to southern Africa with lactase persistence alleles. Curr. Biol. 24, 875–879. ( 10.1016/j.cub.2014.03.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pickrell JK, Patterson N, Loh P-R, Lipson M, Berger B, Stoneking M, Pakendorf B, Reich D. 2014. Ancient west Eurasian ancestry in southern and eastern Africa. Proc. Natl Acad. Sci. USA 111, 2632–2637. ( 10.1073/pnas.1313787111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith AB. 2008. Pastoral origins at the Cape, South Africa: influences and arguments. South Afr. Humanit. 20, 49–60. [Google Scholar]
- 7.Sadr K. 2015. Livestock first reached Southern Africa in two separate events. PLoS ONE 10, e0134215 ( 10.1371/journal.pone.0134215) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlebusch CM, et al. 2012. Genomic variation in seven Khoe-San groups reveals adaptation and complex African history. Science 338, 374–379. ( 10.1126/science.1227721) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Schlebusch C, Jakobsson M. 2014. Genetic variation reveals large-scale population expansion and migration during the expansion of Bantu-speaking peoples. Proc. R. Soc. B 281, 20141448 ( 10.1098/rspb.2014.1448) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phillipson DW. 2005. African archaeology. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 11.Coelho M, Sequeira F, Luiselli D, Beleza S, Rocha J. 2009. On the edge of Bantu expansions: mtDNA, Y chromosome and lactase persistence genetic variation in southwestern Angola. BMC Evol. Biol. 9, 80 ( 10.1186/1471-2148-9-80) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nurse GT, Weiner JS, Jenkins T. 1986. The peoples of Southern Africa and their affinities. New York, NY: Oxford University Press. [Google Scholar]
- 13.Sabeti PC, et al. 2006. Positive natural selection in the human lineage. Science 312, 1614–1620. ( 10.1126/science.1124309) [DOI] [PubMed] [Google Scholar]
- 14.Voight BF, Kudaravalli S, Wen X, Pritchard JK. 2006. A map of recent positive selection in the human genome. PLoS Biol. 4, 0446–0458. ( 10.1371/journal.pbio.0040446) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabeti PC, et al. 2007. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918. ( 10.1038/nature06250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yi X, et al. 2010. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78. ( 10.1126/science.1190371) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. 2011. Bayesian inference of ancient human demography from individual genome sequences. Nat. Genet. 43, 1031–1034. ( 10.1038/ng.937) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veeramah KR, Wegmann D, Woerner A, Mendez FL, Watkins JC, Destro-Bisol G, Soodyall H, Louie L, Hammer MF. 2012. An early divergence of KhoeSan ancestors from those of other modern humans is supported by an ABC-based analysis of autosomal resequencing data. Mol. Biol. Evol. 29, 617–630. ( 10.1093/molbev/msr212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pickrell JK, et al. 2009. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 19, 826–837. ( 10.1101/gr.087577.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnard A. 1992. Hunters and herders of Southern Africa - a comparative ethnography of the Khoisan peoples. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 21.Teshima KM, Coop G, Przeworski M. 2006. How reliable are empirical genomic scans for selective sweeps? Genome Res. 16, 702–712. ( 10.1101/gr.5105206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Bakker PIW, et al. 2006. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 38, 1166–1172. ( 10.1038/ng1885) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guernier V, Hochberg ME, Guégan JF. 2004. Ecology drives the worldwide distribution of human diseases. PLoS Biol. 2, e141 ( 10.1371/journal.pbio.0020141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gommeaux J, Grégoire C, Nguessan P, Richelme M, Malissen M, Guerder S, Malissen B, Carrier A. 2009. Thymus-specific serine protease regulates positive selection of a subset of CD4+ thymocytes. Eur. J. Immunol. 39, 956–964. ( 10.1002/eji.200839175) [DOI] [PubMed] [Google Scholar]
- 25.Abeler-Dörner L, Swamy M, Williams G, Hayday AC, Bas A. 2012. Butyrophilins: an emerging family of immune regulators. Trends Immunol. 33, 34–41. ( 10.1016/j.it.2011.09.007) [DOI] [PubMed] [Google Scholar]
- 26.Dement-Brown J, Newton CS, Ise T, Damdinsuren B, Nagata S, Tolnay M. 2012. Fc receptor-like 5 promotes B cell proliferation and drives the development of cells displaying switched isotypes. J. Leukoc. Biol. 91, 59–67. ( 10.1189/jlb.0211096) [DOI] [PubMed] [Google Scholar]
- 27.Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. 2001. Symptom pathogenesis during acute influenza: Interleukin-6 and other cytokine responses. J. Med. Virol. 64, 262–268. ( 10.1002/jmv.1045) [DOI] [PubMed] [Google Scholar]
- 28.Cheung CY, Poon LLM, Lau AS, Luk W, Lau YL, Shortridge KF, Gordon S, Guan Y, Peiris JSM. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 360, 1831–1837. ( 10.1016/S0140-6736(02)11772-7) [DOI] [PubMed] [Google Scholar]
- 29.Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, Lei HY. 2005. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 75, 185–194. ( 10.1002/jmv.20255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stanford MM, McFadden G, Karupiah G, Chaudhri G. 2007. Immunopathogenesis of poxvirus infections: forecasting the impending storm. Immunol. Cell Biol. 85, 93–102. ( 10.1038/sj.icb.7100033) [DOI] [PubMed] [Google Scholar]
- 31.Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O'Neill LA. 2000. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl Acad. Sci. USA 97, 10 162–10 167. ( 10.1073/pnas.160027697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DiPerna G, et al. 2004. Poxvirus protein N1 L targets the I-κB kinase complex, inhibits signaling to NF-κB by the tumor necrosis factor superfamily of receptors, and inhibits NF-κB and IRF3 signaling by toll-like receptors. J. Biol. Chem. 279, 36 570–36 578. ( 10.1074/jbc.M400567200) [DOI] [PubMed] [Google Scholar]
- 33.Dunlop LR, Oehlberg KA, Reid JJ, Avci D, Rosengard AM. 2003. Variola virus immune evasion proteins. Microbes Infect. 5, 1049–1056. ( 10.1016/S1286-4579(03)00194-1) [DOI] [PubMed] [Google Scholar]
- 34.Seet BT, et al. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21, 377–423. ( 10.1146/annurev.immunol.21.120601.141049) [DOI] [PubMed] [Google Scholar]
- 35.Hilton HG, Norman PJ, Nemat-Gorgani N, Goyos A, Hollenbach JA, Henn BM, Gignoux CR, Guethlein LA, Parham P. 2015. Loss and gain of natural killer cell receptor function in an African hunter-gatherer population. PLoS Genet. 11, e1005439 ( 10.1371/journal.pgen.1005439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Data from Schlebusch et al. 2012. See http://jakobssonlab.iob.uu.se/data/.
- 37.Ortutay C, Siermala M, Vihinen M. 2007. Molecular characterization of the immune system: emergence of proteins, processes, and domains. Immunogenetics 59, 333–348. ( 10.1007/s00251-007-0191-0) [DOI] [PubMed] [Google Scholar]
- 38.Immunome. 2012. See http://structure.bmc.lu.se/idbase/Immunome/index.php (accessed 15 May 2012).
- 39.Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 ( 10.2307/2408641) [DOI] [PubMed] [Google Scholar]
- 40.Busing F, Meijer E, Leeden R. 1999. Delete-m jackknife for unequal m. Stat. Comput. 9, 3–8. ( 10.1023/A:1008800423698) [DOI] [Google Scholar]
- 41.Safran M, et al. 2010. GeneCards Version 3 the human gene integrator. Database 2010, baq020. ( 10.1093/database/baq020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genecards: The Human Gene Database. 2012. See http://www.genecards.org (accessed 30 May 2012).
- 43.Fujita PA, et al. 2011. The UCSC genome browser database: Update 2011. Nucleic Acids Res. 39(Suppl 1), 876–882. ( 10.1093/nar/gkq963) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.UCSC Genome Bioinformatics. 2012. See http://genome.ucsc.edu (accessed 30 May 2012).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study investigates published data [8] for a data description and access. The data can be accessed at: http://jakobssonlab.iob.uu.se/data/.