

Abstract
Atrophy patterns on MRI can reliably predict three neuropathological subtypes of Alzheimer’s disease (AD): typical, limbic-predominant, or hippocampal-sparing. A method to enable their investigation in the clinical routine is still lacking. We aimed to (1) validate the combined use of visual rating scales for identification of AD subtypes; (2) characterise these subtypes at baseline and over two years; and (3) investigate how atrophy patterns and non-memory cognitive domains contribute to memory impairment. AD patients were classified as either typical AD (n = 100), limbic-predominant (n = 33), or hippocampal-sparing (n = 35) by using the Scheltens’ scale for medial temporal lobe atrophy (MTA), the Koedam’s scale for posterior atrophy (PA), and the Pasquier’s global cortical atrophy scale for frontal atrophy (GCA-F). A fourth group with no atrophy was also identified (n = 30). 230 healthy controls were also included. There was great overlap among subtypes in demographic, clinical, and cognitive variables. Memory performance was more dependent on non-memory cognitive functions in hippocampal-sparing and the no atrophy group. Hippocampal-sparing and the no atrophy group showed less aggressive disease progression. Visual rating scales can be used to identify distinct AD subtypes. Recognizing AD heterogeneity is important and visual rating scales may facilitate investigation of AD heterogeneity in clinical routine.
Alzheimer’s disease (AD) is a heterogeneous disease1,2,3,4,5. Current diagnostic criteria recognize this heterogeneity in the form of different cognitive presentations6,7,8. However, there is also neuropathological and structural heterogeneity4,9. Whitwell et al.10 grouped AD patients into amnestic and non-amnestic types. Amnestic patients evidenced atrophy in the medial temporal lobe, while non-amnestic patients showed atrophy in lateral regions of the parietal, temporal, and frontal lobes with relative sparing of the medial temporal lobes10. Subtyping based on the spread of neurofibrillary tangles (NFT) revealed fairly corresponding groups4. The amnestic form was highly represented on both the typical AD subtype (balanced NFT counts in the hippocampus and the associative cortex, i.e. lateral parietal, temporal, and frontal regions) and the limbic-predominant subtype (NFT counts predominantly in the hippocampus). The non-amnestic syndromes were more frequent in the atypical hippocampal-sparing AD subtype (NFT counts predominantly in the associative cortex). In a subsequent study, patterns of atrophy in MRI reliably tracked the distribution of NFT pathology at autopsy9. Hence, evidence suggests a connection between patterns of NFT spread, brain atrophy, and the cognitive presentation.
Recently, Byun et al.11 investigated these three subtypes as well as a fourth AD group with no atrophy by studying brain atrophy patterns on MRI data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI-112,13). Further, longitudinal progression over two years was studied. Limbic-predominant AD and the group with no atrophy showed slower progression than typical AD and hippocampal-sparing AD11. Data-driven approaches using MRI data have largely confirmed these pathologically defined subtypes1,2,14,15. Other authors have also applied data-driven approaches to cognitive data but the resulting subtypes differ noticeably from study to study3,5,16,17. However, data-driven approaches rely on group analysis and sophisticated methods that make them difficult to translate into clinical practice at present. Still, MRI is in a privileged position for studying AD heterogeneity because impairment in a given cognitive function may emerge from heterogeneous underlying neuropathology and atrophy patterns8,9,10,18.
We investigated whether visual rating scales of brain atrophy in MRI might be useful to capture the above-mentioned AD subtypes. Visual rating scales are quick and easy to use, and are the primary method for assessing brain structural changes in clinical settings18,19,20,21. However, visual rating scales are often used individually. Applying them in combination increases their diagnostic capacity and enables the study of patterns of brain atrophy18,19. We propose a way to easily identify patterns of atrophy using three visual rating scales covering the medial temporal, frontal and posterior cortices. We aimed to (1) validate the combined use of visual rating scales for identification of AD subtypes; (2) characterize the resulting subtypes at baseline and longitudinally over two years; and (3) since all the AD patients in our sample were amnestic, we also investigated how atrophy patterns and non-memory cognitive domains contribute to memory impairment, a relevant question not yet investigated in different AD subtypes. Thus, the three aims were addressed to facilitate investigation of the different AD subtypes in the clinical routine using already at-place and widely used clinical diagnostic tools.
Results
Clinical and cognitive characterization of the AD subtypes
Table 1 shows the main demographic and clinical characteristics of the study groups. Visual examples for each group are shown in Fig. 1. The largest group was typical AD (n = 100), as expected, present in 50.5% of the AD patients. The atypical subtypes were less prevalent and showed similar frequency: hippocampal-sparing (n = 35, 17.7%), limbic-predominant (n = 33, 16.7%), and no atrophy group (n = 30, 15.2%). Maps of cortical thickness as well as hippocampal volumes are displayed in Fig. 2.
Table 1. Characteristics of the AD subtypes and healthy controls.
| Healthy controls (n = 230) | Alzheimer’s disease subtypes | |||||
|---|---|---|---|---|---|---|
| Typical AD (n = 100, 50.5%) | Limbic-predominant (n = 33, 16.7%) | Hippocampal-sparing (n = 35, 17.7%) | No atrophy group (n = 30, 15.2%) | p-value | ||
| Age | 75.9 (5.0) | 77.7 (6.4) | 72.3 (7.1)a,b | 78.2 (8.2)c | 69.2 (6.9)a,b,d | <0.001 |
| Gender, % female | 48.7 | 37.0 | 57.6 | 48.6 | 73.3b | 0.016 |
| Years of education | 16.0 (2.9) | 15.0 (3.4)a | 14.6 (2.2) | 14.9 (3.1) | 13.6 (3.7)a | <0.001 |
| MMSE | 29.1 (1.0) | 22.9 (2.1)a | 23.2 (1.9)a | 23.7 (2.2)a | 24.2 (1.5)a,b | <0.001 |
| CDR total | 0 (0) | 0.8 (0.2)a | 0.7 (0.2)a,b | 0.7 (0.3)a | 0.7 (0.2)a,b | <0.001 |
| Age at onsete | — | 73.6 (7.2) | 68.6 (6.9)b | 76.3 (7.6)c | 66.8 (7.1)b,d | <0.001 |
| Disease duratione | — | 3.8 (2.6) | 3.3 (2.2) | 3.2 (2.7) | 2.6 (1.4) | 0.189 |
| ApoE status, % ε4 allele | 26.5 | 69.0a | 69.7a | 51.4a | 73.3a | <0.001 |
| CSF Aß1-42, % abnormalf | 40.0 | 96.0a | 90.0a | 85.0a | 91.7a | <0.001 |
| CSF T-tau, % abnormalg | 21.2 | 56.0a | 75.0a | 75.0a | 66.7a | <0.001 |
The table shows mean (SD) except for Gender, APOE status and the CSF biomarkers, where percentage is reported. aSignificantly different from Healthy controls; bSignificantly different from Typical AD; cSignificantly different from Limbic-predominant; dSignificantly different from Hippocampal-sparing. eN = 154; fN = 217; gN = 215. AD = Alzheimer’s disease; CDR = clinical dementia rating; MMSE = mini-mental state examination; APOE = apolipoprotein E; CSF = cerebrospinal fluid; Aß1-42 = amyloid-ß-peptide 1–42; T-tau = total level of tau protein.
Figure 1. Subtypes of AD based on patterns of brain atrophy from visual rating scales.

Regional atrophy was measured with the MTA, PA and GCA-F visual rating scales based only on T1-weigthed images. In the three visual rating scales, a score of zero denotes no atrophy, whereas scores from one to three (PA and GCA-F) or four (MTA) indicate an increasing degree of atrophy. The typical AD subtype was defined as abnormal MTA together with abnormal PA and/or abnormal GCA-F. The limbic-predominant subtype was defined as abnormal MTA alone with normal PA and GCA-F. The hippocampal-sparing subtype included abnormal PA and/or abnormal GCA-F, but normal MTA. The no atrophy group was defined as normal scores in MTA, PA, and GCA-F. The figure shows examples for each subtype as well as descriptive analysis on the different study groups. AD = Alzheimer’s disease; MTA = medial temporal atrophy scale; PA = posterior atrophy scale; GCA-F = global cortical atrophy scale – frontal subscale; A = anterior part of the brain; P = posterior part of the brain; R = right; L = left.
Figure 2. Cortical thickness and hippocampal volumes.

The brain images show cortical maps of differences in thickness when comparing the different AD subtypes with the healthy controls. A general linear model was fitted at each vertex. Study group was entered as independent variable and TIV as a covariate. Z Monte Carlo simulations were conducted for cluster-forming with a threshold of p ≤ 0.001 (two-sided), yielding clusters corrected for multiple comparisons. Only vertexes belonging to clusters surviving this correction are displayed. Significant clusters were mapped on standard templates depicted in lateral (first two images on each row) and medial (last two images on each row) views, both for left (L) and right (R) hemispheres. The coloured bar illustrates the significance level of the differences (i.e. less cortical thickness in the AD patients) from dark blue (p ≤ 0.05) to light blue (p ≤ 0.00001). All these results stand after controlling for age, gender, years of education, and APOE ε4 status (data not shown). The boxplot represents the averaged hippocampal volume controlling for TIV, age, years of education, and APOE ε4 status. Hence, the y-axis represents adjusted and standardized values. Box values represent median and confidence intervals. The groups’ sizes are specified in Fig. 1 and Table 1. AD = Alzheimer’s disease.
Three random forest models were conducted to characterize the study groups according to (1) demographic-clinical variables, (2) memory variables, and (3) non-memory cognitive variables (see Table 2 for a list of variables included in each analysis as well as summary of results). Results showed great overlap (Fig. 3). Healthy controls and typical AD patients were correctly classified in the three models. Both resulted as the least and the most severe groups, respectively. However, any combination of the different sets of variables did not allow classifying limbic-predominant, hippocampal-sparing, and the no atrophy group better than chance. To note, the four AD subtypes were comparable on disease duration, CSF biomarkers and APOE ε4 distribution.
Table 2. Random Forest models.
| (A) Description of variables included in the models | ||
|---|---|---|
| Model 1 | Model 2 | Model 3 |
| Demographic-clinical variables | Memory variables | Non-memory cognitive variables |
| Age | AVLT Learning trial 1 | WAIS-R Digit Symbol |
| Gender | AVLT Learning trial 2 | WMS-R Digit forward span |
| Years of education | AVLT Learning trial 3 | WMS-R Digit forward length |
| APOE status | AVLT Learning trial 4 | WMS-R Digit backward span |
| GDS | AVLT Learning trial 5 | WMS-R Digit backward length |
| FAQ | AVLT Learning trial 1 errors | TMT-A correct answers |
| CDR | AVLT Learning trial 2 errors | TMT-A commission errors |
| MMSE | AVLT Learning trial 3 errors | TMT-A omission errors |
| CSF Aß42a | AVLT Learning trial 4 errors | TMT-B correct answers |
| CSF Total taua | AVLT Learning trial 5 errors | TMT-B commission errors |
| CSF p-taua | AVLT B (interference) | TMT-B omission errors |
| Disease durationa | AVLT B (interference) errors | Verbal Fluency – vegetables |
| Age of onseta | AVLT immediate free recall | Verbal Fluency – vegetables intrusions |
| AVLT immediate free recall errors | Verbal Fluency – vegetables perseverations | |
| AVLT delayed free recall | Verbal Fluency – animals | |
| AVLT delayed free recall errors | Verbal Fluency – animals intrusions | |
| AVLT recognition | Verbal Fluency – animals perseverations | |
| AVLT recognition errors | BNT total | |
| BNT spontaneous | ||
| BNT number semantic cues | ||
| BNT spontaneous + semantic | ||
| BNT number phonetic cues | ||
| BNT spontaneous + semantic + phonetic | ||
| Clock Test drawing | ||
| Clock Test copy | ||
| (B) Summary of results | ||
| Model 1 | Model 2 | Model 3 |
| N = 415 | N = 408 | N = 398 |
| Error by chance = 80% | Error by chance = 80% | Error by chance = 80% |
| Classification error: | Classification error: | Classification error: |
| Healthy controls: 0% | Healthy controls: 4% | Healthy controls: 3% |
| Typical AD: 16% | Typical AD: 23% | Typical AD: 25% |
| Limbic-predominant: 88% | Limbic-predominant: 100% | Limbic-predominant: 100% |
| Hippocampal-sparing: 82% | Hippocampal-sparing: 100% | Hippocampal-sparing: 97% |
| No atrophy group: 63% | No atrophy group: 100% | No atrophy group: 100% |
aThe same model was repeated including CSF biomarkers in all the sample (model 1’), and CSF biomarkers + disease duration + age of onset only in Alzheimer’s disease patients (model 1”). Results obtained were the same as in model 1. Results presented in the manuscript Fig. 3 are those from model 1 (8 variables). APOE = apolipoprotein E; GDS = geriatric depression scale; FAQ = functional activities questionnaire; CDR = clinical dementia rating; MMSE = mini-mental state examination; CSF = cerebrospinal fluid; Aß1-42 = amyloid-ß-peptide 1–42; T-tau = total level of tau protein; AVLT = Auditory Verbal Learning test; WAIS-R = Wechsler Adult Intelligence Scale – Revised; WMS-R = Wechsler Memory Scale – Revised; TMT-B = Trail Making Test part B; BNT = Boston Naming Test; AD = Alzheimer’s disease.
Figure 3. Demographic-clinical variables and cognitive profile.
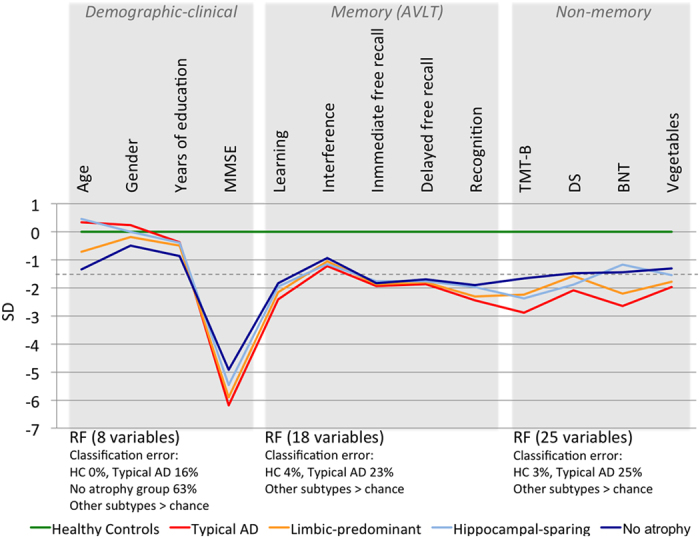
A selection of variables is reported for each random forest (RF) analysis. Classification error is reported only for the correctly classified study groups. The complete list of variables as well as full results from these models are detailed in Table 2. The dashed line shows the clinical cut-off of −1.5 standard deviations (SD). The random forest models reveal great overlap among subtypes with high comparability in demographic, clinical, and cognitive variables at baseline, including cerebrospinal fluid biomarkers, and APOE ε4 distribution. The groups’ sizes are specified in Fig. 1 and Table 1. AD = Alzheimer’s disease; HC = healthy controls; MMSE = mini-mental state examination; AVLT = Auditory Verbal Learning test; TMT-B = Trail Making Test part B; DS = digit symbol from the Wechsler Adult Intelligence Scale – Revised; BNT = Boston Naming Test.
A follow-up mixed ANOVA was performed to further investigate the interaction between five memory components (within-subjects factor: total learning, interference, immediate free recall, delayed free recall, and recognition) and AD subtype (between-subjects factor). This interaction was statistically significant (F(5, 324) = 3.419; p = 0.004). Impairment in the learning component was greater in typical AD than in both hippocampal-sparing (t(86) = −3.427; p = 0.006) and the no atrophy group (t(117) = −3.181; p = 0.010), but this effect was not observed for the other components (Fig. 3). These results hold after controlling for age, Mini-Mental state examination (MMSE), Clinical Dementia Rating (CDR), and disease duration (data not shown).
We then investigated whether these findings in memory could have any clinical relevance by classifying performance as normal or clinically impaired (−1.5 SD) using the healthy controls as reference group. Descriptive analyses confirmed that clinical impairment in learning was more frequent in typical AD than in the other subtypes (Table 3). Clinical impairment in delayed recall and recognition was more frequent in typical AD and limbic-predominant. The hippocampal-sparing group was the subtype having more interference effect. The gain variable (recognition minus delayed free recall) showed that hippocampal-sparing and specially the no atrophy group get more benefit from cues (recognition) than typical AD and limbic-predominant.
Table 3. Percentage of AD patients with clinical impairment (−1.5 SD) across memory components.
| Typical AD (n = 100) | Limbic-predominant (n = 33) | Hippocampal-sparing (n = 35) | No atrophy group (n = 30) | |
|---|---|---|---|---|
| Learninga | 88.0 | 75.8 | 81.8 | 74.1 |
| Interferenceb | 36.6 | 33.3 | 42.6 | 29.6 |
| Immediatec | 78.0 | 66.7 | 63.6 | 66.7 |
| Delayedd | 87.1 | 87.9 | 82.4 | 74.1 |
| Recognitiond | 74.2 | 78.8 | 64.7 | 51.9 |
| Gaind | 12.9 | 9.1 | 17.7 | 22.2 |
Values in the table represent percentage. The healthy control group was used as reference for calculating the clinical cut-off of −1.5 SD. Interference was based on the AVLT list B item in order to estimate interference effects during learning. A higher percentage of individuals showing clinical impairment (−1.5 SD) in this variable reflects greater interference effect, which is frequently interpreted as distortions of existing memories possibly due to source-monitoring deficits. Gain was calculated by subtracting the recognition percentage from the delayed percentage in order to quantify benefit from additional help when retrieving stored information: higher gain values represent more retrieving problems. aN = 411; bN = 412; cN = 408; dN = 414. AD = Alzheimer’s disease.
Contribution of non-memory functions to memory performance
The results above suggest that despite memory impairment looks similar across AD subtypes, the nature of this memory impairment might be different, i.e. non-memory cognitive functions may be contributing differently to performance in memory across subtypes. To test for this we performed separate multiple linear regression models to investigate which non-memory components contributed the most to performance in memory across AD subtypes, and in comparison with the healthy controls. Due to the small sample size for some AD subtypes (n ≈ 30), results that were not stable in the non-parametric multiple regression model (i.e. dominance analysis) were rejected and not reported (Table 4). Only learning, delayed free recall and recognition where tested for simplicity.
Table 4. Contribution of non-memory cognitive functions to different memory measurements.
| TMT-B | DS | BNT | Vegetables | Learning | ||
|---|---|---|---|---|---|---|
| Learning | ||||||
| Multiple regression model | ||||||
| Healthy controls | F(2, 222) = 22.065; p < 0.001; R2adj. = 16% | 0.229 | 0.292 | — | ||
| Typical AD | F(3, 80) = 15.880; p < 0.001; R2adj. = 35% | 0.264 | 0.219 | 0.363 | — | |
| Limbic-predominant | F(2, 30) = 13.526; p < 0.001; R2adj. = 44% | 0.340 | 0.510 | — | ||
| Hippocampal-sparing | non significant | — | ||||
| No atrophy group | non significant | — | ||||
| Dominance analysis (bootstrapping) | ||||||
| Healthy controls | Explained variance = 18% | 1% | 6% | 1% | 10% | — |
| Typical AD | Explained variance = 38% | 4% | 8% | 9% | 17% | — |
| Limbic-predominant | Explained variance = 49% | 10% | 11% | 5% | 23% | — |
| Delayed free recall | ||||||
| Multiple regression model | ||||||
| Healthy controls | F(1, 223) = 276.979; p < 0.001; R2adj. = 55% | 0.744 | ||||
| Typical AD | F(2, 81) = 15.080; p < 0.001; R2adj. = 25% | −0.249* | 0.560 | |||
| Limbic-predominant | F(1, 31) = 9.435; p = 0.008; R2adj. = 21% | 0.483 | ||||
| Hippocampal-sparing | F(2, 27) = 5.225; p = 0.012; R2adj. = 23% | 0.548 | 0.455 | |||
| No atrophy group | F(2, 22) = 17.815; p < 0.001; R2adj. = 58% | —0.336 | 0.725 | |||
| Dominance analysis (bootstrapping) | ||||||
| Healthy controls | Explained variance = 56% | 0% | 3% | 1% | 3% | 49% |
| Typical AD | Explained variance = 28% | 0% | 2%* | 1% | 2% | 23% |
| Limbic-predominant | Explained variance = 26% | 1% | 2% | 0% | 2% | 21% |
| Hippocampal-sparing | Explained variance = 33% | 14% | 5% | 3% | 10% | 1% |
| No atrophy group | Explained variance = 66% | 6% | 8% | 2% | 6% | 44% |
| Recognition | ||||||
| Multiple regression model | ||||||
| Healthy controls | F(1, 223) = 62.858; p < 0.001; R2adj. = 22% | 0.469 | ||||
| Typical AD | F(2, 81) = 11.366; p < 0.001; R2adj. = 20% | 0.253* | 0.486 | |||
| Limbic-predominant | non significant | |||||
| Hippocampal-sparing | non significant | |||||
| No atrophy group | F(2, 22) = 5.890; p = 0.027; R2adj. = 29% | 0.357 | 0.445 | |||
| Dominance analysis (bootstrapping) | ||||||
| Healthy controls | Explained variance = 24% | 1% | 1% | 1% | 1% | 20% |
| Typical AD | Explained variance = 24% | 3%* | 1% | 2% | 1% | 17% |
| No atrophy group | Explained variance = 37% | 1% | 1% | 8% | 10% | 17% |
Values in the table represent standardized beta (for multiple regression model) and the fraction of the variance explained by a predictor (dominance analysis). In addition to multiple regression analyses, confirmatory dominance analyses were performed due to the small sample size in some of the AD subtypes. The asterisk (*) in the table indicates when a significant predictor in the regression models (in bold) was rejected due to lack of stability in the dominance analysis (i.e. the percentage in bold is low, or equal to the one in not-bold). All p-values are corrected with Hochberg’s correction for multiple comparisons. AD = Alzheimer’s disease; adj. = adjusted; TMT-B = Trail Making Test part B; DS = digit symbol from the Wechsler Adult Intelligence Scale – Revised; BNT = Boston Naming Test.
In the healthy controls, learning was significantly associated with semantic abilities (β = 0.292) and processing speed (β = 0.229). The same pattern was obtained for typical AD and limbic-predominant, but for typical AD, lexical access was a significant predictor of learning as well (β = 0.219).
Delayed free recall was significantly predicted by learning alone in healthy controls (β = 0.744), typical AD (β = 0.560), and limbic-predominant (β = 0.483); and by both learning (β = 0.725) and attention/processing speed (β = −0.336) in the no atrophy group. In hippocampal-sparing, delayed free recall was associated to executive functioning (β = 0.548) and semantic abilities (β = 0.455), but not learning.
Finally, recognition was significantly predicted by learning alone in healthy controls (β = 0.469) and typical AD (β = 0.486); and by learning and lexical access in the no atrophy group (β = 0.445 and β = 0.357, respectively). Dominance analysis showed that semantic abilities are also important for recognition in the no atrophy group.
Disease progression over two years
Figure 4 shows the longitudinal progression in the visual rating scores across study groups.
Figure 4. Disease progression over two years.

Longitudinal scores for the visual rating scales (i.e. MTA, GCA-F, and PA) were available for 190 cases (110 healthy controls, 39 typical AD, 15 limbic-predominant, 13 hippocampal-sparing, 13 no atrophy group). Scores from MTA left and MTA right were averaged for simpler representation since longitudinal progression was similar in the two of them. Longitudinal values for CDR were available for 229 cases (147 healthy controls, 41 typical AD, 16 limbic-predominant, 12 hippocampal-sparing, 13 no atrophy group). Mixed effects analysis uses all data available at each time point. MTA = medial temporal atrophy visual rating scale; posterior atrophy visual rating scale; global cortical atrophy visual rating scale – frontal subscale; AD = Alzheimer’s disease; HC = healthy controls; CDR = clinical dementia rating; MMSE = mini-mental state examination.
The mixed effects model showed that clinical progression (CDR) over 2 years was faster in all the AD subtypes (estimates between 0.14 and 0.29) than in the healthy controls (estimate = 0.03) (F(4, 690) = 46.896; p < 0.001) (Fig. 4). Moreover, the slope was greater in typical AD (estimate = 0.29) than in hippocampal-sparing (estimate = 0.16; t(728) = −2.998; p = 0.015) and the no atrophy group (estimate = 0.14; t(680) = −3.443; p < 0.001); and in limbic-predominant (estimate = 0.28) than in hippocampal-sparing (t(708) = −2.374; trend for significance: p = 0.054) and the no atrophy group (t(672) = −2.746; p = 0.024). No significant effects were obtained for the quadratic model, indicating that clinical progression in all the subtypes is lineal rather than quadratic.
Regarding global cognitive decline (MMSE), all the AD subtypes had faster decline over 2 years (estimates between −3.08 and −1.48) than the healthy controls (estimate = −0.01) (F(4, 676) = 57.432; p < 0.001) (Fig. 4). Moreover, the slope was greater in typical AD (estimate = −2.69) than in hippocampal-sparing (estimate = −1.48; t(704) = 3.152; p = 0.010); and in limbic-predominant (estimate = −3.08) than in hippocampal-sparing (t(688) = 3.551; p < 0.001) and the no atrophy group (t(663) = 2.850; p = 0.020). No significant effects were obtained for the quadratic model, indicating that cognitive decline in all the subtypes is lineal rather than quadratic. All these results stand after controlling for age, gender, and years of education (data not shown).
Discussion
The AD subtypes investigated here have consistently been identified in previous studies. However, a method to enable their investigation in the clinical routine has been lacking so far. In the current study, visual rating scales were used as subtyping method because they are the primary method for assessing brain structural changes in clinical settings18,19,20,21. Although grouped data is reported, the visual rating scales can be applied at the individual level for clinical diagnosis. The aims in the current study were to (1) validate the combined use of visual rating scales for identification of AD subtypes; (2) characterize the resulting subtypes; and (3) investigate how atrophy patterns and non-memory cognitive domains contribute to memory impairment. The findings show that visual rating scales help to identify distinct AD subtypes with different disease progression. However, routine cognitive and clinical evaluations, CSF biomarkers and APOE ε4 genotype did not allow such discrimination, which may limit their clinical use for subtypes identification. Typical AD was the most impaired subtype at baseline and together with limbic predominant AD had worse clinical progression. Identification of patients with hippocampal-sparing and no atrophy is also clinically relevant since they showed less aggressive disease progression.
Visual rating scales are feasible and reproducible19,21,22,23,24,25. Intra-rater values (weighted κ) are usually around 0.90 for MTA19,23, and between 0.70 and 0.90 for GCA and PA19,23,25. Inter-rater values (weighted κ) are usually around 0.85 for MTA23, and between 0.60 and 0.80 for GCA and PA23,25. These weighted κ values correspond to substantial and almost perfect agreement26, thus proving their reproducibility.
The subtypes in this study were defined a-priori based on converging evidence suggesting three AD subtypes1,2,3,4,5,9,10,11,14,15. Vertex analyses in the current study showed that visual rating scales can successfully identify patterns of atrophy similar to those depicted by previous sophisticated MRI studies1,2,11,14,15, and coherent with those tracking the spread of NFT9. The prevalence of different subtypes obtained in this study are very similar to those previously reported, where typical AD usually includes 50–75% of the AD patients, limbic-predominant is referred in around 15–35%, and hippocampal-sparing in around 10–25%1,4,9,11,15. In the only study identifying a no atrophy group, the prevalence was 10%11. Since visual rating scales can be easily applied in the clinical routine18, this finding may have significant impact for current diagnosis and management of AD patients in clinical settings. The fact that the four subtypes were rather comparable on cognition, CSF biomarkers and APOE ε4 distribution at baseline further supports the use of MRI to identify clinically relevant subtypes. Different longitudinal progression of these subtypes supports this approach as well.
There was great overlap in the clinical and cognitive profiles, especially among the atypical AD subtypes, which highlights the difficulty in detecting these in routine clinical evaluations by only using clinical or cognitive measurements. Despite this, when comparing our results with other studies, the characteristics of our AD subtypes are largely comparable with what has previously been reported. The typical AD subtype has previously been found to be among the oldest1,2,4,9,11,15, have later onset1,4,9,11,15, include a higher frequency of males2,11, and have similar disease duration to limbic-predominant and hippocampal-sparing1,9,11. Contrary to our finding, hippocampal-predominant AD has been found to be among the oldest groups1,4,9,15 and to have shorter disease duration14. An explanation could be that atrophy in the medial temporal lobe is frequent in normal aging, hence clinical cut-offs for the medial temporal atrophy (MTA) scale are age-corrected19. This could lead to a younger hippocampal-predominant subtype in the present study. Age-corrections were not performed in previous studies except in Byun et al.11 and Varol et al.2, who indeed showed consistent results with our findings. Also contrary to our finding, hippocampal-sparing has been related with younger age1,4,9,15, earlier onset1,4,9,14,15, and shorter disease duration4. These results in previous studies could be explained by higher prevalence of early-onset AD in their samples, known to display predominant posterior atrophy and a more aggressive presentation9,27. However, early-onset AD is not common in ADNI-1, our sample, where posterior atrophy possibly results from a different process perhaps related or amplified by increased age.
To our knowledge, only our study and the one by Byun et al.11 have investigated a group of AD patients with no atrophy. Although data from ADNI-1 was used in both studies, we included 198 AD patients while Byun et al.11 included 163 AD patients. Most of the drops in Byun et al.11 corresponds to the hippocampal-sparing and the no atrophy group groups. Visual inspection of the demographic and clinical characteristics reveals that both groups are largely comparable, although our no atrophy group is younger and has higher frequency of females. An age correction was performed in both studies but the gender correction was not performed in our study because scores in our scales of posterior and frontal atrophy are not influenced by gender, and scores in MTA are only marginaly influenced by gender19. Thus, the gender correction and the fact that most of the droped cases in the no atrophy group from Byun et al.11 were young females, could be the explanation for these differences.
Interactions between memory components and contribution of non-memory cognitive functions were investigated for the first time in a study of this kind. Learning capacity was compromised in the four subtypes, but typical AD and limbic-predominant evidenced more consolidation problems, while hippocampal-sparing and the no atrophy group showed more problems in free retrieval of information. In addition, hippocampal-sparing was the group showing greater vulnerability to interference. Similar results were obtained in another study using ADNI-1 data5. The cognitive profiles discussed above were further confirmed in the regression/dominance analysis, where delayed recall and recognition were more dependent on non-memory cognitive functions in hippocampal-sparing and the no atrophy group than in typical AD and limbic-predominant. These profiles are coherent with the underlying pattern of brain atrophy. Noh et al.1 found prominent memory impairment in their parietal predominant AD subtype (analogous to hippocampal-sparing). The authors suggested that memory deficits might be associated with attention and working memory dysfunction in their parietal predominant subtype1. Hence, despite great overlap among AD subtypes, the nature of memory impairment seems to be different5. In this regard, Whitwell et al.9 made an important observation: a patient with Alzheimer’s dementia dominated by memory impairment can have any of the three subtypes of AD. This is exactly what we demonstrate in the current study, since all our AD patients have memory impairment, and we further demonstrate that the determinants of this memory impairment are varied and correspond with different atrophy patterns. This finding may have clinical utility, both for diagnosis/prognosis and cognitive interventions. Likewise, it is very important to note that these cognitive profiles were obtained using multivariate statistical methods on grouped data. Such information can be very difficult to find at the individual level in clinical routine, not to mention with often used cognitive tests. Thus, the use of visual rating scales seems to be extremely beneficial in this context.
One limitation in the current study is that ADNI-1 is a quite homogeneous sample. All AD patients fulfil the amnestic criteria at entry and aspects such as vascular pathology are excluded. Our results should thus be replicated in a more heterogeneous clinical sample that also includes non-amnestic AD presentations. It could also be argued that the different subtypes identified here reflect AD patients at different stages of the disease (e.g. typical AD being a later stage of limbic-predominant AD), rather than truly distinct AD subtypes. However, it has been demonstrated that these subtypes result from differential spread of NFT4. Further, no differences were observed on scales that stage the disease such as CDR, neither on disease duration. Related to this, it could be argued that the no atrophy group might be an initial stage of the disease but, again, no differences on CDR or disease duration were obtained. Alternatively, it could be argued that visual rating scales might not be sensitive enough to capture subtle atrophy in this no atrophy group. However, automated MRI methods confirmed this lack of atrophy in our vertex analysis, and a similar result was obtained using voxel-based morphometry in another ADNI-1 study11. Finally, we performed group analyses in order to characterize the different AD subtypes. Due to large within-group variability, there was great overlapping between subtypes and some conclusions especially on the non-imaging variables may be difficult to translate to the individual level, where clinical diagnosis takes place.
In conclusion, visual rating scales can be used to identify distinct and clinically relevant AD subtypes. To note, these subtypes could not be properly discriminated based on other common clinical tools such as cognitive tests, CSF biomarkers or APOE ε4 distribution. It has previously been argued that heterogeneity in AD complicates overcoming the two main challenges in the field at present, namely, discovery of disease-modifying treatments and achievement of accurate diagnosis and clinical prognosis1,2,3,10,28. It has also been discussed that advancing in the knowledge on different AD subtypes could shed some light to recently failed clinical trials by enabling tailored treatments in more homogeneous subgroups of patients2,28. The same would also help to better discriminate between highly overlapping clinical phenotypes such as AD with significant frontal involvement and frontotemporal lobe dementia4,10,18,29. Findings from the current study suggest that visual rating scales may facilitate investigation of AD heterogeneity in clinical routine. Implementing AD subtypes based on visual rating scales in the clinical routine should be easy and it is warranted to do as soon as possible in order to test its potential clinical impact. Whether using this method clinically may improve diagnosis and prognosis in “real world” AD patients stills needs to be determined.
Methods
Participants
AD patients and healthy controls from the ADNI-1 (adni.loni.usc.edu, PI Michael M. Weiner) with longitudinal data available at 1 and 2 years follow-up were selected for the current study, giving a total of 198 AD patients and 230 healthy controls. ADNI was launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies, and non-profit organizations12. The project was established to develop standardized imaging techniques and biomarkers in AD research. The AD patients and healthy controls were clinically diagnosed following standard procedures as detailed before19. Of note, memory impairment was required for all the AD patients based on Logical Memory II (Wechsler Memory Scale – Revised, WMS-R30). All diagnoses were made without the use of MRI scans. The study was approved by the institutional review boards of all participating ADNI centres. Written informed consent was obtained from all participants or authorized representatives after extensive description of the ADNI according to the Declaration of Helsinki. All methods were performed in accordance with the relevant guidelines and regulations.
Magnetic resonance imaging, automated image processing, and visual rating scales
A 3D T1-weighted magnetization-prepared rapid gradient-echo (MPRAGE) sequence was acquired on 1.5 T MRI scanners (voxel size 1.1 × 1.1 × 1.2 mm3)12. TheHiveDB Database system31 was used to automatically process the images with FreeSurfer 5.3.0, following previously described standard procedures32. This procedure provides measurements of cortical thickness at the vertex level, as well as a measurement of the total intracranial volume (TIV).
Regional atrophy was measured with visual rating scales based only the T1-weigthed images as detailed elsewhere19. Briefly, atrophy in the medial temporal lobe was evaluated with the MTA scale33; 2) atrophy in the posterior cortex was evaluated with the posterior atrophy (PA) scale23; and 3) atrophy in the frontal lobe was evaluated with the global cortical atrophy scale – frontal subscale (GCA-F)25. The MTA scale scores the degree of atrophy from zero to four in the hippocampus, parahippocampal gyrus, entorhinal cortex and the surrounding cerebrospinal fluid spaces. The PA scale scores the degree of atrophy from zero to three in the posterior cingulate sulcus, precuneus, parieto-occipital sulcus and the parietal cortex. The GCA-F scale scores the degree of atrophy from zero to three in the frontal lobe as delimited by the central sulcus, the frontal bone and the fissure of Sylvius. Therefore, the three scales primarily reflect cortical atrophy. Nonetheless, these scales also indirectly reflect ventricular enlargement since the inferior lateral ventricles are considered in MTA ratings, and GCA and PA ratings are based on widening of sulci, which is frequently correlated with ventricular enlargement. This is relevant because AD patients have more brain atrophy and larger ventricles than controls in the ADNI-1 cohort34. On the other hand, these scales are not designed to capture white matter hyperintensities (WMH), which are commonly regarded as markers of cerebrovascular disease. However, WMH burden in ADNI-1 is minimal relative to other cohorts35 due to exclusion of individuals with a Hachinski Ischemic Score36 of more than or equal to 5.
In the three visual rating scales, a score of zero denotes no atrophy, whereas scores from one to four indicate an increasing degree of atrophy. MTA analysis was based on coronal reconstructions, GCA-F on axial reconstructions and PA on reconstructions from all three planes. The images were rated both at baseline and at two years follow-up. Subtyping is based on baseline ratings, while longitudinal ratings were used to study disease progression over two years.
All cases were rated by an experienced radiologist (L.C.). Intra-rater reliability in 120 random cases achieved a weighted κ of 0.94 and 0.89 for MTA in left and right hemispheres, respectively, 0.88 for PA, and 0.83 for GCA-F. The same 120 random cases were also rated by a newly trained radiologist (C.-J.G.) for inter-rater analysis: weighted κ of 0.71 and 0.70 for MTA in left and right hemispheres, respectively, 0.88 for PA, and 0.79 for GCA-F. Both raters were blind to any information about the participants.
AD subtypes based on patterns of brain atrophy
Deviation from normality was established following a recently proposed list of practical cut-offs19. The MTA scores ≥1.5, ≥1.5, ≥2, ≥2.5 were considered abnormal for the respective age ranges 45–64, 65–74, 75–84, and 85–94 years. A previous study using the same dataset as here demonstrated that an age-correction does not improve PA and GCA-F diagnostic performance19. Therefore, the same cut-off was used for PA and GCA-F. A score ≥1 was considered abnormal irrespectively of the age range19. The three AD subtypes identified in previous literature4,9 were defined based on the combination of MTA, PA, and GCA-F as follows (see also Fig. 1). The typical AD subtype was defined as atrophy in the medial temporal lobe (abnormal MTA) together with atrophy in the posterior cortex (abnormal PA) and/or frontal cortex (abnormal GCA-F). The limbic-predominant subtype was defined as atrophy in the medial temporal lobe alone (abnormal MTA with normal PA and GCA-F). The hippocampal-sparing subtype included atrophy in the posterior cortex (abnormal PA) and/or frontal cortex (abnormal GCA-F), but not in the medial temporal lobe (normal MTA). A group with no atrophy was also identified as in Byun et al.11 when AD patients displayed normal scores in MTA, PA, and GCA-F.
Demographic and clinical variables
Age, gender, and years of education were included as demographic variables. Clinical severity was assessed with the CDR37 scale and global cognition with the MMSE38. FAQ39 was used to measure functional activities of daily living and GDS40 to measure depressive symptomatology. Age at disease onset, disease duration, and APOE ε4 status were also measured. Memory was assessed with the Auditory Verbal Learning test (AVLT)41. The different AVLT items were used to investigate different memory components. In particular, the sum of the five learning trials of the list A reflects learning capacity. Performance in the list B served as an estimate of interference effects during learning. Interference effects are frequently interpreted as distortions of existing memories possibly due to source-monitoring deficits. Free recall of the list A right after recall of the list B measures immediate recall. Free recall of the list A 30 minutes after learning measures delayed recall. Recognition reflects the ability to identify previously learned items from the list A. Finally, gain was calculated by subtracting the recognition percentage from the delayed percentage in order to quantify benefit from additional help when retrieving stored information. Executive functions (Trail Making Test part B, TMT-B)42, attention/processing speed (Digit Symbol, DS, from the Wechsler Adult Intelligence Scale – Revised, WAIS-R)43, language (Boston Naming Test, BNT)44, and semantic abilities (semantic fluency, vegetables)45 were also assessed. Digit span from the WMS-R30 and the Clock Test46 were further included for the random forest models (see statistical analysis). CSF samples were available for 102 AD patients and 115 healthy controls. Complete procedure descriptions are available at www.adni-info.org.
Statistical analysis
Mixed effects models (fixed and random effects) and mixed ANOVA/ANCOVA (split plot) were used to analyse the interaction between a between-subjects factor (study group) and a within-subjects factor (memory component and time). In the mixed effects models, the fixed-effect factors were study group, time, and the study group-by-time interaction. The random effect factor was the participants. When time was included in the model, both linear and quadratic effects were tested in order to investigate whether for example disease progression is linear or gets accelerated/decelerated after a certain time point. Multiple linear regression (backwards) was performed to analyse the contribution of non-memory cognitive functions to different memory components. Confirmatory dominance analyses, an extension of multiple regression, were performed with a non-parametric test based on bootstrapping (1000 iterations), which is less vulnerable to small sample sizes. Random forest analysis (500 trees) was also used to investigate differences between groups in multiple variables while avoiding multiple testing. P-values in all principal and post-hoc analyses were adjusted with the Benjamini-Hochberg’s47 correction for multiple comparisons. Model assumptions were tested in all the cases by visual inspection of residuals and data distribution, as well as by inspecting the pertinent statistical parameters. Results were considered significant when p ≤ 0.05 (two-tailed).
Image analyses based on the vertex across the cortical mantle were carried out using FreeSurfer software as detailed elsewhere48. Briefly, maps were smoothed using a circularly symmetric Gaussian kernel across the surface with a full width at half maximum (FWHM) of 10 mm. A general linear model was fitted at each vertex. Study group was entered as independent variable and TIV as a covariate. Z Monte Carlo simulations were used with a cluster-forming threshold of p ≤ 0.001 (two-sided), yielding results corrected for multiple comparisons.
Additional Information
How to cite this article: Ferreira, D. et al. Distinct subtypes of Alzheimer’s disease based on patterns of brain atrophy: longitudinal trajectories and clinical applications. Sci. Rep. 7, 46263; doi: 10.1038/srep46263 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This project is financially supported by the Swedish Foundation for Strategic Research (SSF). The authors also thank the Strategic Research Programme in Neuroscience at Karolinska Institutet (StratNeuro), Åke Wiberg foundation, Hjärnfonden, the Swedish research council (VR) Alzheimerfonden and Birgitta och Sten Westerberg for additional financial support. Data collection and sharing for the ADNI project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Contributors from the ADNI consortium are detailed in Supplementary Table 1. The above-mentioned funding sources had no involvement in the study design, collection, analysis and interpretation of the data, writing of the report and the decision to submit the article for publication. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Footnotes
The authors declare no competing financial interests.
Author Contributions The study design and concept was done by D.F., C.V., J.A.H.C. and E.W. Acquisition, analysis, and interpretation of data was done by D.F., C.V., J.A.H.C., L.C., C.J.G., U.E., J.S.M., A.S., J.B., L.O.W. and E.W. Drafting of the manuscript was done by D.F. and E.W. Critical revision of the manuscript for important intellectual content was done by D.F., C.V., J.A.H.C., L.C., C.J.G., U.E., J.S.M., A.S., J.B., L.O.W. and E.W. Statistical analyses were carried out by D.F., C.V. and J.A.H.C. L.O.W. and E.W. obtained funding. D.F., C.V., J.A.H.C., L.C., C.J.G., U.E., J.S.M., A.S., J.B., L.O.W. and E.W. provided admin.
References
- Noh Y. et al. Anatomical heterogeneity of Alzheimer disease: based on cortical thickness on MRIs. Neurology 83, 1936–1944 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol E., Sotiras A. & Davatzikos C. HYDRA: revealing heterogeneity of imaging and genetic patterns through a multiple max-margin discriminative analysis framework. Neuroimage pii: S1053-8119(16)00150-6. doi: 10.1016/j.neuroimage.2016.02.041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens N. M. E. et al. The identification of cognitive subtypes in Alzheimer’s disease dementia using latent class analysis. J. Neurol. Neurosurg. Psychiatry 87, 235–243 (2016). [DOI] [PubMed] [Google Scholar]
- Murray M. E. et al. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet. Neurol. 10, 785–96 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J. et al. Subgroups of Alzheimer’s disease: stability of empirical clusters over time. J. Alzheimers. Dis. 42, 651–661 (2014). [DOI] [PubMed] [Google Scholar]
- McKhann G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers. Dement. 7, 263–269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J. C. et al. Harmonized diagnostic criteria for Alzheimer’s disease: recommendations. J. Intern. Med. 275, 204–213 (2014). [DOI] [PubMed] [Google Scholar]
- Dubois B. et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 9, 1118–1127 (2010). [DOI] [PubMed] [Google Scholar]
- Whitwell J. L. et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer’s disease: a case-control study. Lancet. Neurol. 11, 868–877 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell J. L. et al. Temporoparietal atrophy: a marker of AD pathology independent of clinical diagnosis. Neurobiol. Aging 32, 1531–1541 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun M. S. et al. Heterogeneity of regional brain atrophy patterns associated with distinct progression rates in Alzheimer’s disease. PLoS One 10, e0142756 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S. G. et al. The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clin. N. Am. 15, 869–877 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R. et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J. Magn. Reson. Imaging 27, 685–991 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiino A. et al. Four subgroups of Alzheimer’s disease based on patterns of atrophy using VBM and a unique pattern for early onset disease. Neuroimage 33, 17–26 (2006). [DOI] [PubMed] [Google Scholar]
- Hwang J. et al. Prediction of Alzheimer’s disease pathophysiology based on cortical thickness patterns. Alzheimer’s Dement. (Amst) 2, 58–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson J. E. et al. An exploration of cognitive subgroups in Alzheimer’s disease. J. Int. Neuropsychol. Soc. 16, 233–243 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopford C. L., Snowden J. S., Thompson J. C. & Neary D. Variability in cognitive presentation of Alzheimer’s disease. Cortex 44, 185–195 (2008). [DOI] [PubMed] [Google Scholar]
- Harper L. et al. MRI visual rating scales in the diagnosis of dementia: evaluation in 184 post-mortem confirmed cases. Brain 139, 1211–1225 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira D. et al. Practical cut-offs for visual rating scales of medial temporal, frontal, and posterior atrophy in Alzheimer’s disease and mild cognitive impairment. J. Intern. Med. 278, 277–290 (2015). [DOI] [PubMed] [Google Scholar]
- Ferreira D. et al. Electroencephalography is a good complement to currently established dementia biomarkers. Dement. Geriatr. Cogn. Disord. 42, 80–92 (2016). [DOI] [PubMed] [Google Scholar]
- van der Flier W. M. et al. Optimizing patient care and research: the Amsterdam Dementia Cohort. J. Alzheimers. Dis. 41, 313–327 (2014). [DOI] [PubMed] [Google Scholar]
- van de Pol L. A. & Scheltens P. Medial temporal lobe atrophy scores translated to clinical practice: editorial comment on ‘influence of age, disease onset and ApoE4 on visual medial temporal lobe atrophy cut-offs’. J. Intern. Med. 275, 331–333 (2014). [DOI] [PubMed] [Google Scholar]
- Koedam E. L. et al. Visual assessment of posterior atrophy development of a MRI rating scale. Eur. Radiol. 21, 2618–2625 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens P., Pasquier F., Weerts J., Barkhof F. & Leys D. Qualitative assessment of cerebral atrophy on MRI: inter- and intra-observer reproducibility in dementia and normal aging. Eur. Neurol. 37, 95–99 (1997). [DOI] [PubMed] [Google Scholar]
- Ferreira D. et al. Quantitative validation of a visual rating scale for frontal atrophy: associations with clinical status, APOE e4, CSF biomarkers and cognition. Eur. Radiol. 26, 2597–2610 (2015). [DOI] [PubMed] [Google Scholar]
- Landis J. & Koch G. The measurement of observer agreement for categorical data. Biometrics 33, 159–174 (1977). [PubMed] [Google Scholar]
- van der Flier W. M., Pijnenburg Y. Al, Fox N. C. & Scheltens P. Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE ɛ4 allele. Lancet Neurol. 10, 280–288 (2011). [DOI] [PubMed] [Google Scholar]
- Kovacs G. G. Clinical stratification of subtypes of Alzheimer’s disease. Lancet. Neurol. 11, 839–841 (2012). [DOI] [PubMed] [Google Scholar]
- Rivero-Santana A. et al. Cerebrospinal fluid biomarkers for the differential diagnosis between Alzheimer’s disease and frontotemporal lobar degeneration: systematic review, HSROC analysis, and confounding factors. J. Alzheimer’s Dis. 55, 625–644. (2016). [DOI] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Memory Scale - Third Edition. Technical Manual. (The Psychological Corporation, 1997). [Google Scholar]
- Muehlboeck J.-S., Westman E. & Simmons A. TheHiveDB image data management and analysis framework. Front. Neuroinform. 7, 49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira D. et al. Cognitive decline is mediated by gray matter changes during middle age. Neurobiol. Aging 35, 1086–1094 (2014). [DOI] [PubMed] [Google Scholar]
- Scheltens P. et al. Atrophy of medial temporal lobes on MRI in ‘probable’ Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J. Neurol. Neurosurg. Psychiatry 55, 967–972 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana C. et al. Measuring global brain atrophy with the brain volume/cerebrospinal fluid index: normative values, cut-offs and clinical associations. Neurodegener. Dis. 16, 77–86 (2016). [DOI] [PubMed] [Google Scholar]
- Ramirez J., McNeely A. A., Scott C. J. M., Masellis M. & Black S. E. White matter hyperintensity burden in elderly cohort studies. The Sunnybrook Dementia Study, Alzheimer’s Disease Neuroimaging Initiative, and Three-City Study. Alzheimers Dement. 12, 203–210 (2016). [DOI] [PubMed] [Google Scholar]
- Hachinski V. C. et al. Cerebral blood flow in dementia. Arch Neurol 32, 632–637 (1975). [DOI] [PubMed] [Google Scholar]
- Morris J. C. The Clinical Dementia Rating (CDR): current vision and scoring rules Neurology. Neurology 43, 2412–2414 (1993). [DOI] [PubMed] [Google Scholar]
- Folstein M. F., Folstein S. E. & McHugh P. R. Mini-mental- state. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198 (1975). [DOI] [PubMed] [Google Scholar]
- Pfeffer R. I., Kurosaki T. T., Harrah C. H., Chance J. M. & Filos S. Measurement of functional activities in older adults in the community. J. Gerontol. 37, 323–329 (1982). [DOI] [PubMed] [Google Scholar]
- Sheikh J. & Yesavage J. Geriatric Depression Scale (GDS): recent evidence and development of a shorter version. Clin Gerontol 5, 165–173 (1986). [Google Scholar]
- Schmidt M. Rey Auditory Verbal Learning Test: A Handbook. (Western Psychological Services, 1996). [Google Scholar]
- Reitan R. Validity of the trail making test as an indicator of organic brain damage. Percept. Mot. Skills 8, 271–276 (1958). [Google Scholar]
- Wechsler D. Wechsler Adult Intelligence Scale - Administration and Scoring Manual, Third Edition. (The Psychological Corporation, San Antonio, TX., 1997). [Google Scholar]
- Kaplan E., Goodglass H. & Weintraub S. Boston Naming Test. (Lea & Febiger, 1983). [Google Scholar]
- Benton A., Hamsher K. & Sivan A. Multilingual Aphasia Examination- Second Edition. (AJA Associates, University of Iowa., 1989). [Google Scholar]
- Goodglass H. & Kaplan E. The Assessment Of Aphasia And Related Disorders. (Lea & Febiger., 1983). [Google Scholar]
- Hochberg Y. & Benjamin Y. More powerful procedures for multiple significance testing. Stat. Med. 9, 811–818 (1990). [DOI] [PubMed] [Google Scholar]
- Ferreira D. et al. Different reserve proxies confer overlapping and unique endurance to cortical thinning in healthy middle-aged adults. Behav. Brain Res. 311, 375–383 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.