

Abstract
Mercury toxicity mechanisms have the potential to induce DNA damage and disrupt cellular processes, like mitochondrial function. Proper mitochondrial function is important for cellular bioenergetics and immune signaling and function. Impacts of mercury on the nuclear genome (nDNA) are conflicting and inconclusive, and mitochondrial DNA (mtDNA) impacts are relatively unknown. In this study, we assessed genotoxic (mtDNA and nDNA), metabolic, and innate immune impacts of inorganic and organic mercury exposure in Caenorhabditis elegans. Genotoxic outcomes measured included DNA damage, DNA damage repair (nucleotide excision repair, NER; base excision repair, BER), and genomic copy number following MeHg and HgCl2 exposure alone and in combination with known DNA damage-inducing agents ultraviolet C radiation (UVC) and hydrogen peroxide (H2O2), which cause bulky DNA lesions and oxidative DNA damage, respectively. Following exposure to both MeHg and HgCl2, low-level DNA damage (~0.25 lesions/10 kb mtDNA and nDNA) was observed. Unexpectedly, a higher MeHg concentration reduced damage in both genomes compared to controls. However, this observation was likely the result of developmental delay. In co-exposure treatments, both mercury compounds increased initial DNA damage (mtDNA and nDNA) in combination with H2O2 exposure, but had no impact in combination with UVC exposure. Mercury exposure both increased and decreased DNA damage removal via BER. DNA repair after H2O2 exposure in mercury-exposed nematodes resulted in damage levels lower than measured in controls. Impacts to NER were not detected. mtDNA copy number was significantly decreased in the MeHg-UVC and MeHg-H2O2 co-exposure treatments. Mercury exposure had metabolic impacts (steady-state ATP levels) that differed between the compounds; HgCl2 exposure decreased these levels, while MeHg slightly increased levels or had no impact. Both mercury species reduced mRNA levels for immune signaling-related genes, but had mild or no effects on survival on pathogenic bacteria. Overall, mercury exposure disrupted mitochondrial endpoints in a mercury-compound dependent fashion.
Keywords: Caenorhabditis elegans, DNA damage, Copy number, Mercury, Mitochondria, Innate immunity
1. Introduction
Mercury is a heavy metal of great interest because of its pervasive presence in the environment and recognized adverse impacts on multiple systems, including most prominently the nervous system. Nervous system and other impacts, including cell injury and death, are likely derived from multiple mechanisms that could also impact DNA homeostasis and alter mitochondrial functions. Proposed genotoxic mechanisms are mostly indirect and include: altering the balance of antioxidant enzymes in favor of a pro-oxidant environment (Stohs and Bagchi 1995, Valko et al. 2005); impairing DNA repair enzymes (Crespo-Lopez et al. 2009); reactive oxygen species (ROS) generation as a result of the inhibition of electron transport chain (ETC) proteins (Yee and Choi 1996, Ni et al. 2010); and altered calcium homeostasis which has been associated with apoptosis and necrosis (Ceccatelli et al. 2010).
Protein impairment from both inorganic and organic exposure is considered an important mechanism in mercury toxicity. Mercury has a strong affinity for thiol and selenol groups, and by binding to these groups mercury can impair protein function (Stohs and Bagchi 1995). Reductions in antioxidant enzyme activity occur following chronic exposure due to inhibition of enzymes such as glutathione reductase, glutathione peroxidase, and superoxide dismutase (Berntssen et al. 2003, Mori et al. 2007), and in one report persisted beyond mercury elimination after developmental exposures in rodents via an unidentified mechanism (Stringari et al. 2008). Declines in antioxidant function can promote oxidative stress as a result of diminished ability to properly cope with endogenous as well as exogenous oxidants (Stringari et al. 2008, Franco et al. 2009). The promotion of oxidative stress may also be related to mitochondrial impacts. Mercury accumulates in the mitochondria, in addition to the nucleus and lysosomes (Atchison and Hare 1994, Bucio et al. 1999, Ikemoto et al. 2004). In addition, ROS including superoxide anion and hydrogen peroxide are produced at higher than normal levels following methylmercury exposure due to the inhibition of ETC proteins and reduced superoxide dismutase scavenging activity (Yee and Choi 1996). Other reported mitochondrial impairments include increased oxygen consumption and increased membrane permeability from augmented calcium influx (Yee and Choi 1996, Yin et al. 2007, Polunas et al. 2011). The combination of reduced antioxidant capabilities and increased ROS may promote damage to macromolecules, including DNA.
DNA damage following mercury exposure has been observed and is thought to occur through either direct oxidation of DNA (Williams et al. 1987, Ondovcik et al. 2012) and/or reduced repair capacity due to impaired DNA repair enzymes (Cebulska-Wasiewska et al. 2005, Gadhia et al. 2012, Pieper et al. 2014, Ryu et al. 2014). Laboratory, field, and epidemiological studies have reported increased strand breaks, chromosome aberrations, 8-hydroxy-2′-deoxyguanosine (8-OHdG), micronuclei, and reduced DNA repair (Al-Saleh et al. 2012, Gadhia et al. 2012, Ryu et al. 2014). However, there are discrepancies among these studies. In some studies, mercury has a strong dose-response relationship below cytotoxic levels, but in others, increased DNA damage was not observed at exposures below cytotoxic levels or when not in combination with another exposure (radiation, H2O2) (Cebulska-Wasiewska et al. 2005, Al Bakheet et al. 2013, Pieper et al. 2014). Epidemiological studies indicate that DNA damage is possible in fish-eating populations, in children, and most importantly at exposure levels lower than those known to adversely impact the nervous system (Franchi et al. 1994, Amorim et al. 2000, Cebulska-Wasiewska et al. 2005, Di Pietro et al. 2008, Al-Saleh et al. 2012). The focus of previous studies has primarily been on impacts to the nuclear genome, with only one study to date reporting mtDNA impacts (Karouna-Renier et al. 2014). Depending on the kind of damage, increased mtDNA damage and damage accumulation in the mitochondrial genome are possible as there are fewer DNA repair pathways in this organelle (Ledoux et al. 1992, Scheibye-Knudsen et al. 2015, Van Houten et al. 2016). Damage to DNA, mitochondria, and other cellular components can lead to broader impacts.
In addition to neurological impacts, multiple effects of mercury exposure on immune responses have been reported, including both increased and decreased antibody and cytokine concentrations (Bagenstose et al. 2001, Silva et al. 2005, Begam and Sengupta 2015). Immune system impacts may be in part related to impaired mitochondrial function as mitochondria have an important role in immune system function, including ROS signaling and generation of mitochondrial specific damage-associated molecular patterns that activate pattern-recognition receptors (West et al. 2011, Weinberg et al. 2015). Caenorhabditis elegans is a useful model to study innate immunity because it lacks an adaptive immune response, simplifying interpretation of outcomes, and has an innate immune system that shares similarities at the molecular level with that of higher eukaryotes. C. elegans mounts bacterial immune responses through well studied signaling pathways including p38 mitogen-activated protein kinase (MAPK) pathway, an insulin/insulin-like growth factor receptor (IGF) pathway, and transforming growth factor (TGF)-beta pathway (Aballay and Ausubel 2002, Kim et al. 2002, Aballay et al. 2003, Millet and Ewbank 2004, Bolz et al. 2010, TeKippe and Aballay 2010, Sun et al. 2011, Aballay 2013). The p38 MAPK pathway can be affected by ROS (Torres and Forman 2003, Bundy et al. 2005, Mendez-Samperio et al. 2010, Gostner et al. 2013). Additionally, human pathogens including gram-negative Pseudomonas aeruginosa can kill C. elegans using virulence factors required for pathogenicity in mammalian systems (Tan et al. 1999, Aballay and Ausubel 2002, Cai et al. 2014). In this paper, we test potential impacts of mercury on one innate signaling pathway, through a mitogen-activated protein kinase (MAPK), in C. elegans. The impact of one environmental stressor, heat-shock, has been observed to impact innate immunity in C. elegans (Mohri-Shiomi and Garsin 2008, Singh and Aballay 2009), but the effect of other environmental exposures, such as heavy metal exposures, has not been assessed to date in this system.
The aim of this study was to assess genotoxic, mitochondrial, and immunotoxic endpoints following inorganic and organic mercury exposure using the model organism C. elegans, which has increasingly been used as a model for assessing DNA damage and mitochondrial toxicity (Tsang and Lemire 2002, 2003, Reinke et al. 2010) caused by environmental stressors (Leung et al. 2008, Liuzzi et al. 2012, Meyer et al. 2013, Turner et al. 2013, Polli et al. 2014). Our objectives were to assess the impact of mercury exposure on: 1) DNA damage and repair, 2) mitochondrial parameters (DNA copy number and steady-state ATP levels), and 3) innate immunity.
2. Materials and Methods
2.1 C. elegans and bacterial strains
Nematode populations were maintained at 20 °C on K agar plates seeded with OP50 strain Escherichia coli unless otherwise stated (Lewis and Fleming 1995). N2 (wild-type) and JK1107 glp-1(q224) were obtained from the Caenorhabditis Genetics Center (CGC, University of Minnesota). PE255 glp-4 (bn2) strain was generously provided by Dr. Christina Lagido (University of Aberdeen, UK). KU25 pmk-1(km25) were obtained from CGC. glp strains were maintained at 15 °C, until time of experiment, and then shifted to the restrictive temperature of 25 °C to limit germ cell production. Experiments involving DNA damage utilized the germline-deficient mutant (glp-1) to minimize the potential confounding of DNA damage dilution from dividing germ cells, as no cell division occurs in adult the adult life-stage (Sulston 1988). Steady-state ATP levels were determined using the transgenic, firefly luciferase-expressing nematode strain PE255 (glp-4), as previously described (Lagido et al. 2008, Lagido et al. 2009). N2 and pmk-1 were utilized to assess the impact of mercury on both immunocompetent and immune compromised nematodes. PMK-1 is a p38 mitogen-activated protein kinase that has a protective role in infection and is required for immune induction (Kim et al. 2002, Kim and Mylonakis 2012). The pathogenic Pseudomonas aeruginosa strain PA14 was also used for the immune function experiments. For all experiments, synchronized L1 larvae were obtained by treating gravid adults with a 5% sodium hypochlorite solution and hatching eggs in the absence of food (K medium plus MgSO4, CaSO4, and cholesterol) (Lewis and Fleming 1995). A general schematic of the experimental design is presented in Figure 1.
Figure 1.

General experimental schematic
2.2 Experiment 1: DNA damage and genome copy number following mercury exposure
Nematodes (glp-1) were grown to young adult stage (36 hr at 25 °C) and then transferred to 24-well plates (100–230 nematodes per well) and exposed to control conditions, HgCl2 (1 and 5 μM), or MeHgCl (1 and 5 μM) for 24hr and then sampled to assess DNA damage. Liquid exposures were performed in EPA reconstituted moderately hard water (hereafter described as “EPA water”) plus UVC-killed E. coli (UVRA strain), to eliminate the potentially confounding effect of bacterial metabolism on exposures, as previously described (Meyer et al. 2010, Yang et al. 2012). The experiment was repeated three times separated in time (n=9–27).
2.3 Experiment 2: DNA damage repair and removal following mercury exposure
Young adult glp-1 nematodes were exposed to mercury and then two prototypical DNA damage-inducing agents, ultraviolet C radiation (UVC) and hydrogen peroxide (H2O2), to test if mercury would inhibit repair or removal of DNA damage. HgCl2 (5 μM) and MeHgCl (1 μM) concentrations were chosen based on the exposures that caused similar increases in DNA damage in Experiment 1 but did not result in significant growth reductions. We excluded growth-inhibiting exposure levels because delayed development is associated with increased DNA repair gene expression (Boyd et al. 2010), and, in this study, with lower basal levels of DNA damage (Figure 1). These differences would confound our ability to isolate the effect of mercury exposure. Nematodes were then washed and exposed to UVC or H2O2. UVC exposure was chosen to assess nucleotide excision repair (NER) which removes bulky lesions including photodimers in the nucleus, and mitophagy which removes the same types of damage from mtDNA. H2O2 exposure was chosen to evaluate base excision repair (BER) which is responsible for removal of most oxidative damage in both genomes.
Nematodes (glp-1) were raised to young adult stage in the same manner as Experiment 1. Following 36 hr, nematodes were transferred to 24-well plates (100–220 nematodes per well) containing control, 5 μM HgCl2, or 1 μM MeHgCl in EPA water plus UVC-killed E. coli for 24 hr. For UVC exposures, nematodes were transferred to K agar plates without OP50 and exposed to 50 J/m2 UVC using an ultraviolet lamp (UVLMS-38 EL Series 3UV Lamp, UVP, Upland, CA, USA) with peak emission at 254 nm. UVC doses were quantified using a UVX digital radiometer. For H2O2 exposures, nematodes were transferred to 24-well plates and exposed to 5 mM H2O2 for 1 hr without food. Immediately following the DNA damage event nematodes were sampled (0 hr) and then returned to their original medium (control or mercury condition). DNA damage removal was assessed by sampling at 6, 24, and 48 hr following the DNA damage event. The experiment was repeated three times separated in time (n=9–21 for each time-point).
2.4 Experiment 3: ATP determination following mercury exposure and DNA damage
Utilizing a similar experimental design as Experiment 2, steady-state ATP levels were determined at 24 and 48hr following the DNA damage event, essentially as described (Lagido et al. 2008, Lagido et al. 2009). Differences included using a transgenic, luciferase-expressing nematode strain (PE255 glp-4). For each time point 100 nematodes (in 100 μL K-medium) were loaded into each well of a white 96-well plate, such that each treatment was loaded into four separate wells (i.e. four technical replicates). GFP fluorescence, which is used to normalize luminescence readings to GFP-luciferase fusion protein expression levels, was measured using a FLUOstar OPTIMA BMG Labtech plate reader (Ortenberg, Germany; excitation filter: 485 nm; emissions filter: 512 nm). An automated dispenser was then used to deliver 50μl of luminescence buffer (citrate phosphate buffer pH 6.5, 0.1 mM D-luciferin, 1% DMSO, 0.05% Triton X-100) to each well, and luminescence was measured in the visible spectral range of 300–600 nm. All luminescence values were normalized to their corresponding GFP fluorescence value and ATP values are reported as percent of control for each time-point. The experiment was repeated four times (n=4 for each time-point).
2.5 Experiment 4: Innate immunity in wild type and immune deficient nematodes, survival assay
The impact of larval mercury exposure (HgCl2 and MeHgCl) on an innate immunity against pathogenic bacteria was assessed by exposing N2 and pmk-1 nematodes to P. aeruginosa PA14. L1 nematodes were transferred to 24-well plates (800–2500 worms per well) and exposed to control, HgCl2: 0.5, 1, 2.5 μM, or MeHgCl: 0.5 μM for 40 hrs at 20 °C. Developmental stage is an important factor that needs to be considered when utilizing this survival assay, because survival time on PA14 is significantly shorter in younger worms (Tan et al. 1999). To avoid potential confounding, mercury concentrations that did not significantly impair nematode growth were used in this experiment. Note that these experiments involved exposures from the first larval stage, which is more sensitive than the later stages used in experiments reported in Figures 2–7. Of these developmental exposures, only 2.5 μM HgCl2 modestly reduced nematode growth.
Figure 2.

Mitochondrial and nuclear DNA damage (average lesions/10kb ±SE) (A) and relative copy number (average % of control ±SE) (B) in young adult nematodes following 24 hr HgCl2 and MeHg exposure. Red lines indicate the limit of detection for DNA lesions (0.1 lesions/10 kb). * indicates significantly different (p<0.05) from control.
Figure 7.

Interaction plots of steady-state ATP levels (% of control) of young adult nematodes exposed to control, mercury (5 μM HgCl2, 1 μM MeHg), or DNA damage (UVC, H2O2) conditions. Interaction lines and box-plots are dodged for better visual representation.
Following larval exposure, nematodes were washed and then transferred to survival plates containing E. coli OP50 or P. aeruginosa PA14. The bacterial lawns used for C. elegans killing assays were prepared by placing a 20 μL drop of an overnight culture of the bacterial strains on modified NGM agar on plates 3.5 cm in diameter. Full lawn plates used for C. elegans killing assays were prepared by spreading a 25 μL drop of an overnight culture grown at 37 °C of P. aeruginosa on the complete surface of modified NGM agar in 3.5 cm diameter Petri plates. Plates were incubated at 37 °C for 12~16 hrs. Plates were cooled to room temperature for at least one hour before seeding with synchronized young adult animals. The killing assays were performed at 25 °C and live animals were transferred daily to fresh plates. Animals were scored at the times indicated and were considered dead when they failed to respond to touch. The experiment was repeated 3–4 times separated in time (n=97–341).
2.6 DNA damage and genome copy number analysis
For damage and genome copy number assays, six nematodes were pooled and treated as a biological replicate. nDNA and mtDNA damage were assessed using a QPCR-based method as previously described (Gonzalez-Hunt et al. 2015). This assay measures lesion frequency based on decreases in amplification efficiency relative to controls, which are assumed to be undamaged (Meyer 2010). Two nuclear genome targets (unc-2 and small nuclear; 9.3 and 0.2 kb) and two mitochondrial genome targets (nd-1 and small mitochondrial; 10.9 and 0.2 kb) were amplified. The amount of amplified long PCR product provides a measurement of lesion frequency, while the amount of short PCR product provides normalization to DNA concentration and genome copy number (Furda et al. 2012).
2.7 Gene expression analysis
Expression of autophagy, mitophagy, biogenesis, BER-related DNA repair, and p38 MAPK-related genes were measured using the experimental design for Experiment 1. Nematodes were reared to the young adult stage and exposed to control conditions, HgCl2 (1 and 5 μM), or MeHgCl (1 and 5 μM) for 24hr. Total RNA from glp-1 nematodes was extracted using a RNeasy kit (Qiagen, Valencia, CA, USA), quantified with a NanoDrop Fluorospectrometer (NanoDrop Technologies, Wilmington, DE, USA), and converted to cDNA using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA) using methods previously described (Bess et al. 2013). Average fold change of each target gene was calculated by comparing the CT (cycle threshold) of the target gene to two housekeeping genes (tba-1 and pmp-3). Real-time PCR conditions were optimized for previously published and designed primers; the primer sequences and conditions are listed in Table A.9.
2.8 Assessment of autophagic foci
Autophagy was assessed using the QU1 izEx1[Plgg-1::gfp::lgg-1+rol-6] (LGG-1::GFP) reporter strain, where increased or impaired autophagic flux is represented by a change in the number ofLGG-1::GFP foci (Palmisano and Melendez 2016b), using the experimental design for Experiment 1. Nematodes were reared to the young adult stage and exposed to control conditions, HgCl2 (1 and 5 μM), or MeHgCl (1 and 5 μM) for 24hr. Following exposure, nematodes were picked onto 10% agar pads with 10 ml of 150mM sodium azide (Sigma Aldrich) (Bess et al. 2012). Single-plane images of seam cells were taken using a Zeiss 780 confocal microscope at 63× magnification and LGG-1::GFP foci in each seam cell were counted manually. The experiment was repeated two times separated in time. In total, 21–48 seam cells from 8–18 nematodes were analyzed per treatment.
2.9 Statistical analysis
Experiment 1 data were analyzed using a 2-way ANOVA with mercurial type (HgCl2, MeHg) and mercury concentration as factors. Significant interactions were followed up with Tukey’s post-hoc test for pairwise comparisons. Data from experiment 2 was analyzed with a 3-way ANOVA. For DNA damage data, significant 3-way ANOVAs were followed up with 2-way ANOVAs (mercury compound × time) for UVC/H2O2 exposed and non-UVC/H2O2 exposed nematodes. For copy number data, significant 3-way ANOVAs were followed up with 2-way ANOVAs (mercury compound × UVC/H2O2 exposure) for each time-point. Significant 2-way interactions for DNA damage data were followed up with Tukey’s post-hoc test for pairwise comparisons. ANOVA tables for Experiment 2 are presented in the Appendix. Experiment 3 data were analyzed with a 2-way ANOVA and significant interactions were followed up with Tukey’s post-hoc test for pairwise comparisons. Gene expression and LGG1::GFP data was analyzed using a 2-way ANOVA, similar to Experiment 1. Statistics for all experiments except survival were conducted using R (Version 3.2.2, Vienna, Austria). Nematode survival in Experiment 4 was plotted as a non-linear regression curve using the PRISM (version 4.00) computer program. Prism uses the product limit or Kaplan-Meier method to calculate survival fractions and the Mantel-Cox log-rank test to compare survival curves. Significance for all experiments was accepted at a level of p<0.05.
3. Results
3.1 Experiment 1: DNA damage and genome copy number following mercury exposure
To test the impact of mercury on mtDNA and nDNA damage and copy number, young adult nematodes were exposed to two concentrations of HgCl2 and MeHg for 24 hrs. DNA damage profiles were similar between the mitochondrial and nuclear genomes. For both mtDNA and nDNA the direction of the change in damage level from low to high concentration was different for the two mercury compounds (interaction between mercury compound and concentration, p<0.001). Increasing lesions were observed with increasing HgCl2 exposure, but for MeHg exposures there was increased damage at 1 μM and decreased lesions at 5 μM (Figure 2A). Genome copy number following mercury exposure differed between the two genomes. In the mitochondrial genome, there was a significant interaction between mercury compound and concentration (p<0.001), as the 5 μM MeHg treatment resulted in reduced mtDNA copy number, but other treatments did not. In the nuclear genome, mercury compound was a significant factor influencing copy number, with MeHg reducing significantly reducing copy number (main effect, p=0.03) by about 4% (Figure 2B).
To better understand the surprising decrease in mtDNA and nDNA damage at the high (5 μM) MeHg concentration, we carried out additional experiments to test possible hypotheses for this observation. First, we tested whether autophagy was increased, because autophagy can remove damaged mtDNAs (Bess et al. 2012) and may also reduce nDNA damage (Vessoni et al. 2013). Increased autophagy would also be consistent with the reduced mtDNA copy number that we also observed (Fig. 2). Consistent with this hypothesis, we identified increased formation of seam cell autophagic foci (Figure A.1). However, foci number was also increased at 1 μM MeHgCl, where we saw increased rather than decreased DNA damage. Importantly, increased foci may also result from inhibition of the autophagic process (i.e., inability to resolve autophagic foci (Palmisano and Melendez 2016a), so we also measured mRNA levels of inducible autophagy related genes (Zhang et al. 2015). The high MeHg concentration (5 μM) significantly reduced atg-18, dct-1, and hmg-5 and reduced (p=0.08) bec-1 expression, suggestive of decreased, rather than increased, autophagy and mitophagy (Figure A.2). We also tested for a transcriptional signal for induction of biogenesis (Figure A.2) that might offset autophagic removal of mitochondria, but did not find support for this possibility. We also considered whether transcripts for key genes involved in BER, which repairs oxidative damage likely to be caused by mercury exposure, would be increased. While not generally considered a highly inducible process, there is evidence for upregulation of some BER genes after exposures (Christmann and Kaina 2013). Instead, BER genes were expressed at similar or lower levels after both HgCl2 and MeHg (Figure A.3). We finally considered the possibility that BER might have been induced, leading to above-normal repair of endogenous DNA damage (i.e., negative lesions). Our data related to this hypothesis is presented below (Section 3.2.3), but overall did not explain the observation of below-baseline damage only in the high MeHg exposure group.
Finally, we considered the possibility that the developmental delay observed at the high concentration of MeHg (Figure A.4) could explain these results. Minimal growth retardation (< 5% growth reduction) was observed following HgCl2 (1 and 5 μM) and 1μM MeHgCl exposures, while 5 μM MeHgCl reduced growth by ~10% (Figure A.4). While there haven’t been previous reports of baseline changes in DNA damage with development, both nDNA and, more dramatically, mtDNA copy number increase during these lifestaqes (Tsang and Lemire 2002), including in glp-1(q244) nematodes (Rooney et al. 2014), and the decreased mtDNA copy numbers that we observed could well be explained by developmental delay. We measured mtDNA and nDNA damage levels in unexposed, wildtype and glp-1 nematodes at multiple timepoints after age-synchronization at the first larval stage (as previously described: (Bess et al. 2012)). We found that DNA damage in both genomes and strains increased with developmental stage (Figure A.5), supporting the hypothesis that the decreased DNA damage observed at 5 μM MeHg could be explained by the developmental delay that was also observed.
3.2 Experiment 2: DNA damage repair and removal following mercury exposure
Next, we carried out experiments designed to test the influence of prior mercury exposure on the response in both genomes to DNA damage induced by exposure to UVC or H2O2.
3.2.1 Repair and removal of UVC-induced DNA damage
The impact of mercury on removal of UVC induced mtDNA and nDNA damage, assessed using a 3-way ANOVA (mercury compound × UVC exposure × time), was mild. In the mitochondrial genome, the 3-way interaction was not significant (p=0.56); however, mercury altered lesion removal over time (mercury compound × time interaction, p=0.01), although the difference was small (Figure 3).
Figure 3.

Mitochondrial and nuclear DNA damage (average lesions/10kb ±SE) in control (black), 5 μM HgCl2 treated (light-blue), and 1 μM MeHg treated young adult nematodes (dark-blue) that were either exposed (solid lines) or not exposed (dotted lines) to UVC (50J /m2). DNA damage was measured at four time-points following UVC exposure. Red lines indicate the approximate limit of detection for DNA lesions (0.1 lesions/10 kb). In the mitochondrial genome all 3 2-way ANOVAs were significant. In the nuclear genome the 3-way interaction was significant. * indicates a significantly different from the control and HgCl2 treatments at 6 hr. See Results section for further statistical explanation.
For nDNA lesions the 3-way interaction was significant (p=0.02), and so the data was further analyzed as a function of UVC exposure. In UVC exposed nematodes, there was a difference in nDNA lesion removal over time between the three treatments (mercury compound × time interaction, p=0.004), but significant differences in levels of damage between treatments were only observed at one time-point (6hr), and the degree of change was relatively small. In non-UVC exposed nematodes, the MeHg exposure resulted in significantly lower nDNA lesions (main effect, p=0.001) than control or HgCl2 treatments. However, again, the effect size was small (≤0.1 lesions) (Figure 3).
3.2.2 Relative copy number following UVC exposure
. The most dramatic impact was that MeHg in combination with UVC led to a decrease in mtDNA copy number. For relative mitochondrial copy number, the 3-way (mercury compound × UVC exposure × time) ANOVA interaction was significant (p<0.001) and the data was further analyzed by time-point. At 0 hr, copy number was significantly decreased by the MeHg treatment compared to both the control (p<0.001) and HgCl2 (p=0.003) treatments and also by the UV treatment (p=0.03). At 6 hr, copy number was significantly reduced by both MeHg (p<0.001) and HgCl2 (p=0.03) treatments compared to controls and by the UV treatment (p<0.001). At 24 hr, copy number was significantly lower in MeHg (main effect, p<0.001) and UV (main effect, p<0.001) treatments and further reduced in the MeHg-UV co-exposed treatment (interaction, p<0.001). At 48hr, copy number was significantly lower in MeHg (p<0.001), HgCl2 (p<0.001), and UV (p<0.001) treatments and further reduced in the MeHg-UV co-exposed treatment (interaction, p<0.001) (Figure 4).
Figure 4.

Mitochondrial and nuclear relative copy number (average % of control ±SE) in control (black), 5 μM HgCl2 treated (light-blue), and 1 μM MeHg treated young adult nematodes (dark-blue) that were either exposed (solid lines) or not exposed (dotted lines) to UVC (50 J/m2). Relative copy number was measured at four time-points following UVC exposure. For mitochondrial copy number the 3-way interaction was significant. * indicates significant 2-way interaction with the MeHg +UV treatment further reducing mitochondrial copy number. See Results section for further statistical explanation.
We did not observe large effects on nuclear copy number. The 3-way interaction was not significant (p=0.05). Though some main effects were significant, the resulting change in nuclear copy number was small, less than 5% (Figure 4, Table A.4).
3.2.3 Repair of H2O2-induced DNA damage
Mercury exposure significantly increased the DNA damage caused by H2O2, and also affected damage removal in complex fashions. The impact of mercury and H2O2 exposure on nDNA and mtDNA damage was assessed using a 3-way ANOVA (mercury compound × H2O2 exposure × time). For mtDNA lesions, the 3-way interaction was not significant (p=0.08). mtDNA damage depended on both the mercury compound and H2O2 exposure (mercury compound × H2O2 exposure interaction, p=0.007). Across time-points MeHg exposed nematodes had significantly higher damage compared to the HgCl2 treatment after H2O2 exposure (p=0.006). MeHg-exposed nematodes had significantly higher damage compared to controls in non-H2O2 exposed nematodes (p=0.01). We could not formally test whether exposure to either mercury compound resulted in a greater number of lesions immediately after H2O2 exposure because of the lack of a significant 3-way interaction.
mtDNA damage also changed over time following H2O2 exposure (time × H2O2 exposure interaction, p<0.001), where nematodes exposed to H2O2 had significantly more DNA damage than non- H2O2 exposed nematodes at all time-points except at 48hr where there was less damage. Additionally, mtDNA damage changed over time depending on the mercury compound (time × mercury compound interaction, p<0.001), where at 0 hr both MeHg and HgCl2 treatments had higher DNA damage compared to controls, at 6 hr the MeHg treatment had significantly more damage compared to controls, and at 24 and 48 hr HgCl2 treatments had significantly less (in fact, “negative”—lesions i.e., below control baseline) damage compared to control and MeHg treatments (Figure 5).
Figure 5.

Mitochondrial and nuclear DNA damage (average lesions/10 kb ±SE) in control (black), 5 μM HgCl2 treated (light-blue), and 1 μM MeHg treated young adult nematodes (dark-blue) that were either exposed (solid lines) or not exposed (dotted lines) to H2O2 (5 mM). DNA damage was measured at four time-points following UVC exposure. Red lines indicate the approximate limit of detection for DNA lesions (0.1 lesions/10 kb). See Results section for further statistical explanation.
The 3-way interaction for nDNA lesions was also not significant (p=0.76). The change in nDNA damage over time differed between H2O2 exposures (H2O2 exposure × time interaction, p<0.001), with significantly more damage in H2O2 treatments at 0 hr (p<0.001) and significantly less damage at 48 hr (p=0.006). Mercury compound also impacted nDNA lesions at different time-points (mercury compound × time interaction, p<0.001). At 0 hr HgCl2 treated nematodes had significantly more nDNA lesions than control and MeHg treatments, at 6 hr both MeHg and HgCl2 nematodes had significantly more damage compared to controls, at 24 hr HgCl2 nematodes had significantly less damage compared to the control and MeHg treatments, at 24 hr the MeHg treatment also had also had significantly more damage than HgCl2 treatment, and at 48 hr HgCl2 treated nematodes had significantly less damage compared to the control and MeHg treatments (Figure 5).
Below-baseline DNA damage (mitochondrial and nuclear) observed at the last time point (48 hr) in mercury exposed animals suggested that BER was induced by mercury exposure. To test for transcriptional induction of BER, we measured gene expression of BER related genes in young adult animals. However, we did not find evidence for induction; rather, we measured decreased gene expression for some BER related genes. The greatest MeHg and HgCl2 concentrations (5 μM) significantly reduced nth-1, exo-1, and lig-1 expression, while the 5 μM HgCl2 exposure also reduced (p=0.10) rnr-2 expression (Figure A.4).
3.2.4 Relative copy number following H2O2 exposure
The major effects that we observed were a decrease in mtDNA copy number following H2O2 exposure that was exacerbated by prior exposure to MeHg. Again, the impact of mercury and H2O2 exposure on relative copy number was assessed using a 3-way ANOVA (mercury compound × H2O2 exposure × time). For mtDNA copy number, the 3-way interaction was significant (p=0.004), so the data was further considered by time-point. At 0 hr, copy number was significantly lower in the MeHg treatment compared to the HgCl2 treatment (p=0.01). At 6 hr, copy number was significantly reduced in the H2O2 exposure (p<0.001). At 24 hr, copy number was reduced in the MeHg (p<0.001) and H2O2 (p<0.001) treatments and further reduced in the MeHg-H2O2 co-exposure treatment (interaction, p<0.001). At 48 hr, copy number was reduced in the HgCl2 (p=0.01) and H2O2 (p<0.001) treatments and further reduced in the MeHg-H2O2 co-exposure treatment (interaction, p<0.001) (Figure 6).
Figure 6.

Mitochondrial and nuclear relative copy number (average % of control ±SE) in control (black), 5 μM HgCl2 treated (light-blue), and 1 μM MeHg treated young adult nematodes (dark-blue) that were either exposed (solid lines) or not exposed (dotted lines) to H2O2 (5 mM). Relative copy number was measured at four time-points following UVC exposure. Exposure groups are dodged for better visual representation. See Results section for further statistical explanation.
The 3-way interaction for nuclear copy number was insignificant (p=0.37). Mercury affected copy number over time (mercury compound × time interaction, p=0.01), but differences in nuclear copy number between treatments were only observed at one time-point (6hr). At 6 hr, MeHg and HgCl2 treatments had significantly higher nuclear copy number compared to the control treatment. No other significant differences were observed at the other time-points (Figure 6).
3.3 Experiment 3: In vivo ATP levels following exposure to mercury, UVC, and H2O2
To assess the impacts of mercury compound, type of DNA damage, and their interaction on ATP levels, we carried out an experiment similar to that in which we analyzed impacts on DNA. A 2-way ANOVA was performed for each time-point. At the 24 hr time-point, mercury compound (main effect, p=0.007) and DNA damage type (main effect, p<0.001) affected ATP levels. ATP levels in the MeHg treatment were significantly higher than the HgCl2 treatment (p=0.005). Additionally, ATP levels after UVC were significantly higher compared to the control treatment (p<0.001) and higher than the H2O2 treatment (p=0.05) (Figure 7).
At the 48 hr time-point, mercury compound (main effect, p=0.04) and DNA damage type (main effect, p<0.001) affected ATP levels. ATP levels in the HgCl2 treatment were significantly lower compared to the control (p=0.001) and MeHg (p=0.004) treatments. H2O2-exposed nematodes had significantly increased ATP levels compared to controls (p=0.03) (Figure 7).
3.4 Experiment 4: Innate immunity in wild type and immune deficient nematodes
Mitochondria are important immune response signaling organelles and mercury-induced alterations to ROS concentrations and mitochondrial status and function (ATP, copy number) could have downstream impacts on immune response. We hypothesized that mercury-induced changes to these processes could alter p38 MAPK, an innate immune signaling pathway that can be activated by redox changes (Torres and Forman 2003, Bundy et al. 2005, Mendez-Samperio et al. 2010, Gostner et al. 2013). Impacts on this pathway were first assessed by measuring mRNA levels of p38 MAPK pathway genes. sek-1 expression was significantly reduced following 5 μM MeHg exposure and reduced (p=0.07) following 5 μM HgCl2 exposure. The 5 μM concentration of both MeHg and HgCl2 significantly reduced pmk-1 expression (Figure 8).
Figure 8.
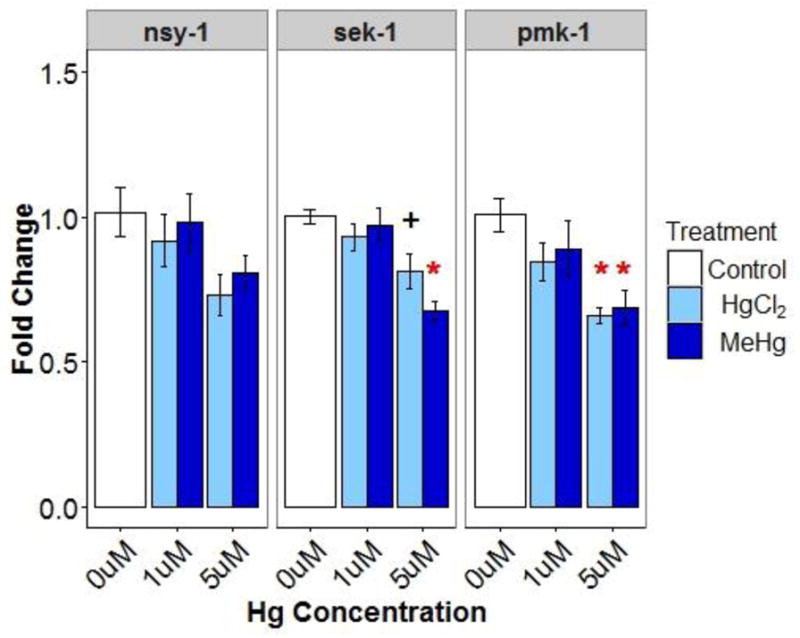
Relative expression (fold change ±SE) of p38 MAPK related genes (nsy-1, sek-1, pmk-1) in young adult nematodes. * indicates significantly different (p<0.05) from control and + indicates 0.05 ≤ p < 0.1.
As we observed that both mercury species impacted this pathway we then assessed the influence of early larval mercury exposure on animals with mutations in the p38/PMK-1 MAPK pathway by measuring survival time of N2 and pmk-1 nematodes on E. coli OP50 or P. aeruginosa PA14. HgCl2 and MeHg treatments were analyzed separately as there was considerable variation in control survival between these trials. There were no survival differences following HgCl2 exposure for either nematode strain on OP50 or PA14 (Figure A.6). MeHg exposure significantly reduced survival time for N2 nematodes on OP50 (median survival difference 21hrs), while no survival differences were observed on PA14. Survival was also not different for the pmk-1 strain on either strain of bacteria (Figure A.7).
4. Discussion
Mercury is well known for having adverse impacts on multiple systems, though the specific mechanisms are not well understood. Proposed mechanisms for injury include increased ROS (Yee and Choi 1996, Crespo-Lopez et al. 2009); reduced cell membrane integrity (Yin et al. 2007, Polunas et al. 2011); altered cell signaling (Worth et al. 2001, Chen et al. 2006); mitochondrial impacts (Yee and Choi 1996, Dreiem et al. 2005, Franco et al. 2007); altered DNA repair (Christie et al. 1986, Asmuß et al. 2000, Cebulska-Wasiewska et al. 2005, Gadhia et al. 2012, Pieper et al. 2014, Ryu et al. 2014); and immunomodulatory impacts (Fortier et al. 2008, Vetvicka and Vetvickova 2009, Gallagher et al. 2011, Li et al. 2014). Here we report the outcomes of HgCl2 and MeHg exposure on DNA damage, DNA repair, mitochondrial endpoints (copy number and ATP), and PMK-1 (p38 mitogen-activated protein kinase) mediated innate immune impacts in C. elegans. Of the outcomes we investigated, mitochondrial impacts were particularly notable.
4.1 DNA damage and damage repair
We found that following exposure to both inorganic and organic mercury species, increased DNA damage and altered DNA repair occurred in both genomes. We predicted DNA damage to both genomes because mercury accumulates in both organelles, but we hypothesized that mitochondria may be more susceptible to damage as mitochondria lack some of the pathways that repair damage to nuclear DNA (Atchison and Hare 1994, Bucio et al. 1999, Ikemoto et al. 2004, Larsen et al. 2005). Until recently studies had primarily focused on nuclear genome impacts following mercury exposure, because the methods used in many studies (e.g. the comet assay) predominantly evaluated nDNA (Cebulska-Wasiewska et al. 2005, Bradfield et al. 2006, Ondovcik et al. 2012, Ryu et al. 2014). mtDNA impacts have been addressed in only a few studies. In an in vitro study, plausibility for mtDNA damage was established through microscopy images that depicted increased ROS in a qualitative manner and DNA damage co-occurring in cellular space with mitochondria (Sharpe et al. 2012). In a field study, some evidence for increased mtDNA damage was obtained in bats from areas with high potential for Hg exposure, but individual-level correlations with Hg levels were poor (Karouna-Renier et al. 2014). Here we report the first quantified impacts of mercury on mtDNA in controlled experiments in an in vivo model.
DNA damage following mercury exposure alone was dependent on life-stage and concentration. Low-level damage in both genomes was only detected at the earliest time-point examined (Figure 2), which suggests that there is increased susceptibility to damage earlier in life. Surprisingly, the high- concentration MeHg resulted in less damage compared to controls. It is important to note that our DNA damage assay defines “control” samples as having no damage, although cells have a normal steady-state level of damage. Negative damage levels are thus interpreted as improved repair or clearance of damaged genomes, relative to standard control conditions. With this assay, younger life-stages appear to have less nDNA and mtDNA damage compared to older life-stages. Our results suggest that the small decrease in lesion number (0.3 lesions/10kb) in the highest MeHg exposure is related to a slight developmental delay, rather than changes damage removal processes (Figures 2 and A.6). Developmental delay in this treatment is also supported by our observation of reduced copy number as both nDNA and, more dramatically, mtDNA copy number increase during these lifestaqes (Tsang and Lemire 2002, Rooney et al. 2014). The increase in DNA damage with developmental stage that we observed is interesting in its own right and consistent with our previous observation of developmental stage decreases in DNA repair genes (Boyd et al., 2010), but also suggests that any exposures that result in developmental rate and also cause DNA damage could lead to DNA damage levels being underestimated, since the developmental delay by itself would result in a lower level of DNA damage.
Our data support findings from previous studies, that nonlethal mercury exposure alone does not induce substantial DNA damage in either genome (Cebulska-Wasiewska et al. 2005, Bradfield et al. 2006). nDNA damage has been observed in some studies, but the weight of the evidence so far seems to support the hypothesis that compromising genetic damage may only occur at cytotoxic exposures (Cantoni and Costa 1983, Pieper et al. 2014). However, although mercury exposure does not appear to directly damage DNA, there is evidence that in combination with other exposures, considerably increased damage can occur.
Elevated DNA damage was observed in both genomes after co-exposure to H2O2 in this study, and has also been observed in vitro with nDNA (Pieper et al. 2014). We did not observe increased damage in conjunction with UVC exposure, confirming a previous report (Cebulska-Wasiewska et al. 2005), though elevated damage has been noted with UVA and also X-ray co-exposures (Cantoni and Costa 1983, Bradfield et al. 2006). Collectively, these findings are consistent with one of mercury’s major proposed mechanisms of action, creating an environment sensitive to oxidative stress due to reduced antioxidant capabilities: H2O2, UVA, and X-ray exposures induce ROS generation, but UVC-induced ROS is very minor (Godar et al. 1993, Agarwal and Sohal 1994, Hao et al. 2004, Manda et al. 2007). Another proposed mechanism for induced DNA damage is the impairment of DNA repair enzymes.
Damage removal is similar between C. elegans and mammals, with NER acting in the nucleus, photodimer removal from mitochondria occurring via mitophagy, and BER acting in both genomes (Costa et al. 2003, Reardon and Sancar 2005, Meyer et al. 2007, Hunter et al. 2012, Bess et al. 2013). Our data indicates that not all types of repair and removal are impacted by mercury exposure, as there were no differences in lesion removal following UVC exposure in either genome. This is the first assessment of photodimer removal in mitochondria after MeHg exposure. Our observation that inorganic mercury exposure does not impact NER is consistent with other studies (Christie et al. 1986, Cebulska-Wasiewska et al. 2005). Although mercury has not been observed to inhibit proteins involved in NER (ex. mammalian XPA protein (Asmuß et al. 2000)), there is evidence for alterations to nonhomologous end joining, homologous recombination, and BER.
Inorganic mercury has been reported in multiple studies to impair BER. Reduced repair of double strand breaks through either nonhomologous end joining and/or homologous recombination repair has been consistently observed with inorganic mercury exposure and gamma radiation (Ryu et al. 2014) and x-ray exposure (Cantoni and Costa 1983, Christie et al. 1986, Cebulska-Wasiewska et al. 2005). In vitro studies indicate that BER impairment occurs from reduced glycosylase expression (ex. OGG1) following inorganic mercury exposure (Gadhia et al. 2012) and reduced repair enzyme recruitment from decreases in the poly(ADP-ribosyl)ation signaling reaction that is induced by strand breaks (Pieper et al. 2014). Our gene expression data are consistent with previous studies in that we observed reduced expression of BER associated-genes, including a glycosylase, an apurinic/apyrimidinic endonuclease, and DNA ligase following exposure to the highest concentrations of HgCl2 and MeHg. Further, these data suggest that at least some of the impairment of BER may be transcriptionally-mediated. However, our in vivo repair kinetics data were not as clear-cut. Impairments the repair rate were not readily evident, as might be expected from decreased gene expression; rather, in some cases, repair appeared to be increased. Of course, in vivo repair integrates transcriptional regulation, protein-level changes (e.g., inhibition by Hg), and other physiological conditions. It is possible that in some cases, elevated ATP levels permitted increased kinetics of DNA repair, despite decreased transcription of repair genes, although we did not test this. In the mitochondria, nematodes co-exposed to H2O2 and HgCl2 or MeHg began repair with a similar level of mtDNA damage and both had similar repair rates. nDNA repair was slower at early time-points in nematodes co-exposed to MeHg and faster in HgCl2 co-exposures. However, a limitation of this study is that actual repair kinetics could not be calculated due to the low number of time-points and the fact that DNA repair was assumed to be nonlinear, as previously observed in C. elegans for repair of photodimers (Meyer et al. 2007).
Despite these limitations, ours is the first report of increased BER following Hg exposure. We observed an apparent overcompensation of repair, evidenced by DNA damage levels in both genomes falling below the damage levels in controls at late time-points after exposure to both mercury and a DNA damaging agent. Overcompensation occurred primarily with HgCl2 in mtDNA and with both HgCl2 and MeHg in nDNA; we speculate that the levels of mercury utilized in this study stimulated a regulatory mechanism that activated DNA repair. For example, methylmercury in particular activates poly(ADP-ribose)polymerase (PARP), well known for the role it plays in DNA damage response signaling (Lu et al. 2011). An overcompensation in repair has also been noticed following moderate oxidative damage in in vitro studies (Driggers et al. 1993, Yakes and VanHouten 1997).
4.2 Mitochondrial impacts
Mitochondria are one target of mercury toxicity and impacts to mitochondria are important to understand because of the important role this organelle has in cellular maintenance (Meyer et al. 2013). Mitochondrial impacts observed in this study included the mtDNA damage and altered repair described in the previous section and altered copy number and steady-state ATP levels. Changes in copy number and ATP content were dependent on mercury compound exposure, supporting the notion that inorganic and organic mercury toxicities can occur through different mechanisms (Tan et al. 1993, Gardner et al. 2010, McElwee et al. 2013). In C. elegans, MeHg and HgCl2 behave differently in terms of uptake and interactions with other co-exposures (e.g. selenium compounds) (Wyatt et al. 2016).
In this study, both MeHg and HgCl2 reduced mitochondrial copy number; however, the greatest copy number reductions (≥25%) occurred with MeHg exposures. In Experiment 1, acute exposure to the high MeHg concentration alone decreased mitochondrial copy number, while in Experiment 2, chronic exposure to a lower concentration diminished copy number only in nematodes that were co-exposed to UVC or H2O2. Following UVC or H2O2 exposure in MeHg-treated nematodes, mitochondrial copy number decreased over time (~40% reduction). These reductions coincide with DNA damage removal which is consistent with mitochondrial dynamics (fission, fusion) and autophagy playing an important role in mtDNA damage removal, and with the removal processes differing between MeHg and HgCl2 treatments (Bess et al. 2012). “Slow” mtDNA removal following H2O2 exposure has also been observed in HeLa cells (Shokolenko et al. 2016).
Declines in mtDNA copy number could result in altered physiology and cell homeostasis. However, the degree to which mitochondrial copy number reductions impact cellular health is not well understood, because many cells have high mtDNA redundancy, copy number varies between tissues, and there is a wide range of apparently normal mtDNA copy number in humans (Shokolenko and Alexeyev 2015). We recently found that small decreases in mtDNA copy number have only mild impacts on ATP levels in C. elegans (Luz and Meyer 2016). Nonetheless, mtDNA depletions around 65% can cause disease in humans (Suomalainen and Isohanni 2010), and a certain level of mtDNA is required to pass some developmental milestones in C. elegans (Tsang and Lemire 2002). Therefore, we also investigated the impacts of Hg on energy metabolism.
In our experiments, ATP levels were increased by MeHg exposure while they were decreased by HgCl2 exposure. In vitro data supports our HgCl2 related findings as respiration was suppressed in fish liver cells following exposure (Mieiro et al. 2015). The finding of increased (or in one case unchanged) ATP levels across treatments following MeHg exposure was a surprise, especially given that MeHg exposures resulted in decreased copy number, particularly in combination with UVC and H2O2 exposures. This increase in ATP levels could be explained by increased production or decreased utilization, possibly in combination with a shift in metabolism; additional work will be required to elucidate the cause.
Again to our surprise, we also observed increased ATP levels after both UVC and H2O2, potentially reflecting a regulated response that devotes energy resources to DNA damage removal. Another explanation is that cellular respiration and resulting ATP levels are correlated to the ratio of functional and dysfunction mitochondria. Reduced mitochondrial copy number and increased ATP levels have been observed with lithium exposure in C. elegans. The authors of that study observed that the metal positively influenced the lifespan of C. elegans and made the argument that mitophagy could benefit mitochondrial energetics by selectively removing damaged mitochondria, thus altering cellular respiration (Tam et al. 2014). Though we observed reduced expression for some mitophagy and autophagy genes, these processes could be upregulated post-translationally.
4.3 Innate immune impacts
Although we observed altered mitochondrial parameters and reduced gene expression of a p38 MAPK (pmk-1) and MAPKK (sek-1) following mercury exposure, effects on innate immunity were mild. Larval mercury exposure to MeHg reduced survival of wildtype N2 nematodes on OP50, which is suggestive of a general impairment of innate immune response. OP50 is generally considered a control food source, but it does have some level of pathogenicity, as animals incubated on this bacteria have a higher expression of an antimicrobial gene, suggesting innate immunity activation (Hahm et al. 2011). Though the reduction in survival time was small (< 24hrs), MeHg does not appear to reduce lifespan from a separate experiment using heat killed OP50 and antibiotics (data not shown). The absence of an observed difference on PA14, a more pathogenic bacteria, seems surprising but may be related to the difficultly in discerning differences where there is a rapid reduction in survival. MeHg exposure appears to affect the PMK-1-mediated immune response, since MeHg had an effect on survival of N2 but not pmk-1 animals. No difference in survival in pmk-1 animals was observed, which also could be related to the difficulty to perceive survival differences when the overall survival time is short. Data from this study supports previous observations of inorganic mercury related innate immune modulation (Silbergeld et al. 2000, Silva et al. 2005, Nyland et al. 2012) and suggests that organic mercury may also have an impact on innate immune signaling.
5. Conclusions
In summary, our data provides evidence that genotoxic, metabolic, and innate immune impacts can result following mercury exposure. Mercury impacts on DNA damage outcomes were similar between genomes, resulting in low-level damage following individual mercury exposures, synergistically increased damage with oxidative co-exposure, and altered DNA damage repair through BER. A major finding was that mercury compounds affect mitochondrial outcomes differently, including genome copy regulation and ATP levels, supporting the hypothesis that MeHg and HgCl2 toxicities operate through different mechanisms. Mitochondrial impacts should be further examined because of the mitochondria’s importance to cellular functions. Additionally, innate immune impacts, though moderate, differed between mercury compounds.
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences (R01-ES017540-01A2 to JNM) and Health Impact Assessment in the Amarakaeri Communal Reserve (to WKP).
Appendix
Table A.1.
mtDNA damage, 3-way ANOVA (mercury compound × UVC exposure × time)
| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.213 | 0.107 | 1.7931 | 0.168226 |
| UV_yn | 1 | 188.326 | 188.326 | 3169.292 | < 2.2e-16 |
| Timepoint | 3 | 1.322 | 0.441 | 7.4159 | 8.30E-05 |
| Hg_type:UV_yn | 2 | 0.545 | 0.273 | 4.5877 | 0.010901 |
| Hg_type:Timepoint | 6 | 1.132 | 0.189 | 3.1762 | 0.004898 |
| UV_yn:Timepoint | 3 | 1.248 | 0.416 | 6.9984 | 0.000145 |
| Hg_type:UV_yn:Timepoint | 6 | 0.286 | 0.048 | 0.8031 | 0.568108 |
| Residuals | 300 | 17.827 | 0.059 |
Table A.2.
nDNA damage, 3-way ANOVA (mercury compound × UVC exposure × time)
| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.48 | 0.24 | 5.3429 | 0.005248 |
| UV_yn | 1 | 53.186 | 53.186 | 1183.051 | < 2.2e−16 |
| Timepoint | 3 | 26.258 | 8.753 | 194.6894 | < 2.2e−16 |
| Hg_type:UV_yn | 2 | 0.131 | 0.066 | 1.4584 | 0.234261 |
| Hg_type:Timepoint | 6 | 0.619 | 0.103 | 2.295 | 0.035055 |
| UV_yn:Timepoint | 3 | 20.351 | 6.784 | 150.8947 | < 2.2e−16 |
| Hg_type:UV_yn:Timepoint | 6 | 0.643 | 0.107 | 2.3845 | 0.028878 |
| Residuals | 300 | 13.487 | 0.045 |
| +UVC | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.1 | 0.05 | 0.9392 | 0.393235 |
| Timepoint | 3 | 46.362 | 15.4539 | 289.9922 | < 2.2e−16 |
| Hg_type:Timepoint | 6 | 1.041 | 0.1734 | 3.2547 | 0.004898 |
| Residuals | 150 | 7.994 | 0.0533 |
| −UVC | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.5114 | 0.255716 | 6.9823 | 0.001261 |
| Timepoint | 3 | 0.2477 | 0.082566 | 2.2544 | 0.084406 |
| Hg_type:Timepoint | 6 | 0.2216 | 0.036929 | 1.0084 | 0.422097 |
| Residuals | 150 | 5.4935 | 0.036623 |
Table A.3.
Mitochondrial copy number, 3-way ANOVA (mercury compound × UVC exposure × time)
| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.98592 | 0.49296 | 96.088 | < 2.2e−16 |
| UV_yn | 1 | 0.68226 | 0.68226 | 132.9868 | < 2.2e−16 |
| Timepoint | 3 | 0.43753 | 0.14584 | 28.4276 | 3.31E−16 |
| Hg_type:UV_yn | 2 | 0.20756 | 0.10378 | 20.229 | 5.74E−09 |
| Hg_type:Timepoint | 6 | 0.25561 | 0.0426 | 8.3039 | 2.47E−08 |
| UV_yn:Timepoint | 3 | 0.13169 | 0.0439 | 8.5565 | 1.81E−05 |
| Hg_type:UV_yn:Timepoint | 6 | 0.13842 | 0.02307 | 4.4968 | 0.000222 |
| Residuals | 300 | 1.5391 | 0.00513 |
| 0hr | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.09549 | 0.047745 | 8.2053 | 0.000583 |
| UV_yn | 1 | 0.02814 | 0.028142 | 4.8364 | 0.030827 |
| Hg_type:UV_yn | 2 | 0.00799 | 0.003993 | 0.6862 | 0.506499 |
| Residuals | 78 | 0.45387 | 0.005819 |
| 6hr | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.075866 | 0.037933 | 9.4618 | 0.000209 |
| UV_yn | 1 | 0.106285 | 0.106285 | 26.5112 | 1.91E−06 |
| Hg_type:UV_yn | 2 | 0.010047 | 0.005024 | 1.253 | 0.291319 |
| Residuals | 78 | 0.312707 | 0.004009 |
| 24hr | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.49831 | 0.24915 | 48.066 | 2.50E−14 |
| UV_yn | 1 | 0.34159 | 0.34159 | 65.897 | 5.57E−12 |
| Hg_type:UV_yn | 2 | 0.1049 | 0.05245 | 10.118 | 0.000124 |
| Residuals | 78 | 0.40432 | 0.00518 |
| 48hr | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.54443 | 0.27221 | 48.795 | 9.80E−14 |
| UV_yn | 1 | 0.34525 | 0.34525 | 61.887 | 4.59E−11 |
| Hg_type:UV_yn | 2 | 0.21574 | 0.10787 | 19.336 | 2.46E−07 |
| Residuals | 66 | 0.3682 | 0.00558 |
Table A.4.
Nuclear copy number, 3-way ANOVA (mercury compound × UVC exposure × time)
| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.0824 | 0.041222 | 3.3721 | 0.035624 |
| UV_yn | 1 | 0.1046 | 0.104558 | 8.5531 | 0.003712 |
| Timepoint | 3 | 0.0404 | 0.013482 | 1.1029 | 0.348196 |
| Hg_type:UV_yn | 2 | 0.0082 | 0.0041 | 0.3354 | 0.715342 |
| Hg_type:Timepoint | 6 | 0.0259 | 0.004317 | 0.3531 | 0.907819 |
| UV_yn:Timepoint | 3 | 0.0142 | 0.004744 | 0.3881 | 0.761665 |
| Hg_type:UV_yn:Timepoint | 6 | 0.156 | 0.025998 | 2.1267 | 0.050227 |
| Residuals | 300 | 3.6674 | 0.012225 |
Table A.5.
mtDNA damage, 3-way ANOVA (mercury compound × H2O2 exposure × time)
| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| Hg_type | 2 | 2.7149 | 1.3575 | 24.1444 | 4.43E−10 |
| H2O2_yn | 1 | 1.9902 | 1.9902 | 35.3987 | 1.25E−08 |
| Timepoint | 3 | 15.8078 | 5.2693 | 93.7218 | < 2.2e−16 |
| Hg_type:H2O2_yn | 2 | 0.5632 | 0.2816 | 5.0086 | 0.007579 |
| Hg_type:Timepoint | 6 | 3.7738 | 0.629 | 11.1872 | 1.07E−10 |
| H2O2_yn:Timepoint | 3 | 4.1309 | 1.377 | 24.4915 | 1.83E−13 |
| Hg_type:H2O2_yn:Timepoint | 6 | 0.6278 | 0.1046 | 1.861 | 0.089453 |
| Residuals | 192 | 10.7947 | 0.0562 |
Table A.6.
nDNA damage, 3-way ANOVA (mercury compound × H2O2 exposure × time)
| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.3199 | 0.15996 | 4.4674 | 0.01269 |
| H2O2_yn | 1 | 0.1168 | 0.11676 | 3.2609 | 0.07252 |
| Timepoint | 3 | 6.9804 | 2.32681 | 64.9858 | < 2.2e−16 |
| Hg_type:H2O2_yn | 2 | 0.0212 | 0.0106 | 0.296 | 0.74415 |
| Hg_type:Timepoint | 6 | 2.8791 | 0.47986 | 13.402 | 1.13E−12 |
| H2O2_yn:Timepoint | 3 | 1.028 | 0.34268 | 9.5707 | 6.40E−06 |
| Hg_type:H2O2_yn:Timepoint | 6 | 0.12 | 0.02 | 0.5587 | 0.76281 |
| Residuals | 192 | 6.8745 | 0.0358 |
Table A.7.
Mitochondrial copy number, 3-way ANOVA (mercury compound × H2O2 exposure × time)
| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.10879 | 0.0544 | 6.748 | 0.001472 |
| H2O2_yn | 1 | 1.17798 | 1.17798 | 146.1282 | < 2.2e−16 |
| Timepoint | 3 | 1.43665 | 0.47888 | 59.4053 | < 2.2e−16 |
| Hg_type:H2O2_yn | 2 | 0.20676 | 0.10338 | 12.8244 | 5.92E−06 |
| Hg_type:Timepoint | 6 | 0.21496 | 0.03583 | 4.4444 | 0.00031 |
| H2O2_yn:Timepoint | 3 | 0.45123 | 0.15041 | 18.6584 | 1.16E−10 |
| Hg_type:H2O2_yn:Timepoint | 6 | 0.16015 | 0.02669 | 3.311 | 0.004 |
| Residuals | 192 | 1.54777 | 0.00806 |
| 0hr | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.10618 | 0.053088 | 4.1621 | 0.02153 |
| H2O2_yn | 1 | 0.01128 | 0.011277 | 0.8841 | 0.3518 |
| Hg_type:H2O2_yn | 2 | 0.00047 | 0.000234 | 0.0183 | 0.98182 |
| Residuals | 48 | 0.61225 | 0.012755 |
| 6hr | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.030473 | 0.015236 | 2.739 | 0.07475 |
| H2O2_yn | 1 | 0.156475 | 0.156475 | 28.1288 | 2.85E−06 |
| Hg_type:H2O2_yn | 2 | 0.031038 | 0.015519 | 2.7898 | 0.07142 |
| Residuals | 48 | 0.267014 | 0.005563 |
| 24hr | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.11907 | 0.05953 | 8.8583 | 0.000532 |
| H2O2_yn | 1 | 0.42122 | 0.42122 | 62.6739 | 2.94E−10 |
| Hg_type:H2O2_yn | 2 | 0.18299 | 0.09149 | 13.6135 | 2.07E−05 |
| Residuals | 48 | 0.3226 | 0.00672 |
| 48hr | Df | Sum Sq | Mean Sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.06804 | 0.03402 | 4.7209 | 0.013437 |
| H2O2_yn | 1 | 1.04024 | 1.04024 | 144.3521 | 4.47E−16 |
| Hg_type:H2O2_yn | 2 | 0.15242 | 0.07621 | 10.5753 | 0.000157 |
| Residuals | 48 | 0.3459 | 0.00721 |
Table A.8.
Nuclear copy number, 3-way ANOVA (mercury compound × H2O2 exposure × time)
| Df | Sum Sq | Mean Sq | F value | Pr(>F) | |
|---|---|---|---|---|---|
| Hg_type | 2 | 0.02318 | 0.011588 | 1.5732 | 0.21003 |
| H2O2_yn | 1 | 0.01852 | 0.018517 | 2.5139 | 0.11449 |
| Timepoint | 3 | 0.56815 | 0.189382 | 25.7104 | 5.01E−14 |
| Hg_type:H2O2_yn | 2 | 0.00167 | 0.000833 | 0.1131 | 0.89315 |
| Hg_type:Timepoint | 6 | 0.12291 | 0.020485 | 2.781 | 0.01289 |
| H2O2_yn:Timepoint | 3 | 0.03457 | 0.011523 | 1.5643 | 0.19942 |
| Hg_type:H2O2_yn:Timepoint | 6 | 0.04762 | 0.007937 | 1.0775 | 0.37737 |
| Residuals | 192 | 1.41427 | 0.007366 |
Table A.9.
Primer sequences and RT-PCR conditions for gene expression analysis
| Gene target | Primer sequence | Temp.(°C) |
|---|---|---|
| tba-1 | R - CCGACCTTGAATCCAGTTGG | 60 |
| F - TGATCTCTGCTGACAAGGCTT | ||
| pmp-3 | R - ACACCGTCGAGAAGCTGTAGA | 60 |
| F - GTTCCCGTGTTCATCACTCAT | ||
| dct-1 | R - GGACAGTCTTTGGAGGTGTATT | 58 |
| F - ATCGCACAATCTCCTCACGT | ||
| polg-1 | R - TTGGAGCCGTCCGGATT | 60 |
| F - CTGCCTAATACCGTTGCCTTCTT | ||
| hmg-5 | R - GCTTCTTCGCTTCGTCTGTG | 60 |
| F - TGTCTGGAGCTGGAATGGAA | ||
| bec-1 | R - TTCGATCCTTGAGCTCTTTCA | 60 |
| F - ATGGCATCGACATGGAAACG | ||
| lgg-1 | R - CCTCGTGATGGTCCTGGTAG | 60 |
| F - GCACCAAAGTCAAAGCTCCA | ||
| atg-18 | R - CCAAGATGTGTAAGATTTTCGCC | 58 |
| F - TGGGGCACAAAGATGGCTA | ||
| nth-1 | R -GGCATTGCATCTGACCGAAT | 60 |
| F - AATCGACTCGGCTGGATCAA | ||
| exo-1 | R - CGAGGGTTTTGGTGTAGTTCTTA | 60 |
| F - AGCTGGAAGTTTGTGTGCTG | ||
| apn-1 | R - GCCACCCACTTCACCGAAAT | 60 |
| F - ATTTAAGTTCTCGAGACAATGGC | ||
| lig-1 | R - TGAGGAGTGCACTAATGGCA | 60 |
| F - ATTCGCTGATCAAGGCTGTT | ||
| rnr-1 | R - AGTCTTTCAGCGAACGAAGC | 60 |
| F - TGAGAACTTGTGCGAACGAT | ||
| nsy-1 | R - TCTTGAGCATGAAGTAGGAGAGA | 60 |
| F - TGTTCCCCGTCGCCTTGATA | ||
| sek-1 | R - AATTATCCCGCTCTGCCTGT | 60 |
| F - ATGGAGCGAAAAGGACGTGA | ||
| pmk-1 | R -TCGATGTGATCAGATCCAGGG | 58 |
| F - TGGATTGGCACGTCAAACTG |
Figure A.1.

Average number of LGG-1::GFP foci per seam cell under the following experimental conditions A) control, B) 1 μM HgCl2, C) 5 μM HgCl2, D) 1 μM MeHg, and E) 5 μM MeHg.
Figure A.2.

Relative expression (fold change ±SE) of mitophagy (dct-1), autophagy (bec-1, lgg-1, atg-18), and biogenesis (polg-1, hmg-5) related genes in young adults. * indicates significantly different (p<0.05) from control and + indicates 0.05 ≤ p < 0.1
Figure A.3.

Relative expression (fold change ±SE) of BER related genes (nth-1, exo-1, apn-1, lig-1, rnr-2) in young adults. * indicates significantly different (p<0.05) from control and + indicates 0.05 ≤ p < 0.1
Figure A.4.

glp-1 nematode size (μM) following treatment with either control, HgCl2, or MeHg conditions at the young adult life-stage.
Figure A.5.

mtDNA and nDNA damage in glp-1 and N2 nematodes using the earliest time-point as the reference for the comparison. Nematodes were cultured on plates under control conditions at 25°C and sampled at 60, 66, 84, and 108 hrs post placement on food after overnight hatch for synchronization for glp-1 nematodes and at 34, 52, and 64 hrs for N2 nematodes.
Figure A.6.

Survival curves for N2 and pmk-1 nematodes exposed from the L1 stage to control (black), 0.5 (red), 1 (green), and 2.5 μM (blue) HgCl2 on OP50 and PA14 survival plates. More than 40 animals were used for each curve
Figure A.7.

Survival curves for N2 and pmk-1 nematodes exposed from the L1 stage to control (black), 0.5 μM (red) MeHg on OP50 and PA14 survival plates. More than 40 animals were used for each curve. MeHg significantly reduced survival of N2 nematodes on OP50 (p<0.05, Mantel-Cox log-rank test)
Footnotes
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aballay A. Role of the Nervous System in the Control of Proteostasis Innate Immune Activation: Insights from C. elegans. Plos Pathogens. 2013:9. doi: 10.1371/journal.ppat.1003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aballay A, Ausubel FM. Caenorhabditis elegans as a host for the study of host-pathogen interactions. Current Opinion in Microbiology. 2002;5:97–101. doi: 10.1016/s1369-5274(02)00293-x. [DOI] [PubMed] [Google Scholar]
- Aballay A, Drenkard E, Hilbun LR, Ausubel FM. Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Current Biology. 2003;13:47–52. doi: 10.1016/s0960-9822(02)01396-9. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Sohal RS. DNA OXIDATIVE DAMAGE AND LIFE EXPECTANCY IN HOUSEFLIES. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:12332–12335. doi: 10.1073/pnas.91.25.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Saleh I, Al-Sedairi A, Elkhatib R. Effect of mercury (Hg) dental amalgam fillings on renal and oxidative stress biomarkers in children. Science of the Total Environment. 2012;431:188–196. doi: 10.1016/j.scitotenv.2012.05.036. [DOI] [PubMed] [Google Scholar]
- Al Bakheet SA, Attafi IM, Maayah ZH, Abd-Allah AR, Asiri YA, Korashy HM. Effect of long-term human exposure to environmental heavy metals on the expression of detoxification and DNA repair genes. Environmental Pollution. 2013;181:226–232. doi: 10.1016/j.envpol.2013.06.014. [DOI] [PubMed] [Google Scholar]
- Amorim AIM, Mergler D, Bahia MO, Dubeau H, Miranda D, Lebel J, Burbano RR, Lucotte M. Cytogenetic damage related to low levels of methyl mercury contamination in the Brazilian Amazon. Anais Da Academia Brasileira De Ciencias. 2000;72:497–507. doi: 10.1590/s0001-37652000000400004. [DOI] [PubMed] [Google Scholar]
- Asmuß M, Mullenders LHF, Hartwig A. Interference by toxic metal compounds with isolated zinc finger DNA repair proteins. Toxicology Letters. 2000;112–113:227–231. doi: 10.1016/s0378-4274(99)00273-8. [DOI] [PubMed] [Google Scholar]
- Atchison WD, Hare MF. MECHANISMS OF METHYLMERCURY-INDUCED NEUROTOXICITY. Faseb Journal. 1994;8:622–629. doi: 10.1096/fasebj.8.9.7516300. [DOI] [PubMed] [Google Scholar]
- Bagenstose LM, Mentink-Kane MM, Brittingham A, Mosser DM, Monestier M. Mercury enhances susceptibility to murine leishmaniasis. Parasite Immunology. 2001;23:633–640. doi: 10.1046/j.1365-3024.2001.00427.x. [DOI] [PubMed] [Google Scholar]
- Begam M, Sengupta M. Immunomodulation of intestinal macrophages by mercury involves oxidative damage and rise of pro-inflammatory cytokine release in the fresh water fish Channa punctatus Bloch. Fish & Shellfish Immunology. 2015;45:378–385. doi: 10.1016/j.fsi.2015.04.017. [DOI] [PubMed] [Google Scholar]
- Berntssen MHG, Aatland A, Handy RD. Chronic dietary mercury exposure causes oxidative stress, brain lesions, and altered behaviour in Atlantic salmon (Salmo salar) parr. Aquatic Toxicology. 2003;65:55–72. doi: 10.1016/s0166-445x(03)00104-8. [DOI] [PubMed] [Google Scholar]
- Bess AS, Crocker TL, Ryde IT, Meyer JN. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Research. 2012;40:7916–7931. doi: 10.1093/nar/gks532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess AS, I, Ryde T, Hinton DE, Meyer JN. UVC-Induced Mitochondrial Degradation via Autophagy Correlates with mtDNA Damage Removal in Primary Human Fibroblasts. Journal of Biochemical and Molecular Toxicology. 2013;27:28–41. doi: 10.1002/jbt.21440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolz DD, Tenor JL, Aballay A. A Conserved PMK-1/p38 MAPK Is Required in Caenorhabditis elegans Tissue-specific Immune Response to Yersinia pestis Infection. Journal of Biological Chemistry. 2010;285:10832–10840. doi: 10.1074/jbc.M109.091629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd WA, Crocker TL, Rodriguez AM, Leung MCK, Lehmann DW, Freedman JH, Van Houten B, Meyer JN. Nucleotide excision repair genes are expressed at low levels and are not detectably inducible in Caenorhabditis elegans somatic tissues, but their function is required for normal adult life after UVC exposure. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 2010;683:57–67. doi: 10.1016/j.mrfmmm.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradfield W, Pye A, Clifford T, Salter L, Gould D, Campbell S, Curnow A. Hg(II) exposure exacerbates UV-induced DNA damage in MRC5 fibroblasts: A comet assay study. Journal of Environmental Science and Health Part a-Toxic/Hazardous Substances & Environmental Engineering. 2006;41:143–148. doi: 10.1080/10934520500349243. [DOI] [PubMed] [Google Scholar]
- Bucio L, Garcia C, Souza V, Hernandez E, Gonzalez C, Betancourt M, Gutierrez-Ruiz MC. Uptake, cellular distribution and DNA damage produced by mercuric chloride in a human fetal hepatic cell line. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 1999;423:65–72. doi: 10.1016/s0027-5107(98)00226-7. [DOI] [PubMed] [Google Scholar]
- Bundy RE, Hoare GS, Kite A, Beach J, Yacoub M, Marczin N. Redox regulation of p38 MAPK activation and expression of ICAM-1 and heme oxygenase-1 in human alveolar epithelial (A549) cells. Antioxidants & Redox Signaling. 2005;7:14–24. doi: 10.1089/ars.2005.7.14. [DOI] [PubMed] [Google Scholar]
- Cai Y, Cao X, Aballay A. Whole-Animal Chemical Screen Identifies Colistin as a New Immunomodulator That Targets Conserved Pathways. Mbio. 2014:5. doi: 10.1128/mBio.01235-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni O, Costa M. CHARACTERIZATION AND MECHANISM OF DNA DAMAGE INDUCED BY CHROMIUM(VI) AND MERCURY(II) IN CULTURED MAMMALIAN-CELLS. Proceedings of the American Association for Cancer Research. 1983;24:74–74. [Google Scholar]
- Cebulska-Wasiewska A, Panek A, Zabinski Z, Moszczynski P, Au WW. Occupational exposure to mercury vapour on genotoxicity and DNA repair. Mutation Research-Genetic Toxicology and Environmental Mutagenesis. 2005;586:102–114. doi: 10.1016/j.mrgentox.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Ceccatelli S, Dare E, Moors M. Methylmercury-induced neurotoxicity and apoptosis. Chemico-Biological Interactions. 2010;188:301–308. doi: 10.1016/j.cbi.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Chen YW, Huang CF, Tsai KS, Sen Yang R, Yen CC, Yang CY, Lin-Shiau SY, Liu SH. The role of phosphoinositide 3-kinase/Akt signaling in low-dose mercury-induced mouse pancreatic beta-cell dysfunction in vitro and in vivo. Diabetes. 2006;55:1614–1624. doi: 10.2337/db06-0029. [DOI] [PubMed] [Google Scholar]
- Christie NT, Cantoni O, Sugiyama M, Cattabeni F, Costa M. DIFFERENCES IN THE EFFECTS OF HG(II) ON DNA-REPAIR INDUCED IN CHINESE-HAMSTER OVARY CELLS BY ULTRAVIOLET OR X-RAYS. Molecular Pharmacology. 1986;29:173–178. [PubMed] [Google Scholar]
- Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Research. 2013;41:8403–8420. doi: 10.1093/nar/gkt635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RMA, Chigancas V, Galhardo RD, Carvalho H, Menck CFM. The eukaryotic nucleotide excision repair pathway. Biochimie. 2003;85:1083–1099. doi: 10.1016/j.biochi.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Crespo-Lopez ME, Macedo GL, Pereira SID, Arrifano GPF, Picanco-Diniz DLW, do Nascimento JLM, Herculano AM. Mercury and human genotoxicity: Critical considerations and possible molecular mechanisms. Pharmacological Research. 2009;60:212–220. doi: 10.1016/j.phrs.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Di Pietro A, Visalli G, La Maestra S, Micale R, Aluce BB, Matarese G, Cingano L, Coglio MES. Biomonitoring of DNA damage in peripheral blood lymphocytes of subjects with dental restorative fillings. Mutation Research-Genetic Toxicology and Environmental Mutagenesis. 2008;650:115–122. doi: 10.1016/j.mrgentox.2007.10.023. [DOI] [PubMed] [Google Scholar]
- Dreiem A, Gertz CC, Seegal RF. The Effects of Methylmercury on Mitochondrial Function and Reactive Oxygen Species Formation in Rat Striatal Synaptosomes Are Age-Dependent. Toxicological Sciences. 2005;87:156–162. doi: 10.1093/toxsci/kfi224. [DOI] [PubMed] [Google Scholar]
- Driggers WJ, Ledoux SP, Wilson GL. REPAIR OF OXIDATIVE DAMAGE WITHIN THE MITOCHONDRIAL-DNA OF RINR-38 CELLS. Journal of Biological Chemistry. 1993;268:22042–22045. [PubMed] [Google Scholar]
- Fortier M, Omara F, Bernier J, Brousseau P, Fournier M. Effects of physiological concentrations of heavy metals both individually and in mixtures on the viability and function of peripheral blood human leukocytes in vitro. Journal of Toxicology and Environmental Health-Part a-Current Issues. 2008;71:1327–1337. doi: 10.1080/15287390802240918. [DOI] [PubMed] [Google Scholar]
- Franchi E, Loprieno G, Ballardin M, Petrozzi L, Migliore L. CYTOGENETIC MONITORING OF FISHERMEN WITH ENVIRONMENTAL MERCURY EXPOSURE. Mutation Research. 1994;320:23–29. doi: 10.1016/0165-1218(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Franco JL, Braga HC, Stringari J, Missau FC, Posser T, Mendes BG, Leal RB, Santos ARS, Dafre AL, Pizzolatti MG, Farina M. Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: Protective effects of quercetin. Chemical Research in Toxicology. 2007;20:1919–1926. doi: 10.1021/tx7002323. [DOI] [PubMed] [Google Scholar]
- Franco JL, Posser T, Dunkley PR, Dickson PW, Mattos JJ, Martins R, Bainy ACD, Marques MR, Dafre AL, Farina M. Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radical Biology and Medicine. 2009;47:449–457. doi: 10.1016/j.freeradbiomed.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Furda AM, Bess AS, Meyer JN, Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods in molecular biology (Clifton, NJ) 2012;920:111–132. doi: 10.1007/978-1-61779-998-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadhia SR, Calabro AR, Barile FA. Trace metals alter DNA repair and histone modification pathways concurrently in mouse embryonic stem cells. Toxicology Letters. 2012;212:169–179. doi: 10.1016/j.toxlet.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Gallagher CM, Smith DM, Meliker JR. Total blood mercury and serum measles antibodies in US children, NHANES 2003–2004. Science of the Total Environment. 2011;410:65–71. doi: 10.1016/j.scitotenv.2011.09.038. [DOI] [PubMed] [Google Scholar]
- Gardner RM, Nyland JF, Silbergeld EK. Differential immunotoxic effects of inorganic and organic mercury species in vitro. Toxicology Letters. 2010;198:182–190. doi: 10.1016/j.toxlet.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godar DE, Thomas DP, Miller SA, Lee W. LONG-WAVELENGTH UVA RADIATION INDUCES OXIDATIVE STRESS, CYTOSKELETAL DAMAGE AND HEMOLYSIS. Photochemistry and Photobiology. 1993;57:1018–1026. doi: 10.1111/j.1751-1097.1993.tb02965.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hunt CP, Rooney JP, Ryde IT, Anbalagan C, Joglekar R, Meyer JN. PCR-based analysis of mitochondrial DNA copy number, mitochondrial DNA damage, and nuclear DNA damage. Current Protocols in Toxicology. 2015;66:20.11.21–20.11.25. doi: 10.1002/0471140856.tx2011s67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostner JM, Becker K, Fuchs D, Sucher R. Redox regulation of the immune response. Redox Report. 2013;18:88–94. doi: 10.1179/1351000213Y.0000000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm JH, Kim S, Paik YK. GPA-9 is a novel regulator of innate immunity against Escherichia coli foods in adult Caenorhabditis elegans. Aging Cell. 2011;10:208–219. doi: 10.1111/j.1474-9726.2010.00655.x. [DOI] [PubMed] [Google Scholar]
- Hao OY, Stamatas G, Saliou C, Kollias N. A chemiluminescence study of UVA-induced oxidative stress in human skin in vivo. Journal of Investigative Dermatology. 2004;122:1020–1029. doi: 10.1111/j.0022-202X.2004.22405.x. [DOI] [PubMed] [Google Scholar]
- Hunter SE, Gustafson MA, Margillo KM, Lee SA, Ryde IT, Meyer JN. In vivo repair of alkylating and oxidative DNA damage in the mitochondrial and nuclear genomes of wild-type and glycosylase-deficient Caenorhabditis elegans. DNA Repair. 2012;11:857–863. doi: 10.1016/j.dnarep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto T, Kunito T, Tanaka H, Baba N, Miyazaki N, Tanabe S. Detoxification mechanism of heavy metals in marine mammals and seabirds: Interaction of selenium with mercury, silver, copper, zinc, and cadmium in liver. Archives of Environmental Contamination and Toxicology. 2004;47:402–413. doi: 10.1007/s00244-004-3188-9. [DOI] [PubMed] [Google Scholar]
- Karouna-Renier NK, White C, Perkins CR, Schmerfeld JJ, Yates D. Assessment of mitochondrial DNA damage in little brown bats (Myotis lucifugus) collected near a mercury-contaminated river. Ecotoxicology (London, England) 2014;23:1419–1429. doi: 10.1007/s10646-014-1284-9. [DOI] [PubMed] [Google Scholar]
- Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, Ausubel FM. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- Kim Y, Mylonakis E. Caenorhabditis elegans Immune Conditioning with the Probiotic Bacterium Lactobacillus acidophilus Strain NCFM Enhances Gram-Positive Immune Responses. Infection and Immunity. 2012;80:2500–2508. doi: 10.1128/IAI.06350-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagido C, McLaggan D, Flett A, Pettitt J, Glover LA. Rapid Sublethal Toxicity Assessment Using Bioluminescent Caenorhabditis elegans, a Novel Whole-Animal Metabolic Biosensor. Toxicological Sciences. 2009;109:88–95. doi: 10.1093/toxsci/kfp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagido C, Pettitt J, Flett A, Glover LA. Bridging the phenotypic gap: real-time assessment of mitochondrial function and metabolism of the nematode Caenorhabditis elegans. BMC Physiol. 2008;8:7. doi: 10.1186/1472-6793-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Ledoux SP, Wilson GL, Beecham EJ, Stevnsner T, Wassermann K, Bohr VA. REPAIR OF MITOCHONDRIAL-DNA AFTER VARIOUS TYPES OF DNA DAMAGE IN CHINESE-HAMSTER OVARY CELLS. Carcinogenesis. 1992;13:1967–1973. doi: 10.1093/carcin/13.11.1967. [DOI] [PubMed] [Google Scholar]
- Leung MCK, Williams PL, Benedetto A, Au C, Helmcke KJ, Aschner M, Meyer JN. Caenorhabditis elegans: An emerging model in biomedical and environmental toxicology. Toxicological Sciences. 2008;106:5–28. doi: 10.1093/toxsci/kfn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Fleming JT. Basic culture methods. Methods in Cell Biology. 1995;48:3–29. [PubMed] [Google Scholar]
- Li X, Yin DQ, Li J, Wang R. Protective Effects of Selenium on Mercury Induced Immunotoxic Effects in Mice by Way of Concurrent Drinking Water Exposure. Archives of Environmental Contamination and Toxicology. 2014;67:104–114. doi: 10.1007/s00244-014-0001-2. [DOI] [PubMed] [Google Scholar]
- Liuzzi VC, Daresta BE, de Gennaro G, De Giorgi C. Different effects of polycyclic aromatic hydrocarbons in artificial and in environmental mixtures on the free living nematode C. elegans. Journal of Applied Toxicology. 2012;32:45–50. doi: 10.1002/jat.1634. [DOI] [PubMed] [Google Scholar]
- Lu TH, Hsieh SY, Yen CC, Wu HC, Chen KL, Hung DZ, Chen CH, Wu CC, Su YC, Chen YW, Liu SH, Huang CF. Involvement of oxidative stress-mediated ERK1/2 and p38 activation regulated mitochondria-dependent apoptotic signals in methylmercury-induced neuronal cell injury. Toxicology Letters. 2011;204:71–80. doi: 10.1016/j.toxlet.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Luz AL, Meyer JN. Effects of reduced mitochondrial DNA content on secondary mitochondrial toxicant exposure in Caenorhabditis elegans. Mitochondrion. 2016;30:255–264. doi: 10.1016/j.mito.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manda K, Ueno M, Anzai K. AFMK, a melatonin metabolite, attenuates X-ray-induced oxidative damage to DNA, proteins and lipids in mice. Journal of Pineal Research. 2007;42:386–393. doi: 10.1111/j.1600-079X.2007.00432.x. [DOI] [PubMed] [Google Scholar]
- McElwee MK, Ho LA, Chou JW, Smith MV, Freedman JH. Comparative toxicogenomic responses of mercuric and methyl-mercury. Bmc Genomics. 2013:14. doi: 10.1186/1471-2164-14-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Samperio P, Perez A, Alba L. Reactive Oxygen Species-activated p38/ERK 1/2 MAPK Signaling Pathway in the Mycobacterium bovis Bacillus Calmette Guerin (BCG)-induced CCL2 Secretion in Human Monocytic Cell Line THP-1. Archives of Medical Research. 2010;41:579–585. doi: 10.1016/j.arcmed.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Meyer JN. QPCR: a tool for analysis of mitochondrial and nuclear DNA damage in ecotoxicology. Ecotoxicology. 2010;19:804–811. doi: 10.1007/s10646-009-0457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JN, Boyd WA, Azzam GA, Haugen AC, Freedman JH, Van Houten B. Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biology. 2007:8. doi: 10.1186/gb-2007-8-5-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JN, Leung MCK, Rooney JP, Sendoel A, Hengartner MO, Kisby GE, Bess AS. Mitochondria as a Target of Environmental Toxicants. Toxicological Sciences. 2013;134:1–17. doi: 10.1093/toxsci/kft102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JN, Lord CA, Yang XYY, Turner EA, Badireddy AR, Marinakos SM, Chilkoti A, Wiesner MR, Auffan M. Intracellular uptake and associated toxicity of silver nanoparticles in Caenorhabditis elegans. Aquatic Toxicology. 2010;100:140–150. doi: 10.1016/j.aquatox.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Mieiro CL, Pardal M, Duarte A, Pereira E, Palmeira CM. Impairment of mitochondrial energy metabolism of two marine fish by in vitro mercuric chloride exposure. Marine Pollution Bulletin. 2015;97:488–493. doi: 10.1016/j.marpolbul.2015.05.054. [DOI] [PubMed] [Google Scholar]
- Millet ACM, Ewbank JJ. Immunity in Caenorhabditis elegans. Current Opinion in Immunology. 2004;16:4–9. doi: 10.1016/j.coi.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Mohri-Shiomi A, Garsin DA. Insulin signaling and the heat shock response modulate protein homeostasis in the Caenorhabditis elegans intestine during infection. Journal of Biological Chemistry. 2008;283:194–201. doi: 10.1074/jbc.M707956200. [DOI] [PubMed] [Google Scholar]
- Mori N, Yasutake A, Hirayama K. Comparative study of activities in reactive oxygen species production/defense system in mitochondria of rat brain and liver, and their susceptibility to methylmercury toxicity. Archives of Toxicology. 2007;81:769–776. doi: 10.1007/s00204-007-0209-2. [DOI] [PubMed] [Google Scholar]
- Ni MW, Li X, Yin ZB, Jiang HY, Sidoryk-Wegrzynowicz M, Milatovic D, Cai JY, Aschner M. Methylmercury Induces Acute Oxidative Stress, Altering Nrf2 Protein Level in Primary Microglial Cells. Toxicological Sciences. 2010;116:590–603. doi: 10.1093/toxsci/kfq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyland JF, Fairweather D, Shirley DL, Davis SE, Rose NR, Silbergeld EK. Low-Dose Inorganic Mercury Increases Severity and Frequency of Chronic Coxsackievirus-Induced Autoimmune Myocarditis in Mice. Toxicological Sciences. 2012;125:134–143. doi: 10.1093/toxsci/kfr264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondovcik SL, Tamblyn L, McPherson JP, Wells PG. Oxoguanine Glycosylase 1 (OGG1) Protects Cells from DNA Double-Strand Break Damage Following Methylmercury (MeHg) Exposure. Toxicological Sciences. 2012;128:272–283. doi: 10.1093/toxsci/kfs138. [DOI] [PubMed] [Google Scholar]
- Palmisano NJ, Melendez A. Detection of Autophagy in Caenorhabditis elegans. Cold Spring Harb Protoc. 2016a;2016 doi: 10.1101/pdb.top070466. pdb.top070466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmisano NJ, Melendez A. Detection of Autophagy in Caenorhabditis elegans Using GFP::LGG-1 as an Autophagy Marker. Cold Spring Harb Protoc. 2016b;2016 doi: 10.1101/pdb.prot086496. pdb.prot086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper I, Wehe CA, Bornhorst J, Ebert F, Leffers L, Holtkamp M, Hoseler P, Weber T, Mangerich A, Burkle A, Karst U, Schwerdtle T. Mechanisms of Hg species induced toxicity in cultured human astrocytes: genotoxicity and DNA-damage response. Metallomics. 2014;6:662–671. doi: 10.1039/c3mt00337j. [DOI] [PubMed] [Google Scholar]
- Polli JR, Zhang YQ, Pan XP. Dispersed crude oil amplifies germ cell apoptosis in Caenorhabditis elegans, followed a CEP-1-dependent pathway. Archives of Toxicology. 2014;88:543–551. doi: 10.1007/s00204-014-1198-6. [DOI] [PubMed] [Google Scholar]
- Polunas M, Halladay A, Tjalkens RB, Philbert MA, Lowndes H, Reuhl K. Role of oxidative stress and the mitochondrial permeability transition in methylmercury cytotoxicity. Neurotoxicology. 2011;32:526–534. doi: 10.1016/j.neuro.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Sancar A. Nucleotide excision repair. In: Moldave K, editor. Progress in Nucleic Acid Research and Molecular Biology. Vol. 79. 2005. pp. 183–235. [DOI] [PubMed] [Google Scholar]
- Reinke SN, Hu X, Sykes BD, Lemire BD. Caenorhabditis elegans diet significantly affects metabolic profile, mitochondrial DNA levels, lifespan and brood size. Molecular Genetics and Metabolism. 2010;100:274–282. doi: 10.1016/j.ymgme.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Rooney JP, Luz AL, Gonzalez-Hunt CP, Bodhicharla R, Ryde IT, Anbalagan C, Meyer JN. Effects of 5′-fluoro-2-deoxyuridine on mitochondrial biology in Caenorhabditis elegans. Exp Gerontol. 2014;56:69–76. doi: 10.1016/j.exger.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu TH, An KG, Kim JK. Genotoxicity in earthworm after combined treatment of ionising radiation and mercury. Radiation Protection Dosimetry. 2014;159:111–117. doi: 10.1093/rpd/ncu172. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM, Bohr VA. Protecting the mitochondrial powerhouse. Trends in Cell Biology. 2015;25:158–170. doi: 10.1016/j.tcb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe MA, Livingston AD, Baskin DS. Thimerosal-Derived Ethylmercury Is a Mitochondrial Toxin in Human Astrocytes: Possible Role of Fenton Chemistry in the Oxidation and Breakage of mtDNA. Journal of Toxicology. 2012:373678. doi: 10.1155/2012/373678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokolenko IN, Alexeyev MF. Mitochondrial DNA: A disposable genome? Biochimica Et Biophysica Acta-Molecular Basis of Disease. 2015;1852:1805–1809. doi: 10.1016/j.bbadis.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokolenko IN, Wilson GL, Alexeyev MF. The “fast” and the “slow” modes of mitochondrial DNA degradation. Mitochondrial DNA. 2016;27:490–498. doi: 10.3109/19401736.2014.905829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbergeld EK, Sacci JB, Azad AF. Mercury exposure and murine response to Plasmodium yoelii infection and immunization. Immunopharmacology and Immunotoxicology. 2000;22:685–695. doi: 10.3109/08923970009016432. [DOI] [PubMed] [Google Scholar]
- Silva IA, Graber J, Nyland JF, Silbergeld E. In vitro HgCl2 exposure of immune cells at different stages of maturation: Effects on phenotype and function. Environmental Research. 2005;98:341–348. doi: 10.1016/j.envres.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Singh V, Aballay A. Regulation of DAF-16-mediated Innate Immunity in Caenorhabditis elegans. Journal of Biological Chemistry. 2009;284:35580–35587. doi: 10.1074/jbc.M109.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohs SJ, Bagchi D. OXIDATIVE MECHANISMS IN THE TOXICITY OF METAL-IONS. Free Radical Biology and Medicine. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- Stringari J, Nunes AKC, Franco JL, Bohrer D, Garcia SC, Dafre AL, Milatovic D, Souza DO, Rocha JBT, Aschner M, Farina M. Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicology and Applied Pharmacology. 2008;227:147–154. doi: 10.1016/j.taap.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J. Caenorhabditis elegans. Cold Spring Harbor Laboratory Press; 1988. Cell Lineage. The Nematode. [Google Scholar]
- Sun JR, Singh V, Kajino-Sakamoto R, Aballay A. Neuronal GPCR Controls Innate Immunity by Regulating Noncanonical Unfolded Protein Response Genes. Science. 2011;332:729–732. doi: 10.1126/science.1203411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes - Many genes, common mechanisms. Neuromuscular Disorders. 2010;20:429–437. doi: 10.1016/j.nmd.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Tam ZY, Gruber J, Ng LF, Halliwell B, Gunawan R. Effects of Lithium on Age-related Decline in Mitochondrial Turnover and Function in Caenorhabditis elegans. Journals of Gerontology Series a-Biological Sciences and Medical Sciences. 2014;69:810–820. doi: 10.1093/gerona/glt210. [DOI] [PubMed] [Google Scholar]
- Tan MW, Mahajan-Miklos S, Ausubel FM. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan XX, Tang C, Castoldi AF, Manzo L, Costa LG. EFFECTS OF INORGANIC AND ORGANIC MERCURY ON INTRACELLULAR CALCIUM LEVELS IN RAT LYMPHOCYTES-T. Journal of Toxicology and Environmental Health. 1993;38:159–170. doi: 10.1080/15287399309531709. [DOI] [PubMed] [Google Scholar]
- TeKippe M, Aballay A. C. elegans Germline-Deficient Mutants Respond to Pathogen Infection Using Shared and Distinct Mechanisms. Plos One. 2010:5. doi: 10.1371/journal.pone.0011777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M, Forman HJ. Redox signaling and the MAP kinase pathways (Reprinted from Thiol Metabolism and Redox Regulation of Cellular Functions) Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- Tsang WY, Lemire BD. Mitochondrial genome content is regulated during nematode development. Biochemical and Biophysical Research Communications. 2002;291:8–16. doi: 10.1006/bbrc.2002.6394. [DOI] [PubMed] [Google Scholar]
- Tsang WY, Lemire BD. The role of mitochondria in the life of the nematode, Caenorhabditis elegans. Biochimica Et Biophysica Acta-Molecular Basis of Disease. 2003;1638:91–105. doi: 10.1016/s0925-4439(03)00079-6. [DOI] [PubMed] [Google Scholar]
- Turner EA, Kroeger GL, Arnold MC, Thornton BL, Di Giulio RT, Meyer JN. Assessing Different Mechanisms of Toxicity in Mountaintop Removal/Valley Fill Coal Mining-Affected Watershed Samples Using Caenorhabditis elegans. Plos One. 2013;8:1–12. doi: 10.1371/journal.pone.0075329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Morris H, Cronin MTD. Metals, toxicity and oxidative stress. Current Medicinal Chemistry. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- Van Houten B, Hunter SE, Meyer JN. Mitochondrial DNA damage induced autophagy, cell death, and disease. Frontiers in Bioscience-Landmark. 2016;21:42–54. doi: 10.2741/4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessoni AT, Filippi-Chiela EC, Menck CF, Lenz G. Autophagy and genomic integrity. Cell Death Differ. 2013;20:1444–1454. doi: 10.1038/cdd.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetvicka V, Vetvickova J. Effects of Glucan on Immunosuppressive Actions of Mercury. Journal of Medicinal Food. 2009;12:1098–1104. doi: 10.1089/jmf.2008.0273. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, Chandel NS. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunity. 2015;42:406–417. doi: 10.1016/j.immuni.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nature Reviews Immunology. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MV, Winters T, Waddell KS. INVIVO EFFECTS OF MERCURY(II) ON DEOXYURIDINE TRIPHOSPHATE NUCLEOTIDOHYDROLASE, DNA-POLYMERASE (ALPHA,BETA), AND URACIL-DNA GLYCOSYLASE ACTIVITIES IN CULTURED HUMAN-CELLS - RELATIONSHIP TO DNA DAMAGE, DNA-REPAIR, AND CYTOTOXICITY. Molecular Pharmacology. 1987;31:200–207. [PMC free article] [PubMed] [Google Scholar]
- Worth RG, Esper RM, Warra NS, Kindzelskii AL, Rosenspire AJ, Todd RF, Petty HR. Mercury inhibition of neutrophil activity: Evidence of aberrant cellular signalling and incoherent cellular metabolism. Scandinavian Journal of Immunology. 2001;53:49–55. doi: 10.1046/j.1365-3083.2001.00834.x. [DOI] [PubMed] [Google Scholar]
- Wyatt L, Diringer S, Rodgers L, Hsu-Kim H, Pan W, Meyer J. Antagonistic effects of mercury and selenium on growth in Caenorhabditis elegans are species-dependent, occur at high aqueous concentrations of Se, and are not dependent on the internal Hg/Se ratio. Environmental Science & Technology. 2016 doi: 10.1021/acs.est.5b06044. (in prep) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, VanHouten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XY, Gondikas AP, Marinakos SM, Auffan M, Liu J, Hsu-Kim H, Meyer JN. Mechanism of Silver Nanoparticle Toxicity Is Dependent on Dissolved Silver and Surface Coating in Caenorhabditis elegans. Environmental Science & Technology. 2012;46:1119–1127. doi: 10.1021/es202417t. [DOI] [PubMed] [Google Scholar]
- Yee S, Choi BH. Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology. 1996;17:17–26. [PubMed] [Google Scholar]
- Yin ZB, Milatovic D, Aschner JL, Syversen T, Rocha JBT, Souza DO, Sidoryk M, Albrecht J, Aschner M. Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Research. 2007;1131:1–10. doi: 10.1016/j.brainres.2006.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chang JT, Guo B, Hansen M, Jia K, Kovacs AL, Kumsta C, Lapierre LR, Legouis R, Lin L, Lu Q, Melendez A, O’Rourke EJ, Sato K, Sato M, Wang X, Wu F. Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy. 2015;11:9–27. doi: 10.1080/15548627.2014.1003478. [DOI] [PMC free article] [PubMed] [Google Scholar]