

ABSTRACT
Fractures heal predominantly through the process of endochondral ossification. The classic model of endochondral ossification holds that chondrocytes mature to hypertrophy, undergo apoptosis and new bone forms by invading osteoprogenitors. However, recent data demonstrate that chondrocytes transdifferentiate to osteoblasts in the growth plate and during regeneration, yet the mechanism(s) regulating this process remain unknown. Here, we show a spatially-dependent phenotypic overlap between hypertrophic chondrocytes and osteoblasts at the chondro-osseous border in the fracture callus, in a region we define as the transition zone (TZ). Hypertrophic chondrocytes in the TZ activate expression of the pluripotency factors [Sox2, Oct4 (Pou5f1), Nanog], and conditional knock-out of Sox2 during fracture healing results in reduction of the fracture callus and a delay in conversion of cartilage to bone. The signal(s) triggering expression of the pluripotency genes are unknown, but we demonstrate that endothelial cell conditioned medium upregulates these genes in ex vivo fracture cultures, supporting histological evidence that transdifferentiation occurs adjacent to the vasculature. Elucidating the cellular and molecular mechanisms underlying fracture repair is important for understanding why some fractures fail to heal and for developing novel therapeutic interventions.
KEY WORDS: Endochondral ossification, Fracture repair, Pluripotency programs, Chondrocyte transformation
Summary: During fracture repair in mice, chondrocytes at the chondro-osseous border adjacent to the invading vasculature transdifferentiate into osteoblasts by activating pluripotency programs and initiating cell division.
INTRODUCTION
Fracture healing begins with an acute inflammatory response that produces a hematoma to isolate the damaged tissue, and then new bone arises by both intramembranous and endochondral ossification. Differentiation of the progenitor cells into chondrocytes or osteoblasts depends on the local mechanical microenvironment (Isaksson et al., 2006; Miclau et al., 2007; Miller et al., 2015). Progenitor cells undergo direct bone formation, or intramembranous ossification, along the periosteal and endosteal surfaces. In the fracture gap, where there is motion, the progenitors differentiate into chondrocytes and healing occurs through endochondral ossification. During healing, these chondrocytes undergo maturation towards hypertrophy and become highly angiogenic. The chondrocytes induce vascular invasion of the cartilage callus and this corresponds to mineralization of the cartilage matrix and formation of the trabecular bone. The newly formed bone is remodeled into cortical bone through the coordinated actions of osteoblasts and osteoclasts.
The source of osteoblasts that give rise to bone during endochondral ossification remains controversial. Two models have been proposed: one in which the hypertrophic chondrocytes undergo programmed cell death and the progenitor cells enter the cartilage callus with the invading vasculature and replace the cartilage with bone. Alternatively, some hypertrophic chondrocytes do not undergo programmed cell death and instead transdifferentiate into osteoblasts. While almost 200 years of dogma has fallen in favor of the model of hypertrophic chondrocyte apoptosis (Kronenberg, 2003; Shapiro et al., 2005), recent studies using contemporary genetic lineage tracing methods provide strong evidence that chondrocyte transdifferentiation is the dominant pathway for endochondral bone formation during development, growth and repair (Bahney et al., 2014; Yang et al., 2014a,b; Zhou et al., 2014).
Very few details regarding the process by which chondrocytes convert into osteoblasts are currently known. In this paper, we focus on endochondral fracture repair. We examine the cellular and molecular properties of the cells located at the chondro-osseous junction of the fracture callus to understand the mechanism(s) by which chondrocytes become osteoblasts during fracture healing.
RESULTS
Mature hypertrophic chondrocytes in the fracture callus express canonical bone markers
We created closed mid-shaft tibia fractures that remained unstabilized to generate a robust endochondral healing response. In previous studies using this model we detailed the sequence of endochondral repair as follows; cartilage condensation (days 3-5), maturation and mineralization of the cartilage (days 5-14), trabecular bone formation (days 10-14), bone remodeling (day 14+) (Lu et al., 2005; Thompson et al., 2002). We focus on the transformation of cartilage into bone during days 7 to 14 of fracture healing. We focus on a specific histological region, the ‘transition zone’ (TZ), which represents the chondro-osseous junction in the fracture callus. This region can be visualized in the day 10 fracture callus using standard histological staining to distinguish cartilage and bone (Fig. 1). Importantly, histology shows that the transitional phenotype occurs around the invading blood vessels (Fig. 1G-I). Here, proteoglycans are lost from matrix surrounding the hypertrophic chondrocytes and these cells start producing a bone matrix (Fig. 1G-I, white arrows).
Fig. 1.

Visualization of the chondro-osseous transition zone in a fracture callus. (A-C) Low magnification of a murine fracture callus, outlined with black dashed line, stained with (A) Safranin-O/Fast Green (SO/FG), (B) Modified Milligan's Trichrome (TC) or (C) Hall and Brunt Quadruple Stain (HBQ). (D-F) A magnified region of cartilage and bone from the fracture callus, outlined with a red box (A-C), with the TZ indicated by black brackets. (G-I) High magnification images of the TZ show the invading vasculature and the chondro-osseous junction. BV, blood vessel. Scale bars: 1 mm (A-C) and 100 µm (D-I).
To provide a detailed characterization of the cellular phenotype in the TZ, we analyzed the spatial expression of the canonical markers of chondrocytes and osteoblasts (Figs 2 and 3; Figs S1 and S2). The cartilaginous region of the fracture callus was observed after Safranin-O staining (Fig. 2A) along with expression of the canonical chondrocyte markers collagen II (Col2a1; Fig. 2B) and Sox9 (Fig. 3A). As the chondrocytes mature, the cells enlarge to a hypertrophic state (Fig. 2A,K) and express collagen X (Col10a1; Fig. 2H,M). In the TZ, chondrocytes lose chondrogenic signatures (Sox9, Col2a1, Col10a1) and begin expressing bone-specific collagen I (Col1a1) despite maintaining a hypertrophic morphology (Fig. 2L-N, Fig. 3A,E). Through the use of thin (3-5 µm) adjacent sections, we tracked individual cells to demonstrate that expression of Col10a1 and Col1a1 are mutually exclusive (Fig. 2E,J,O,T).
Fig. 2.

Maturation of cartilage in the transition zone. Chondrocytes away from the TZ (A-D), compared with hypertrophic chondrocytes (HCs) in the TZ of murine fracture callus (E-O,T) or newly formed bone (P-S). Left column shows cartilage and bone histology stained with either SO/FG (A,F,K) or TC (P). In situ hybridization with Col2a1 (B,G,L,Q), Col10a1 (C,H,M,R) or Col1a1 (D,I,N,S). (E,J,O,T) Col10a1 and Col1a1 staining on adjacent sections 3-5 µm apart. Individual cells were tracked (cells 1-6) to demonstrate that staining does not overlap. Scale bars: 100 µm.
Fig. 3.

Hypertrophic chondrocytes adjacent to vasculature in the transition zone lose their chondrocyte phenotype and acquire an osteoblast phenotype. Immunohistochemistry in the cartilage away from the TZ (A-D), compared with HCs in the TZ (E-L) or new bone (NB) (M-P). (I-L) Black arrows indicate HCs in TZ that are Sox9 negative (I), and positive for Runx2 (J), β-catenin (K) or Oc (L). (M-P) Red arrows in NB tissue indicate Runx2+ (N) and Oc+ (P) cells. Scale bars: 100 µm.
Transcriptional regulation of these ‘hypertrophic osteoblasts’ has switched from chondrogenic programming (loss of Sox9: Fig. 3I, black arrows), to osteogenic (appearance of Runx2: Fig. 3J, black arrows). Expression of Runx2 correlates with nuclear localization of β-catenin, indicating activation of canonical Wnt signaling in hypertrophic chondrocytes in the TZ adjacent to the vasculature (Fig. 3C,G,K). Runx2 and Wnt are required for osteogenesis. (Day et al., 2005; Ducy et al., 1997; Gaur et al., 2005; Hill et al., 2005; Komori et al., 1997; Otto et al., 1997; Topol et al., 2009).
Downstream canonical bone programs – osteocalcin (Oc, also known as BGLAP), osteopontin [Opn (Spp1)] and osterix [Osx (Sp7)] – also appear in these cells that would morphologically be identified as hypertrophic chondrocytes (Fig. 3; Fig. S1-S2). Away from the TZ, osteocalcin expression begins as intracellular staining (Fig. 3H), with protein accumulating in the matrix around hypertrophic cells within the TZ adjacent to the vasculature and in the newly formed bone (Fig. 3H,L). Similarly, as shown by in situ hybridization, expression of Opn is initially absent from the immature cartilage but becomes robustly expressed in hypertrophic cells adjacent to the vasculature (Fig. S1). Lastly, we evaluate Osx expression using an Osterix(Sp7)-CreERT mouse crossed to the R26R reporter line. For all lineage-tracing experiments, animals were allowed to heal without intervention for 6 days, at which point there is a robust cartilage callus. Recombination is induced from days 6 to 10 by daily intraperitoneal tamoxifen injections and fractured legs harvested at day 14 for analysis. Osx is expressed in the hypertrophic chondrocytes in the TZ in areas around the vasculature (Fig. S2A-C), in the osteoblasts and osteocytes of the new bone (Fig. S2D, red arrows), and in the bone lining cells of the newly formed trabeculae (Fig. S2D, black arrows).
Hypertrophic chondrocytes re-enter the cell cycle or undergo apoptosis
Hypertrophic chondrocytes have traditionally been considered as a terminally differentiated, post-mitotic cell. To visualize the pattern of cell division during endochondral fracture repair, we administered BrdU 2 hours prior to the harvest of tibiae 14 days after fracture (Fig. 4). Proliferation occurs in immature chondrocytes in the central portion of the fracture, suggesting that these cells are responsible for expansion of the cartilage callus (Fig. 4B). Consistent with the dogma that hypertrophic chondrocytes are post-mitotic, no cell division is observed in the hypertrophic chondrocyte away from the TZ (Fig. 4C). However, hypertrophic cells directly adjacent to the vasculature incorporate BrdU, indicating that they re-enter the cell cycle (Fig. 4D,E). Ki-67 (MKI67), a marker of proliferation, was also expressed by hypertrophic cells at the TZ (Fig. 4F). Co-staining for collagen X (Fig. 4G) and BrdU, shows that BrdU+ cells can be either positive (Fig. 4H, arrows) or negative (Fig. 4H, arrowheads) for collagen X. To compare cell proliferation between areas of the fracture callus, the populations of BrdU+ cells within immature cartilage (C), hypertrophic cartilage (HC), TZ and new bone (NB) tissues was determined using stereology (Fig. 4I). There are significantly more dividing cells in the TZ than in either C (P=0.0022) or HC (P=0.0001).
Fig. 4.

Hypertrophic chondrocytes adjacent to vasculature in the transition zone re-enter the cell cycle. BrdU detection for entire fracture callus (A), immature chondrocytes away from the TZ (B), HCs away from the TZ (C), HCs in the TZ (D,E). (F,G) Ki67 and collagen X immunohistochemistry in HCs at the TZ. (H) Collagen X is observed to overlap with some of the BrdU+ cells (arrows), but not all (arrowheads). (I) Quantification of BrdU+/BrdU− cells in fracture callus. Scale bars: 100 µm.
To test the classically accepted model that hypertrophic chondrocytes undergo apoptosis, we perform TUNEL staining to identify cells with fragmented DNA and activated caspase-3 immunohistochemistry to indicate cells fated for apoptosis. Minimal evidence of programmed cell death is observed in the immature or hypertrophic cartilage within the fracture callus by either TUNEL (Fig. 5A-C) or caspase-3 staining (Fig. 5E-G). Those cells that are dying are found in close proximity to osteoclasts (Fig. 5I-L), suggesting cell death may be necessary for remodeling of the cartilage to give rise to marrow space. Conversion of cartilage to bone is a continual process that occurs at the edge of the cartilage callus, so the amount of cell death at any given time is expected to be variable; consequently, we included a region of maximal cell death from our analysis of over 20 animals (Fig. 5D,H,L).
Fig. 5.

Cell death is not the predominant fate of hypertrophic chondrocytes during endochondral fracture repair. TUNEL staining (A-D), caspase-3 immunohistochemistry (E-H) or co-staining with TUNEL (green; I-L) to detect dying cells (black arrows) and TRAP to detect osteoclasts (purple/red; I-L) within immature chondrocytes (A,E,I), HCs away from the TZ (B,F,J), HCs within the TZ (C,G,K) and a region of maximal cell death at the TZ (D,H,L). Scale bars: 100 µm.
Chondrocytes transdifferentiate to give rise to new bone in the fracture callus
Collagen II and aggrecan, the most abundant proteoglycan in the cartilage matrix, are the two classic markers of the chondrocyte. To understand the fate of chondrocytes during endochondral fracture repair, we performed lineage-tracing experiments on cells expressing collagen II and aggrecan by crossing the Ai9 reporter strain (Madisen et al., 2010) to tamoxifen-inducible Col2CreERT (Nakamura et al., 2006) and Agc1CreERT (Henry et al., 2009) mice.
Chondrocytes are stochastically labeled in the growth plate (Fig. 6A-C) and fracture callus (Fig. 6D-F) of both the Col2CreERT::Ai9 (Fig. 6B,E,H) and Agc1CreERT::Ai9 mice (Fig. 6C,F,I) indicating effective recombination. We obtained significantly better recombination in chondrocytes in the Agc1CreERT::Ai9 mice (Fig. 6D-F), indicating that it is a more robust genetic tool for analysis of adult regeneration. Together, these mice show chondrocyte-derived cells evident within the matrix (Fig. 6G-I) and lining the surface (Fig. 6I) of the newly formed bone. These data support similar lineage tracing experiments showing that chondrocytes survive and give rise to osteoblasts and osteocytes in the growth plate and bone regenerate (Bahney et al., 2014; Yang et al., 2014a,b; Zhou et al., 2014).
Fig. 6.

Chondrocytes give rise to osteoblasts and bone lining cells in the newly formed bone during fracture repair. Adult growth plate (A-C), cartilage (D-F) and newly formed bone (G-I) in fracture callus. Col2CreERT::Ai9 (B,E,H) or Agc1CreERT::Ai9 (C,F,I) mice 14 days post-fracture. Scale bars: 100 µm (B,C,E,F,H,I).
Hypertrophic chondrocytes at the transition zone express OCT4, SOX2 and NANOG
Hypertrophic chondrocytes could give rise to osteoblasts by a number of different mechanisms. One possibility is that hypertrophic chondrocytes de-differentiate, or acquire a stem-cell-like state, to facilitate a lineage fate switch. Induction of the pluripotency factor Nanog was previously noted during a transcriptional analysis of fracture healing (Bais et al., 2009) and Oct4A expression was found by immunohistochemistry in hypertrophic chondrocytes during fracture repair (Bahney et al., 2014).
Here, we aimed to determine the extent to which activation of the core pluripotency-inducing programs [Oct4 (Pou5f1), Sox2, Nanog] is associated with the transformation of chondrocytes to osteoblasts during endochondral repair. Using immunohistochemistry on WT mice (n>12), we find expression of Oct4 and Nanog are low or absent in the immature chondrocytes away from the TZ, whereas Sox2 was expressed in some immature chondrocytes (Fig. 7A-D). Expression of these pluripotency factors becomes more widespread in the hypertrophic chondrocytes around the vasculature in the TZ (Fig. 7I-L). Co-localization of Oct4 and Sox2 is observed within the nuclei of these hypertrophic chondrocytes near the TZ (Fig. S3). Within the newly formed trabecular bone, expression of the pluripotent factors is apparent in cells that maintain a hypertrophic morphology (Fig. 7M-P, red arrows), but is not observed in cells with osteoblast/osteocyte morphology. Bone lining cells also stain positively for these markers (Fig. 7M-P, black arrows). Expression is not observed in bone outside the TZ, cortical bone or muscle (not shown); but can be observed in the growth plate of adult mice (Fig. S5). To understand the lineage of cells expressing pluripotency factors, we performed Oct4, Sox2 and Nanog immunohistochemistry on fractures made in the Agc1CreERT::Ai9 mice (Fig. 7R-T, Fig. S4). The pluripotency programs are active in Ai9+ cells, indicating that these cells were aggrecan-expressing chondrocytes at one point.
Fig. 7.

Expression of pluripotent stem cell programs in the transition zone of the fracture callus. Pluripotent stem cell protein is found in few cells at immature chondrocytes (A-D) but more frequently in HCs near the TZ (E-H) and within the TZ (I-L). (M-P) Within the NB, expression is observed in the HC encased in bone matrix and in bone-lining cells, but not in cells morphologically resembling osteoblasts/osteocytes. (Q) Low magnification of the representative fracture callus on which staining was performed on adjacent sections. (R-T) Cells from the chondrocyte lineage in Agc1CreERT::Ai9 mice (red) co-stained for Oct4 (R), Sox2 (S), Nanog (T) using an Alexa Fluor 488 secondary antibody (green); co-expression appears yellow. Green arrows indicate negative cells; red arrows, positive hypertrophic chondrocytes; black arrows, positive bone lining cells. Scale bar: 100 µm.
To map the fate of cells expressing Oct4 or Sox2, we mated the tamoxifen-inducible Oct4-CreERT (Greder et al., 2012) and Sox2-CreERT (Arnold et al., 2011) mice to the R26R or ROSAmT/mG reporter strains, respectively (Fig. 8). Lineage analysis of Nanog-expressing cells could not be performed because a transgenic mouse with Cre-recombinase under control of the Nanog promoter is not available. At 14 days post-fracture, β-galactosidase staining of calluses harvested from Oct4-CreERT::R26R mice shows no Oct4-expressing cells present in the immature cartilage (Fig. 8A), but Oct4-positive cells are found at the TZ around the new blood vessels and embedded in the newly formed bone (Fig. 8B,C, red arrows). In the bone matrix further from the TZ, Oct4-expressing cells with both chondrocyte and osteoblast morphology are apparent (Fig. 8B,C, red arrows).
Fig. 8.

Transgenic reporter mice for Oct4 and Sox2 demonstrate transformation of chondrocytes into osteoblasts. Oct4-CreERT::R26R (A-D) and Sox2-CreERT::ROSAmT/mG (E-H) mice 14 days post-fracture in cartilage away from the TZ (A,D), TZ (B,E) and NB (C,F). (D) X-gal staining of C57B6. (H) Control with only secondary antibody. Red arrows indicate positive hypertrophic chondrocytes; black arrows, positive bone lining cells. Scale bars: 100 µm.
Similarly, the Sox2-CreERT mouse crossed to the ROSAmT/mG reporter confirms the Sox2 immunohistochemistry results shown in Fig. 7. Owing to strong autofluorescence, GFP-positive cells in Sox2-CreERT::ROSAmT/mG mice are detected by DAB immunohistochemistry. Sox2-CreERT::ROSAmT/mG demonstrates sporadic Sox2 expression in chondrocytes away from the TZ (Fig. 8D). The frequency of Sox2 expression in hypertrophic chondrocytes increases near the TZ (Fig. 8E). In the new bone matrix, Sox2 expression is observed in cells with both hypertrophic chondrocyte and osteoblast morphology (Fig. 8G, red arrows) and in bone lining cells (Fig. 8G, black arrows).
SOX2 has a functional role in fracture healing
To test whether expression of pluripotency factors has a functional role in fracture healing we completed gain- and loss-of-function experiments. To test gain-of-function we transfected OCT4 and SOX2 into explanted fracture callus cartilage. Strong expression of Sox2 is observed in the transfected samples when compared with controls, with a more modest, but significant increase in Oct4 expression (Fig. 9). Osteocalcin gene expression was significantly upregulated by both the osteogenic medium and transfection of SOX2 and OCT4, but not chondrogenic medium.
Fig. 9.
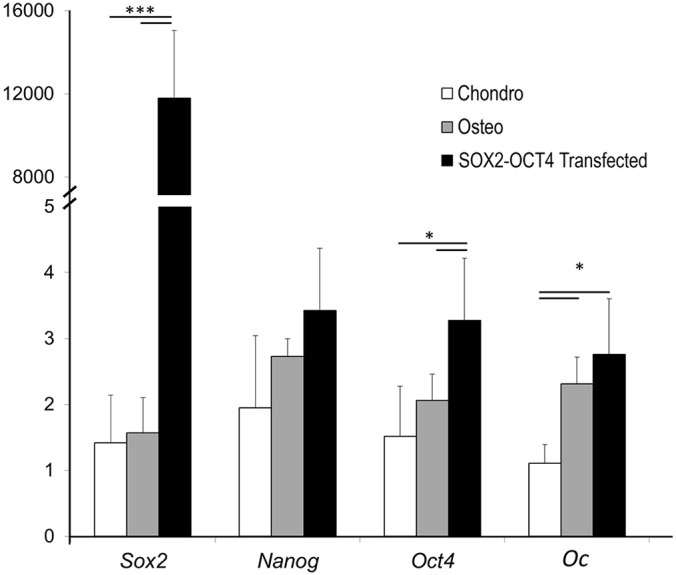
Transfection of OCT4 and SOX2 induces osteocalcin expression in fracture callus cartilage. Relative gene expression analysis of cartilage callus explants cultured in vitro in chondrogenic medium (white), osteogenic medium (gray), or chondrogenic medium with the OCT4-SOX2 transgene (black). Values are means±95% confidence. of n=6.
For loss-of-function studies, we compared fracture healing in inducible Sox2 knockout mice (Sox2CreERT/fl) with C57B6 controls, both receiving tamoxifen injections from day 6. Sox2 and Oct4 are effectively lost from the fracture callus of the knockout mice (Fig. 10A,B). Deletion of Sox2 results in significantly decreased total callus size, bone and vasculature volume (Fig. 10C). Similarly, we find a compositional switch from a lower percentage of bone and a higher percentage of cartilage in the Sox2CreERT/fl mice compared with the control (Fig. 10D).
Fig. 10.

Conditional deletion of Sox2 compromises fracture healing. (A,B) µCT shows smaller callus size and immunohistomistry shows reduced expression of Sox2 and Oct4, but not Nanog. Tamoxifen was administered on days 4-7, 10 and 12 days post-fracture in (A) C57B6 control or (B) Sox2-CreER/fl mice. Histomorphometeric quantification of total tissue volumes (C) or tissue composition (D). Values are means±95% confidence of n=5. *P<0.05, **P<0.01. Scale bars: 100 µm.
The vasculature coordinates chondrocyte to osteoblast transdifferentiation
In hypertrophic chondrocytes, conversion from a chondrogenic to osteogenic genotype (Figs 1–3) and expression of the pluripotency factors (Fig. 7) is spatially correlated with vascular invasion (Fig. 11). This is illustrated histologically in the fracture callus by localizing expression of the highly angiogenic vascular endothelial growth factor (Vegf) in the hypertrophic chondrocytes, followed by subsequent invasion of vascular endothelial cells into the TZ. The immature cartilage in the fracture callus is avascular and the chondrocytes do not express Vegf (Fig. 11A,B). As the chondrocytes mature and become hypertrophic, they express Vegf (Fig. 11C) and stimulate vascular invasion [Fig. 11F,I; Pecam1 (CD31) in black]. Vegf is also expressed by the endothelial cells from the invading vasculature and becomes retained in the matrix at the TZ (Fig. 11D). Based on the physical proximity of the blood vessels to the phenotypic changes occurring during the chondrocyte to osteoblast transformation, we hypothesized that the vasculature might serve as a trigger for transdifferentiation.
Fig. 11.

Hypertrophic chondrocytes recruit vasculature through VEGF expression and vasculature coordinates activation of pluripotent programs. (A-D) Vegf immunohistochemistry (brown) of entire fracture callus (A), immature chondrocytes (B), HCs away from the TZ (C) or HCs in the TZ (D). HBQ (E,H) and Pecam1 (CD31) (F,I) immunohistochemistry (black) on adjacent sections. (G) Relative gene expression of pluripotent, chondrogenic and osteogenic genes for cartilage callus explants cultured in vitro with chondrogenic (white), osteogenic (gray), or HUVEC-CM (black). Values are means±95% confidence of n=4. *P<0.05, ***P<0.0005, ****P<0.0001. Scale bars: 100 µm.
To test this, we conditioned media with human umbilical vascular endothelial cells (HUVECs) and applied it to fracture callus explants cultured ex vivo as described (Bahney et al., 2014). The HUVEC conditioned medium strongly stimulated expression of Oct4, Sox2 and Nanog relative to explants remaining in chondrogenic medium (Fig. 11G). Osteogenic medium does not lead to induction of the pluripotency genes, but both osteogenic and HUVEC conditioned media stimulate osteocalcin gene expression (Fig. 9 and Fig. 11G).
DISCUSSION
Fate of chondrocytes in the fracture callus
The established model of endochondral ossification suggests that chondrocytes undergo apoptosis and new bone is formed by invading osteoprogenitors. Despite classical support, the only experimental evidence for this model comes from Maes et al. who conclude that perivascular Osx-CreERT::R26R cells give rise to trabecular osteoblasts, osteocytes and stromal cells during endochondral ossification (Maes et al., 2010). However, as shown in this paper (Fig. S2), Osx is robustly expressed by hypertrophic chondrocytes in the fracture callus, indicating that osterix-derived bone may not come exclusively from the vasculature, but could represent a chondrocyte-derived population.
While some early scientific data suggested that chondrocytes could transform into bone-forming cells, this pathway never gained traction (Holtrop, 1972; Pritchard and Ruzicka, 1950; Roach, 1992, 1997). In addition to the data shown in this manuscript (Fig. 6), recent studies using contemporary genetic methods of lineage tracing have similarly demonstrated that chondrocytes give rise to osteoblasts and osteocytes in the growth plate during bone formation, post-natal growth and repair (Bahney et al., 2014; Scotti et al., 2010; Yang et al., 2014a,b; Zhou et al., 2014). Taken together, strong evidence that hypertrophic chondrocytes directly contribute to the new bone challenges the traditional model that hypertrophy is the terminal differentiation state of chondrocytes.
The argument for chondrocyte apoptosis during endochondral ossification comes largely from the data in the growth plate (reviewed in Shapiro et al., 2005) and the suggestion that fracture repair recapitulates aspects of skeletal development (Gerstenfeld et al., 2003). However, since both anatomical structure and microenvironment at the chondro-osseous junction in the growth plate is very different than the fracture callus, it is important to consider that cellular fates may also be different. In this study we show minimal evidence of apoptosis in the TZ during fracture repair using both TUNEL staining and immunohistochemistry for activated caspase-3 (Fig. 4). Importantly, cell death adjacent to osteoclast activity suggests apoptosis aids in conversion of the solid cartilaginous tissue to trabecular bone with marrow space.
In contrast to apoptosis, TZ chondrocytes re-entered the cell cycle, as evident by incorporation of BrdU and expression of Ki67 (Fig. 4). The potential for hypertrophic chondrocytes to undergo cell division in the growth plate has been observed in the chick femur by Roach et al., where they proposed subpopulations of hypertrophic chondrocytes undergo asymmetric cell division to give rise to osteoblasts (Roach and Clarke, 2000; Roach and Erenpreisa, 1996). More recently, hypertrophic chondrocytes in the growth plate of mice were observed to incorporate BrdU prior to transdifferentiation into osteoblasts (Yang et al., 2014a). Division of the hypertrophic chondrocyte may explain how these enlarged cells (Cooper et al., 2013) lose volume to acquire the size of an osteoblast. In addition to incorporating BrdU, we provide evidence that TZ chondrocytes activate pluripotency programs, which may help to explain why these cells, unlike the non-dividing hypertrophic chondrocytes away from the TZ, are capable of cell division (Fig. 7).
Chondrocytes transdifferentiate to osteoblasts by activating a stem cell-like state
Plasticity of chondrocytes has been demonstrated in a number of studies and, cumulatively, these data suggest that hypertrophic chondrocytes represent a pivotal state between the opposing programs that regulate the chondrogenic versus osteogenic phenotype. Sox9 is essential for transcriptional regulation of chondrogenesis (Bi et al., 1999; Lefebvre et al., 1997; Zhao et al., 1997). Osteogenesis is induced by expression of Runx2 (Ducy et al., 1997; Komori et al., 1997; Otto et al., 1997) and activation of canonical Wnt (Clevers, 2006; Gaur et al., 2005). Co-expression of Runx2 and β-catenin specifically prevents differentiation toward the chondrogenic lineage by suppressing Sox9 expression (Day et al., 2005; Hill et al., 2005). Conversely, ablation of Sox9 in pre-hypertrophic growth plate chondrocytes resulted in acquisition of an osteoblastic phenotype (Dy et al., 2012), and overexpression of Sox9 leads to expansion of the hypertrophic zone and delayed ossification (Hattori et al., 2010; Tsang et al., 2007).
Downstream, the canonical bone-related genes, osterix (Osx) and osteopontin (Opn), are strongly dependent on Runx2 activity. Homozygous deletion of Runx2 or Osx leads to the complete absence of osteoblasts in mouse embryos, and Osx expression is absent following deletion of Runx2, suggesting that Osx is downstream (Nakashima et al., 2002; Otto et al., 1997). Opn, in turn, is transcriptionally regulated by Runx2 and Osx (Ducy et al., 1997; Nakashima et al., 2002). In the fracture callus, we find the hypertrophic chondrocytes in the TZ lose the chondrogenic phenotype (Sox9 expression) and activate osteogenic programs (Runx2, nuclear β-catenin, OC, Opn, Osx, Col1a1) suggesting that these cells are primed for bone formation.
The mechanisms that enable chondrocytes to become osteoblasts remain unexplored. In this study, we investigated the hypothesis that the hypertrophic chondrocytes acquire plasticity by activating canonical stem-cell programs: Oct4, Sox2 and Nanog. We utilized immunostaining techniques (Fig. 7, Fig. S3) complemented by both Oct4 and Sox2 murine reporter systems (Fig. 8) to demonstrate their expression in cells in the TZ. Importantly, by performing immunohistochemistry on fracture calluses harvest from Agc1-CreERT::Ai9 mice, we confirmed that these proteins are observed in cells that were chondrocytes (Fig. 7R-T, Fig. S4). We have shown selective hypertrophic chondrocytes in the TZ exhibit positive immunostaining for Oct4A (Bahney et al., 2014). In this paper, we again show expression of Oct4 in the TZ of the fracture callus, but we used an Oct4 antibody that does not distinguish between isoforms (Fig. 7) because this reflects data obtained with the Oct4-CreERT::R26R transgenic system used for lineage tracing (Fig. 8). However, Oct4 is localized in the nucleus along with Sox2 (Fig. S3), and current understanding of Oct4 biology is that only Oct4A localizes to the nucleus (Lee et al., 2006; Liedtke et al., 2008).
Within the literature, it is well established that Oct4, Sox2 and Nanog maintain pluripotency in embryonic stem cells and can reprogram terminally differentiated somatic cells into a stem-cell-like state. Their role in normal adult tissues remains largely unknown and their activation is currently associated with cancer pathogenesis. Tumors positively genotyped for Oct4, Sox2, or Nanog are associated with a poor prognosis and increased metastasis by enabling cancerous cells to undergo sustained proliferation and evade cell death, suggesting that these genes may confer a stem-like state onto the cancer cells (Gong et al., 2015; Samardzija et al., 2012; Weina and Utikal, 2014).
Oct4 expression and function in healthy adult tissue is somewhat contentious because of the gene's multiple transcripts and alternative splicing. Oct4A is the isoform associated with pluripotency (Lee et al., 2006). Oct4B, another well-known isoform, is not believed to be involved in canonical pluripotency programs and, instead, has been reported to be associated with the cell stress response (Wang et al., 2009). One study suggested that Oct4 does not play a role in normal homeostasis since deletion had no effect on adult stem cell populations in the intestinal epithelium, bone marrow, hair follicle, brain or liver (Lengner et al., 2007). However, fracture healing is not a homeostatic process. Instead, it is a regenerative process that is now understood to involve transformation of chondrocytes to osteoblasts.
Interestingly, in contrast to Oct4, Sox2 has been localized to various adult stem cell niches and suggested to have a functional role in maintaining multipotency. In this study, we demonstrate a functional role of Sox2 during fracture healing through a significant decrease in fracture callus and bone formation following conditional deletion of the protein from days 4 to 12 during endochondral repair (Fig. 10). Sox2 was first suggested to play a role in maintaining the multipotency and self-renewal capacity of stem cells in the adult hippocampus (Suh et al., 2007). Subsequently, Sox2-positive cells have been identified in several epithelial tissues, including stomach, cervix, anus, testes, lens and multiple glands. Lineage tracing experiments demonstrated that these Sox2-expressing cells represent an adult stem cell population with both self-renewal and differentiation potentials (Arnold et al., 2011). Sox2 has also been associated with continuous eruption of the murine incisor. The labial cervical loop of the mouse incisor contains a pool of Sox2-expressing cells that are capable of giving rise to all the epithelial lineages of the tooth, including ameloblasts, which produce the enamel (Juuri et al., 2012).
Less is known about the post-natal role of Nanog outside tumorigenesis, but it was previously identified as a transiently expressed gene in a transcriptional analysis of fracture healing (Bais et al., 2009) and is involved in bone regeneration by its contribution to marrow stromal cell maintenance and differentiation (Bais et al., 2012). More recently, Nanog was also shown to regulate cell division in stratified epithelia in adult organisms (Piazzolla et al., 2014).
Role of the vasculature in the transformation of chondrocytes to osteoblasts
An intact vasculature is essential for bone fracture healing. The delayed and non-union rate is 10-20% in the overall population (Aspenberg et al., 2010), but it increases to almost 50% in the presence of vascular injuries (Brownlow et al., 2002). However, the reason for this is unknown. Vascular invasion of the cartilage callus is an important step during endochondral repair (reviewed in Bahney et al., 2015) and fracture healing is significantly delayed when the vasculature is disrupted (Lu et al., 2007). Previous studies suggest that the endothelial cells enable degradation of the cartilage matrix by producing MMP-9 (Colnot et al., 2003; Wang et al., 2013), facilitate mineralization of the cartilage matrix by secreting BMPs (Bahney et al., 2014; Matsubara et al., 2012; Yu et al., 2010) and regulate deposition of collagen I (Ben Shoham et al., 2016).
Results from our study suggest the vasculature may have a signaling role in regulating transformation of chondrocytes to osteoblasts. Paracrine factors secreted from the vascular endothelial cells may trigger the chondrocyte to osteoblast transformation by activating the pluripotent stem cell programs, initiating cell division and/or stimulating the bone phenotype. This model is supported by immunohistochemical analysis of the expression of Sox2, Oct4 and Nanog within the TZ and in vitro data demonstrating upregulation of these genes in cartilage explants cultured with HUVEC conditioned medium (Figs 4,7,11). However, Yao et al. have shown that Sox2, Oct4 and Nanog can be induced in endothelial cells by elevated Bmp signaling, so it is possible that endothelial cells are also being induced to express the pluripotency programs (Yao et al., 2013); but lineage-tracing experiments demonstrate that the vast majority of the expression is in chondrocyte-derived cells (Fig. 7R-T).
A new model of endochondral ossification during bone fracture healing
Taken together with recent studies (Bahney et al., 2014; Yang et al., 2014b; Zhou et al., 2014), this work provides evidence that chondrocytes are a major precursor of the osteoblasts/osteocytes during endochondral bone repair. Importantly, our work begins to explore mechanisms through which chondrocyte-to-osteoblast transformation occurs, and we propose a new model for endochondral fracture repair in which the hypertrophic chondrocytes regain some stem cell-like functionality to facilitate the lineage change (Fig. 12). The ability of chondrocytes to de-differentiate was previously suggested by Song and Tuan following in vitro transdifferentiation assays with human mesenchymal stem cells (hMSCs) (Song and Tuan, 2004). More recently, serial transplantation assays in vivo demonstrated that chondrogenically differentiated hMSCs regained self-renew properties and multi-lineage potential (Serafini et al., 2014). While our data does not demonstrate that chondrocytes regain multipotency, we do show that activation of pluripotency programs has a functional role during fracture healing and propose that this allows for epigenetic remodeling to switch off chondrogenic programs and induce osteogenic programs. Mechanistic studies of chondrocyte-to-bone transformation during endochondral ossification at the growth plate have not yet been completed. Based on the many parallel mechanisms that occur between fracture healing and development, it is possible that hypertrophic chondrocytes at the chondro-osseous border in the growth plate may also take on a stem cell-like state prior to transformation to osteoblasts or osteocytes (Fig. S5).
Fig. 12.

A new model for endochondral ossification during fracture healing. Local osteochondral progenitors from the periosteum and endosteum are the stem cells that differentiate to form bone and cartilage in the fracture site. To generate the cartilage callus, osteochondral progenitors differentiate into chondrocytes (blue) that proliferate to generate the early soft callus. Chondrocytes within the callus mature to hypertrophy and express angiogenic factors that result in vascular invasion. Mineralization of the hypertrophic cartilage (red) occurs in the TZ where blood vessels have invaded. Some hypertrophic chondrocytes undergo cell death to facilitate remodeling of the solid cartilage callus and create marrow space within the trabecular bone. Other hypertrophic chondrocytes regain some stem cell-like properties by expressing the pluripotent transcription factors OCT4, SOX2 and NANOG. These large cells re-enter the cell cycle, divide and then transform into osteoblasts. The identity and origin of the signal that triggers the chondrocyte-to-osteoblast transformation remains unclear. Data from this paper and others suggest that both the vasculature and cell autonomous signals from the hypertrophic chondrocyte may facilitate this change (dotted arrows). This model does not exclude previously proposed systems in which osteoblasts in the newly formed bone are derived from osteoprogenitors that are brought in by the invading vasculature.
MATERIALS AND METHODS
Fractures
All studies were approved by the UCSF Institutional Animal Care and Use Committee. Adult (10-14 weeks) male mice were anesthetized, and closed non-stable fractures were made mid-diaphysis of the tibia via three-point bending (Thompson et al., 2002). Fractures were not stabilized to promote robust endochondral repair. Animals were provided with post-operative analgesics and allowed to ambulate freely (Table S1).
For lineage tracing, induction of Cre recombination was achieved by intraperitoneal administration of tamoxifen (75 mg tamoxifen/kg), daily from days 6-10 post-fracture. For Sox2 knockout, tamoxifen was administered on days 4-7, 10 and 12. Tibiae (n≥5) were harvested 14 days post-fracture for analysis.
Bone tissue embedding and histology
Fractured tibiae were fixed in 4% paraformaldehyde (PFA, pH 7.2-7.4) for 24 h, then decalcified in 19% EDTA (pH 7.4) for 14 days at 4°C. Unless otherwise noted, mice were processed for paraffin histology. Tissues from mice crossed to R26R or Ai9 reporter strains were embedded in OCT and sectioned using a cryostat. Serial sections were cut at 8-10 µm for histological and immunohistochemical analysis or 3-5 µm for cell tracing analysis. Standard histological protocols to visualize bone and cartilage were used: Modified Milligan's Trichrome (bone stains blue), Safranin O/Fast Green (cartilage stains red) or Hall and Brunt Quadruple stain (HBQ, bone stains red, cartilage stains blue).
Immunohistochemistry on fracture callus
In R26R transgenic tissues, detection of β-galactosidase activity was performed on frozen sections post-fixed in 0.2% glutaraldehyde for 15 min and then exposed to X-gal staining solution with 50 mg/ml 5-Bromo-4-chloro-3-indolyl β-D-galactoside (Sigma, B4252) overnight at 37°C.
Immunohistochemistry (IHC) was performed on paraffin sections from fractured tibiae of wild-type mice (>12 mice per antibody) 10 days post-fracture unless otherwise indicated. The basic protocol included antigen retrieval in 10 mM sodium citrate buffer (20 min, 100°C), endogenous peroxidase blocking in 3% H2O2 in phosphate buffered saline (PBS) (30 min) and non-specific epitope blocking with 5% bovine serum albumin (BSA, 1 h). Primary antibodies were applied to the sections overnight at 4°C (full antibody details are shown in Table S2). Species-specific secondary antibodies (1:500 in PBS with 1% BSA for 1 h at room temperature) were detected using the VectaStain ABC Kit (Vector, PK-4000) and 3,3′-diaminobenzidine (DAB) colorimetric reaction, or by immunofluorescence using species-specific Alexa-Fluor-488 or -594 secondary antibodies.
In situ hybridization
In situ hybridization was performed on paraffin wax embedded sections as previously described (Hu and Marcucio, 2009; Rumballe et al., 2008). Subclones of mouse collagen II (Col2a1), collagen X (Col10a1), collagen I (Col1a1) and osteopontin (Opn) were linearized for transcription of DIG-labeled anti-sense riboprobes.
Cell proliferation detection
Bromodeoxyurindine (BrdU, 5 mg) was diluted in PBS and delivered by an intraperitoneal injection 2 h prior to euthanasia. Positive cells were detected by an anti-BrdU staining kit (Invitrogen, 93-3943). To quantify BrdU+ cells, a central section representative of the largest cross-section of the fracture callus of 4 mice, was evaluated using an Olympus CAST system and software by Visiopharm. Tissues of interest (C, HC, TZ, NB) were outlined using low magnification (20×), and cell counting was performed under high magnification (200×) using a count frame probe that covered 50% of the area within a field of view. Following stereology counting frame rules, a minimum of 200 cells for each tissue were acquired using uniform random sampling. The percentage of BrdU+ versus BrdU− cells was estimated by applying the fractionator method (Howard and Reed, 1998). Graphs represent mean±s.d. from four biological replicates. Statistical analysis was completed using JMP v.12.1.0 software to perform an ANOVA followed by a post hoc comparison of all pairs using Tukey–Kramer HSD, P<0.05 was considered significant (*P<0.05, **P<0.005, ***P<0.0005).
TUNEL
The Roche In Situ Cell Death Detection Kit (Roach, 116847959) was used according to the manufactures protocol. Sections were deparaffinized, treated with proteinase K (20 µg/ml in 10 mM Tris-HCl, pH 8, 15 min), and then reacted with the kit for 1 h at 37°C in the dark. Positive controls were treated with DNase I prior to TUNEL reaction, while negative controls were not treated with the TUNEL reaction mixture. Slides were mounted in VectaShield with DAPI (Vector, H-1200) and visualized using an epifluorescence microscope.
In vitro cartilage explant culture
Cartilage explants were isolated from the central portion of the day 7 fracture callus using a dissecting microscope to remove any adherent non-cartilaginous tissues and the perichondrium. This method reliably yields tissue that is highly cartilaginous, with no evidence of bone or stem cell markers (Bahney et al., 2014). Explants were grown in vitro for 1 week in serum-free chondrogenic medium to promote hypertrophic maturation [high glucose DMEM, 1% penicillin-streptomycin, 1% ITS+ Premix (BD Biosciences Cat #354352), 1 mM sodium pyruvate, 100 ng/ml ascorbate-2-phosphate and 10−7 M dexamethasone (Bahney et al., 2014)]. Explants were kept in chondrogenic medium, or transferred into either osteogenic medium or a human vascular endothelial cell conditioned medium (HUVEC-CM) for an additional week. Osteogenic medium is chondrogenic medium plus 10 nM bone morphogenetic protein 2 (BMP2) and 10 mM β-glycerol phosphate. HUVEC-CM was collected from confluent plates of early passage HUVEC cells, without addition of growth factors (Bahney et al., 2014).
Transfection of pluripotent genes
An OCT4-SOX2 specific construct was generated from the commercially available pEP4-E02S-EN2L (Addgene, 20922) by excising NANOG and LIN28 with BamHI. These genes were transfected into the fracture callus cartilage explants ex vivo using Lipofectamine2000 reagent (Thermo Fisher, 11668019). Cartilage was dissected from the day 7 fracture callus of 10 mice as described above, combined, and then finely minced into small pieces. After 24 h in chondrogenic medium, fracture callus explants were transfected with 0.8 µg plasma DNA using 2 µl lipofectamine in 100 µl Opti-MEM medium for 6 h at 37°C (Foppiano et al., 2007). The explants receiving OCT4-SOX2 genes were then transferred back to chondrogenic medium and compared with constructs that remained in chondrogenic medium as a control, or those cultured in osteogenic medium. Cartilage was then harvested 48 h later for mRNA isolation.
Stereology
Quantification of graft composition (cartilage, bone, fibrous, marrow space) was determined using an Olympus CAST system (Center Valley, PA) and software by Visiopharm (Hørsholm, Denmark) according to established methodologies (Howard and Reed, 1998). For quantification of the bone and cartilage, 10 µm serial sections, three sections per slide, were taken through the entire leg. Tissue was stained with Safranin-O as described above, and the first section from every 10th slide analyzed such that sections were 300 µm apart. The fracture callus was outlined using low magnification (2×), then tissue composition was quantified in 15% of fracture callus using automated uniform random sampling to meet or exceeds the basic principles of stereology (Howard and Reed, 1998). Cell identity within each random sampling domain was determined at high magnification (20×) according to histological staining patterns and cell morphology. Volume of the specific tissue type (e.g. bone or cartilage composition) was determined in reference to the total fracture callus volume by summing the individual compositions relative to the whole. Marrow space was considered as tissue that fell within a blood vessel or marrow cavity of the new bone. Graphs plot mean±95% confidence interval; JMP software tested for significance using the non-parametric Wilcoxon/Kruskal–Wallis Test (*P<0.05).
mRNA isolation and quantitative RT-PCR
mRNA was isolated from cartilage grafts in 100 µl Trizol. cDNA was reverse transcribed with Superscript III (Invitrogen, 18080), and quantitative RT-PCR was performed using SYBR Green and primers as listed in Table S3. Relative gene expression was calculated by normalizing to GAPDH (ΔCT), then to explants cultured under chondrogenic conditions (ΔΔCT). Fold change was calculated as 2−ΔΔCT. Graphs represent mean ±95% confidence interval of biological replicates for the cartilage grafts (n=5). Significance was determined using an ANOVA followed by a post hoc multiple comparison using Tukey-Kramer HSD. (*P<0.05, **P<0.005, ***P<0.0005).
Acknowledgements
We would like to acknowledge Dr Sarah Know and Noel Cruz-Pacheco, UCSF Department of Cell & Tissue Biology, who generously bred, genotyped and supplied us with the Sox2CreERT/fl mice used in this study. We would also like to thank Alfred Li at the UCSF VA hospital for assistance with the Col2CreERT::Ai9 mice, and Gina Baldoza and AnnaLissa Wi at the UCSF Orthopaedic Trauma Institute for daily lab function and grants administration.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
R.S.M., C.S.B., and D.P.H. devised the experiments and performed analysis of data. D.P.H., F.F., F.Y. and A.J.T. executed the experiments. R.S.M., C.S.B., T.M., W.C. provided financial support and materials for the studies. All authors contributed to writing and editing the manuscript.
Funding
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (NIH) (5F32AR062469 to C.S.B.; AR053645 and AR057344 to T.M.; and AR067291 to W.C.). Additional research support was provided by the Musculoskeletal Transplant Foundation (MTF Junior Investigator Award to C.S.B.), the AO Foundation (Career Development Award, S-14-114B to C.S.B.), the UCSF Clinical and Translational Science Institute (UL1TR000004 to C.S.B.), UCSF Core Center for Musculoskeletal Biology and Medicine (P30AR066262 to C.S.B.), the US Bone and Joint Initiative Young Investigator Program (to C.S.B.) and Department of Veterans Affairs Program Project Award (IPIBX001599 to W.C.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.130807.supplemental
References
- Arnold K., Sarkar A., Yram M. A., Polo J. M., Bronson R., Sengupta S., Seandel M., Geijsen N. and Hochedlinger K. (2011). Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9, 317-329. 10.1016/j.stem.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspenberg P., Genant H. K., Johansson T., Nino A. J., See K., Krohn K., García-Hernández P. A., Recknor C. P., Einhorn T. A., Dalsky G. P. et al. (2010). Teriparatide for acceleration of fracture repair in humans: a prospective, randomized, double-blind study of 102 postmenopausal women with distal radial fractures. J. Bone Miner. Res. 25, 404-414. 10.1359/jbmr.090731 [DOI] [PubMed] [Google Scholar]
- Bahney C. S., Hu D. P., Taylor A. J., Ferro F., Britz H. M., Hallgrimsson B., Johnstone B., Miclau T. and Marcucio R. S. (2014). Stem cell-derived endochondral cartilage stimulates bone healing by tissue transformation. J. Bone Miner. Res. 29, 1269-1282. 10.1002/jbmr.2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahney C. S., Hu D. P., Miclau T. III and Marcucio R. S. (2015). The multifaceted role of the vasculature in endochondral fracture repair. Front. Endocrinol. 6, 4 10.3389/fendo.2015.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bais M., McLean J., Sebastiani P., Young M., Wigner N., Smith T., Kotton D. N., Einhorn T. A. and Gerstenfeld L. C. (2009). Transcriptional analysis of fracture healing and the induction of embryonic stem cell-related genes. PLoS ONE 4, e5393 10.1371/journal.pone.0005393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bais M. V., Shabin Z. M., Young M., Einhorn T. A., Kotton D. N. and Gerstnefeld L. C. (2012). Role of Nanog in the maintenance of marrow stromal stem cells during post natal bone regeneration. Biochem. Biophys. Res. Commun. 417, 211-216. 10.1016/j.bbrc.2011.11.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Shoham A., Rot C., Stern T., Krief S., Akiva A., Dadosh T., Sabany H., Lu Y., Kadler K. E. and Zelzer E. (2016). Deposition of collagen type I onto skeletal endothelium reveals a new role for blood vessels in regulating bone morphology. Development 143, 3933-3943. 10.1242/dev.139253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W., Deng J. M., Zhang Z., Behringer R. R. and de Crombrugghe B. (1999). Sox9 is required for cartilage formation. Nat. Genet. 22, 85-89. 10.1038/8792 [DOI] [PubMed] [Google Scholar]
- Brownlow H. C., Reed A. and Simpson A. H. R. W. (2002). The vascularity of atrophic non-unions. Injury 33, 145-150. 10.1016/S0020-1383(01)00153-X [DOI] [PubMed] [Google Scholar]
- Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469-480. 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- Colnot C. (2009). Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J. Bone Miner. Res. 24, 274-282. 10.1359/jbmr.081003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C., Thompson Z., Miclau T., Werb Z. and Helms J. A. (2003). Altered fracture repair in the absence of MMP9. Development 130, 4123-4133. 10.1242/dev.00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper K. L., Oh S., Sung Y., Dasari R. R., Kirschner M. W. and Tabin C. J. (2013). Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 495, 375-378. 10.1038/nature11940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day T. F., Guo X., Garrett-Beal L. and Yang Y. (2005). Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739-750. 10.1016/j.devcel.2005.03.016 [DOI] [PubMed] [Google Scholar]
- Ducy P., Zhang R., Geoffroy V., Ridall A. L. and Karsenty G. (1997). Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89, 747-754. 10.1016/S0092-8674(00)80257-3 [DOI] [PubMed] [Google Scholar]
- Dy P., Wang W., Bhattaram P., Wang Q., Wang L., Ballock R. T. and Lefebvre V. (2012). Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell 22, 597-609. 10.1016/j.devcel.2011.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foppiano S., Hu D. and Marcucio R. S. (2007). Signaling by bone morphogenetic proteins directs formation of an ectodermal signaling center that regulates craniofacial development. Dev. Biol. 312, 103-114. 10.1016/j.ydbio.2007.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur T., Lengner C. J., Hovhannisyan H., Bhat R. A., Bodine P. V. N., Komm B. S., Javed A., van Wijnen A. J., Stein J. L., Stein G. S. et al. (2005). Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 280, 33132-33140. 10.1074/jbc.M500608200 [DOI] [PubMed] [Google Scholar]
- Gerstenfeld L. C., Cullinane D. M., Barnes G. L., Graves D. T. and Einhorn T. A. (2003). Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J. Cell. Biochem. 88, 873-884. 10.1002/jcb.10435 [DOI] [PubMed] [Google Scholar]
- Gong S., Li Q., Jeter C. R., Fan Q., Tang D. G. and Liu B. (2015). Regulation of NANOG in cancer cells. Mol. Carcinog. 54, 679-687. 10.1002/mc.22340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greder L. V., Gupta S., Li S., Abedin M. J., Sajini A., Segal Y., Slack J. M. W. and Dutton J. R. (2012). Analysis of endogenous Oct4 activation during induced pluripotent stem cell reprogramming using an inducible Oct4 lineage label. Stem Cells 30, 2596-2601. 10.1002/stem.1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori T., Muller C., Gebhard S., Bauer E., Pausch F., Schlund B., Bosl M. R., Hess A., Surmann-Schmitt C., von der Mark H. et al. (2010). SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 137, 901-911. 10.1242/dev.045203 [DOI] [PubMed] [Google Scholar]
- Henry S. P., Jang C. W., Deng J. M., Zhang Z., Behringer R. R. and de Crombrugghe B. (2009). Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis 47, 805-814. 10.1002/dvg.20564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill T. P., Spater D., Taketo M. M., Birchmeier W. and Hartmann C. (2005). Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727-738. 10.1016/j.devcel.2005.02.013 [DOI] [PubMed] [Google Scholar]
- Holtrop M. E. (1972). The ultrastructure of the epiphyseal plate. II. The hypertrophic chondrocyte. Calcif. Tissue Res. 9, 140-151. 10.1007/BF02061952 [DOI] [PubMed] [Google Scholar]
- Howard C. and Reed M. (1998). Unbiased Stereology: Three-Dimensional Measurement in Microscopy. New York: Springer-Verlag New York Inc. [Google Scholar]
- Hu D. and Marcucio R. S. (2009). A SHH-responsive signaling center in the forebrain regulates craniofacial morphogenesis via the facial ectoderm. Development 136, 107-116. 10.1242/dev.026583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson H., Wilson W., van Donkelaar C. C., Huiskes R. and Ito K. (2006). Comparison of biophysical stimuli for mechano-regulation of tissue differentiation during fracture healing. J. Biomech. 39, 1507-1516. 10.1016/j.jbiomech.2005.01.037 [DOI] [PubMed] [Google Scholar]
- Juuri E., Saito K., Ahtiainen L., Seidel K., Tummers M., Hochedlinger K., Klein O. D., Thesleff I. and Michon F. (2012). Sox2+ stem cells contribute to all epithelial lineages of the tooth via Sfrp5+ progenitors. Dev. Cell 23, 317-328. 10.1016/j.devcel.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R. T., Gao Y.-H., Inada M. et al. (1997). Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755-764. 10.1016/S0092-8674(00)80258-5 [DOI] [PubMed] [Google Scholar]
- Kronenberg H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332-336. 10.1038/nature01657 [DOI] [PubMed] [Google Scholar]
- Lee J., Kim H. K., Rho J.-Y., Han Y.-M. and Kim J. (2006). The human OCT-4 isoforms differ in their ability to confer self-renewal. J. Biol. Chem. 281, 33554-33565. 10.1074/jbc.M603937200 [DOI] [PubMed] [Google Scholar]
- Lefebvre V., Huang W., Harley V. R., Goodfellow P. N. and de Crombrugghe B. (1997). SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol. Cell. Biol. 17, 2336-2346. 10.1128/MCB.17.4.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner C. J., Camargo F. D., Hochedlinger K., Welstead G. G., Zaidi S., Gokhale S., Scholer H. R., Tomilin A. and Jaenisch R. (2007). Oct4 expression is not required for mouse somatic stem cell self-renewal. Cell Stem Cell 1, 403-415. 10.1016/j.stem.2007.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke S., Stephan M. and Kögler G. (2008). Oct4 expression revisited: potential pitfalls for data misinterpretation in stem cell research. Biol. Chem. 389, 845-850. 10.1515/BC.2008.098 [DOI] [PubMed] [Google Scholar]
- Lu C., Miclau T., Hu D., Hansen E., Tsui K., Puttlitz C. and Marcucio R. S. (2005). Cellular basis for age-related changes in fracture repair. J. Orthop. Res. 23, 1300-1307. 10.1016/j.orthres.2005.04.003.1100230610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Miclau T., Hu D. and Marcucio R. S. (2007). Ischemia leads to delayed union during fracture healing: a mouse model. J. Orthop. Res. 25, 51-61. 10.1002/jor.20264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R. et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133-140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C., Kobayashi T., Selig M. K., Torrekens S., Roth S. I., Mackem S., Carmeliet G. and Kronenberg H. M. (2010). Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329-344. 10.1016/j.devcel.2010.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara H., Hogan D. E., Morgan E. F., Mortlock D. P., Einhorn T. A. and Gerstenfeld L. C. (2012). Vascular tissues are a primary source of BMP2 expression during bone formation induced by distraction osteogenesis. Bone 51, 168-180. 10.1016/j.bone.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miclau T., Lu C., Thompson Z., Choi P., Puttlitz C., Marcucio R. and Helms J. A. (2007). Effects of delayed stabilization on fracture healing. J. Orthop. Res. 25, 1552-1558. 10.1002/jor.20435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. J., Gerstenfeld L. C. and Morgan E. F. (2015). Mechanical microenvironments and protein expression associated with formation of different skeletal tissues during bone healing. Biomech. Model. Mechanobiol. 14, 1239-1253. 10.1007/s10237-015-0670-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura E., Nguyen M.-T. and Mackem S. (2006). Kinetics of tamoxifen-regulated Cre activity in mice using a cartilage-specific CreER(T) to assay temporal activity windows along the proximodistal limb skeleton. Dev. Dyn. 235, 2603-2612. 10.1002/dvdy.20892 [DOI] [PubMed] [Google Scholar]
- Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J. M., Behringer R. R. and de Crombrugghe B. (2002). The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17-29. 10.1016/S0092-8674(01)00622-5 [DOI] [PubMed] [Google Scholar]
- Otto F., Thornell A. P., Crompton T., Denzel A., Gilmour K. C., Rosewell I. R., Stamp G. W., Beddington R. S., Mundlos S., Olsen B. R. et al. (1997). Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765-771. 10.1016/S0092-8674(00)80259-7 [DOI] [PubMed] [Google Scholar]
- Piazzolla D., Palla A. R., Pantoja C., Canamero M., de Castro I. P., Ortega S., Gómez-López G., Dominguez O., Megías D., Roncador G. et al. (2014). Lineage-restricted function of the pluripotency factor NANOG in stratified epithelia. Nat. Commun. 5, 4226 10.1038/ncomms5226 [DOI] [PubMed] [Google Scholar]
- Pritchard J. J. and Ruzicka A. J. (1950). Comparison of fracture repair in the frog, lizard and rat. J. Anat. 84, 236-261. [PMC free article] [PubMed] [Google Scholar]
- Roach H. I. (1992). Trans-differentiation of hypertrophic chondrocytes into cells capable of producing a mineralized bone matrix. Bone Miner. 19, 1-20. 10.1016/0169-6009(92)90840-A [DOI] [PubMed] [Google Scholar]
- Roach H. I. (1997). New aspects of endochondral ossification in the chick: chondrocyte apoptosis, bone formation by former chondrocytes, and acid phosphatase activity in the endochondral bone matrix. J. Bone Miner. Res. 12, 795-805. 10.1359/jbmr.1997.12.5.795 [DOI] [PubMed] [Google Scholar]
- Roach H. I. and Clarke N. M. P. (2000). Physiological cell death of chondrocytes in vivo is not confined to apoptosis. New observations on the mammalian growth plate. J. Bone Joint Surg. Br. 82, 601-613. 10.1302/0301-620X.82B4.9846 [DOI] [PubMed] [Google Scholar]
- Roach H. I. and Erenpreisa J. E. (1996). The phenotypic switch from chondrocytes to bone-forming cells involves asymmetric cell division and apoptosis. Connect. Tissue Res. 35, 85-91. 10.3109/03008209609029178 [DOI] [PubMed] [Google Scholar]
- Rumballe B., Georgas K., Little M. H. (2008). High-throughput paraffin section in situ hybridization and dual immunohistochemistry on mouse tissues. CSH Protoc. 2008, pdb prot5030 10.1101/pdb.prot5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samardzija C., Quinn M., Findlay J. K. and Ahmed N. (2012). Attributes of Oct4 in stem cell biology: perspectives on cancer stem cells of the ovary. J. Ovarian Res. 5, 37 10.1186/1757-2215-5-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti C., Tonnarelli B., Papadimitropoulos A., Scherberich A., Schaeren S., Schauerte A., Lopez-Rios J., Zeller R., Barbero A. and Martin I. (2010). Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. Proc. Natl. Acad. Sci. USA 107, 7251-7256. 10.1073/pnas.1000302107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti C., Piccinini E., Takizawa H., Todorov A., Bourgine P., Papadimitropoulos A., Barbero A., Manz M. G. and Martin I. (2013). Engineering of a functional bone organ through endochondral ossification. Proc. Natl. Acad. Sci. USA 110, 3997-4002. 10.1073/pnas.1220108110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini M., Sacchetti B., Pievani A., Redaelli D., Remoli C., Biondi A., Riminucci M. and Bianco P. (2014). Establishment of bone marrow and hematopoietic niches in vivo by reversion of chondrocyte differentiation of human bone marrow stromal cells. Stem Cell Res. 12, 659-672. 10.1016/j.scr.2014.01.006 [DOI] [PubMed] [Google Scholar]
- Shapiro I. M., Adams C. S., Freeman T. and Srinivas V. (2005). Fate of the hypertrophic chondrocyte: microenvironmental perspectives on apoptosis and survival in the epiphyseal growth plate. Birth Defects Res. C Embryo Today 75, 330-339. 10.1002/bdrc.20057 [DOI] [PubMed] [Google Scholar]
- Song L. and Tuan R. S. (2004). Transdifferentiation potential of human mesenchymal stem cells derived from bone marrow. FASEB J. 18, 980-982. 10.1096/fj.03-1100fje [DOI] [PubMed] [Google Scholar]
- Suh H., Consiglio A., Ray J., Sawai T., D'Amour K. A. and Gage F. H. (2007). In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell 1, 515-528. 10.1016/j.stem.2007.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson Z., Miclau T., Hu D. and Helms J. A. (2002). A model for intramembranous ossification during fracture healing. J. Orthop. Res. 20, 1091-1098. 10.1016/S0736-0266(02)00017-7 [DOI] [PubMed] [Google Scholar]
- Topol L., Chen W., Song H., Day T. F. and Yang Y. (2009). Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J. Biol. Chem. 284, 3323-3333. 10.1074/jbc.M808048200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang K. Y., Chan D., Cheslett D., Chan W. C. W., So C. L., Melhado I. G., Chan T. W. Y., Kwan K. M., Hunziker E. B., Yamada Y. et al. (2007). Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 5, e44 10.1371/journal.pbio.0050044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhao Y., Xiao Z., Chen B., Wei Z., Wang B., Zhang J., Han J., Gao Y., Li L. et al. (2009). Alternative translation of OCT4 by an internal ribosome entry site and its novel function in stress response. Stem Cells 27, 1265-1275. 10.1002/stem.58 [DOI] [PubMed] [Google Scholar]
- Wang X., Yu Y. Y., Lieu S., Yang F., Lang J., Lu C., Werb Z., Hu D., Miclau T., Marcucio R. et al. (2013). MMP9 regulates the cellular response to inflammation after skeletal injury. Bone 52, 111-119. 10.1016/j.bone.2012.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weina K. and Utikal J. (2014). SOX2 and cancer: current research and its implications in the clinic. Clin. Transl. Med. 3, 19 10.1186/2001-1326-3-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G., Zhu L., Hou N., Lan Y., Wu X.-M., Zhou B., Teng Y. and Yang X. (2014a). Osteogenic fate of hypertrophic chondrocytes. Cell Res. 24, 1266-1269. 10.1038/cr.2014.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Tsang K. Y., Tang H. C., Chan D. and Cheah K. S. (2014b). Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 111, 12097-12102. 10.1073/pnas.1302703111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y., Jumabay M., Ly A., Radparvar M., Cubberly M. R. and Bostrom K. I. (2013). A role for the endothelium in vascular calcification. Circ. Res. 113, 495-504. 10.1161/CIRCRESAHA.113.301792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. Y., Lieu S., Lu C., Miclau T., Marcucio R. S. and Colnot C. (2010). Immunolocalization of BMPs, BMP antagonists, receptors, and effectors during fracture repair. Bone 46, 841-851. 10.1016/j.bone.2009.11.005 [DOI] [PubMed] [Google Scholar]
- Zhao Q., Eberspaecher H., Lefebvre V. and De Crombrugghe B. (1997). Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 209, 377-386. [DOI] [PubMed] [Google Scholar]
- Zhou X., von der Mark K., Henry S., Norton W., Adams H. and de Crombrugghe B. (2014). Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 10, e1004820 10.1371/journal.pgen.1004820 [DOI] [PMC free article] [PubMed] [Google Scholar]