

In the last decade, the non-coding transcriptome in normal and pathological conditions has been a focus of intensive research.1 The most well-studied non-coding RNAs, microRNAs, are of critical importance in the post-transcriptional regulation in the cell and were shown to play a role in biologically and clinically heterogeneous diseases such as acute myeloid leukemia (AML).2 Along with the insights into AML pathogenesis, microRNA expression profiling proved to be of clinical relevance as specific microRNA expression signatures were shown to be associated with distinct AML subtypes and with patients’ prognosis.3,4 However, so far most studies have focused mainly on individual microRNAs and only a few have developed prognostic scores, which were limited to cytogenetically normal AML (CN-AML) or intermediate risk AML.5,6 In accordance with a recent study,7 we aimed to develop and validate a microRNA expression-based prognostic score in adult AML applicable not only to a specific AML subtype but rather to any AML subtype. For this purpose, we used a microarray-based training expression dataset from 91 AML patients (Ulm dataset; Online Supplementary Appendix),8 and validated findings using the RNA sequencing (RNA-Seq) data from 177 patients available through The Cancer Genome Atlas (TCGA) project.9 The analysis used the expression data for 168 microRNAs common for both datasets (Online Supplementary Appendix).
The microRNAs to be included in the model were determined using the Robust Likelihood-Based Survival Modeling with Microarray Data method. This technique utilizes the partial likelihood of the Cox model and functions through the generation of multiple gene (microRNAs in this case) models (Online Supplementary Appendix). The optimal model included 7 microRNAs (miR-100, miR-132, miR-185, miR-186, miR-302a, miR-330, and miR-422a) (Online Supplementary Appendix). A total continuous score was calculated for each patient sample using the Cox regression coefficients obtained for the Ulm dataset. The total score was calculated for each patient sample in the training and the validation dataset. To build a binary score classifying the sample to either a high or low score group, we defined a cut-off value specific for each dataset. This was achieved through Receiver Operating Characteristics (ROC) analysis and optimal cut-off selection based on the log-rank test (Online Supplementary Appendix). Notably, the microRNAs included in our prognostic score did not overlap with those used by Chuang et al.,7 a phenomenon also frequently seen for mRNA-based outcome prediction signatures. Depending on the approach, different surrogates for outcome are chosen by the model. This can be explained by two main differences between the studies. First, our approach used a direct selection via a multivariate microRNA expression model, while Chuang et al. focused only on microRNAs significantly associated with overall survival (OS) in univariate analysis. Second, the Chuang et al. dataset included more patients aged over 60 years (41%) and, therefore, many had not undergone intensive treatment.7
Univariate and multivariate analyses were performed for the training and validation datasets with either the continuous or discrete scores to determine their prognostic power on OS. For the training set, both continuous and discrete scores were significant prognostic factors in the univariate analyses (Figure 1A, B, E and F and Online Supplementary Appendix). In multivariate models for the training set, the discrete score was a significant prognostic factor and appeared to modify the FLT3-ITD associated risk (Figure 1C, D, G and H and Online Supplementary Appendix). In accordance with this, we focused on the discrete score, which performed almost identically in the univariate analysis when applied to all validation dataset cases (P=0.0024), and it was a significant factor in multivariate analysis (P=0.047; including FLT3-ITD and NPM1 mutational status, cytogenetic risk group, age and sex) (Figure 1C, D, G and H and Online Supplementary Appendix).
Figure 1.
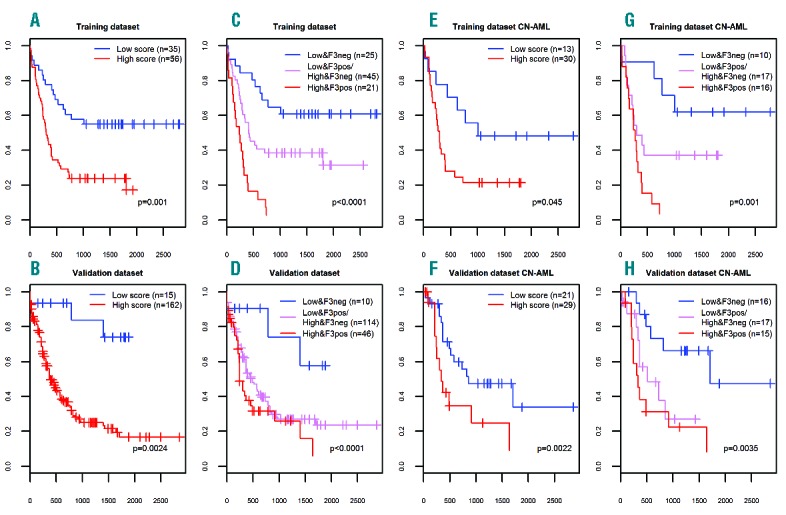
Overall survival (OS) in the training and validation sets based on the discrete microRNA expression-based score. (A, C, E, G) training dataset; (B, D, F, H) validation dataset; (A, B, C, D) included all patients from both datasets; (E, F, G, H) included only cytogenetically normal acute myeloid leukemia (CN-AML) patients aged 60 years or under from both datasets. P-values are from Cox regression, unless stated otherwise in the plot. Horizontal axes represent the time in days and the vertical axes represent the probability for OS.
In intermediate risk cytogenetic cases, the discrete score also was shown to be a significant prognostic factor in the univariate analysis in the training dataset (P=0.032), and showed a trend in multivariate models (P=0.12) (Online Supplementary Appendix). In accordance with this, in the validation dataset the discrete score was a significant prognostic factor and retained significance also in multivariate models (P=0.017 and P=0.022, respectively) (Online Supplementary Appendix). Finally, analysis restricted to cytogenetically normal (CN) AML also showed a trend to significance for the discrete score (Online Supplementary Appendix) and the log-rank test showed a significant adverse prognostic impact for the miRNA score (Figure 1E). In the TCGA validation subset of younger CN-AML patients, the discrete scores were significant in the univariate analysis, and appeared as independent prognostic factors in multivariate models (P=0.0022 and P=0.0014, respectively) (Figure 1D and H and Online Supplementary Appendix).
The microRNAs of our prognostic score had so far not been frequently reported to be associated with AML subtypes.10 Thus, we used a systems biology approach to see whether this set of microRNAs might account for leukemia-relevant mechanisms further supporting the model. A computational network including the seven microRNAs and their predicted or validated targets (mirTarBase, TargetScan and MicroCosm) showed a significant enrichment for nucleic acids binding proteins, thereby pointing to a general impact on transcriptional deregulation (Online Supplementary Appendix). The 479 target genes included in the network were enriched for several cancer-related pathways (Online Supplementary Appendix). Using the TCGA gene expression data, we identified 850 probe sets (corresponding to 624 genes) that were differentially expressed between Low and High Score patients at the level of P<0.01 (Figure 2A). GO analysis on the list of the top 200 differentially expressed probe sets (corresponding to 148 genes) notably also revealed “General transcription regulation” to be the most significantly over-represented pathway (Online Supplementary Appendix). Twenty genes were common with the target genes included in the network analysis (Online Supplementary Appendix). Within CN-AML, the respective analysis identified a total of 171 differentially expressed probe sets corresponding to 137 genes. The unsupervised clustering based on these probe sets showed three different groups that correlated with the discrete score subgrouping (Figure 2A). In accordance to the findings above, GO analysis revealed over-represented GO classes related to RNA metabolism, as well as the cell cycle pathway (Online Supplementary Appendix).
Figure 2.
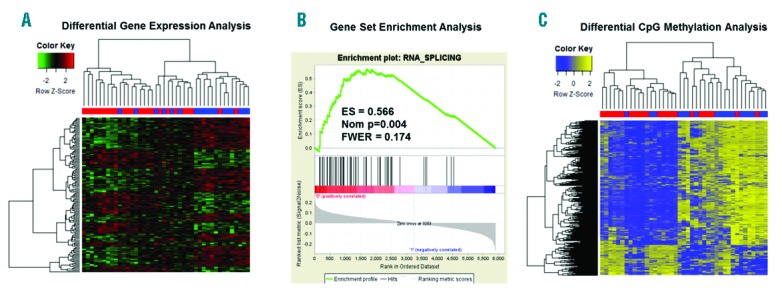
Gene expression and epigenetic profiles discriminating Low and High Score validation dataset cytogenetically normal acute myeloid leukemia (CN-AML) cases (≤60 years). (A) Hierarchical clustering of differentially expressed probe sets discriminating Low and High Score CN-AML patients. (B) Enrichment plot for the “RNA_SPLICING” signature identified in GSEA of CN-AML patients aged 60 years or under. (C) Unsupervised clustering of differentially methylated CpG sites in CN-AML patients. Samples’ annotation in (A) and (C): blue: Low Score patients; red: High Score patients. ES: enrichment score; FWER: familywise error rate.
To further investigate the biological relevance of the difference in gene expression pattern between the Low and High Score subgroups, we performed a Gene Set Enrichment Analysis (GSEA) using the Broad Institute GSEA bioinformatics platform. The GSEA of the entire validation set cohort showed a positive enrichment in the Low Score group for genes deregulated after beta-catenin overexpression (e.g. “BCAT.100_UP.V1_DN” gene signature; P=0.009) (Online Supplementary Appendix). This is in accordance with the subgroup of Low Score patients being enriched for t(15;17) cases (Online Supplementary Appendix), in which the Wnt/beta-catenin signaling pathway was reported hyperactive. The GSEA within the subset of CN-AML aged 60 years or under showed the seven top scoring gene signatures to be associated with RNA metabolism and processing (Online Supplementary Appendix). The top scoring gene set was the “RNA SPLICING” signature (Figure 2B). This observation suggests that deregulated miRNA expression might affect RNA splicing patterns and contribute to the pathogenesis of myeloid malignancies. For example, two of the genes that we found over-expressed in Low versus High Score cases, SRSF2 and U2AF1, are also found recurrently mutated in the myeloid malignancies, thereby further highlighting the importance of altered splicing in leukemogenesis.11 In AML, mutations were also found in other splicing factors (SFPQ), but also in other non-canonical RNA metabolism regulators such as CTCF and RAD21,12 and recently variable expression of U2AF1 was shown to influence alternative splicing events.13 Our findings further support the idea that the level of expression of splicing factors genes is associated with leukemia pathogenesis.
Thus, the observation of the over-representation and enrichment of RNA splicing-related gene signatures prompted us to investigate differential exon usage (DEU) between Low and High Score patients. To do this we used the level 3 RNA-Seq data from the TCGA server. We filtered out tags (exons) with low expression, analyzed 45264 tags, and identified 7500 differentially expressed tags (P<0.05), with 151 tags showing a log fold change greater than 2. Furthermore, DEU reliably classified Low versus High Score patients (Online Supplementary Appendix), thereby further supporting the hypothesis that there is a significant difference in the RNA splicing between the two groups.
Finally, we questioned whether the observed differences between the Low and High Score CN-AML cases would also be reflected on the epigenetic level. We obtained the TCGA DNA methylation data (Infinium II platform) and found a total of 1218 CpG sites (corresponding to 574 genes) differentially methylated between the Low and High Score patients, and hierarchical clustering showed a very good correlation with the miRNA score, with only 5 cases being discordantly grouped (Figure 2C). This finding is consistent with the previous reports of AML subtypes being associated with differential DNA methylation profiles of prognostic relevance.9,14,15
In conclusion, our data show a rational and feasible approach to combine microarray and RNA-Seq data to derive prognostic scores in AML (or other cancers) and to integrate additional omics data levels to explore the potential underlying biological features. Together with the recent report by Chuang et al.,7 our report further demonstrates that meaningful microRNA expression-based prognostic scores can be developed on heterogeneous training and validation datasets across a heterogeneous disease such as AML. The respective signatures warrant further clinical testing, especially the newly identified miRNAs that have not been previously reported. Most importantly, this work provides further evidence of the role of the RNA splicing machinery deregulation in the pathogenesis of AML. This warrants further studies to understand the aberrant mechanisms and to translate findings into clinical practice.
Supplementary Material
Footnotes
Funding: this study was supported in part by the Deutsche Forschungsgemeinschaft (SFB 1074 project B3 to KD and LB and Heisenberg-Professur BU 1339/8-1 to LB).
The online version of this letter has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Shivarov V, Bullinger L. Expression profiling of leukemia patients: Key lessons and future directions. Exp Hematol. 2014;42(8):651–660. [DOI] [PubMed] [Google Scholar]
- 2.Schotte D, Pieters R, Den Boer ML. MicroRNAs in acute leukemia: from biological players to clinical contributors. Leukemia. 2012;26(1):1–12. [DOI] [PubMed] [Google Scholar]
- 3.Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJ, Lowenberg B. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood. 2008;111(10):5078–5085. [DOI] [PubMed] [Google Scholar]
- 4.Garzon R, Volinia S, Liu CG, et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008;111(6):3183–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marcucci G, Radmacher MD, Maharry K, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1919–1928. [DOI] [PubMed] [Google Scholar]
- 6.Diaz-Beya M, Brunet S, Nomdedeu J, et al. MicroRNA expression at diagnosis adds relevant prognostic information to molecular categorization in patients with intermediate-risk cytogenetic acute myeloid leukemia. Leukemia. 2014;28(4):804–812. [DOI] [PubMed] [Google Scholar]
- 7.Chuang MK, Chiu YC, Chou WC, et al. A 3-microRNA scoring system for prognostication in de novo acute myeloid leukemia patients. Leukemia. 2015;29(5):1051–1059. [DOI] [PubMed] [Google Scholar]
- 8.Russ AC, Sander S, Luck SC, et al. Integrative nucleophosmin mutation-associated microRNA and gene expression pattern analysis identifies novel microRNA - target gene interactions in acute myeloid leukemia. Haematologica. 2011;96(12):1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marcucci G, Mrozek K, Radmacher MD, Garzon R, Bloomfield CD. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood. 2011;117(4):1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Visconte V, Makishima H, Maciejewski JP, Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia. 2012;26(12):2447–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012;120(18):e83–92. [DOI] [PubMed] [Google Scholar]
- 13.Przychodzen B, Jerez A, Guinta K, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122(6):999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bullinger L, Ehrich M, Dohner K, et al. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood. 2010;115(3):636–642. [DOI] [PubMed] [Google Scholar]
- 15.Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.