

Abstract
The clinical and histopathological distinctions between inherited versus acquired bone marrow failure and myelodysplastic syndromes are challenging. The identification of inherited bone marrow failure/myelodysplastic syndromes is critical to inform appropriate clinical management. To investigate whether a subset of pediatric and young adults undergoing transplant for aplastic anemia or myelodysplastic syndrome have germline mutations in bone marrow failure/myelodysplastic syndrome genes, we performed a targeted genetic screen of samples obtained between 1990–2012 from children and young adults with aplastic anemia or myelodysplastic syndrome transplanted at the Fred Hutchinson Cancer Research Center. Mutations in inherited bone marrow failure/myelodysplastic syndrome genes were found in 5.1% (5/98) of aplastic anemia patients and 13.6% (15/110) of myelodysplastic syndrome patients. While the majority of mutations were constitutional, a RUNX1 mutation present in the peripheral blood at a 51% variant allele fraction was confirmed to be somatically acquired in one myelodysplastic syndrome patient. This highlights the importance of distinguishing germline versus somatic mutations by sequencing DNA from a second tissue or from parents. Pathological mutations were present in DKC1, MPL, and TP53 among the aplastic anemia cohort, and in FANCA, GATA2, MPL, RTEL1, RUNX1, SBDS, TERT, TINF2, and TP53 among the myelodysplastic syndrome cohort. Family history or physical examination failed to reliably predict the presence of germline mutations. This study shows that while any single specific bone marrow failure/myelodysplastic syndrome genetic disorder is rare, screening for these disorders in aggregate identifies a significant subset of patients with inherited bone marrow failure/myelodysplastic syndrome.
Introduction
The overlap in clinical presentation and bone marrow features of inherited and acquired bone marrow failure syndromes and pediatric myelodysplastic syndromes (MDS) can pose a significant diagnostic challenge. Idiopathic acquired aplastic anemia (AA) is typically a diagnosis of exclusion after inherited disorders, acquired MDS, infections, metabolic diseases, or nutritional deficiencies have been ruled out. Childhood MDS is emerging as clinically and biologically distinct from MDS in the elderly.1,2 Importantly, and in contrast to MDS in the elderly, the majority of cases of refractory cytopenia of childhood, the most common subtype of pediatric MDS, are hypocellular with a normal karyotype.3 If MDS-related cytogenetic abnormalities are absent, the distinction between refractory cytopenia of childhood and AA or an inherited bone marrow failure or myelodysplastic predisposition syndrome (inherited BMF/MDS) is challenging. Furthermore, patients with inherited BMF/MDS can initially present with MDS or acute leukemia, obscuring the diagnosis of their underlying disorder.
The clinical and histopathological distinctions between germline genetic versus acquired disorders have important therapeutic implications. The major treatments for AA currently consist of either hematopoietic stem cell transplant (HSCT) or immunosuppressive therapy with antithymocyte globulin and cyclosporine. The choice of therapy is largely based on the patient’s age, availability of a matched sibling donor, and co-morbidities. The success of immunosuppressive therapy is limited by lack of response in approximately 30% of patients, relapse in 30% of patients who initially respond, and clonal evolution in 10–20% of patients.4 Progressive improvement in outcomes of matched unrelated donor transplants raises the question of whether this should be considered in lieu of immunosuppression as first-line therapy for a subset of patients. In contrast, patients with inherited bone marrow failure syndromes generally have poor or transient responses to immunosuppressive therapy.5–7 In childhood MDS, the indications for HSCT are currently based on the severity of cytopenias, the degree of marrow dysplasia,8 the peripheral blood and marrow blast percentages, and cytogenetic aberrations. In addition to informing the timing and indication for HSCT, accurate distinction of these entities informs HSCT approaches. Many inherited BMF/MDS are associated with excessive transplant regimen-related toxicities and may require specialized reduced intensity conditioning regimens for optimal outcomes.9,10 Additionally, the careful evaluation of a related stem cell donor is critical in the context of a familial genetic disease.
The objectives of this study were to investigate whether pediatric and young adult patients undergoing transplant for AA and MDS harbored pathogenic constitutional mutations in inherited BMF/MDS genes and to examine whether family history or physical findings were associated with mutation status. We employed a multiplexed targeted capture gene panel of known inherited and acquired BMF/MDS genes coupled with next generation sequencing to query 208 pediatric and young adult patients referred for HSCT for AA and MDS for mutations in known inherited BMF/MDS genes.
Methods
Subjects
The study was conducted in accordance with a protocol approved by the Institutional Review Board of the Fred Hutchinson Cancer Research Center and the Declaration of Helsinki. Genomic DNA was obtained from peripheral blood mononuclear cells from the Fred Hutchinson Cancer Research Center Transplant Genomics Biorepository. This repository contains tissue samples from Fred Hutchinson Cancer Research Center and Seattle Cancer Care Alliance HSCT patients, donors, and family members. Study inclusion criteria were patients transplanted at the Seattle Cancer Care Alliance for whom a pre-transplant sample was available and who were: ≤40 years old and transplanted for AA or ≤45 years old and transplanted for MDS. Patients <20 years of age with monosomy 7 acute myeloid leukemia were also included. Patients presented between 1990 and 2012. As the majority of adult MDS patients present after the age of 60 years (SEER Cancer Statistics Review 1975–2012), an age cut-off of ≤45 years-old was selected to study the genetic features of MDS presenting at an unusually young age. Clinical data were obtained by retrospective chart review of available pre- and post-transplant medical records. Genomic DNA was isolated from peripheral blood mononuclear cells banked in the Genomics Biorepository and from paraffin-embedded clinical tissue biopsy samples (ReliaPrep™ FFPE gDNA Miniprep System, Promega) with the patients’ informed consent in accordance with a protocol approved by the Fred Hutchinson Cancer Research Center. Additional details of the retrospective chart review are provided in the Online Supplementary Methods section.
Genomics
Targeted gene capture and massively parallel sequencing were performed as previously described using a capture assay targeting mutations in known inherited and acquired BMF/MDS genes.11 The genes included on the targeted capture panel are listed in Online Supplementary Table S1. Reads were aligned to the human reference genome (hg19) using the Burrows-Wheeler aligner12 and single nucleotide and small insertion-deletion variants called with GATK, using best practice guidelines, as previously described.13 Alignment to the whole genome facilitated exclusion of variants that fell in pseudogenes. Copy number variants were identified as previously described.14
Mutations were identified by a variant allele fraction consistent with heterozygosity (0.3–0.7). Somatic mutations defined by a variant allele fraction of less than 30% were excluded from the analysis as were recessive mutations inconsistent with a Mendelian pattern of inheritance. Variants were classified as pathogenic by predicted effect on protein function, as previously described.15,16 Pathogenic variants were validated by Sanger sequencing. Compound heterozygous variants were confirmed to be in trans by subcloning and Sanger sequencing. Variants were confirmed as constitutional by sequencing DNA isolated from paraffin-embedded biopsy samples of skin, lip, or gastrointestinal tissue or by chromosome fragility testing of patient-derived lymphoblasts, performed as previously described.17 More detailed descriptions are given in the Online Supplementary Methods section.
Results
Characteristics of the patients with aplastic anemia
The study group transplanted for AA included 53 pediatric patients (≤18 years old) and 45 young adult patients (>18 years old) (range, 1–40 years old). A family history of a first- or second-degree relative with malignancy or cytopenias was present in 40% (39/98) of patients. Physical anomalies were noted in 11% (11/98) of patients. An HLA-matched sibling transplant was performed for 38 patients, six of whom had received immunosuppressive therapy prior to transplantation. Fifty-four patients received alternative donor HSCT due to refractory or relapsed disease following immunosuppressive therapy and the remaining six patients received alternative donor HSCT after other medical therapies had failed (Online Supplementary Table S2).
Characteristics of the patients with myelodysplastic syndrome
Among the 135 patients transplanted for MDS, 25 patients with a current or antecedent history of a myeloproliferative disorder were excluded from further analyses, leaving a total of 110 patients for study (Online Supplementary Table S3). The final study group consisted of 46 pediatric patients (≤18 years old) and 64 young adult patients (>18 years old) (range, 1–46 years old). A family history of a first- or second-degree relative with malignancy or cytopenias was present in 52% (57/110) of patients. Physical anomalies were noted in 24% (26/110) of patients.
Analysis of inherited bone marrow failure or myelodysplastic predisposition syndrome genes
Given the diagnostic challenge of discriminating between inherited versus acquired BMF/MDS, we hypothesized that a subset of pediatric and young adult patients who underwent HSCT at our center for AA or MDS had cryptic inherited disorders. This hypothesis was addressed using a targeted capture panel coupled to high-throughput, next-generation sequencing of genes known to contribute to inherited and acquired BMF/MDS (Online Supplementary Table S1).11 For all samples evaluated, the median coverage across the 712 kb targeted region was 444X, with 99.4% of bases having >50X coverage and 99.6% of bases having >10X coverage. This depth of coverage enabled identification of point mutations, small indels, and copy number variants spanning three exons.
Aplastic anemia genetics
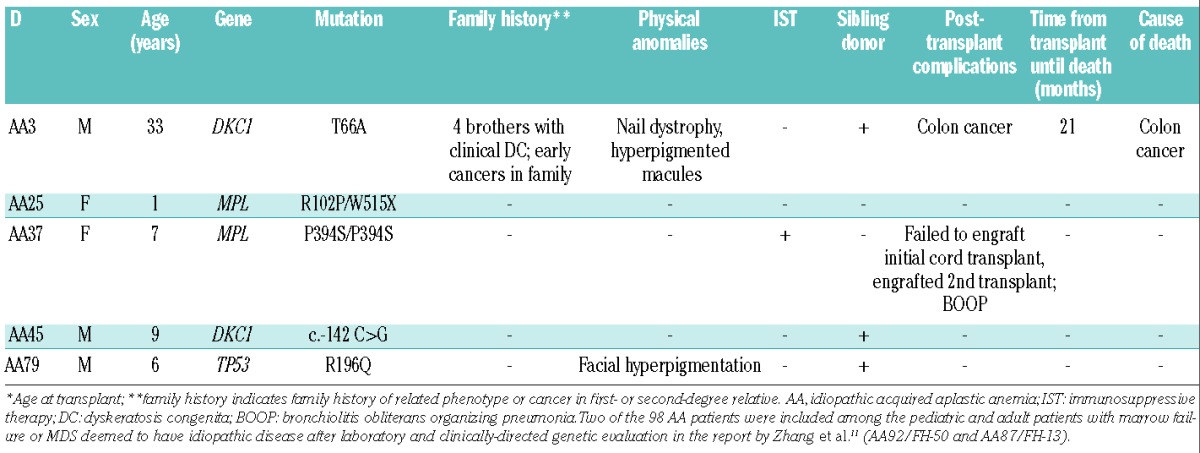
Pathological constitutional mutations in known inherited BMF/MDS genes were identified in five of 98 (5.1%) pediatric and young adult patients with AA (Table 1 and Online Supplementary Table S4). Hemizygous mutations in DKC1 were identified in patients AA3 (T66A) and AA45 (c.-142 C>G). Compound heterozygous and homozygous mutations in MPL were identified in patients AA25 (R102P and W515X) and AA37 (P394S), respectively, and a heterozygous mutation in TP53 was identified in patient AA79 (R196Q). Constitutional mutations in DKC1, MPL, and TP53 cause X-linked dyskeratosis congenita, autosomal recessive congenital amegakaryocytic thrombocytopenia, and the autosomal dominant familial cancer predisposition syndrome, Li Fraumeni, respectively.7 Patients with germline TP53 mutations have an increased risk of developing acute myeloid leukemia and MDS.18–20 The genetics are described in more detail in the Online Supplementary Results.
Table 1.
Clinical and genetic features of AA patients.
Aplastic anemia genotype-phenotype analysis
Among the five AA patients in whom constitutional mutations in known inherited BMF/MDS genes were found, two patients were clinically suspected to have a constitutional disorder prior to their initial HSCT. Patient AA3 was diagnosed with dyskeratosis congenita at 10 years old based on the findings of his physical examination (nail dystrophy and a hyperpigmented macule), a family history of four brothers with clinical dyskeratosis congenita and several family members with early cancers. Patient AA25 presented with an intracranial hemorrhage and severe thrombocytopenia at birth and was diagnosed with congenital amegakaryocytic thrombocytopenia which was genetically confirmed. Of the five patients with constitutional mutations, three received an upfront transplant from an HLA-matched sibling donor. One patient (AA37) received immunosuppressive therapy prior to an alternative donor transplant and a second patient, AA25, received granulocyte colony-stimulating factor and erythropoietin prior to undergoing an alternative donor transplant. Patient AA3 died of colon cancer 21 months after transplantation (Table 1). Colon cancer is a known complication of dyskeratosis congenita.21
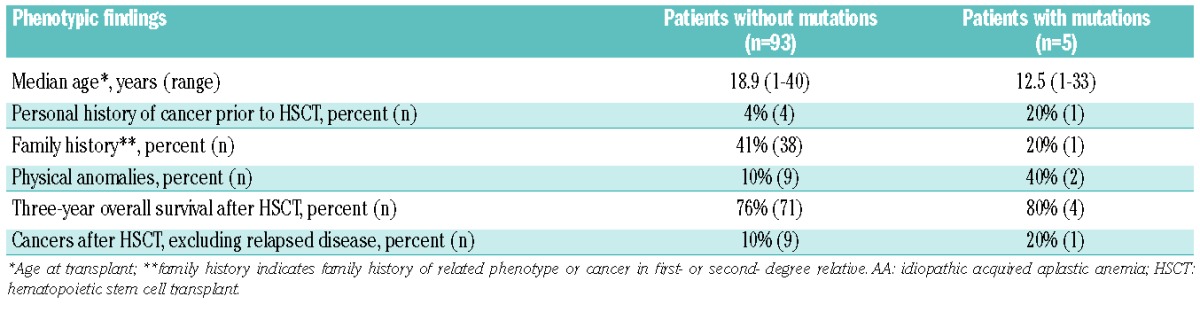
A negative family history did not reliably distinguish those patients lacking mutations in inherited BMF/MDS genes (Table 2). Absence of physical anomalies also failed to distinguish patients with acquired versus germline genetic forms of AA (Table 2).
Table 2.
Genotype-phenotype analyses in AA patients without or with constitutional mutations.
The presence of a paroxysmal nocturnal hemoglobinuria clone was assessed by flow cytometry using four-color or six-color flow cytometry of marrow or peripheral blood, or, in one patient, by the Ham test. A paroxysmal nocturnal hemoglobinuria clone was detected in five of 50 patients tested (10%), none of whom harbored a constitutional mutation in an inherited BMF/MDS gene.
Myelodysplastic syndrome genetics
Pathological mutations in known inherited BMF/MDS genes were identified in 15 out of 110 (13.6%) pediatric and young adult patients with MDS (Table 3 and Online Supplementary Table S4). The mutations were constitutional in 14 out of these 15 patients. Compound heterozygous mutations were identified in FANCA (HIP12286: p.Q549X and c.3349−1G>A), MPL (HIP05737: p.L79Efs* and c.393+5G>C), RTEL1 (HIP02696: p.M492I and R1264H; HIP17561: p.T56M and H116R), and SBDS (HIP02099: p.K62X and c.258+2T>C); constitutional mutations in these genes cause autosomal recessive Fanconi anemia, congenital amegakaryocytic thrombocytopenia, dyskeratosis congenita, and Shwachman-Diamond syndrome, respectively.
Table 3.
Clinical and genetic features of the MDS patients.
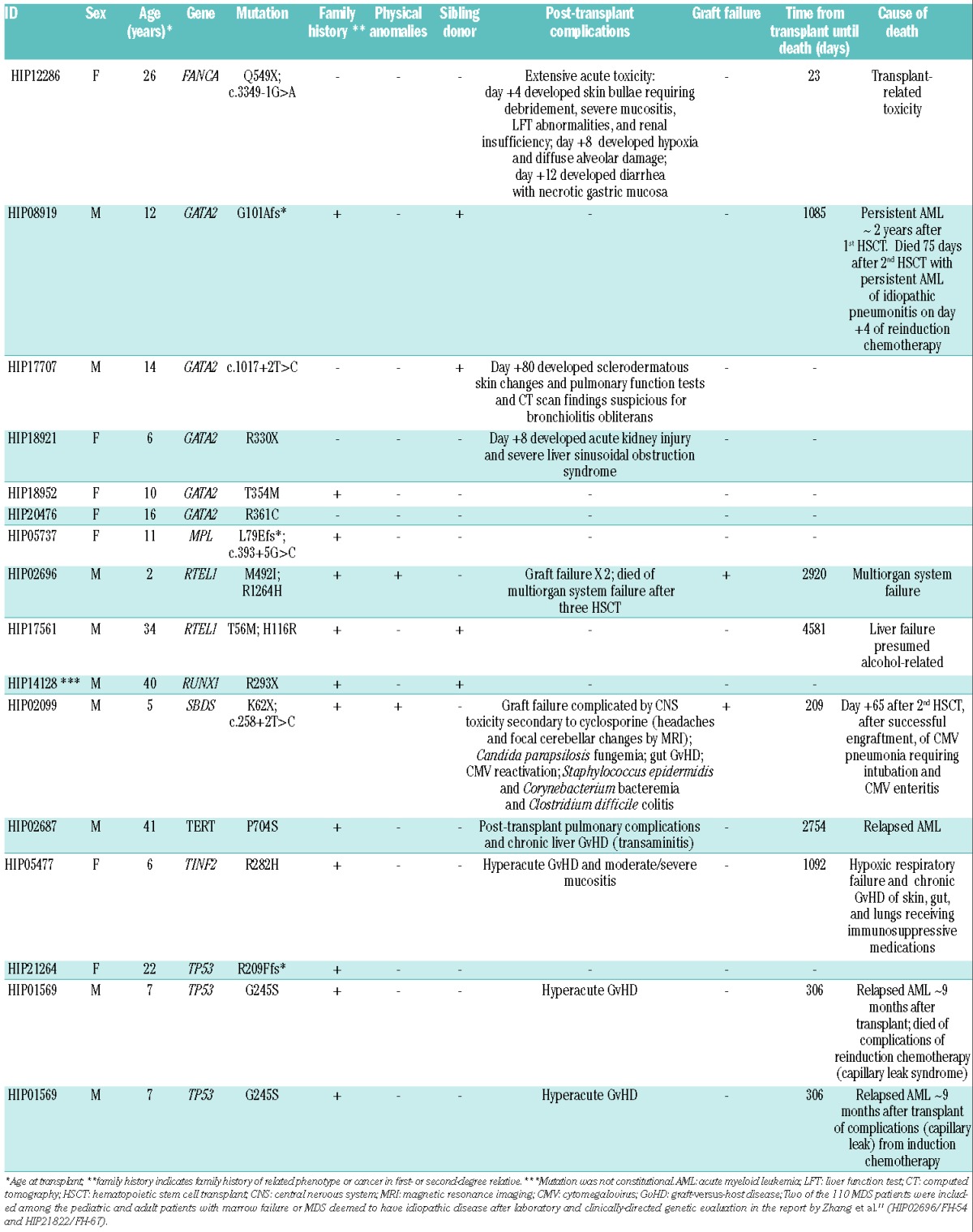
Heterozygous mutations were identified in GATA2 (HIP08919: p.G101Afs*; HIP17707: c.1017+2T>C; HIP18921: p.R330X; HIP18952: p.T354M; HIP20476: p.R361C), RUNX1 (HIP14128: p.R293X), TERT (HIP02687: P704S), TINF2 (HIP05477: R282H), and TP53 (HIP21264: R209Ffs*; HIP01569: G245S); constitutional mutations in these genes cause autosomal dominant inherited predisposition to leukemia and MDS (GATA2, RUNX1), dyskeratosis congenita (TERT, TINF2), and Li Fraumeni syndrome (TP53).
Patient HIP14128 is a particularly instructive clinical case. This patient carried a heterozygous nonsense mutation in RUNX1 (c.958C>T [p.R293X]) in DNA isolated from pre-transplant peripheral blood. This mutation was previously reported as a somatic variant in de novo acute myeloid leukemia22 and occurs in a highly conserved amino acid in the transactivation domain of the protein. Importantly, in DNA isolated from the patient’s small bowel and skin, the patient was wild-type at this base pair, consistent with a somatically acquired, rather than inherited mutation. This distinction is critical, as germline mutations in RUNX1 cause an autosomal dominant familial platelet disorder with a high risk of developing a myeloid malignancy23 necessitating close clinical monitoring of the patient and appropriate genetic counseling of family members. Distinguishing somatic from germline mutations is critical for clinical management decisions. Additional details are available in the Online Supplementary Results.
Myelodysplastic syndrome genotype-phenotype analysis
Among the 14 MDS patients in whom constitutional mutations in known inherited BMF/MDS genes were found, only three patients were clinically suspected to have a constitutional disorder prior to their initial HSCT. Patient HIP05737 was diagnosed with congenital amegakaryocytic thrombocytopenia during her pre-transplant evaluation and patient HIP02099 was suspected to have Shwachman-Diamond syndrome before transplant based on a history of marrow failure and pancreatic insufficiency presenting in a 3.5-month old boy (without genetic confirmation of the diagnosis). Patient HIP21264 was a member of a known Li-Fraumeni family whose TP53 mutation was formerly defined.
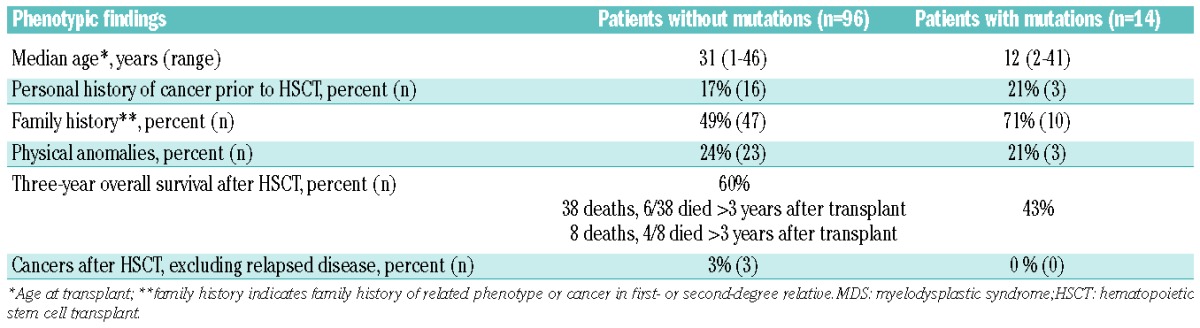
Additionally, while patients with constitutional mutations presented for HSCT at a younger age than those lacking mutations (median age 12.1 years old versus 31.2 years old), the absence of a prior personal history of cancer, family history of a first- or second-degree relative with cancer or a related phenotype, or the presence of physical anomalies did not reliably rule out mutations in inherited BMF/MDS genes (Table 4). Only two of the five patients carrying heterozygous pathogenic mutations in GATA2 had a family history suspicious of an inherited disorder (HIP08919 and HIP18921, Table 3).
Table 4.
Genotype-phenotype associations in MDS patients without or with constitutional mutations.
Standard bone marrow transplant conditioning regimens used for MDS can cause excessive and life-threatening toxicities in patients with certain inherited BMF/MDS.7 Some patients, for example those with dyskeratosis congenita,9 may also face a higher risk of graft failure. The clinical course of patient HIP12286, who carried compound heterozygous deleterious mutations in FANCA, illustrates this issue. She initially presented at the age of 20 years (5 years prior to transplantation) with epistaxis and was found to have mild thrombocytopenia and macrocytosis in the context of a hypocellular marrow for her age. Her family history was unremarkable and she lacked physical or structural anomalies suggestive of an inherited BMF/MDS. She developed progressive cytopenias and monosomy 7 and received an unrelated donor bone marrow transplant with busulfan and cyclophosphamide conditioning. She died 23 days after transplantation of severe transplant-related toxicity, including whole body blistering and a desquamative rash requiring surgical debridement, hypoxic respiratory failure with evidence of diffuse alveolar damage, and severe diarrhea with endoscopic findings of necrotic gastric mucosa. Patients with Fanconi anemia are exquisitely sensitive to genotoxic agents (e.g., alkylators and ionizing radiation) and face potentially severe regimen-related toxicity and high mortality rates with standard conditioning regimens containing these agents.
Patient HIP02696, who carried compound heterozygous mutations in RTEL1, provides a second example of transplant outcomes in a patient with an unrecognized inherited BMF/MDS at the time of their initial transplant, specifically, Hoyeraal-Hreidarsson syndrome. His transplant courses were marked by recurrent graft failure – initially after a non-myeloablative HLA-matched unrelated donor transplant utilizing a preparative regimen containing fludarabine and 200 cGy total body irradiation and again after a second non-myeloablative peripheral blood stem cell transplant from the same donor utilizing a preparative regimen containing fludarabine, antithymocyte globulin, and cyclophosphamide. He ultimately engrafted after infusion of bone marrow hematopoietic stem cells without conditioning from the same original donor and died 95 months after his original transplant from multi-organ system failure. Of note, among the six patients with damaging mutations in dyskeratosis congenita genes in this study (Online Supplementary Table S5), pulmonary complications developed in one, about 6 months after transplantation.
Discussion
Inherited bone marrow failure syndromes, idiopathic AA, and hypoplastic MDS share significant overlap in clinical presentation and bone marrow findings, which often pose a diagnostic challenge. In the present study we determined whether pediatric and young adults referred to our center for HSCT for either AA or MDS harbor mutations in inherited BMF/MDS genes. We found that 5.1% of AA and 13.6% of MDS patients carried mutations in known inherited BMF/MDS genes.
Prior studies have sought to define this incidence by analyzing single genes or small sets of genes. Genetic analyses were confined to single genes or limited gene sets. Yamaguchi et al. screened patients with apparently acquired AA for mutations in TERT, DKC1, NHP2, and NOP10, and reported heterozygous mutations in TERT in ~3.4% (7/205) of patients with AA unresponsive to immunosuppressive therapy.5 Their study included patients aged 2–83 years old with a median age of 34 years old, which is older than our series of AA patients (median age 18 years old). A second study of TERC mutations in 210 patients with bone marrow failure syndromes, including 150 AA and 55 MDS patients, identified heterozygous mutations in only 3/210 (1.4%) cases.6 Field et al. found only two TERC mutations in 284 blood samples obtained from pediatric patients with AA (n=109), MDS (n=137), or juvenile myelomonocytic leukemia (n=38) under 18 years of age who underwent a stem cell transplant for marrow failure from the National Donor Marrow Program Research Sample Repository.24 The incidence of inherited BMF/MDS appeared to be low based on these studies. A recent German study, limited to GATA2 sequencing, demonstrated a 7% (28/426) incidence of germline GATA2 mutations among children and adolescents with de novo MDS.25
Our study here demonstrated, by expanding the genetic screen to include a large set of inherited BMF/MDS genes, that a significant subset of patients carried germline mutations. These results extend a previous study in which we found that 11% (8/71) of patients, primarily from the pediatric age group, harbored germline BMF/MDS gene mutations.11 While the majority of mutations identified in our previous study affected the GATA2 gene, our current study identified mutations in a wide variety of genes. A subsequent study utilized a targeted next-generation sequencing panel of 72 inherited bone marrow failure genes to screen ten patients with severe AA unresponsive to immunosuppressive therapy.26 Mutations were found in three of ten patients (30%). One patient carried compound heterozygous mutations in RTEL1, one patient had a heterozygous TERT mutation, and a third patient had a heterozygous mutation in microtubule-associated serine/threonine kinase like (MASTL), which had previously been reported in autosomal dominant thrombocytopenia. This group additionally identified mutations involving CXCR4, GATA2, G6PC3, MAST, MYH9, RPL5, RTEL1, TERT, TERC, TINF2, or WAS in 18.1% (15/83) of patients with unclassified inherited bone marrow failure syndromes.26 These studies demonstrate that while mutations in any single gene are rare, mutations in BMF/MDS genes as a group are found in a significant subset of patients.
The results of this study underscore the limitations of relying on clinical stigmata or family history to identify patients with an underlying constitutional disorder. Only two of the five AA patients with mutations in an inherited BMF/MDS gene had a physical anomaly and only one patient had a family history of cancer or a related phenotype. Among the MDS cohort, only three of the 14 patients with mutations in an inherited BMF/MDS gene had a physical anomaly and while patients with mutations were more likely to have a positive family history of cancer or a related phenotype than those patients without mutations, the absence of a family history did not exclude the possibility of a mutation. Although the retrospective design of this study limits the available physical anomaly and family history data to those captured in the clinical records and further studies are warranted to confirm the generalizability of our findings, our conclusions are consistent with other published work. Our earlier study identified mutations in eight of 71 (11%) pediatric and adult patients (<40 years old at presentation or with a family history of BMF/MDS regardless of age) with idiopathic bone marrow failure, with all eight patients lacking the classical clinical stigmata of these syndromes and only four having a suggestive family history despite extensive longitudinal evaluation.11 Our study both complements and expands on a recent large study of 1,120 pediatric cancer patients which found that 8.5% of patients had germline mutations in inherited cancer predisposition genes and more than half of the children with germline mutations lacked any family history of cancer.27
Our study also highlights the importance of recognizing inherited BMF/MDS to inform appropriate medical care. One of the five AA patients with mutations in inherited BMF/MDS genes received immunosuppressive therapy without response. Patients with inherited BMF/MDS respond poorly or transiently to immunosuppressive therapy and are at increased risk of relapse and clonal evolution with reduced survival.28 Additionally, sibling mutation status would inform transplant donor choice. For relapsed MDS after a sibling donor transplant, it would be informative to determine whether relapsed MDS involved donor or recipient-derived cells. Published reports suggest a high transplant-related mortality and organ toxicity in patients with certain inherited BMF/MDS transplanted using standard preparatory regimens for AA or MDS.7,9,10 The severe and immediate post-transplant complications observed in the Fanconi anemia, subtype A patient in this study (HIP12866) provides an instructive example of this risk. Additionally, dyskeratosis congenita appears to be associated with early and late graft failure and pulmonary toxicity with standard transplant preparatory regimens,9 which was observed in this series (HIP02696 and HIP05477, respectively).
Given the increased risks of hematologic and solid tumors associated with inherited BMF/MDS, a clinical surveillance protocol for patients has the potential to improve outcomes in individuals and affected family members of genetically defined probands. These patients may face a particularly high risk of tumors following HSCT.
One of the challenges in genetic studies is the ascertainment of pathogenicity for a given variant. As our understanding evolves, reassessment of previously described variants is continually warranted. For example, we observed the TERT c.C1234T (H412Y) variant in a 10-year old girl with MDS, a 2-year old boy with AA and a 27-year old man with AA and have also observed this variant in four other patients among the 1042 patients with bone marrow failure or MDS sequenced on our targeted gene sequencing platform. This missense mutation affects an amino acid that is not highly conserved across species and is reported in 121 alleles (among a total of 19080) on the Exome Aggregation Consortium, including two homozygous individuals (ExAC, Cambridge, MA, USA). The lack of amino acid conservation across species and frequent observation in our laboratory and on ExAC suggest that while the variant reduces telomerase activity in vitro,5 it might be clinically benign in humans in the heterozygous state. Du et al. described a family in which the proband had clinical dyskeratosis congenita, very short telomeres, and a homozygous TERT P704S mutation resulting in reduced telomerase activity. The proband’s father carried compound heterozygous P704S and H412Y TERT mutations and had very short telomeres. The proband’s mother had neither clinical evidence of dyskeratosis congenita nor very short telomeres in peripheral blood mononuclear cells and was heterozygous for the P704S allele. Since both mutations are hypomorphic alleles, impairing but not eliminating telomerase activity,29 these data suggest that hypomorphic TERT mutations might contribute to disease when present together in the compound heterozygous or homozygous state. Further studies are warranted to clarify whether a single heterozygous TERT H412Y variant is pathogenic in humans.
Several limitations of this study must be considered. While the rarity of AA and MDS in children and young adults precluded a prospective study, the retrospective study design limits the available clinical history. Further studies with comprehensive clinical data, including a detailed family history and physical examination findings with attention to subtle features associated with bone marrow failure, to accompany the genetic evaluation, are warranted. The frequency of germline mutations in our study group may be underestimated since our variant pipeline filtered out mutations with a variant allele fraction of less than 0.30, which may have eliminated patients with somatic mosaicism.27
In current practice, testing for an underlying genetic disorder is most commonly considered in pediatric and young adult patients presenting with bone marrow failure. Genetic testing is rarely pursued in pediatric and young adult patients presenting with MDS. While screening BMF/MDS patients for mutations in a single gene or small set of genes reveals rare cases of germline mutations, we found that comprehensive screening of BMF/MDS genes in aggregate revealed a significant subset of previously unrecognized patients. As we restricted our AA analysis to those patients with severe AA requiring HSCT, it will be interesting to determine the frequency of constitutional mutations in inherited BMF/MDS genes among patients diagnosed with moderate AA. Since many of these disorders require specialized evaluation of family donors and counseling, reduced intensity conditioning regimens, and surveillance for disease-specific extra-hematopoietic complications, diagnosis prior to transplantation is critical. Our findings suggest that genetic screening to evaluate the inherited BMF/MDS genes collectively should be considered in pediatric and young adult patients presenting with AA or MDS.
Supplementary Material
Acknowledgments
The authors would like to thank Sharon Savage, Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD, and Keith Loeb, Department of Pathology, University of Washington, Seattle, WA and J. Arturo Londoño-Vallejo, Institut Curie, Paris, France and Simon Boulton, Francis Crick Institute, UK for helpful discussions. This work was supported by NIH/NIDDK grant R24DK099808 (AS, MCK, and JA), the Aplastic Anemia & Myelodysplastic Syndrome International Foundation, the Ghiglione Aplastic Anemia Fund, and the Julian’s Dinosaur Guild at Seattle Children’s Hospital (AS).
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/11/1343
References
- 1.Hirabayashi S, Flotho C, Moetter J, et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood. 2012;119(11):e96–99. [DOI] [PubMed] [Google Scholar]
- 2.Hasle H, Niemeyer CM, Chessells JM, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17(2):277–282. [DOI] [PubMed] [Google Scholar]
- 3.Niemeyer CM, Baumann I. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematology Am Soc Hematol Educ Program. 2011;2011:84–89. [DOI] [PubMed] [Google Scholar]
- 4.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamaguchi H, Calado RT, Ly H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352(14):1413–1424. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi H, Baerlocher GM, Lansdorp PM, et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102(3):916–918. [DOI] [PubMed] [Google Scholar]
- 7.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alter BP, Elghetany MT. The CCC system: is it really the answer to pediatric MDS¿ J Pediatr Hematol Oncol. 2003;25(5):426–427; author reply 427–428. [DOI] [PubMed] [Google Scholar]
- 9.Gadalla SM, Sales-Bonfim C, Carreras J, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant. 2013;19(8):1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gluckman E, Devergie A, Schaison G, et al. Bone marrow transplantation in Fanconi anaemia. Br J Haematol. 1980;45(4):557–564. [DOI] [PubMed] [Google Scholar]
- 11.Zhang MY, Keel SB, Walsh T, et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica. 2015;100(1):42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gulsuner S, Walsh T, Watts AC, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154(3):518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh T, Lee MK, Casadei S, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA. 2010;107(28):12629–12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108(44):18032–18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimamura A, Montes de Oca R, Svenson JL, et al. A novel diagnostic screen for defects in the Fanconi anemia pathway. Blood. 2002;100(13):4649–4654. [DOI] [PubMed] [Google Scholar]
- 18.Kuribayashi K, Matsunaga T, Sakai T, et al. A patient with TP53 germline mutation developed Bowen’s disease and myelodysplastic syndrome with myelofibrosis after chemotherapy against ovarian cancer. Intern Med. 2005;44(5):490–495. [DOI] [PubMed] [Google Scholar]
- 19.Felix CA, Hosler MR, Provisor D, et al. The p53 gene in pediatric therapy-related leukemia and myelodysplasia. Blood. 1996;87(10):4376–4381. [PubMed] [Google Scholar]
- 20.Anensen N, Skavland J, Stapnes C, et al. Acute myelogenous leukemia in a patient with Li-Fraumeni syndrome treated with valproic acid, theophyllamine and all-trans retinoic acid: a case report. Leukemia. 2006;20(4):734–736. [DOI] [PubMed] [Google Scholar]
- 21.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang JL, Hou HA, Chen CY, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114(26):5352–5361. [DOI] [PubMed] [Google Scholar]
- 23.Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175. [DOI] [PubMed] [Google Scholar]
- 24.Field JJ, Mason PJ, An P, et al. Low frequency of telomerase RNA mutations among children with aplastic anemia or myelodysplastic syndrome. J Pediatr Hematol Oncol. 2006;28(7):450–453. [DOI] [PubMed] [Google Scholar]
- 25.Wlodarski MW, Hirabayashi S, Pastor V, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–1397. [DOI] [PubMed] [Google Scholar]
- 26.Ghemlas I, Li H, Zlateska B, et al. Improving diagnostic precision, care and syndrome definitions using comprehensive next-generation sequencing for the inherited bone marrow failure syndromes. J Med Genet. 2015;52(9):575–584. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Walsh MF, Wu G, et al. Germline Mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheinberg P, Cooper JN, Sloand EM, Wu CO, Calado RT, Young NS. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. JAMA. 2010;304(12):1358–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du HY, Pumbo E, Manley P, et al. Complex inheritance pattern of dyskeratosis congenita in two families with 2 different mutations in the telomerase reverse transcriptase gene. Blood. 2008;111(3):1128–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.