

Despite the significant clinical consequences of red blood cell (RBC) alloimmunization, our understanding of the fundamental molecular and cellular mechanisms regulating anti-RBC antibody generation is limited. Relative to infectious stimuli,1 transfused RBCs induce a less robust antibody response as measured both by individual response rates2 and resulting antibody half-lives.3 We therefore hypothesized that anti-RBC alloantibodies are driven by less-redundant signaling pathways, and should be particularly sensitive to perturbations of individual innate cytokine signals. To test this hypothesis, we combined a recently developed mouse model of RBC alloimmunization with mice harboring a conditional allele for the receptor of the cytokine IL-6 (IL-6Rα). Mice with a deletion of IL-6Rα in the germ-line or only on their T cells generated significantly lower levels of anti-RBC alloantibodies in response to transfusion. Furthermore, relative to their wild-type (WT) counterparts, IL-6Rα deficient naive CD4+ T cells demonstrated a decrease in both their maximal expansion and subsequent differentiation into T-follicular helper (TFH) cells in response to transfusion. Thus, we have identified for the first time a specific molecule (IL-6Rα) and its downstream cellular target (CD4+ T cells) that co-ordinately act to enhance RBC alloimmunization.
In order to investigate the molecular and cellular mechanisms regulating RBC alloimmunization, we turned to a recently developed in vivo mouse model of RBC alloimmunization that has provided important clues into how transfused allogeneic RBCs drive antibody responses. HOD transgenic mice express a protein on the surface of RBCs that is a triple fusion construct of Hen Egg Lysozyme, Ovalbumin, and the human Duffy red cell antigen.4 These mice serve as blood donors wherein mouse blood is collected and stored in a manner directly analogous to modern human blood banking practices. In the HOD system, transfusion of fresh, leukoreduced RBCs into non-transgenic C57BL/6J (B6) mice leads to low anti-HOD antibody levels. Alternatively, transfusion of mouse RBCs stored under conditions similar to those employed in modern blood banks resulted in significant increases in alloantibody production.5 Thus, stored RBCs can enhance alloimmune antibody responses. However, it remains unclear which cells and molecules drive alloantibody production in response to either fresh or stored RBC transfusion.
Given our lack of knowledge of which innate stimuli and resulting cytokines are functionally important in driving RBC alloimmunization, we decided to focus on a cytokine that has been shown to be induced early on by stored blood transfusion, namely IL-6.6 In order to determine both the global and cell-specific role of IL-6 signaling, we studied mice that were either germ-line IL-6Rα deficient (hereafter referred to as IL-6RαKO) or lacked IL-6Rα expression only in T cells (hereafter referred to as IL-6RαTKO).7 After confirming both germline and T-cell specific deletion of IL-6 signaling capability in these mice (Online Supplementary Figures S1 and S2), we characterized their alloantibody production in response to fresh and stored HOD blood transfusion (Figure 1). Antibody responses to transfused HOD RBCs are directed against the HEL antigen,4 and anti-HEL antibody levels were measured via limiting dilution titers on high protein binding ELISA plates (details of methods used are available in the Online Supplementary Appendix). In response to transfusion with fresh HOD RBCs, WT mice demonstrated titers well above background, while both IL-6RαKO and IL-6RαT-KO mice showed a significant reduction in anti-HEL antibody titers compared to wild-type mice. Transfusion with stored HOD RBCs led to a significant enhancement in anti-HEL antibody titers in wild-type mice relative to fresh blood. In response to stored blood transfusion, IL-6RKO and IL-6RαTKO mice demonstrated a significant reduction in anti-HEL antibody titers relative to wild-type mice. Flow crossmatch assays confirmed the reduction in anti-HOD alloantibody levels in IL-6RαKO and IL-6RαTKO mice (Online Supplementary Figure S3). These results demonstrate that IL-6Rα in general, and IL-6Rα expression on T cells specifically, is required for maximal generation of anti-HOD alloantibodies in response to both fresh and stored blood transfusion. This occurred despite the fact that no circulating IL-6 was detected in response to fresh blood transfusion (data not shown), suggesting that local IL-6 signaling is sufficient to drive alloantibody responses.
Figure 1.
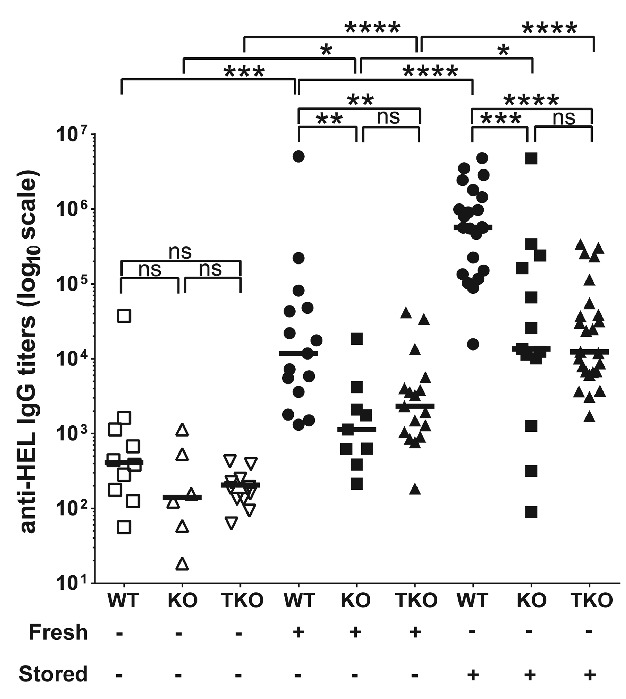
Interleukin-6 receptor signaling on T cells regulates generation of anti-HOD alloantibodies in response to transfused fresh and stored HOD red blood cell. WT, IL-6RαKO (KO), and IL-6RTKO (TKO) C57BL/6J mice (10–12 weeks of age) were transfused with leukoreduced fresh (Fresh) or stored HOD red blood cells (RBC) (Stored). Serum was collected 14 days post transfusion and levels of anti-HOD alloantibody generation was measured through anti-HEL IgG ELISA. Anti-HEL IgG endpoint titers are depicted on a log10 scale, and bars on scatter plots represent median values. Figure shows combined results from 3 independent experiments. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; ns=P>0.05.
Given that IL-6 is known to be an activator of innate immune cells and an initiator of the generalized acute phase response, we next determined whether IL-6 was required to drive the general pro-inflammatory cytokine response to transfused RBCs. Though we observed significant increases in multiple cytokines with stored transfusion, only IL-6 was decreased in IL-6KOs (Figure 2 and Online Supplementary Figure S4). Thus, IL-6 does not interfere with the initial generation of the general inflammatory response to stored blood by innate immune cells, and prompted us to look at later stages and other cell types for IL-6 function.
Figure 2.
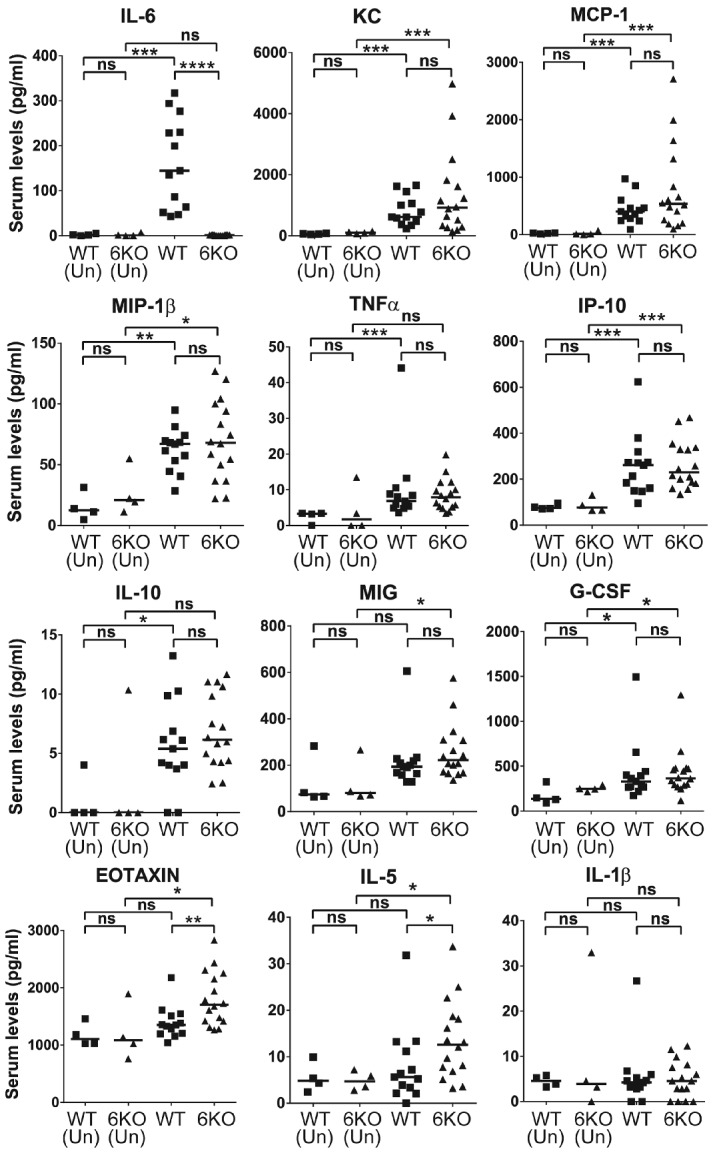
IL-6 signaling is not required for the initial pro-inflammatory cytokine response to stored red blood cell (RBC) transfusion in recipient mice. The 12 cytokines (from a total of 32 cytokines measured) whose expression was induced in response to transfusion with stored HOD RBCs in recipient mice are shown. Serum levels of inflammatory cytokines (as labeled) in WT and IL-6KO (6KO) C57BL/6J mice (aged 10–12 weeks) 90-min post transfusion with stored HOD RBCs compared to untransfused mice (Un) measured through bead-based Luminex assay. Combined data from 3 independent experiments. Bars on scatter plots represent median values. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; ns=P>0.05.
In in vitro assays, IL-6 has been shown to support CD4 differentiation into a specialized class of helper T cells (TFH) that are essential for the generation of T-dependent antibody production.8 We were, therefore, interested in determining whether the T-cell specific IL-6Rα phenotype we observed in alloantibody production correlated with TFH generation in vivo. We, therefore, took advantage of the fact that T-cell responses to the HOD antigen in B6 mice are directed against OVA and antigen specific T-cell responses can be monitored using OVA-specific OTII TCR transgenic T cells.9 By adoptively transferring a mixed population of congenically marked IL-6RαKO and WT naïve OTII cells into wild-type animals, RBC-specific T-cell responses to both fresh or stored HOD transfusion could be measured in the same animal (see detailed gating strategy in Online Supplementary Figure S5). We chose four days post transfusion to detect early T-cell responses as this is one of the earliest time points that antigen specific TFH can be observed.10 Though there appeared to be a small expansion of OTII cells in response to fresh transfusion, this did not reach statistical significance (Figure 3A and B). In contrast, the observed OTII expansion was much greater in response to stored blood transfusion, demonstrating that stored blood acts as much stronger expansion stimulus. Interestingly, IL-6Rα deficient OTII T cells expanded less than their wild-type counterparts (Figure 3E), demonstrating that T-cell intrinsic IL-6Rα expression provides a competitive expansion advantage in response to both fresh and stored blood.
Figure 3.
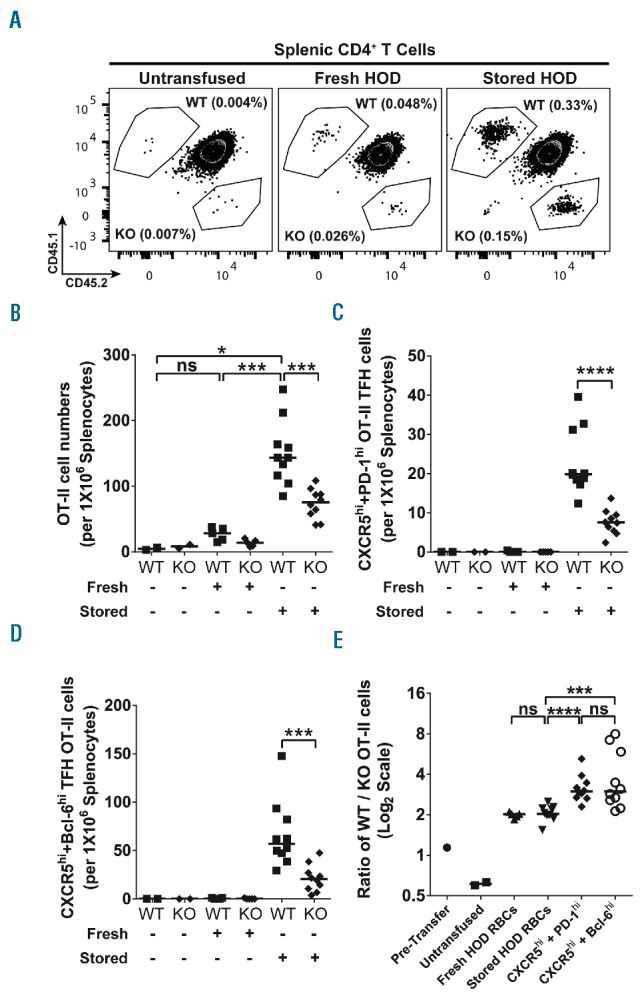
IL-6Rα signaling in T cells directly regulates T-follicular helper cell (TFH) differentiation in response to stored HOD red blood cell (RBC) transfusions. To determine whether direct IL-6Rα signaling in T cells regulates their differentiation into antigen specific TFH cells in response to transfusions with stored HOD RBCs, TFH differentiation in IL-6RαKO OT-II and WT OT-II cells was directly compared in wild-type (WT) recipients transfused with HOD RBCs. (A) Representative flow cytometry contour plots depicting the net expansion of WT and IL-6RαKO (KO) OT-II cells in response to fresh or stored HOD RBC transfusions. Panels show total gated CD4+ population in splenocytes, with frequencies indicated for WT OT-II (CD45.1+CD45.2- gate) and IL-6RαKO OT-II (CD45.1-CD45.2+ gate) populations within the splenic CD4+ T cells. (B) Number of WT or IL-6RαKO (KO) OT-II cells per 1×106 splenocytes in mice that were untransfused or transfused with either fresh (Fresh) or stored HOD RBCs (Stored). (C) Number of WT and IL-6RαKO (KO) OT-II CXCR5hiPD-1hi TFH cells per 1×106 splenocytes in mice transfused with either fresh (Fresh) or stored HOD RBCs (Stored) or left untransfused. (D) WT and IL-6RαKO (KO) OT-II CXCR5hiBcl-6-1hi TFH cell numbers per 1×106 splenocytes in recipients that were untransfused or transfused with fresh (Fresh) or stored HOD RBCs (Stored). (E) Ratios of WT to IL-6RαKO OT-II cell numbers in each recipient animal. Pre-Transfer indicates the initial ratio of total WT OT-II to IL-6RαKO OT-II cells in the OT-II cell mixed population adoptively transferred into recipient animals. Untransfused, Fresh HOD RBCs, and Stored HOD RBCs indicate the ratio of total splenic WT OT-II to IL-6RαKO OT-II cell numbers in mice that were untransfused, transfused with fresh HOD RBCs, or transfused with stored HOD RBCs, respectively. CXCR5hi + PD-1hi and CXCR5hi + Bcl-6hi indicate ratios of the number of WT to IL-6RαKO CXCR5hiPD-1hi TFH OT-II and WT to IL-6RαKO CXCR5hiBcl-6hi TFH OT-II cell number respectively within each recipient transfused with stored HOD RBCs. Bars on scatter plots show median values. *P<0.05; ***P<0.001; ****P<0.0001; ns=P>0.05.
We next measured TFH differentiation of OTII cells in response to fresh and stored blood via either surface expression of CXCR5 and PD-1 (Figure 3C) or surface expression of CXCR5 along with intracellular expression of the canonical TFH transcription factor BCL6 (Figure 3D). TFH differentiation was undetectable in response to fresh blood, yet robust in response to stored blood. Most importantly, in response to stored blood, we observed significantly fewer TFH cells among IL-6RαKO OTII T cells compared to WT OT-II cells. The lack of TFH numbers was not solely due to differences in underlying OTII expansion, as the ratio of WT/KO cells was roughly 4-fold among TFH versus 2-fold for all OTII cells (Figure 3B and E). Thus, T-cell intrinsic IL-6Rα expression contributes to both T-cell expansion as well as TFH differentiation in response to stored blood transfusion.
Overall, our data with IL-6Rα deficient mice clearly demonstrate that IL-6Rα expression on T cells enhances the alloantibody responses and T-cell expansion of RBC-specific naïve T cells in response to both fresh and stored RBC transfusions. In response to stored blood, IL-6Rα also controls TFH differentiation. However, we failed to detect TFH differentiation in response to fresh blood, despite the fact that it is capable of inducing alloantibodies that are sensitive to IL-6Rα signaling on T cells. We believe the simplest explanation for this apparent discrepancy is that TFH differentiation is either delayed or occurs below the level of detection in response to fresh blood transfusion. Alternatively, TFH may not play a role in alloantibody production in response to fresh blood. Though IL-6 is induced via transfusion, the role of exogenous sources of IL-6 has yet to be determined. Given that different types of inflammation are differentially correlated with both IL-6 production and alloantibody production in patients, it will be interesting to further investigate how the timing and context of various inflammatory stimuli modulate transfusion-generated alloantibody production.
Collectively, our data have uncovered a previously unappreciated role for IL-6R expression in regulating RBC alloimmunization. Of note, our findings in the transfusion setting stand in sharp contrast to the majority of studies that have looked at the role of IL-6 in supporting TFH production in response to vaccination or infection.10–14 In infectious and vaccine models, antigen-specific TFH develop normally in vivo in the absence of isolated IL-6 deficiency, presumably due to the activation of redundant signaling pathways such as IL-21. Indeed, the recent report of Nish et al. found much smaller defects in TFH numbers in response to adjuvant vaccination of T-cell specific IL-6Rα-deficient animals than we observed in response to transfusion.15 Collectively, our data support the hypothesis that transfusion initiates less redundant immune signaling pathways, and provides a potential molecular mechanism for the low overall immunization rates and short alloantibody half-lives observed in alloimmunized patients.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Angela Drew for kindly providing mice critical for this study, and members of the Luckey lab for helpful discussions.
Footnotes
Funding: this research was supported by 1R21HL111866-01 (CJL; PI; JCZ; Co-I). AA was supported by 2T32HL007627-31 from the Brigham & Women’s Hospital Department of Pathology. JES was supported by 5P30CA044579-24 (Loughran PI) from the University of Virginia Cancer Center.
The online version of this letter has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357(19):1903–1915. [DOI] [PubMed] [Google Scholar]
- 2.Heddle NM, Soutar RL, O’Hoski PL, et al. A prospective study to determine the frequency and clinical significance of alloimmunization post-transfusion. Br J Haematol. 1995;91(4):1000–1005. [DOI] [PubMed] [Google Scholar]
- 3.Tormey CA, Stack G. The persistence and evanescence of blood group alloantibodies in men. Transfusion. 2009;49(3):505–512. [DOI] [PubMed] [Google Scholar]
- 4.Desmarets M, Cadwell CM, Peterson KR, Neades R, Zimring JC. Minor histocompatibility antigens on transfused leukoreduced units of red blood cells induce bone marrow transplant rejection in a mouse model. Blood. 2009;114(11):2315–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hendrickson JE, Hod EA, Spitalnik SL, Hillyer CD, Zimring JC. Storage of murine red blood cells enhances alloantibody responses to an erythroid-specific model antigen. Transfusion. 2010;50(3):642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hod EA, Zhang N, Sokol SA, et al. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood. 2010;115(21):4284–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McFarland-Mancini MM, Funk HM, Paluch AM, et al. Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J Immunol. 2010;184(12):7219–7228. [DOI] [PubMed] [Google Scholar]
- 8.Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209(7):1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudson KE, Lin E, Hendrickson JE, Lukacher AE, Zimring JC. Regulation of primary alloantibody response through antecedent exposure to a microbial T-cell epitope. Blood. 2010;115(19):3989–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi YS, Kageyama R, Eto D, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34(6):932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eto D, Lao C, DiToro D, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One. 2011;6(3):e17739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karnowski A, Chevrier S, Belz GT, et al. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med. 2012;209(11):2049–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nurieva RI, Chung Y, Hwang D, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29(1):138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poholek AC, Hansen K, Hernandez SG, et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol. 2010;185(1):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nish SA, Schenten D, Wunderlich FT, et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. Elife. 2014;3(e01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.