

Abstract
ETV6-related thrombocytopenia is an autosomal dominant thrombocytopenia that has been recently identified in a few families and has been suspected to predispose to hematologic malignancies. To gain further information on this disorder, we searched for ETV6 mutations in the 130 families with inherited thrombocytopenia of unknown origin from our cohort of 274 consecutive pedigrees with familial thrombocytopenia. We identified 20 patients with ETV6-related thrombocytopenia from seven pedigrees. They have five different ETV6 variants, including three novel mutations affecting the highly conserved E26 transformation-specific domain. The relative frequency of ETV6-related thrombocytopenia was 2.6% in the whole case series and 4.6% among the families with known forms of inherited thrombocytopenia. The degree of thrombocytopenia and bleeding tendency of the patients with ETV6-related thrombocytopenia were mild, but four subjects developed B-cell acute lymphoblastic leukemia during childhood, resulting in a significantly higher incidence of this condition compared to that in the general population. Clinical and laboratory findings did not identify any particular defects that could lead to the suspicion of this disorder from the routine diagnostic workup. However, at variance with most inherited thrombocytopenias, platelets were not enlarged. In vitro studies revealed that the maturation of the patients’ megakaryocytes was defective and that the patients have impaired proplatelet formation. Moreover, platelets from patients with ETV6-related thrombocytopenia have reduced ability to spread on fibrinogen. Since the dominant thrombocytopenias due to mutations in RUNX1 and ANKRD26 are also characterized by normal platelet size and predispose to hematologic malignancies, we suggest that screening for ETV6, RUNX1 and ANKRD26 mutations should be performed in all subjects with autosomal dominant thrombocytopenia and normal platelet size.
Introduction
Until the end of the last century, only a few forms of inherited thrombocytopenia were known, all of which were extremely rare and characterized by a severe bleeding tendency. Since then, knowledge of these thrombocytopenias has improved greatly and we presently recognize at least 26 disorders caused by mutations in 30 genes.1,2 This advancement of knowledge revealed that most patients with inherited thrombocytopenias have only mild or moderate thrombocytopenia, with trivial bleeding episodes or no bleeding at all. However, it also became apparent that many patients are exposed to a threat of acquiring additional defects that worsen their quality of life or can even be fatal. Subjects with MYH9-related disease are predisposed to proteinuric nephropathy evolving into end-stage renal failure, those with congenital amegakaryocytic thrombocytopenia always develop bone marrow aplasia, while patients with ANKRD26-related thrombocytopenia (ANKRD26-RT) or familial platelet disorder with predisposition to acute myeloid leukemia (AML) due to RUNX1 mutations (FPD/AML) have increased risk of AML and myelodysplastic syndromes. Thus, bleeding is no longer the unique problem of inherited thrombocytopenia patients.
In 2015, four independent studies showed that mutations in the ETV6 gene are responsible for a new form of inherited thrombocytopenia and suggested that ETV6-related thrombocytopenia (ETV6-RT) predisposes to acute lymphoblastic leukemia (ALL).3–6 However, only a few families have been reported so far and the clinical and laboratory features of ETV6-RT remain poorly defined.
In order to gain further information on this disorder, we screened 130 consecutive unrelated propositi with inherited thrombocytopenia of unknown origin for ETV6 mutations and identified seven affected families. Two of these pedigrees have been briefly reported in a previous paper.4 Here we describe the features of 20 affected subjects, who form the largest cohort of ETV6-RT patients collected so far. As these patients were identified by screening a series of consecutive, unselected probands with familial thrombocytopenia, we could estimate the relative frequency of ETV6-RT among inherited thrombocytopenias and the risk of hematologic malignancies associated with this condition. By reporting the clinical and laboratory features of these patients in detail, we provide indications to raise the level of suspicion of the presence of this disorder from the findings of routine diagnostic workup of probands with inherited thrombocytopenia. Finally, we discuss the pathogenesis of ETV6-RT, having investigated, for the first time, megakaryocytes differentiated from hematopoietic progenitors of patients with ETV6-RT and functionally characterized the patients’ platelets.
Methods
Patients
Between 2003 and 2014, we analyzed at the IRCCS Policlinico San Matteo Foundation of Pavia (Italy) 274 consecutive unrelated probands with familial thrombocytopenia. By applying a well-defined diagnostic algorithm for inherited thrombocytopenias,1 we made a molecular diagnosis in 144 of these families, whereas 130 probands remained without a definite diagnosis as they did not fit the criteria for any known inherited thrombocytopenia. These 130 consecutive propositi with inherited thrombocytopenia of unknown origin have been screened for mutations in ETV6. Whenever ETV6 mutations were identified, the available relatives of probands were also investigated.
Bleeding tendency was measured using the International Society on Thrombosis and Haemostasis bleeding assessment tool.7
The institutional review board of San Matteo Foundation approved the study and all subjects or their legal guardians signed written informed consent in accordance with the Declaration of Helsinki.
Mutation screening and reverse transcriptase polymerase chain reaction analysis
Genomic DNA and RNA were extracted from peripheral blood. The ETV6 gene was analyzed using Sanger and whole exome sequencing. Methods of mutation screening and reverse transcriptase polymerase chain reaction analysis are detailed in the Online Supplementary Information.
Bioinformatic tools and analysis of ETV6 structure
The bioinformatic tools used to evaluate missense variants together with the methods used to analyze ETV6 structure are reported in the Online Supplementary Information.
Basic blood cell studies
Blood cell counts were evaluated by electronic counters. Parameters relative to platelet diameter were measured by software-assisted image analysis on blood smears, as reported elsewhere.8 The following previously defined parameters were computed: mean platelet diameter, platelet diameter distribution width, platelet diameter large cell ratio, and platelet diameter small cell ratio.8 The percentage of large platelets was also estimated empirically, as previously reported8 and detailed in the Online Supplementary Information. Surface expression of platelet glycoproteins (GP) was investigated by flow cytometry as reported, whereas platelet aggregation was evaluated using the densitometric method described by Born.9 The antibodies and platelet agonists used are listed in the Online Supplementary Information.
Platelet activation
Platelet activation in response to ADP or TRAP was investigated by flow cytometry as reported previously.10 The protocol is described in detail in the Online Supplementary Information.
Platelet adhesion and spreading
Platelet adhesion and spreading on the subendothelium components of the extracellular matrix, type I collagen, von Willebrand factor, or fibrinogen, were investigated as previously described11,12 and as detailed in the Online Supplementary Information.
Investigation of megakaryocytes
Megakaryocytes were differentiated in vitro from peripheral blood CD45+ cells as previously reported.13,14 Morphological analysis of megakaryocytes was performed by phase-contrast and fluorescence microscopy, while the percentage of fully differentiated megakaryocytes and megakaryocyte ploidy at the end of the culture were investigated by flow cytometry.14,15 Proplatelet yields were evaluated both in suspension and following adhesion on fibrinogen at the end of the culture, as previously described.13,16 Methods are reported in the Online Supplementary Information.
Statistical analysis
Data are presented as means and standard deviations or ranges. Statistical comparisons were performed by the two-tailed Student t test. Incidences of hematologic malignancies (per 100,000 person-years) together with their exact 95% confidence intervals (95% CI) were computed.
Results
Mutation screening
Analysis of the ETV6 gene allowed us to identify five different heterozygous variants in seven unrelated pedigrees. Two variants (c.641C>T/p.P214L and c.1252A>G/p.R418G+p.N385Vfs*7) have been reported previously in two families (Figure 1A, families B and G).4 The remaining three novel variants are two missense alterations and one deletion.
Figure 1.
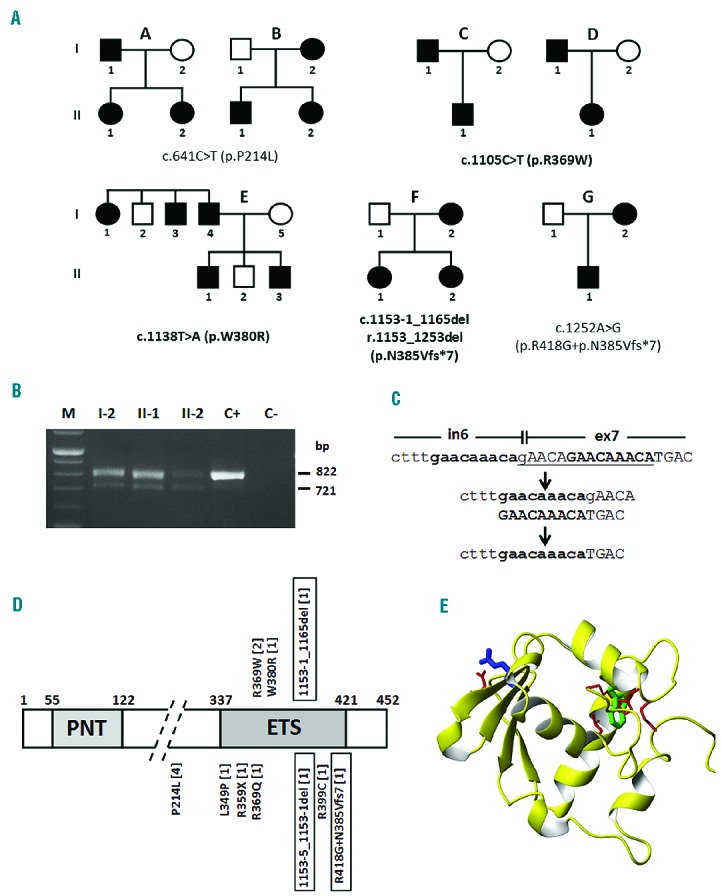
Mutations identified in the ETV6 gene and their effect on protein structure. (A) Pedigrees of families enrolled in this study carrying different mutations as indicated (novel mutations in bold). Nucleotide numbering reflects the ETV6 cDNA with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence (RefSeq NM_001987.4). Therefore, the initiation codon is residue 1 in the amino acid sequence. Families B and G have been previously reported (Noetzli et al.4). (B) RT-PCR in affected members (I-2, II-1, and II-2) of family F to determine the consequence of the c.1153-1_1165del mutation on splicing. C+, wild-type control; C−, negative control. The analysis shows two fragments, the wild-type (822 bp) and the exon 7 skipping (721 bp) products. (C) The deletion of the 14 bp (gAACAGAACAAACA) of c.1153-1_1165del is likely due to non-allelic homologous recombination between the two GAACAAACA repeats located at the intron 6 and exon 7 boundary. (D) Domain structure of ETV6 (XP_011518909.1) based on Pfam annotation at http://www.ncbi.nlm.nih.gov/gene/2120 (PNT, pointed N-terminal domain; ETS, C-terminal DNA binding domain), with mutations identified in ETV6, already reported or identified in this study (top). The numbers of families carrying each mutation are in brackets. Mutations leading to skipping of exon 7 are boxed. (E) Structural modeling of the ETS domain with residues R369 (blue) and W380 (green) affected by the p.R369W and p.W380R mutations.
The two missense variants, c.1105C>T (p.R369W) and c.1138T>A (p.W380R) segregate in the affected family members and are not present in healthy relatives (Figure 1A). They are absent in public genomic databases, such as dbSNP (www.ncbi.nlm.nih.gov/SNP), 1000 genomes (www.1000genomes.org), and Exome Aggregation Consortium (www.exac.broadinstitute.org). Multiple-sequence alignment indicated that they affect highly conserved amino acid residues (data not shown). They are predicted to be deleterious for protein function according to different tools (Online Supplementary Table S1). Moreover, the CADD scores were 26.1 and 23.1 for p.R369W and p.W380R, respectively. Both mutations map in the E26 transformation-specific (ETS) domain which is in the C-terminal half of the protein (residues 337–421). Analysis of the coordinates of the ETS domain of ETV6 (2DAO) (numbered in pdb C8 and L112, so that R369 and W380 correspond to R39 and W50) showed that W380 is well buried in the hydrophobic core and surrounded by a number of hydrophobic residues, such as L341 and M394 (L11 and M64 in the structure) (Figure 1E). W380 is also close to the side chains of H383 and K384 (H53 and K54). Its substitution to an arginine will greatly destabilize the structure by creating both an uncompensated cavity in the hydrophobic core and electrostatic repulsion of nearby positively charged residues. Residue R369 is well exposed on the protein surface and is predicted to form an electrostatic interaction with the spatially nearby E361 (E31). Its substitution by a tryptophan could destabilize the fold by abolishing this interaction. Alternatively, this residue could be implicated in protein-protein interactions. In this case, its substitution by a much bulkier and uncharged residue could be deleterious.
The c.1153-1_1165del deletion variant removes the last “G” nucleotide of intron 6 and the first 13 nucleotides of exon 7. To investigate the effect of this deletion, we carried out reverse transcriptase polymerase chain reaction analysis on the three affected individuals of family F. Sequencing analysis of the altered 721 bp product showed skipping of exon 7 (r.1153_1253del/p.N385Vfs*7; Figure 1B) resulting in truncation of the ETS domain. Since the 721 bp band was fainter than the wild-type product (822 bp), we cannot exclude that the alternatively spliced mRNA was partially degraded. Inspection of the intron 6/exon 7 genomic boundary revealed repeats that are likely to be involved in non-allelic homologous recombination leading to micro-deletions/duplications (Figure 1C).
The seven families reported in Figure 1A formed our cohort of 20 affected individuals who have been studied to characterize the phenotype of ETV6-RT.
Clinical picture
A mild bleeding tendency was present in 12 patients, whereas eight subjects did not have any significant bleeding diathesis (Table 1). The more common bleeding symptoms were petechiae, ecchymoses, gum bleeding, epistaxis, and menorrhagia. Thrombocytopenia was discovered in adulthood in five patients, whereas it was identified at birth in six patients because of the family history of low platelet count. One patient (E/I-3) was initially misdiagnosed with immune thrombocytopenia, and received steroids and underwent splenectomy at the age of 9 years without this producing an increase in his platelet count. Ten patients had undergone 17 operations and six had had teeth extracted without excessive bleeding. Four women had given birth to six children, three vaginally and three by Cesarean section. Prophylactic platelet transfusion was deemed necessary to cover one vaginal delivery. Excessive bleeding (800 mL) was reported in another woman (patient F/I-2) who had given birth vaginally. We have no information on her platelet count or function at the time of the delivery; however, it is interesting to note that she had a defective platelet response to low doses of collagen and ADP when she was investigated at our institution (see below).
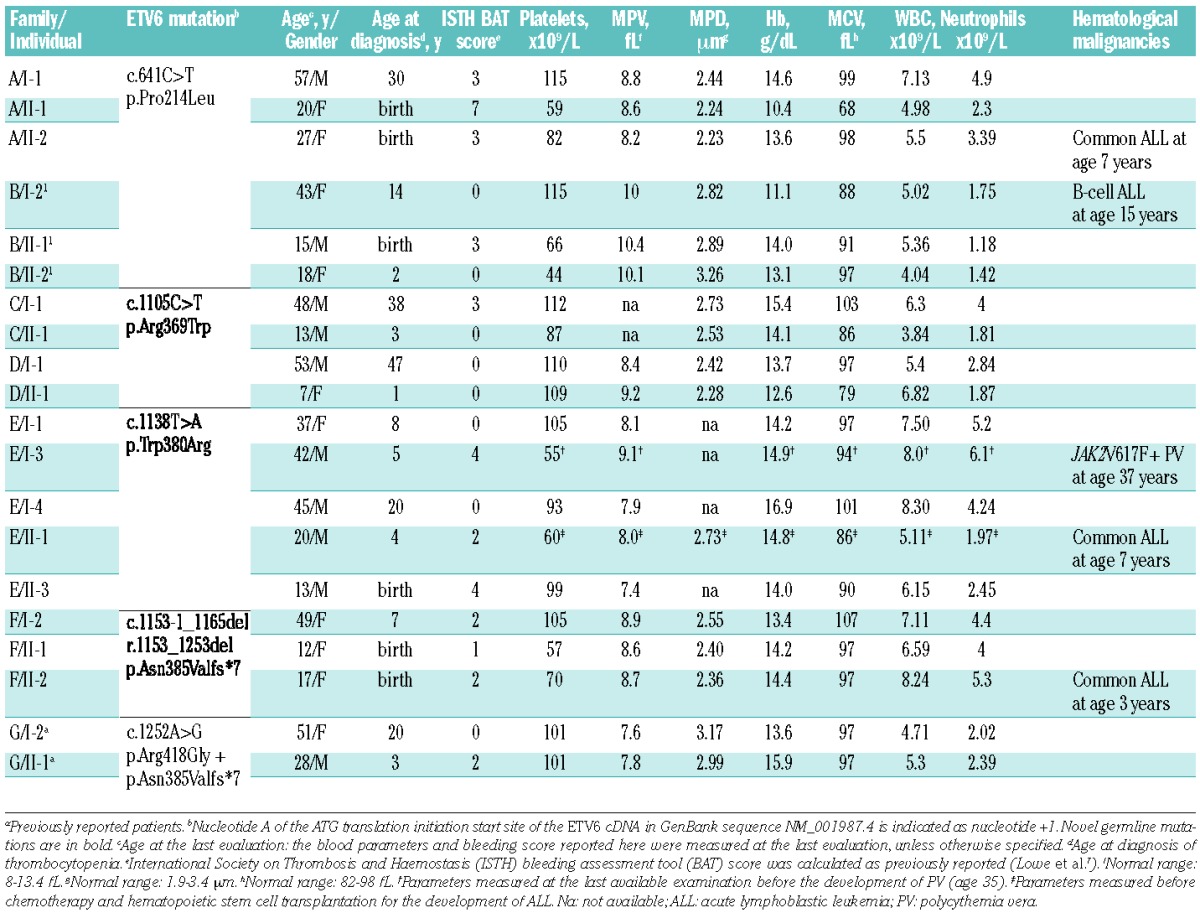
Table 1.
Main characteristics of the investigated patients.
Unilateral polydactyly was observed in one patient, mitral valve prolapse in two subjects, and renal ectopia in one. So, no recurrent extra-hematologic abnormalities have been identified.
Four patients from four families developed B-cell ALL during childhood (common ALL in three cases, not better defined in one). Conventional cytogenetic analysis resulted normal in three cases, while patient E/II-1 had hyperdiploid ALL; the search for the ETV6-AML1 transcript was performed in one patient (E/II-1) with normal findings. The incidence of ALL in our case series was 731.3 per 100,000 (95% CI, 274.5–1948.4), while it is 1.4 per 100,000 in the general population according to the National Cancer Institute.17 Three patients obtained remission after conventional chemotherapy, and one after hematopoietic stem cell transplantation from an unrelated donor. Patient E/I-3, who had a history of isolated thrombocytopenia since childhood (Table 1), at the age of 37 developed an increased hemoglobin level (19.0 g/dL, hematocrit 56%), with mild leukocytosis and thrombocytosis. The JAK2V617F mutation was identified and a diagnosis of polycythemia vera was made.
A history of non-hematologic neoplasms was present in three patients. Patient B/II-1 had breast fibroadenoma at 35 years old and meningioma at the age of 42. Patient G/I-2 had breast carcinoma at the age of 49, while patient F/II-2 developed breast fibroadenoma when she was 14 years old.
Blood cell counts and peripheral blood film examination
Table 1 reports the blood cell counts obtained at the last examination for 18 patients, and at the last available examination before the development of polycythemia vera and before hematopoietic stem cell transplantation for patients E/I-3 and E/II-1, respectively. Eleven patients had fewer than 100×109 platelets/L and only one fewer than 50×109/L. For most patients we had platelet counts measured at different ages prior to the evaluation for this study (Online Supplementary Table S2). There was some fluctuations in the patients’ platelet counts over time, but none of the patients showed a definite trend toward improvement or worsening of thrombocytopenia during their life.
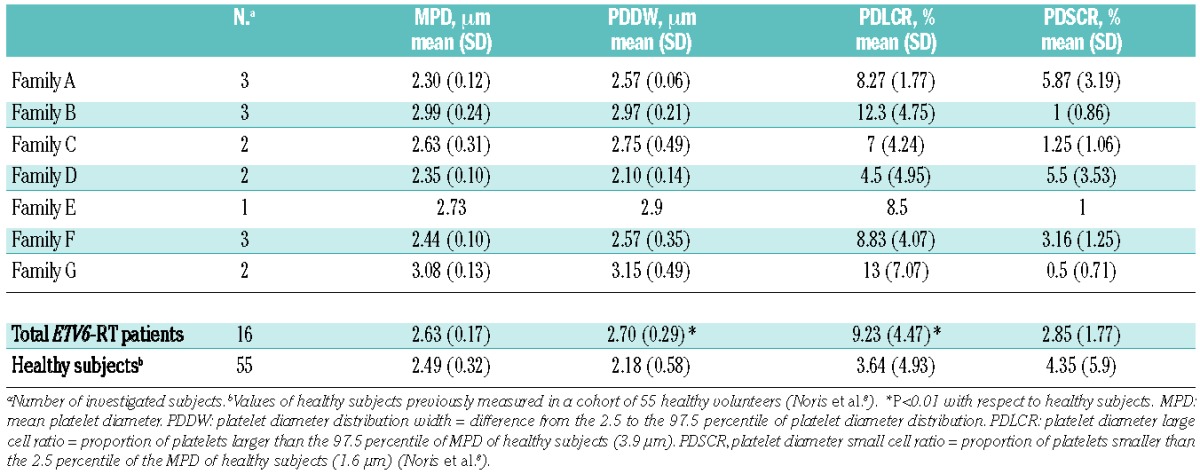
The mean platelet volume was slightly reduced in four cases and normal in the other 14 evaluable patients (Table 1). Peripheral blood film examination in 16 patients showed that mean platelet diameter was similar to that of healthy subjects, confirming that average platelet size is consistently normal in ETV6-RT patients (Table 2). We found very mild but significant increases in platelet diameter distribution width and platelet diameter large cell ratio, which indicate that a mild platelet anisocytosis and a slightly increased proportion of large platelets were frequent features of the investigated patients. In agreement with previous findings,8 empirical measurement of the percentage of platelets larger than half an erythrocyte gave similar results to the assessment of platelet diameter large cell ratio by image analysis (data not shown). Conversely, the increased mean platelet diameter distribution width detected by image analysis did not correspond to increased mean platelet distribution width values obtained by automated cell counts (data not shown).
Table 2.
Parameters of platelet diameters measured on peripheral blood films in investigated patients.
Mild anemia was observed in one patient with iron deficiency (A/II-1). Mean corpuscular volume was reduced in this subject, increased without any apparent cause in five subjects, and within the normal range in the remaining patients. White blood cell count was normal in all the cases.
In vitro platelet studies
Platelet aggregation
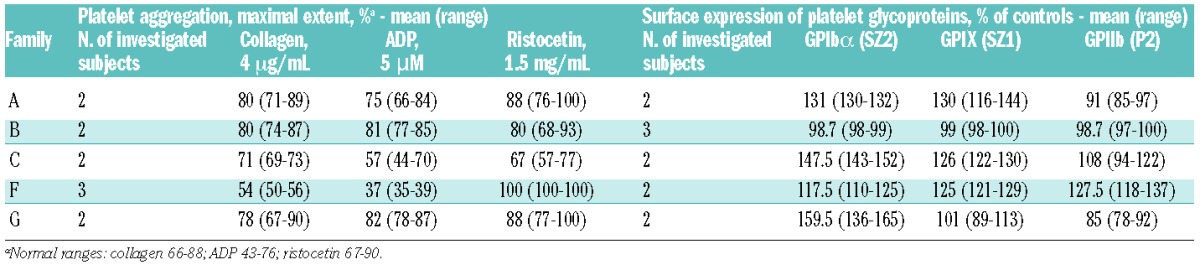
Among the 11 investigated patients, the three patients from family F had mildly reduced platelet aggregation after stimulation with collagen 4 μg/mL and ADP 5 μM, while individual C/II-1 showed a slightly reduced response to ristocetin 1.5 mg/mL (Table 3). However, all patients had completely normal responses to higher concentrations of these agonists (collagen 20 μg/mL, ADP 20 μM, ristocetin 3 mg/mL, data not shown), indicating that, if present, the aggregation defects were mild.
Table 3.
In vitro platelet aggregation and surface expression of major platelet glycoproteins in investigated patients.
Platelet flow cytometry
As shown in Table 3, flow cytometry performed in 11 patients did not identify any consistent defect of the major glycoproteins of the platelet surface.
Platelet activation
Overall, the surface expression of activated GPIIb-IIIa and P-selectin and the reduction of GPIbα upon stimulation of platelets with ADP or TRAP, were not significantly different in 11 ETV6-RT patients with respect to those in controls (Online Supplementary Figure S1). A mild reduction of activated GPIIb-IIIa expression after stimulation with TRAP (52% to 65% of the expression in controls) was observed in three patients.
Platelet adhesion and spreading
In vitro adhesion of platelets from seven patients to subendothelium components of the extracellular matrix was not different from that of controls. However, the ability of ETV6-RT platelets to spread on fibrinogen was consistently and significantly reduced, while spreading on collagen and von Willebrand factor was normal (Table 4).
Table 4.
In vitro platelet interaction with subendothelium molecules in seven ETV6-RT patients.
In vitro culture of megakaryocytes and assessment of proplatelet formation
Megakarocytes from eight patients and eight healthy subjects were cultured in vitro. After 14 days of culture, expression levels of the major megakaryocyte differentiation surface markers (GPIIIa, GPIIb and GPIbα) were similar to those of healthy controls (Figure 2A,B). Conversely, megakaryocyte ploidy was significantly lower in patients than in controls (Figure 2C), and this was paralleled by differences in megakaryocyte diameters (Figure 2D). The analysis of proplatelet formation revealed that, compared to megakaryocytes from controls, megakaryocytes from patients had elongated proplatelet shafts of shorter length and with decreased number of branches. Furthermore, the percentage of proplatelet-forming megakaryocytes was significantly reduced in patients. In contrast, the size of proplatelet tips was similar in patients and in controls (data not shown). Similar results were obtained with megakaryocytes in suspension (Figure 3A,B) and following adhesion on fibrinogen (Figure 3C,D).
Figure 2.
Normal differentiation but decreased ploidy of ETV6-RT megakaryocytes. Hematopoietic progenitors from peripheral blood samples of healthy controls (CTRL) and patients (ETV6-RT) were differentiated in vitro into megakaryocytes in the presence of thrombopoietin, interleukin-6 and interleukin-11. (A) Representative immunofluorescence staining of plasma membrane GPIIIa in CTRL and ETV6-RT megakaryocytes (red=GPIIIa; blue=nuclei; scale bar=20 μm). (B) Flow cytometry analysis of GPIIb and GPIbα expression revealed comparable percentages of double-stained populations in CTRL and ETV6-RT at the end of the culture. (C) Ploidy of megakaryocytes at the end of the culture was significantly reduced in cells from ETV6-RT patients (*P<0.05). (D) Diameters of megakaryocytes were also significantly lower in ETV6-RT patients (total number of cells analyzed: 1,100, *P<0.01).
Figure 3.
Aberrant proplatelet formation by ETV6-RT megakaryocytes. (A) Representative light microscopy analysis of proplatelet formation and structure from control (CTRL, i) and patient (ETV6-RT, ii–v) megakaryocytes cultured for 16 h in suspension (scale bar=50 μm). (B) The percentage of proplatelet-forming megakaryocytes was calculated as the number of megakaryocytes displaying at least one filamentous pseudopod with respect to total number of round megakaryocytes per analyzed field (*P<0.01). (C) Representative fluorescence microscopy analysis of proplatelet formation and structure from CTRL (i–ii) and ETV6-RT (iii–vi) megakaryocytes cultured for 16 h with adhesion on fibrinogen. The pictures clearly show defective proplatelet elongation in ETV6-RT (red=β1-tubulin; blue=nuclei; scale bar=30 μm). (D) The percentage of proplatelet-forming megakaryocytes was calculated as the number of β1-tubulin-positive cells displaying at least one pseudopod with respect to total number of round megakaryocytes per analyzed field (*P<0.01).
Discussion
Here we report the molecular and phenotypic characterization of seven families with germline mutations in ETV6. In addition to the variants previously reported,4 we identified three novel alterations, which are likely to be pathogenic. The two novel missense variants (p.R369W and p.W380R) segregated within the families, are absent in public genomic databases, and are expected to be deleterious for protein function according to bioinformatic tools and analysis of protein conformational structure. ETV6 is a modular protein which contains a PNT and an ETS domain sandwiched between regions of potential intrinsically unstructured nature. Both p.R369W and p.W380R affect the ETS domain, a conserved region that interacts directly with DNA consensus sequences. We have shown that the role of W380 is structural, it being surrounded by hydrophobic residues in the domain hydrophobic core. Its substitution by an arginine will therefore severely destabilize the domain structure. Residue R369 is involved in an electrostatic interaction and possibly in protein-protein interactions.
It is important to note that the somatic p.R369W has previously been associated with chronic myelomonocytic leukemia, colorectal cancer, and childhood leukemia.5,6 Moreover, Zhang et al. previously reported one ETV6-RT pedigree carrying a different germline missense variant affecting the same residue (p.R369Q). Similarly to our patients with the p.R369W, the subjects with p.R369Q had mild thrombocytopenia and normal platelet morphology.6 Among the eight members of the p.R369Q pedigree, one had chronic myelomonocytic leukemia at the age of 82 and one had colorectal cancer at the age of 43,6 whereas we did not observe neoplasms in our p.R369W patients at a median age at evaluation of 30 years. Finally, both p.R369Q and p.R369W have been associated with genetic predisposition to childhood ALL.3 These observations suggest that arginine 369 is a mutational hot spot.
With regards to the c.1153-1_1165del variant, reverse transcriptase polymerase chain reaction analysis demonstrated that it affects the splicing process leading to skipping of exon 7 (p.N385Vfs*7). The same alternative splicing was also caused by the c.1153-5_1153-1del mutation,5 which, as c.1153-1_1165del, is likely to derive from non-allelic homologous recombination between repetitive sequences present at the intron 6/exon 7 boundary. The skipping of exon 7 is also determined by the c.1252A>G substitution.4 Affecting the second to last nucleotide of exon 7, this allele is associated with both correctly (p.R418G) and alternatively (p.N385Vfs*7) spliced mRNA.4
Of the ten different mutant forms identified so far in ETV6-RT families, p.P214L is the only one that does not affect the ETS domain (Figure 1D), but instead alters a less conserved central domain that interacts with several transcription repressors further controlling expression of the target genes. Unlike the other germline mutations, which are mainly private, this substitution was responsible for ETV6-RT in four of the 14 families characterized so far, indicating that it represents another potential mutational hot spot.
ETV6 is a transcriptional repressor involved in embryonic development and hematopoietic regulation.18 In particular, animal studies suggested that ETV6 has two independent roles in mouse hematopoiesis: on the one hand it is required for survival of hematopoietic stem cells, on the other it promotes the late phases of megakaryopoiesis. Interest in ETV6 increased greatly at the end of the last century after demonstration that its deregulation due to rearrangements, fusions or deletions is involved in hematologic malignancies.19,20 Moreover, somatic mutations in ETV6 were recently found in a variety of hematologic neoplasm, including AML, T- and B-cell ALL, mixed-phenotype acute leukemia, myelodysplastic syndromes, chronic lymphocytic leukemia and chronic myelogenous leukemia.21 Even more recently, targeted sequencing of ETV6 in 4405 childhood ALL cases identified 31 germline variants potentially related to leukemia in 35 cases.3 Based on this evidence, it is not surprising that the four studies that identified ETV6-RT in 41 subjects from nine families found that 16 patients (39%) had hematologic malignancies, with 12 patients (29%) developing ALL.3–6 Of note, 11 of the 12 subjects with ALL were children. The other blood neoplasms observed in ETV6-RT patients were mixed-phenotype acute leukemia, multiple myeloma, myelodysplastic syndromes and chronic myelomonocytic leukemia.
We found that four of 20 consecutive patients with ETV6-RT (20%) developed ALL during childhood, thus confirming that early leukemic transformation is a major risk in these patients. Moreover, we observed that one patient developed JAK2-positive polycythemia vera at the age of 37, supporting the previous hypothesis that ETV6-RT predisposes not only to ALL, but also to other blood neoplasms. The frequency of hematologic malignancies is lower in our study than in the previous ones (25% versus 39%). This is explained by the fact that, in the previous investigations,3–6 the occurrence of hematologic malignancies was one of the criteria for the recruitment of patients, while we examined a series of consecutive, unselected patients with inherited thrombocytopenia of unknown origin. This approach appears more suitable for providing a reliable estimation of the incidence of hematologic neoplasms among ETV6-RT patients. Of course, the analysis of a larger series of patients is needed to confirm our figure.
Similarly to this study, we previously searched a large series of unselected patients for ANKRD26 mutations and discovered that ten of 118 (8%) subjects with ANKRD26-RT had developed myeloid malignancies.22 Thus, hematologic malignancies seem much more frequent in ETV6-RT than in ANKRD26-RT. The risk of malignancies appears even higher in FPD/AML, since over 40% of such patients had myeloid neoplasms.23 However, as discussed for ETV6-RT, the RUNX1 mutational screening was also generally performed in pedigrees with hematologic malignancies,24 and it is therefore likely that the incidence of transformation has been overestimated. However, each patient with an inherited thrombocytopenia caused by mutations in ETV6, RUNX1 or ANKRD26 has a relevant risk of hematologic malignancies, and recognizing these patients is important not only to provide effective genetic counseling and appropriate follow-up, but also to give appropriate treatment to patients who develop blood neoplasms and need hematopoietic stem cell transplantation. In fact, as shown in different disorders predisposing to myeloid malignancies,25 the use of an affected family member as the donor would entail the risk of developing malignancies once again.
ETV6-RT is a relatively frequent form of inherited thrombocytopenia. In fact, in our series of 274 consecutive propositi, ETV6-RT was identified in seven families and had, therefore, a relative prevalence of 2.6% in the whole case series, and of 4.6% in the series of probands with known inherited thrombocytopenia (7/151). In our cohort, the frequency of ETV6-RT was lower only to that of monoallelic Bernard-Soulier syndrome (12.2% in the whole series), MYH9-related disease (11.4%), ANKRD26-RT (9.4%), and biallelic Bernard-Soulier syndrome (5.7%). Since most of our patients with monoallelic Bernard-Soulier syndrome had the Ala156Val mutation of GPIbα (Bolzano mutation), which is exclusive to the Italian population,26 it is expected that the relative frequency of ETV6-RT is even higher in other countries.
Our study did not identify any peculiar feature that can be used to raise the suspicion of ETV6-RT from the routine diagnostic workup and the diagnosis does, therefore, remain difficult. A previous investigation, which reported five patients who have been re-evaluated in this study, suggested that red blood cell macrocytosis is a feature of the ETV6-RT phenotype.4 In that investigation, the percentage of patients with increased mean corpuscular volume was 40%,4 whereas it was 25% in the present study. With regards to the five patients reported in both studies, red blood cell macrocytosis was found in two individuals in the previous examination but was not confirmed in the present evaluation. Of note, the absolute mean corpuscular volumes were similar in the two studies (mean 94.5 fL with SD 3.8 versus mean 93.3 fL with SD 8.9) and the discrepancy in the percentage of patients with red cell macrocytosis resulted from the different upper limits of normal range used in the two investigations (95 fL in the previous study and 98 fL in the present one, according to the normal ranges of the different laboratories). On the whole, these findings indicate that red blood cell macrocytosis is present in a minority of patients with ETV6-RT, and suggest that it may be inconstantly found in the same patients over time. Thus, red cell macrocytosis seems to have limited diagnostic value for recognizing this condition. Moreover, we did not identify any distinguishing defect of major platelet glycoproteins or in vitro platelet aggregation and evaluation of peripheral blood films did not reveal any morphological abnormalities, except for mild platelet anisocytosis. However, at variance with most inherited thrombocytopenias, mean platelet diameter and mean platelet volume were consistently normal in ETV6-RT, and it is precisely the normal size of platelets that should raise suspicion of this condition in subjects with an autosomal dominant thrombocytopenia. The other dominant inherited thrombocytopenias with this feature are FDP/AML, ANKRD26-RT, and CYCS-RT. Of note, CYCS-RT is a very rare condition described so far in only two pedigrees,1 whereas the other two disorders are more frequent and, like ETV6-RT, predispose to hematologic malignancies. Thus, we suggest that all subjects with a dominant inherited thrombocytopenia and normal platelet size should be tested for mutations in ETV6, RUNX1, and ANKRD26, in order to identify one of these predisposition syndromes.
The psychological impact of receiving a diagnosis of ETV6-RT, as well as of FPD-AML or ANKRD26-RT, should be carefully considered by physicians. We suggest that all patients are correctly informed, before undergoing diagnostic workup for thrombocytopenia of suspected genetic origin, about the possibility of receiving a diagnosis that implicates the risk of malignancies, and have the chance to state in advance whether they want to receive information about the risk of neoplasms, for themselves as well as for their progeny.
In this study, the in vitro megakaryopoiesis of ETV6-RT patients was investigated for the first time. We showed that ETV6 pathogenic variants impair megakaryocyte maturation, as demonstrated by the production of smaller megakaryocytes with decreased ploidy. The ability of these immature megakaryocytes to extend fully developed proplatelets was impaired, providing an explanation for the patients’ thrombocytopenia. These findings seem consistent with the results of studies in mice, which suggested a role for ETV6 in terminal megakaryocyte maturation,18 and with the findings obtained with megakaryocyte differentiated from human CD34+ cells transduced with some ETV6 variants.4 We also had the possibility to study platelet function in detail in a substantial number of patients. Although we did not identify any consistent defect of in vitro platelet aggregation, activation or adhesion, we found that the ability of platelets to spread on fibrinogen was reduced in all the investigated patients. As the platelet expression of GPIIb-IIIa was normal, this finding suggests that mutations in the ETV6 transcription factor alter the expression of one or more proteins involved in the GPIIb-IIIa-mediated platelet outside-in signaling after interaction with fibrinogen. Moreover, this defect could contribute to the bleeding diathesis observed in some ETV6-RT individuals. In fact, although the degree of bleeding was always mild, the proportion of patients with spontaneous bleeding (60%) appeared globally high with respect to the very mild degree of thrombocytopenia.
In conclusion, our study showed that monoallelic ETV6 mutations cause a relatively frequent form of inherited thrombocytopenia and confirmed that affected subjects have a mild bleeding tendency but propensity to hematologic malignancies, in particular ALL. Since ETV6-RT is one of the few autosomal dominant forms of inherited thrombocytopenia without platelet macrocytosis, screening for ETV6 mutations is recommended in all patients with these characteristics.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Carmine Tinelli for his contribution to the statistical analysis, Prof. Federica Meloni for technical assistance with the flow cytometry analysis, Prof. Joseph Italiano for providing β1-tubulin antibody, and Prof. Enrica Tira for providing purified type I collagen.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/11/1333
Funding
This study was supported by the ERA-Net for Research Program on Rare Diseases (EUPLANE), Telethon Foundation (grant GGP13082), Cariplo Foundation (2012-0529), Italian Ministry of Health (RF-2010-2309222), the Ministry of Education, Youth and Sports of the Czech Republic under the project CEITEC 2020 (LQ1601), and Czech Ministry of Health (grant AZV 16-29447A). MF receives a fellowship from the Associazione Italiana per la Ricerca sul Cancro (n. 18024/16).
References
- 1.Pecci A. Diagnosis and treatment of inherited thrombocytopenias. Clin Genet. 2016;89(2):141–153. [DOI] [PubMed] [Google Scholar]
- 2.Savoia A. Molecular basis of inherited thrombocytopenias. Clin Genet. 2016:89(2): 154–162. [DOI] [PubMed] [Google Scholar]
- 3.Moriyama T, Metzger ML, Wu G, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 2015;16(16):1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noetzli L, Lo RW, Lee-Sherick AB, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015;47(5):535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topka S, Vijai J, Walsh MF, et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PLoS Genet. 2015;11(6): e1005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowe GC, Lordkipanidzé M, Watson SP. Utility of the ISTH bleeding assessment tool in predicting platelet defects in participants with suspected inherited platelet function disorders. J Thromb Haemost. 2013;11(9):1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noris P, Biino G, Pecci A, et al. Platelet diameters in inherited thrombocytopenias: analysis of 376 patients with all known disorders. Blood. 2014;124(6):e4–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noris P, Guidetti GF, Conti V, et al. Autosomal dominant thrombocytopenias with reduced expression of glycoprotein Ia. Thromb Haemost. 2006;95(3):483–489. [DOI] [PubMed] [Google Scholar]
- 10.Psaila B, Bussel JB, Linden MD, et al. In vivo effects of eltrombopag on platelet function in immune thrombocytopenia: no evidence of platelet activation. Blood. 2012;119(17):4066–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pecci A, Bozzi V, Panza E, et al. Mutations responsible for MYH9-related thrombocytopenia impair SDF-1-driven migration of megakaryoblastic cells. Thromb Haemost. 2011;106(4):693–704. [DOI] [PubMed] [Google Scholar]
- 12.Canobbio I, Catricalà S, Di Pasqua LG, et al. Immobilized amyloid Aβ peptides support platelet adhesion and activation. FEBS Lett. 2013; 587(16):2606–2611. [DOI] [PubMed] [Google Scholar]
- 13.Pecci A, Malara A, Badalucco S, et al. Megakaryocytes of patients with MYH9-related thrombocytopenia present an altered proplatelet formation. Thromb Haemost. 2009;102(1):90–96. [DOI] [PubMed] [Google Scholar]
- 14.Bluteau D, Balduini A, Balayn N, et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J Clin Invest. 2014;124(2):580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balduini A, Di Buduo CA, Malara A, et al. Constitutively released adenosine diphosphate regulates proplatelet formation by human megakaryocytes. Haematologica. 2012;97(11):1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Buduo CA, Moccia F, Battiston M, et al. The importance of calcium in the regulation of megakaryocyte function. Haematologica. 2014;99(4):769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.National Cancer Institute. Cancer statistics. Available at: www.seer.cancer.gov/statistics/ Accessed December 20, 2015.
- 18.Hock H, Meade E, Medeiros S, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 2004;18(19):2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohlander SK. ETV6: a versatile player in leukemogenesis. Semin Cancer Biol. 2005;15(3):162–174. [DOI] [PubMed] [Google Scholar]
- 20.De Braekeleer E, Douet-Guilbert N, Morel F, Le Bris MJ, Basinko A, De Braekeleer M. ETV6 fusion genes in hematological malignancies: a review. Leuk Res. 2012;36(8):945–961. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Dong S, Yao H, et al. ETV6 mutation in a cohort of 970 patients with hematologic malignancies. Haematologica. 2014;99(10):e176–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noris P, Favier R, Alessi MC, et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood. 2013;122(11):1987–1989. [DOI] [PubMed] [Google Scholar]
- 23.Liew E, Owen C. Familial myelodysplastic syndromes: a review of the literature. Haematologica. 2011;96(10):1536–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Owen CJ, Toze CL, Koochin A, et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood. 2008;112(12):4639–4645. [DOI] [PubMed] [Google Scholar]
- 25.Churpek JE, Artz A, Bishop M, Liu H, Godley LA. Correspondence regarding the consensus statement from the Worldwide Network for Blood and Marrow Transplantation Standing Committee on Donor Issues. Biol Blood Marrow Transplant. 2016;22(1):183–184. [DOI] [PubMed] [Google Scholar]
- 26.Noris P, Perrotta S, Bottega R, et al. Clinical and laboratory features of 103 patients from 42 Italian families with inherited thrombocytopenia derived from the monoallelic Ala156Val mutation of GPIbα (Bolzano mutation). Haematologica. 2012; 97(1):82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.