

The epidermal growth factor receptor (EGFR) inhibitor erlotinib has demonstrated significant EGFR-independent activity against acute myeloid leukemia (AML) cell lines and primary AML blasts in preclinical studies; however, these findings have not been reproduced in clinical trials.1,2 Combining erlotinib with other antineoplastic agents has been proposed as a strategy for improving the clinical activity of erlotinib in AML.1,2 With the goal of identifying erlotinib combination candidates, we screened erlotinib against several chemical libraries in the erlotinib-insensitive AML cell lines TEX and OCI-AML2, identifying the poly(ADP-ribose) glycohydrolase (PARG) inhibitor ethacridine lactate as the top synergistic hit common to both cell lines. The erlotinib-ethacridine combination synergized to induce lethal levels of reactive oxygen species (ROS), resulting from erlotinib-mediated potentiation of intracellular ethacridine accumulation. Thus, we have identified that erlotinib promotes the accumulation of select drugs, thereby leading to synergism. In addition, the potential anti-AML activity of PARG inhibitors warrants further study.
Erlotinib has well-documented preclinical activity against AML cells, where it induces differentiation,3 cell cycle arrest,3–5 and apoptosis,3,6 yet EGFR expression in these cells is absent.3,7 To evaluate erlotinib sensitivity in the AML cell lines TEX and OCI-AML2, we subjected these cells to treatment with increasing concentrations of erlotinib for 72 hours, and subsequently measured relative growth and viability using the sulforhodamine-B (SRB) assay. The average erlotinib IC50s were 8.99 μM and 15.61 μM in TEX and OCI-AML2, respectively (Online Supplementary Figure S1). These erlotinib IC50 values were significantly higher than clinically achievable concentrations,8 and far greater than the IC50 of the EGFR-mutant NSCLC cell line HCC827 determined using the Cell Titer Fluor growth and viability assay (Online Supplementary Figure S1).
Given the relative insensitivity of TEX and OCI-AML2 cells to erlotinib, we sought to identify compounds that sensitize these cells to erlotinib-induced killing. We therefore screened this drug against three chemical libraries—the MicroSource Discovery Systems International Drug and Natural Product (“NatProd”) collections, and a 312-compound library from Sequoia Research Products—in TEX and OCI-AML2 cells. Erlotinib at its IC10 and IC25 was combined with increasing concentrations of 1,352 drugs for a total of 16,230 different assays in the two cell lines. Treated cells were incubated for 72 hours and relative growth and viability was measured with the SRB assay. Synergy was calculated based on the excess over Bliss additivism (EOBA) formula, as previously described,9 and compounds were plotted in order of increasing positive EOBA scores for each drug library (Figure 1A). Ethacridine lactate, a PARG inhibitor9 and abortifacient,10 was validated as the top synergistic hit common to both TEX and OCI-AML2 cells, generating EOBA scores of up to 0.79 in TEX and 0.69 in OCI-AML2 (Figure 1B).
Figure 1.
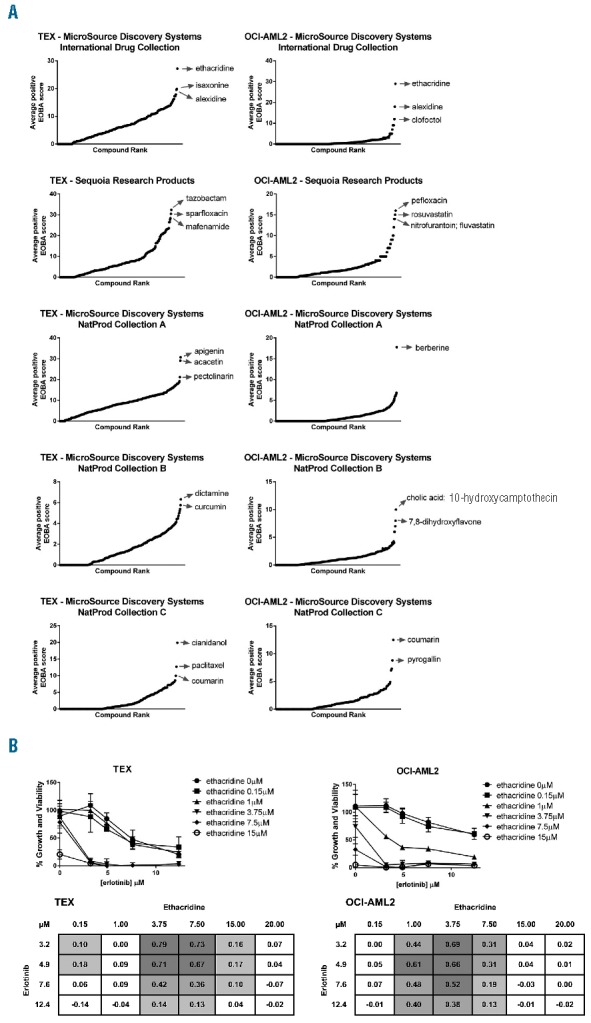
Identification of erlotinib sensitizers in TEX and OCI-AML2 cells. (A) Erlotinib was screened against 1,352 drugs from three chemical libraries (Sequoia, International, and Natural Products A, B and C). Following a 72h incubation, EOBA synergy scores were calculated from relative growth and viability values determined by the SRB assay. Compounds were ranked in order of increasing positive EOBA score. (B) Top synergistic hit ethacridine lactate was validated in TEX and OCI-AML2 cells using broader concentration ranges. Cells were combination-treated for 72h and percent growth and viability was measured with the SRB assay (graphs). Synergy was calculated according to EOBA criteria (tables), with EOBA values >0.1 (lightest grey) denoting a synergistic combination, and values >0.5 (darkest grey) denoting a profoundly synergistic combination. Results depict mean percent growth and viability ± SD (graphs) or mean EOBA scores (tables) from a single experiment performed in triplicate.
Further validation of the erlotinib-ethacridine combination revealed profound synergistic cytotoxicity in the AML cell lines U937 and K562 at clinically relevant erlotinib concentrations (Figure 2A). This combination also synergized in all 5 primary AML samples evaluated (Figure 2B), reaching EOBA scores in the >0.30 range in AML130208 and AML130237 (Online Supplementary Figure S2, see Online Supplementary Table S1 for patient characteristics). In contrast, the erlotinib-ethacridine combination was not synergistic in either of the normal hematopoietic cell samples derived from healthy donors of GCSF-mobilized peripheral blood stem cells (Figure 2B, Online Supplementary Figure S2). Evaluation of the erlotinib-ethacridine combination in an in vivo model of AML where SCID mice were subcutaneously injected with OCI-AML2 cells demonstrated reduced tumor growth (****P<0.0001) (Figure 2C), with minimal change in bodyweight (Figure 2D).
Figure 2.
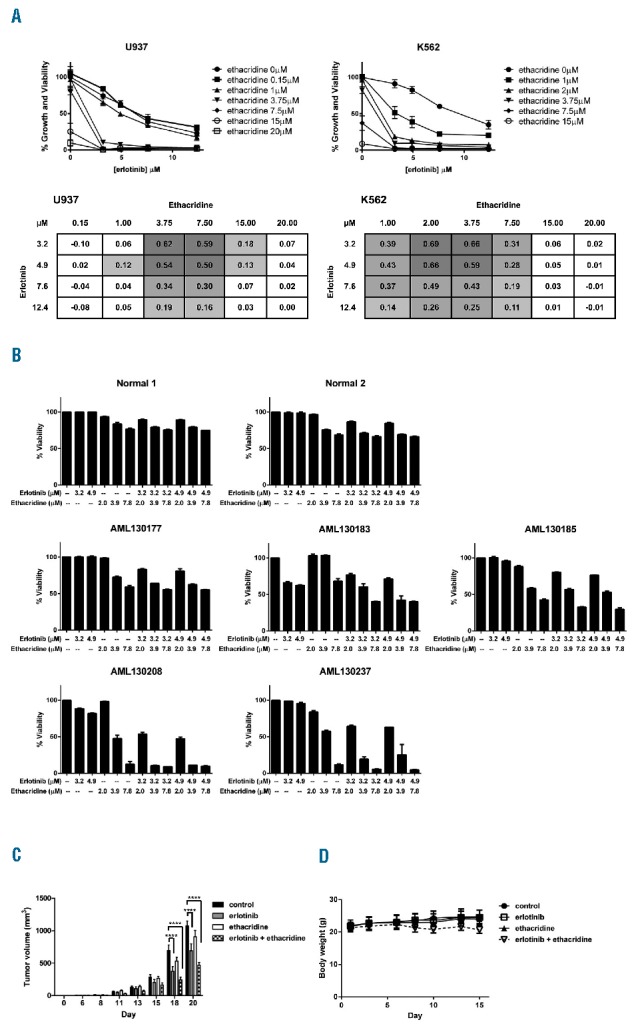
Preclinical evaluation of the erlotinib-ethacridine combination. (A) U937 and K562 cells were combination-treated with erlotinib and ethacridine for 72h; percent growth and viability was determined by the SRB assay. Graphs depict mean percent growth and viability ± SD from a single experiment performed in triplicate. Tables represent mean EOBA values from the same experiment. (B) Primary AML blasts and peripheral blood stem cells from GCSF-treated stem cell donors were collected from consenting patients, with the approval of the University Health Network (Toronto, Canada) institutional review board. Samples were combination-treated with erlotinib and ethacridine for 48h; viability was determined by Annexin V and PI staining. Graphs depict mean percent viability ± SD from a single experiment performed in triplicate. (C) 1×106 OCI-AML2 cells were subcutaneously injected into SCID mice. Starting on day 6 post-injection, mice were treated by intraperitoneal injection with 75mg/kg of erlotinib, 20mg/kg ethacridine, both in combination, or vehicle control (10% DMSO, 10% Cremophor, 80% 0.9% NaCl) daily, 5×/week for two weeks (n = 10/group). Tumor volume (C) and body weight (D) were assessed on the indicated days, and are depicted as mean ± SEM (C) or mean ± SD (D). ****P<0.0001, from a two-way ANOVA with Bonferroni posttest comparing all treatment groups at day 18 and 20.
Having validated this synergistic combination in preclinical AML models, we sought to characterize the mechanism responsible for its lethality. We quantified intracellular reactive oxygen species (ROS) production in TEX and OCI-AML2 cells following a 24-hour incubation with erlotinib, ethacridine, or both drugs in combination. ROS was measured using carboxy-H2DCFDA (FITC) staining on flow cytometry, and the absence of ethacridine autofluorescence in this channel was confirmed (Online Supplementary Figure S3). We observed a striking increase in ROS production: up to a 2-fold increase in TEX and a 4-fold increase in OCI-AML2 cells, respectively (Figure 3A). ROS induction appeared functionally important for combination-induced synergistic cytotoxicity, as pre-treatment with the antioxidant α-tocopherol significantly increased viability as shown by Annexin V and PI staining following a 48-hour treatment with the erlotinib-ethacridine combination (Figure 3B). Interestingly, the source of ROS was likely not mitochondrial in origin, as MitoSOX staining was not increased following combination erlotinib-ethacridine treatment of TEX and OCI-AML2 cells (Online Supplementary Figure S4).
Figure 3.
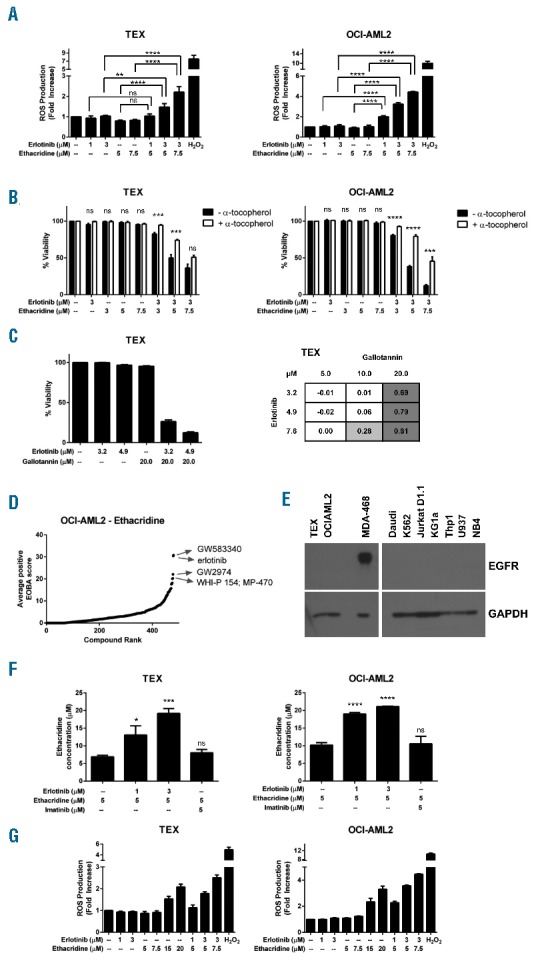
Mechanistic evaluation of the erlotinib-ethacridine combination. (A) TEX and OCI-AML2 cells were combination-treated with erlotinib and ethacridine for 24h. Intracellular ROS was measured with carboxy-H2DCFDA staining and dead cells were excluded by PI staining. Fold increase in ROS production was calculated relative to the geometric mean fluorescence intensity (GMFI) of vehicle-treated cells. H2O2 was included as a positive intracellular ROS control. Results depict mean fold increase in GMFI ± SD from a representative experiment performed in triplicate. Data are representative of two independent experiments. (B) TEX and OCI-AML2 cells were treated with 3mM α-tocopherol for 24h, then treated with the erlotinib-ethacridine combination for the following 48h (with α-tocopherol maintained at 2.4mM). Viability was measured with Annexin V and PI staining on flow cytometry, and calculated relative to respective (± α-tocopherol) controls. Data represent mean percent viability ± SD from an experiment performed in triplicate. These data are representative of two independent experiments. (C) TEX cells were combination-treated with erlotinib and gallotannin for 48h. Viability was determined by Annexin V and PI staining. Graph depicts mean percent viability ± SD from a single experiment performed in triplicate. Table represents mean EOBA values from the same experiment. Data are representative of at least three independent experiments. (D) Ethacridine was screened against a 480-compound kinase inhibitor library in OCI-AML2 cells. Cells were treated for 72h. Growth and viability was measured with the SRB assay and synergy scores were calculated with the EOBA formula. Compounds were ranked in order of increasing positive synergy score. (E) EGFR expression in a panel of AML cell lines was detected by immunoblot, with the MDA-468 cell line included as a positive control. (F) TEX and OCI-AML2 cells were treated with ethacridine in the presence and absence of erlotinib for 1h. Cells were lysed and analyzed by mass spectrometry. Imatinib was included as a negative (non-synergizing) control. Data depict mean ethacridine accumulation ± SD from an experiment performed in triplicate. Data are representative of three (TEX) or two (OCI-AML2) independent experiments. (G) TEX and OCI-AML2 cells were treated with high-dose ethacridine or erlotinib and ethacridine in combination for 24h. Intracellular ROS was measured with carboxy-H2DCFDA staining (with PI exclusion of dead cells) and calculated relative to GMFI of vehicle-treated cells. Data depict mean fold increase in GMFI ± SD from a representative experiment performed in triplicate, and are representative of two independent experiments. In all panels, *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; ns=not significant, as determined by one-way ANOVA (A, F) or unpaired Student’s t test (B), with Sidak (A), Holm-Sidak (B), or Dunnett’s (F) tests for multiple comparisons.
Given that ethacridine is a known PARG inhibitor, we investigated whether PARG inhibition by ethacridine might explain its synergy with erlotinib. PARG hydrolyzes poly(ADP-ribose) (PAR) polymers, which are synthesized by poly(ADP-ribose) polymerases (PARPs) in response to DNA damage and other cellular stresses. PARG-deficient cells have increased reported sensitivity to genotoxic and oxidative stress-inducing agents by failing to reduce cellular PAR levels following PARP activation.11 Excessive PAR accumulation, due to either PARP1 activation or PARG inhibition, can induce cell death by parthanatos.12 We noted that erlotinib synergized with gallotannin, another chemical PARG inhibitor (Figure 3C). Thus, PARG inhibition is a possible target of ethacridine explaining its synergy with erlotinib, and further evaluation of this target in AML cells is warranted.
We next sought to identify the target(s) of erlotinib responsible for its synergy with ethacridine. Given that several kinase targets, such as SRC family kinases, BTK, SYK, and JAK2,3,5,13 have been proposed or reported to account for erlotinib-mediated anti-AML activity, we hypothesized that erlotinib was inhibiting one or more kinases to synergize with ethacridine. We therefore screened ethacridine against an in-house kinase inhibitor library, comprising 480 kinase inhibitors with a broad range of kinase targets and varying degrees of kinase specificity. OCI-AML2 cells were incubated with ethacridine along with two different concentrations of each kinase inhibitor for 72 hours, for a total of 2,883 different assays. Relative growth and viability were subsequently measured with the SRB assay and synergy scores were again calculated with the EOBA formula and plotted in order of increasing synergy score (Figure 3D, see Online Supplementary Table S2 for screen data). Erlotinib was the second most synergistic hit, which served as further validation for our initial combination screen. Interestingly, 4 of the 5 top synergistic hits (GW583340, erlotinib, GW2974, and WHI-P 154) were reported EGFR inhibitors. Furthermore, the clinically approved EGFR inhibitors gefitinib and lapatinib were also identified as synergistic hits (Online Supplementary Table S2).
The observed synergy between ethacridine and multiple EGFR inhibitors prompted us to investigate whether erlotinib might inhibit EGFR to synergize with ethacridine in TEX and OCI-AML2 cells. We evaluated total EGFR expression in these cell lines by immunoblot, and were unable to detect expression of this kinase in either cell line (Figure 3E). Likewise, EGFR expression was absent in K562 and U937 cells, which had also demonstrated erlotinib-ethacridine synergy (Figure 2A). Thus, these chemical EGFR inhibitors likely synergize with ethacridine via a common, EGFR-independent mechanism.
One previously reported effect of erlotinib (and other small molecule EGFR inhibitors) is its capacity to inhibit ATP-binding cassette (ABC) transporter efflux activity, and thus enhance the cellular accumulation of, and sensitivity to, ABC transporter substrates, which include drugs.14 We therefore investigated whether erlotinib potentiates ethacridine accumulation in AML cells. We treated TEX and OCI-AML2 cells with 5 μM ethacridine in the presence and absence of erlotinib for one hour. Cells were lysed and contents were quantified by LC-MS/MS. Ethacridine concentrations increased nearly 2-fold in the presence of as little as 1 μM erlotinib in both cell lines (Figure 3F). In contrast, imatinib [which did not synergize with ethacridine (Online Supplementary Figure S5)] did not potentiate ethacridine accumulation in either cell line. While this experiment did not address the mechanism responsible for erlotinib-mediated ethacridine accumulation (for example, whether erlotinib impairs ethacridine extrusion or promotes its uptake), these observations highlight ethacridine accumulation as a potential key explanation for this drug combination’s striking synergy.
To determine whether erlotinib-mediated intracellular ethacridine accumulation could be responsible for excessive ROS production and thus synergistic cytotoxicity, we treated TEX and OCI-AML2 cells with high concentrations of ethacridine and measured changes in intracellular ROS production, using the method described above. Cells were incubated for 24 hours with 15 μM and 20 μM ethacridine to mimic the increase in ethacridine accumulation observed in the presence of 3 μM erlotinib, alongside combination-treated cells. High-dose ethacridine increased ROS production more than 2-fold in TEX cells, and greater than 3-fold in OCI-AML2 cells (Figure 3G), which was comparable to the increase in ROS production observed with combination erlotinib-ethacridine treatment. These findings suggest that erlotinib-mediated ethacridine accumulation is the mechanism that explains synergistic cell death caused by excessive ROS production.
In summary, we have identified the PARG inhibitor ethacridine as a novel combination candidate for erlotinib in AML. Erlotinib synergizes with ethacridine by potentiating its intracellular accumulation; however, the mechanism by which ethacridine accumulates in TEX and OCI-AML2 cells remains to be elucidated. The potential impact of PARG inhibition as a therapeutic strategy in AML warrants further investigation.
Supplementary Material
Footnotes
The online version of this letter has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Thepot S, Boehrer S, Seegers V, et al. A phase I/II trial of Erlotinib in higher risk myelodysplastic syndromes and acute myeloid leukemia after azacitidine failure. Leuk Res. 2014;38(12):1430–1434. [DOI] [PubMed] [Google Scholar]
- 2.Sayar H, Czader M, Amin C, Cangany M, Konig H, Cripe L. Pilot study of erlotinib in patients with acute myeloid leukemia. Leuk Res. 2015;39(2):170–172. [DOI] [PubMed] [Google Scholar]
- 3.Boehrer S, Adès L, Braun T, et al. Erlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical study. Blood. 2008;111(4):2170–2180. [DOI] [PubMed] [Google Scholar]
- 4.Lainey E, Thépot S, Bouteloup C, et al. Tyrosine kinase inhibitors for the treatment of acute myeloid leukemia: delineation of antileukemic mechanisms of action. Biochem Pharmacol. 2011;82(10):1457–1466. [DOI] [PubMed] [Google Scholar]
- 5.Boehrer S, Galluzzi L, Lainey E, et al. Erlotinib antagonizes constitutive activation of SRC family kinases and mTOR in acute myeloid leukemia. Cell Cycle. 2011;10(18):3168–3175. [DOI] [PubMed] [Google Scholar]
- 6.Boehrer S, Adès L, Galluzzi L, et al. Erlotinib and gefitinib for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a preclinical comparison. Biochem Pharmacol. 2008;76(11):1417–1425. [DOI] [PubMed] [Google Scholar]
- 7.Stegmaier K, Corsello S, Ross K, Wong J, DeAngelo D, Golub T. Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood. 2005;106(8):2841–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hidalgo M, Siu L, Nemunaitis J, et al. Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol. 2001;19(13):3267–3279. [DOI] [PubMed] [Google Scholar]
- 9.Rotin L, Gronda M, MacLean N, et al. Ibrutinib synergizes with poly(ADP-ribose) glycohydrolase inhibitors to induce cell death in AML cells via a BTK-independent mechanism. Oncotarget. 2016;7(3):2765–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou S, Chen Q, Zhang L, Fang A, Cheng L. Mifepristone combined with misoprostol versus intra-amniotic injection of ethacridine lactate for the termination of second trimester pregnancy: a prospective, open-label, randomized clinical trial. Eur J Obstet Gynecol Reprod Biol. 2010;151(2):149–153. [DOI] [PubMed] [Google Scholar]
- 11.Koh D, Lawler A, Poitras M, et al. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A. 2004;101(51):17699–17704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Virág L, Robaszkiewicz A, Rodriguez-Vargas J, Oliver F. Poly(ADP-ribose) signaling in cell death. Mol Aspects Med. 2013;34(6):1153–1167. [DOI] [PubMed] [Google Scholar]
- 13.Weber C, Schreiber T, Daub H. Dual phosphoproteomics and chemical proteomics analysis of erlotinib and gefitinib interference in acute myeloid leukemia cells. J Proteomics. 2012;75(4):1343–1356. [DOI] [PubMed] [Google Scholar]
- 14.Lainey E, Sébert M, Thépot S, et al. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle. 2012;11(21):4079–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.