

Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disorder characterized by fever, hepatosplenomegaly, cytopenia, hypertriglyceridemia, hypercytokinemia, and hemophagocytosis. The genetically determined forms of HLH, also referred to as familial hemophagocytic lymphohistiocytosis, commonly manifest in infants or in young children and are characterized by genetic abnormalities of natural killer (NK)-cell function. HLH may also develop secondary to inborn errors of metabolism.1,2 Heme oxygenases are the first and rate-limiting enzymes of heme degradation. Heme oxygenase-1 (HO-1) is ubiquitously expressed at low levels, but is highly inducible in response to stress.3 The cytoprotective, antioxidant and anti-inflammatory roles of HO-1 are mainly attributed to the elimination of free heme, thus limiting the generation of reactive oxygen species (ROS), but also to the generation of the protective products of heme degradation carbon monoxide, biliverdin, and bilirubin.4
We describe a patient carrying a G139V mutation of the active center of HO-1 resulting in loss of normal HO-1 function and gain in pathological peroxidase activity. The patient showed increased urinary excretion of in vivo markers for lipid peroxidation, oxidative protein and DNA damage, oxidative glycation and oxidative bilirubin metabolites. Functional studies of peripheral blood mononuclear cells revealed exquisite oxidative stress sensitivity and lipopolysaccharide hyper-responsiveness. Clinically, the mutation was associated with hemophagocytic lymphohistiocytosis (HLH), a severe inflammatory disorder hallmarked by excessive macrophage activation and abnormal NK-cell function. This distinguishes the phenotype from two published pediatric cases caused by HO-1 null-mutations with loss of enzyme function, which are not associated with HLH.5,6 We show that the disease is clinically modulated by limiting heme challenge through calculated iron depletion, thus highlighting the importance of HO-1 function in oxidative stress defense and in the regulation of the macrophage-dependent inflammatory response.
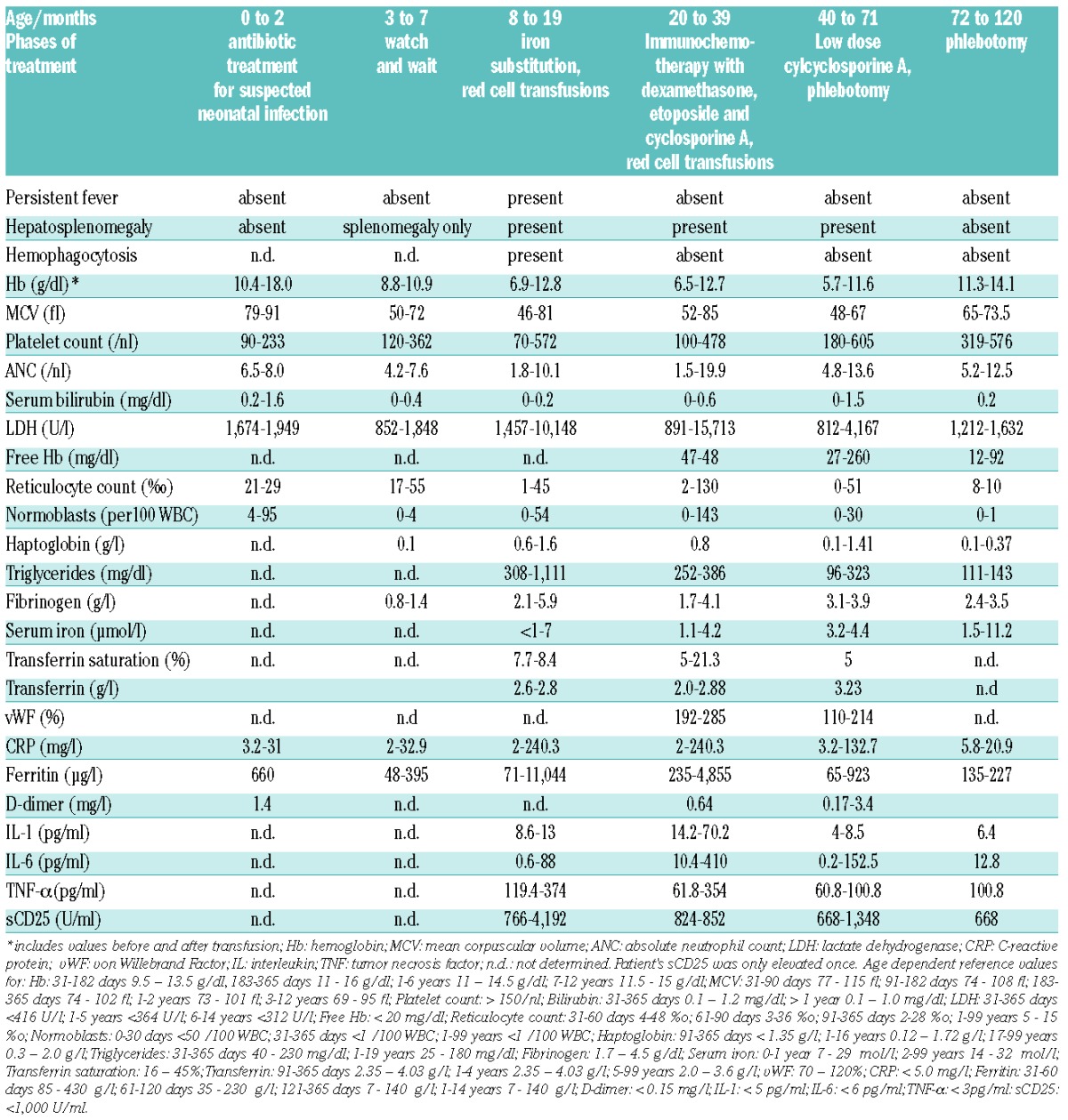
The patient is the son of consanguineous Turkish parents. At the age of three months, he developed microcytic anemia and progressive splenomegaly. At 8 months, oral iron treatment was started, but the anemia did not resolve. On the contrary, the anemia worsened and repeated red cell transfusions were required. The parents and the healthy brother of the patient showed hemoglobin-concentrations and red cell indices at the lower normal limit of the normal range or slightly below (father: Hb 12.7 g/dl, MCV 81 fl; mother Hb 14.4 g/dl, MCV 79 fl; brother age 7 years Hb 11.4 g/dl, MCV 65 fl). At 18 months, the patient developed persistent fever and progressive hepatosplenomegaly (see Table 1 for results of clinical laboratory assessment). Liver histology showed severe hemophagocytosis and Kupffer cell siderosis plus extramedullary hematopoiesis and slight hemophagocytosis was observed in the bone marrow. At 20 months, immunochemotherapy (HLH 2004 protocol7) was started and resulted in sustained remission of the clinical and laboratory signs of HLH. Remarkably, IL-1β, IL-6, TNF-α, ferritin and CRP remained highly elevated indicating ongoing inflammation, whilst the increased sCD25 level normalized rapidly (Table 1). DNA sequence analysis excluded known mutations as potential causes of familial HLH.
Table 1.
Clinical and laboratory features of the patient with HO-1 G139V.
In vitro NK-cell function, measured at age 39 and 45 months, showed decreased killing activity in unstimulated cells, but appropriate killing following IL-2 stimulation (Figure 1A). We tapered immunochemotherapy after 14 months, but four weeks later the patient showed inflammatory activity flare-up (CRP 122 mg/l). CSA was therefore re-started. With improved understanding of the disease pathophysiology, iron depletion was started by periodical low volume phlebotomy; this resulted in the improvement of anemia and complete resolution of liver fibrosis, Kupffer cell siderosis, and hemophagocytosis. At age 10 years, the patient is currently off immunosuppression with no clinically apparent hyperinflammation. However, elevation of vWF, CRP and D-dimers indicate an ongoing inflammatory vascular process.
Figure 1.
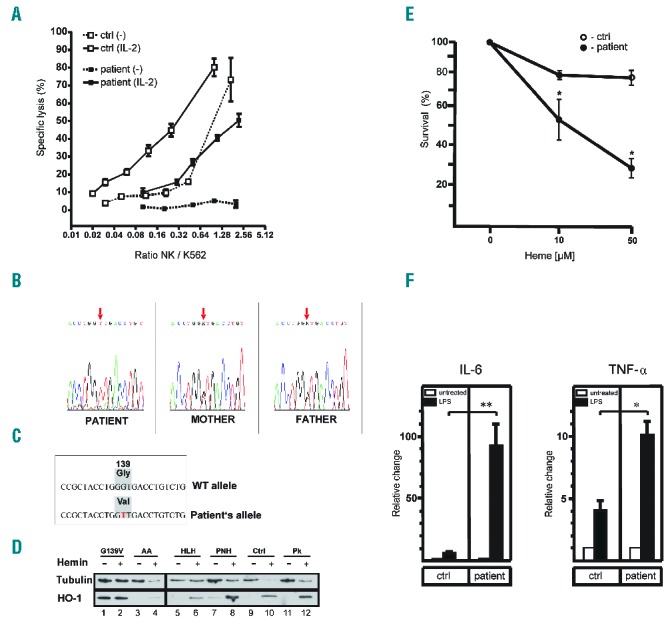
Genetic and functional characterisation of HO-1 G139V. (A) Impaired cytotoxic activity of the patient’s NK cells. Whole blood samples of healthy individuals and the patient were treated with or without 100 U / ml IL-2 overnight at 37 °C. NK-cell activity was then determined by a 4 h 51Cr release assay against the NK-cell sensitive target cell K562. All experiments were performed in triplicate. Data are presented as mean ± SD. Ctrl, control; NK, natural killer cell. (B) DNA sequence of nucleotides 405 to 427 of the HO-1 coding sequence including the GGT to GTT mutation of codon 139, which was found homozygously in the patient and heterozygously in his parents. (C) The mutation identified in the patient causes the replacement of a glycine by a valine at position 139 in the HO-1 catalytic center. (D) HO-1 and tubulin immunoblot of protein isolated from PBMC of the G139V mutated patient, a normal control (Ctrl) and control patients with aplastic anemia (AA), genetic HLH with underlying immunodeficiency (HLH), paroxysmal nocturnal hemoglobinuria (PNH), and pyruvate kinase deficiency (Pk). The PBMC were cultured for 4 hours post-isolation, in the absence (lanes 1, 3, 5, 7, 9, 11) and in the presence (lanes 2, 4, 6, 8, 10, 12) of 100μM hemin. (E) PBMC of the patient (closed circles) and healthy controls (open circles) were isolated and cultured with the indicated concentrations of heme (10 μM and 50 μM). Dead or apoptotic cells were identified by annexin V staining. Percent survival was determined and compared to cells cultured in medium without heme. Each point represents the mean ± SD of 3 independent experiments. Statistics, Student’s t-test for unpaired values: significant differences patient vs. control, * =P<0.05. (F) PBMC of the patient and healthy controls were isolated and cultured as described in the Methods section. After overnight culture, medium was replaced with medium containing either LPS (1 μg/ml; solid bars) or no LPS (open bars) for another 24 h. Concentrations of IL-6 and TNF-α were measured in the supernatant using an ELISA and were normalized to cell number. Values are means ± SEM of duplicates from three independent experiments; a value of 1 was assigned to the cytokine concentration in untreated cells and the relative change after LPS treatment is expressed as -fold induction. Statistics, Student’s t-test for unpaired values: significant differences LPS vs. control, * =P< 0.05; ** = P<0.001. ctrl, control; IL-6, interleukin 6; TNF-α, tumor necrosis factor-α.
The persistently low bilirubin, despite excessively elevated LDH, led us to sequence the HO-1 gene; this revealed a homozygous G139V mutation in the patient which was heterozygous in both parents (Figure 1B and 1C). The mutation is located directly in the enzyme’s catalytic domain.8 Replacements of glycine at this position with larger aliphatic residues cause loss of oxygenase activity but, importantly, also result in gain in peroxidase activity.8,9 This mutation was not identified in an additional analysis we performed on 20 HLH patients with unknown genetics, indicating that HO-1 G139V is not a common cause of familial HLH.
We then confirmed the defective enzyme activity by documenting bilirubin synthesis in PBMC. Control cells showed the expected >4-fold heme-induced increase of bilirubin from 18.8 & 23.6 to 95.9 & 84.6 pmol/mg protein/60min following heme treatment. In contrast, patient cells showed a marginal increase from 21.9 & 26.1 to 33.1 & 27.0 pmol/mg protein/60min.
We next assessed HO-1 protein levels in cultured PBMC and found the expected low constitutive and strongly heme-inducible level in healthy controls and in controls with different hemolytic anemias. In contrast, the patient’s HO-1 G139V cells showed a strong constitutive HO-1 expression, which did not further increase following hemin treatment (Figure 1D).
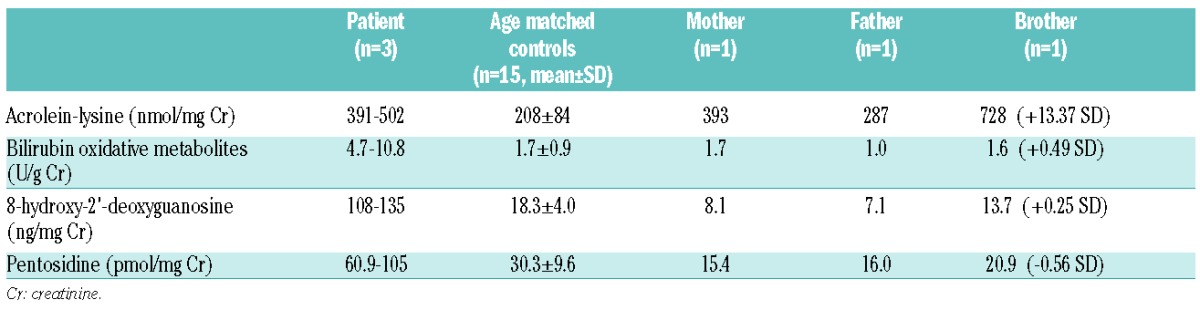
The strong constitutive expression of HO-1 G139V with defective normal but gained abnormal peroxidase function was paralleled by increased excretion of peroxidation products in the urine of the patient and, to some degree, in his heterozygous parents and heterozygous brother (Table 2).
Table 2.
Quantitative determination of ROS metabolites in urine samples.
We then analysed the effect of HO-1 G139V on immunological function in PBMC and showed that exposure to heme led, in contrast to controls, to increased cell death in cytotoxicity assays (Figure 1E), which implies a confounding effect of cell death on HO-1 expression.
Next we determined inflammatory cytokine levels in cultured PBMC in the presence or absence of LPS. In HO-1 G139V cells, the basal IL-6 and TNF-α levels were elevated and responses to LPS were much stronger than in control cells (Figure 1F); this documents a remarkable inflammatory hyperresponsiveness of the patient’s PBMC. Considering potential mechanisms of how the mutation may induce hyperinflammation, it is notable that CO, a stoichiometric by-product of heme degradation and bilirubin biosynthesis, also stimulates the interaction between caveolin-1 (cav-1) and TLR4, thus inhibiting TLR4 function.10 Furthermore, excess heme is known to stimulate TLR4 function.11 Deregulated TLR4 function may therefore play a role in the pathogenesis of hyperinflammation in this patient.
With the broader availability of whole exome sequencing, we searched for other mutations which could plausibly explain the phenotype of the patient. De novo homozygous mutations in the SPON2 and IBTK genes, which bind LPS and regulate B-cell function, respectively, were detected in the patient and were found to be heterozygous in the parents. These mutations may contribute to the phenotype. Additionally, in the absence of whole genome sequencing, significant non-exonic mutations might have been missed.
It is interesting to compare the features of this patient carrying the G139V mutation with those of two children with absent HO-1 expression.5,6,12,13 Both null-mutations and G139V cause hemolytic anemia with almost completely abolished bilirubin synthesis. Interestingly, in HO-1−/− mice splenic macrophages are eliminated by the toxic effect of intracellular heme, resulting in splenic fibrosis and intravascular hemolysis.14 Consistent with this phenotype in mice, asplenia occurred in previously described patients with HO-1 null-mutations. In contrast, the G139V-mutated patient had splenomegaly. Both the null-mutations and G139V resulted in signs of endothelial damage; though this was much more severe in the patients with null-mutations. Abnormal red cell morphology was also observed in both types of mutation; however, the patients with null-mutants suffered the most severe transfusion-dependent hemolytic anemia, whilst the G139V-patient showed only transient severe anemia and mild anemia subsequently. In remarkable contrast to the patient with the deletion mutant, iron supplementation and red-cell transfusion in the G139V-patient were associated with the development of life-threatening inflammation. Additionally, iron depletion by calculated phlebotomy was associated with continuous improvement of anemia and long-term remission of HLH. These important differences can be explained by the gain of abnormal peroxidase function of the strong, constitutive expression of HO-1 G139V, which was predicted by previous biochemical analyses8,9 and confirmed by the increased excretion of urinary peroxidation products in the patient’s urine. Despite the fact that we cannot provide a genetic model for this disease, it is intriguing that this abnormal biochemical function distinguishes this novel clinical entity from that caused by HO-1 null-mutations, although there is overlap caused by loss of HO-1 function in both types of mutants.
These clinical and functional observations confirm the pathogenetic role of the iron and heme inducible mutated HO-1 protein in this disorder. In principle, one may consider complementing this treatment with other antioxidant or iron chelating medication; this has not been done here due to the current stable condition of the patient without further intervention.
The case reported here demonstrates the clinical importance of HO-1 function in oxidative stress defense and in the regulation of macrophage-dependent inflammatory responses.
Supplementary Material
Acknowledgments
We acknowledge Viktoria Jeney for expert technical assistance and The Dietmar Hopp Stiftung and The German Ministry of Science and Technology (E-rare) for financial support. JB is supported by the Hungarian Academy of Sciences (11003).
Footnotes
This study is dedicated to the late Hermann Heimpel who significantly contributed to this study.
Funding: JB is supported by the Hungarian Academy of Sciences (11003).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Janka GE, Schneider EM. Modern management of children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2004;124(1):4–14. [DOI] [PubMed] [Google Scholar]
- 2.Menasche G, Feldmann J, Fischer A, de Saint Basile G. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev. 2005;203:165–179. [DOI] [PubMed] [Google Scholar]
- 3.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. [DOI] [PubMed] [Google Scholar]
- 4.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–354. [DOI] [PubMed] [Google Scholar]
- 5.Yachie A, Niida Y, Wada T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 1999;103(1):129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radhakrishnan N, Yadav SP, Sachdeva A, et al. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J Pediatr Hematol Oncol. 2011;33(1):74–78. [DOI] [PubMed] [Google Scholar]
- 7.Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Koenigs Lightning L, Huang H, et al. Replacement of the distal glycine 139 transforms human heme oxygenase-1 into a peroxidase. J Biol Chem. 2000;275(44):34501–34507. [DOI] [PubMed] [Google Scholar]
- 9.Lad L, Koshkin A, de Montellano PR, Poulos TL. Crystal structures of the G139A, G139A-NO and G143H mutants of human heme oxygenase-1. A finely tuned hydrogen-bonding network controls oxygenase versus peroxidase activity. J Biol Inorg Chem. 2005;10(2):138–146. [DOI] [PubMed] [Google Scholar]
- 10.Wang XM, Kim HP, Nakahira K, Ryter SW, Choi AM. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J Immunol. 2009;182(6):3809–3818. [DOI] [PubMed] [Google Scholar]
- 11.Vinchi F, Costa da Silva M, Ingoglia G, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127(4):473–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawashima A, Oda Y, Yachie A, Koizumi S, Nakanishi I. Heme oxygenase-1 deficiency: the first autopsy case. Hum Pathol. 2002;33(1): 125–130. [DOI] [PubMed] [Google Scholar]
- 13.Saikawa Y, Kaneda H, Yue L, et al. Structural evidence of genomic exon-deletion mediated by Alu-Alu recombination in a human case with heme oxygenase-1 deficiency. Hum Mutat. 2000;16(2):178–179. [DOI] [PubMed] [Google Scholar]
- 14.Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood. 2010;116(26):6054–6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.