

Abstract
Multiple sclerosis (MS) is a neuroinflammatory, demyelinating disease of the CNS. Fibrinogen deposition at sites of blood–brain barrier breakdown is a prominent feature of neuroinflammatory disease and contributes to disease severity. Plasminogen, the primary fibrinolytic enzyme, also modifies inflammatory processes. We used a murine model of MS, experimental autoimmune encephalomyelitis (EAE), to evaluate the hypothesis that the loss of plasminogen would exacerbate neuroinflammatory disease. However, contrary to initial expectations, EAE-challenged plasminogen-deficient (Plg−) mice developed significantly delayed disease onset and reduced disease severity compared with wild-type (Plg+) mice. Similarly, pharmacologic inhibition of plasmin activation with tranexamic acid also delayed disease onset. The T-cell response to immunization was similar between genotypes, suggesting that the contribution of plasminogen was downstream of the T-cell response. Spinal cords from EAE-challenged Plg− mice demonstrated significantly decreased demyelination and microglial/macrophage accumulation compared with Plg+ mice. Although fibrinogen-deficient mice or mice with combined deficiencies of plasminogen and fibrinogen had decreased EAE severity, they did not exhibit the delay in EAE disease onset, as seen in mice with plasminogen deficiency alone. Together, these data suggest that plasminogen and plasmin-mediated fibrinolysis is a key modifier of the onset of neuroinflammatory demyelination.
SIGNIFICANCE STATEMENT Multiple sclerosis is a severe, chronic, demyelinating disease. Understanding the pathobiology related to the autoreactive T-cell and microglial/macrophage demyelinating response is critical to effectively target therapeutics. We describe for the first time that deficiency of plasminogen, the key fibrinolytic enzyme, delays disease onset and protects from the development of the paralysis associated with a murine model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). Administration of a widely used, pharmacologic inhibitor of plasminogen activation, tranexamic acid, also delays the onset of neuroinflammation associated with EAE.
Keywords: fibrinogen, fibrinolysis, mice, neuroinflammation, plasminogen
Introduction
Multiple sclerosis (MS) is a chronic demyelinating disease of the CNS. A primary pathophysiologic feature of this disease process is autoreactive CD4+ T cells driving microglial/macrophage-mediated destruction and phagocytosis of the myelin sheath (Frohman et al., 2006). As the principal effector cell of the neuroinflammatory demyelination response, microglial cells and recruited macrophages play a significant role in the development of MS (Gehrmann et al., 1993; Lassmann et al., 1993; Heppner et al., 2005). While the adaptive immune response is important in the initiation of neuroinflammatory disease, microglial/macrophage activation and function are critical for the development of neuropathologies associated with MS (Davalos et al., 2012).
As macrophage function is critical in the development of MS, interference with activation or recruitment of macrophages to lesions in the CNS is an attractive target in this disease process. Indeed, alteration of fibrinogen, which is known to be central to macrophage activation events, modifies disease severity in a murine model of MS, experimental autoimmune encephalomyelitis (EAE; Akassoglou et al., 2004; Adams et al., 2007). Diminution of fibrinogen or mutation of the αMβ2 binding motif on the γ chain of fibrinogen both ameliorates the paralysis associated with EAE and reduces CNS demyelination. This diminution of disease is mechanistically tied to diminished macrophage activation/phagocytosis in the CNS (Adams et al., 2007).
Plasmin is the primary enzyme that degrades and clears fibrin matrices. Beyond fibrinolysis, plasmin has a broader proteolytic specificity and is capable of modifying other extracellular matrix proteins (Liotta et al., 1981; Chain et al., 1991), directly and through the activation of other proteases (He et al., 1989; Makowski and Ramsby, 1998), as well as growth factors (Houck et al., 1992; Campbell and Andress, 1997; Shanmukhappa et al., 2009), complement (Barthel et al., 2012), and other proteins (Ellis et al., 1968; Pepinsky et al., 1988). Given this wide array of proteolytic targets, plasmin has been implicated in multiple cellular processes beyond its well characterized role in fibrinolysis. Plasmin-mediated proteolysis is key in wound healing (Romer et al., 1996), host defense (Degen et al., 2007; Guo et al., 2011), and cell migration (Ploplis et al., 1998). Frequently, the importance of plasminogen has been linked to fibrinogen/fibrinolysis. However, fibrinogen-independent roles of plasminogen have been unambiguously recognized as well, including hepatic repair (Shanmukhappa et al., 2009).
One of the most intriguing recognized functions of plasmin, both fibrinolysis dependent and fibrinolysis independent, is the control of inflammatory events. Plasmin plays a critical role in the trafficking of macrophages to the peritoneal cavity in peritonitis challenges (Ploplis et al., 1998). Further, plasminogen supports the development of asthma in mice (Swaisgood et al., 2007). Additionally, both plasminogen and plasminogen activators promote the development of autoimmune inflammatory arthritis (Cook et al., 2010; De Nardo et al., 2010; Raghu et al., 2014).
Given the key role of fibrinogen in the setting of neuroinflammation, we investigated the potential for plasminogen to influence inflammatory disease. Here we establish for the first time that plasminogen deficiency results in a major delay in disease onset and protection from paralysis in myelin oligodendrocyte protein (MOG) peptide-induced EAE. Further, fibrinogen deficiency alone reduces EAE severity, while fibrinogen deficiency combined with plasminogen deficiency abrogates the early delay to disease onset conferred by plasminogen deficiency alone. These findings establish an unanticipated role for plasminogen as a regulator of disease onset in autoimmune neuroinflammatory disease.
Materials and Methods
Mice and induction of experimental autoimmune encephalomyelitis.
Plasminogen-deficient mice (Plg−; RRID:IMSR_JAX:002830), fibrinogen-deficient mice (Fib−; RRID:MGI:3663779), and combined Plg−/Fib− mice have been previously described (Bugge et al., 1995, 1996; Suh et al., 1995). Both Plg− and Fib− animals have a known normal adaptive immune response (Kitching et al., 1997; Drew et al., 2001; Flick et al., 2007; Munks et al., 2010; Berri et al., 2013). All animals used were backcrossed a minimum of six generations to C57BL/6J and, consistent with the known sex bias of disease, only females were used. All animals used were generated from the same colony at Cincinnati Children's Research Foundation. EAE was induced in 10-week-old animals by subcutaneous injection of MOG35–55 (MEVGWYRSPFSRVVHLYRNGK) emulsified in complete Freund's adjuvant on day 0 (Hooke Laboratories). Animals received 230 ng of pertussis toxin, i.p. (Hooke Laboratories), in PBS on days 0 and 1 of the induction period. An observer blinded to genotype performed all clinical scoring. Mice were scored daily as follows, based on motor symptoms: 0, no disease; 1, loss of tail tone; 2, hindlimb ataxia; 3, hindlimb paralysis; 4, forelimb ataxia or paralysis; 5, moribund or death. The Cincinnati Children's Research Foundation Institutional Animal Care and Use Committee approved all experiments.
Rotarod treadmill assay.
Mice were challenged on a rotarod treadmill specifically designed for mouse usage (Med Associates) to evaluate motor function and stamina. Studies on the rotarod were first initiated before the induction of EAE to acclimatize the animals to the instrument and to remove learning as a variable and then were repeated in the same cohorts over time. Mice with any motor symptoms were closely monitored while using the rotarod. An acceleration setting was used, the initial speed started at 4 rotations per minute (rpm) with gradual acceleration to a maximum of 40 rpm. Analyses are reported as the mean of three separate trials on the rotarod apparatus with a maximum time limit of 360 s.
Quantitative PCR.
Spinal cords were isolated from either unchallenged animals or challenged animals at the peak of disease after first perfusing the killed animals with PBS through the left ventricle of the heart. Whole spinal cords were then obtained by flushing the spinal canal with PBS. The spinal cords were then homogenized in TRIzol (Life Technologies) using a TissueLyser II (Qiagen), and RNA was isolated according to the manufacturer directions. Total RNA was used to make cDNA with a high-fidelity RNA-to-cDNA kit (Life Technologies) and quantitative PCR (qPCR) was performed. Primer sets for matrix metalloprotease 9 (MMP9), F4/80, interleukin (IL)-6, IL-17α, interferon-γ (IFN-γ), IL-1β, chemokine (C-C motif) ligand 5 (CCL5), and chemokine (C-X-C motif) ligand (CXCL1) were used in conjunction with a primer set for Polr2a as a reference gene (TaqMan). Relative expression of each gene was calculated using the Pfaffl method (Pfaffl, 2001).
Isolation of splenocytes, splenocyte proliferation assay, and FACS analysis.
Spleens were removed at the peak of EAE disease and a splenocyte proliferation assay was performed as previously described (Ma et al., 1998). Briefly, splenocytes were mechanically dissociated via passage through a 40 μm sieve, washed with an erythrocyte lysing solution (Invitrogen), and then placed into RPMI (Invitrogen) containing 2% C57BL/6J mouse serum using 96-well plates at 2 × 105 cells/well. The cultured cells were incubated in media alone or stimulated with either 0.1 mg/ml MOG35–55 (Hooke Laboratories) or anti-CD3 antibody (antibody-2C11, American Type Culture Collection; RRID:CVCL_7234) as a positive control. After 48 h in culture, the supernatant was harvested, and then the cells were pulsed with 1 μCi of [methyl-3H]-thymidine (PerkinElmer) for 24 h. Incorporation of [methyl-3H]-thymidine per well was determined. Splenocytes (1 × 106) were also immunostained for separate flow cytometry analyses using fluorescence-conjugated antibodies to CD3 (fluorescein isothiocyanate clone 145–2C11, BD Biosciences; RRID:AB_394594), CD19 (phycoerythrin, clone 1D3, BD Biosciences; RRID:AB_395050), F4/80 (allophycocyanin, clone BM8, eBiosciences; RRID:AB_469452), and CD25 (allophycocyanin, clone PC61.5, eBiosciences; RRID:AB_469366). FACS analysis was performed with a BD FACS Canto system (BD Biosciences) and analyzed with BD FACS Diva 8.0 (BD Biosciences). Supernatant cytokine levels (IFN-γ, IL-1β, IL-2, IL-4, IL-6, IL-12, and IL-17α) were determined by a multiplex Luminex assay (Millipore).
Treatment of mice with tranexamic acid.
The drinking water of experimental cohorts was supplemented with 50 mg/ml tranexamic acid (TXA). Similar doses of TXA have previously been found to be biologically effective in mice (Hattori et al., 2000; Bruno et al., 2008). Control animals were maintained on standard water without supplement. Three days after the initiation of TXA treatment, mice were immunized with MOG, as described above. Mice were evaluated for clinical scores for 4 weeks following EAE induction. An additional cohort of TXA-treated mice had spinal cords harvested at the time of peak disease for histological analysis.
Passive EAE induction.
For adoptive transfer of EAE in SJL/J mice, wild-type donor SJL/J mice were immunized with 100 μg of proteolipid protein 139–151 (PLP139–151) (HSLGKWLGHPDKF, Auspep) in complete Freund's adjuvant (Sigma-Aldrich) supplemented with 400 μg of heat-inactivated mycobacterium tuberculosis H37Ra (Difco; Adams et al., 2007). Ten days after immunization, cells from draining lymph nodes and spleen were isolated. Lymphocytes were restimulated with 20 μg/ml PLP139–151 peptide and 10 ng/ml IL-12 (eBioscience) for 4 d in complete RPMI-1640, and 3 × 107 cells were transferred into healthy SJL/J recipients. For the treatment of mice with TXA, the drinking water was supplemented with 50 mg/ml TXA 3 d before cell transfer. We monitored mice daily for clinical signs as described above.
Blood–brain barrier leakage assay.
Unchallenged and EAE-challenged mice were injected with 150 μl of 1% Evans Blue dye in PBS. Two hours later, mice were killed and perfused with 10 ml of PBS. Whole spinal cords were obtained by flushing the spinal column with PBS. Subsequently, Evans Blue dye was extracted from the spinal cord tissue using formamide overnight at 55°C and quantified using a fluorescence reader (SpectraMax M2, Molecular Devices) with excitation at 620 nm and emission at 680 nm. Evans Blue dye was expressed as the total dye per gram of tissue.
Zymography.
Spinal cords were harvested from both Plg+ and Plg− animals at the peak of disease. The cords were homogenized in PBS and total protein concentrations were determined by BCA assay (ThermoFisher Scientific). Gelatin zymography (10% gel; Bio-Rad) was performed with 20 μg of total protein, as previously described (Raghu et al., 2014). Gel images were captured with a Canoscan LIDE 30 Scanner (Canon). Densitometry of the zymography gels was performed using the Fiji configuration of ImageJ (National Institutes of Health).
Histopathology.
Histopathologic examination of mouse spinal cords was performed on spinal cord sections obtained from animals harvested at the time of peak disease. Spinal cords were removed from the spinal canal of perfusion-fixed mice and processed into paraffin. Tissue sections were stained with Luxol fast blue (Electron Microscopy Services) and counterstained with hematoxylin and eosin (Sigma-Aldrich) to evaluate demyelination. Macrophages were immunostained using a rabbit anti-Iba1 primary antibody (Wako Chemicals) together with a biotinylated goat anti-rabbit antibody (Vector Laboratories), the ABC Streptavidin/horseradish peroxidase detection system (Vectastain, Vector Laboratories), and diaminobenzidine (Sigma-Aldrich). Photomicrographs were captured using an Axioplan 2 microscope and an AxiocamHR camera (Carl Zeiss). Determination of the percentage area of spinal cord demyelination was performed by an observer blinded to mouse genotype by manual measurement of areas of demyelination on six cross-sections of spinal cord per mouse using ImageJ. Area of Iba1 staining was established by applying manual thresholding to the image using the Threshold Color ImageJ Plugin (Dr. G. Landini, University of Birmingham, Birmingham, UK) on ImageJ. The percentage of total Iba1 area was averaged for six spinal cord sections per mouse.
Statistical analysis.
For endpoints measured serially, differences between groups at specified time points were two-sided tests obtained from a linear mixed model using all measurements taken throughout the experiment. Within-subject correlation was accounted for using an order one autoregressive error structure. These analyses were performed using SAS version 9.3. All other statistical analysis was accomplished using GraphPad Prism version 6 for Windows (GraphPad Software). Time until onset of disease and time until onset of hindlimb/forelimb paralysis were compared using log-rank analysis. All remaining analyses were performed using a two-tailed Mann–Whitney U test. SE is expressed as SEM.
Results
Plasminogen deficiency delays the onset and decreases severity of paralysis associated with EAE
To test the hypothesis that plasminogen deficiency modifies neuroinflammation in EAE, cohorts of Plg+ (n = 19) and Plg− (n = 21) mice were challenged with MOG35–55-induced EAE. Contrary to expectations, based on evidence that fibrinogen is a potent driver of neuroinflammatory disease in mice, and despite the fact that the loss of the key fibrinolytic protease plasmin would favor fibrin persistence, EAE-challenged Plg− mice exhibited significantly decreased clinical disease scores relative to control Plg+ mice. In contrast to Plg+ mice, Plg− mice exhibited improved motor function over the entire observation period of >5 weeks (Fig. 1A). While EAE disease penetrance was 100% in both groups of animals, loss of plasminogen also resulted in a significantly prolonged time to EAE disease onset (i.e., any symptom of EAE, loss of tail tone, or ataxia) with a median of 14 d for Plg+ mice versus 16 d for Plg− mice (Fig. 1B). Additionally, time to more advanced motor function loss (e.g., hindlimb paralysis) was also significantly prolonged in Plg− mice, with a median of 15 d for Plg+ mice and 19 d for Plg− animals (Fig. 1C). At no point of time during the >5 weeks that the mice were followed did the Plg− animals develop a similar level of disease as the Plg+ mice. At the peak of disease (day 19), the loss of motor function was typically limited to only mild ataxia in Plg− mice yielding a mean clinical score of 2.3 ± 0.3, whereas Plg+ animals challenged in parallel typically exhibited hindlimb paralysis and/or partial forelimb paralysis, yielding a mean score of 3.4 ± 0.2 (p < 0.001). A similar highly significant benefit in motor function capability was observed in Plg− mice relative to Plg+ cohorts late in the disease course. At day 38, the mean clinical score for Plg− animals was 2.3 ± 0.3 compared with a mean clinical score of 3.3 ± 0.3 for Plg+ animals (p < 0.01). This difference is not only statistically significant, it is also quite functionally significant, as a score of 2 is defined by ataxia alone, compared with paralysis associated with a score of 3. Further, the total days of paralysis (clinical scores equal to or >3) were significantly increased in the Plg+ cohort versus in the Plg− cohort (Fig. 1D). Plg− animals were also spared from the severe body weight loss typically associated with developing EAE. At the peak of disease, Plg− mice (n = 10) had a median body mass of 94.0 ± 3.2% of their body mass before disease onset, compared with a median of 79.9 ± 2.4% of initial body mass in Plg+ animals (n = 10; p < 0.005).
Figure 1.

Mice lacking plasminogen exhibit diminished EAE disease severity and paralysis. A, Cohorts of Plg− (n = 19) and Plg+ (n = 21) animals were challenged with MOG-induced EAE. Plg− animals exhibited significant protection from both disease onset and paralysis throughout the study period. These data reflect two combined experiments with similar results. *p < 0.05; ∧p < 0.01; §p < 0.005; #p < 0.001. B, Kaplan-Meier analysis of time until onset of any EAE disease-related symptoms. Plg− animals have a statistically increased time until disease onset; however, all of the Plg− animals developed symptoms of EAE. C, Time to onset of hindlimb or forelimb paralysis (a clinical score of ≥3). Plg− mice were found to have significantly prolonged time to the onset of hindlimb paralysis. Indeed, >20% of the Plg− mice did not exhibit any paralysis during the study period. D, Plg− animals also exhibited fewer days of paralysis per mouse during the study period. The majority of Plg− mice had <10 d of paralysis, while the majority of Plg+ mice had >20 d of paralysis. E, Cohorts of Plg− and Plg+ mice were challenged with EAE and tested with the rotarod apparatus. These animals were challenged on the rotarod before the MOG injection, at 6 d after the injection (before the onset of disease), and again at day 15 (after disease onset, but before peak disease). While the Plg− animals had no significant difference in their ability to use the rotarod at day 15 compared with day 6, the Plg+ mice had a significant decrease in the capability to walk on the apparatus both compared with themselves on day 6 and the Plg− mice at day 15. F, EAE-challenged wild-type animals were found to have significantly increased BBB permeability (23-fold) over nonchallenged wild-type mice. However, Plg− animals challenged with EAE did not demonstrate a statistically significant increase compared with unchallenged mice. EAE-challenged Plg− animals exhibited a twofold reduction in BBB leakage compared with EAE-challenged Plg+ mice.
A subsequent cohort of Plg− (n = 11) and Plg+ (n = 9) mice were challenged with EAE to assess motor function on a rotarod treadmill. On day 6, before the onset of clinical disease, no statistical difference was observed in the time that Plg+ or Plg− animals were able to actively remain on the slowly accelerating treadmill. However, by day 15 after the induction of EAE, there was a statistically significant twofold diminution in the time that Plg+ animals could remain on the treadmill relative to the initial analysis at day 6. In stark contrast, at day 15, there was no discernible decrease in the time EAE-challenged Plg− mice could actively remain on the rotarod treadmill relative to initial times, before disease onset, at day 6 (Fig. 1E).
Decreased blood–brain barrier leakage in Plg− animals
To further define any genotype-dependent differences in early pathological changes associated with the development of EAE, we assessed blood–brain barrier (BBB) leakage using the well described method of Evans Blue extravasation. Here, cohorts of EAE-challenged mice were administered Evans Blue dye at the peak of disease (day 15) and the extent of dye leakage into the CNS tissues evaluated. Consistent with prior results, a statistically significant difference in clinical scores between Plg+ (n = 12) and Plg− (n = 8) animals was observed at day 15 (mean scores, 3.6 ± 0.2 vs 0.8 ± 0.4, respectively; p < 0.0001). Consistent with the notion that plasminogen is mechanistically coupled to vascular leakage in the CNS in the context of MOG-induced neuroinflammatory disease, EAE-challenged Plg− mice had a statistically significant twofold diminution in vascular leakage relative to EAE-challenged Plg+ mice (Fig. 1F; p < 0.05). EAE-challenged Plg+ mice had a robust increase of vascular permeability compared with unchallenged Plg+ animals (Fig. 1F; p < 0.005), while EAE-challenged Plg− mice had only modest vascular leakage over unchallenged Plg− mice, but the difference was not statistically significant (Fig. 1F; p > 0.25).
Pharmacological suppression of plasminogen activation ameliorates EAE pathology
To complement the genetic studies in Plg− mice, EAE studies were performed in mice treated with a pharmacologic inhibitor of plasminogen activation. Here, EAE-challenged wild-type mice were treated with a pharmacologic suppressor of plasminogen activation, TXA, which is widely used and clinically approved for the suppression of bleeding events. Compared with wild-type mice treated with water alone, mice treated with TXA exhibited a significantly improved early disease course (Fig. 2A). Additionally, mice treated with TXA had a significant prolongation until the onset of any EAE disease-related symptoms compared with water-treated controls (Fig. 2A,B). Furthermore, as an additional objective metric of the benefits of TXA in limiting the disease progression, TXA-treated mice were significantly protected from the body mass loss associated with EAE compared with mice that received only water (Fig. 2C).
Figure 2.
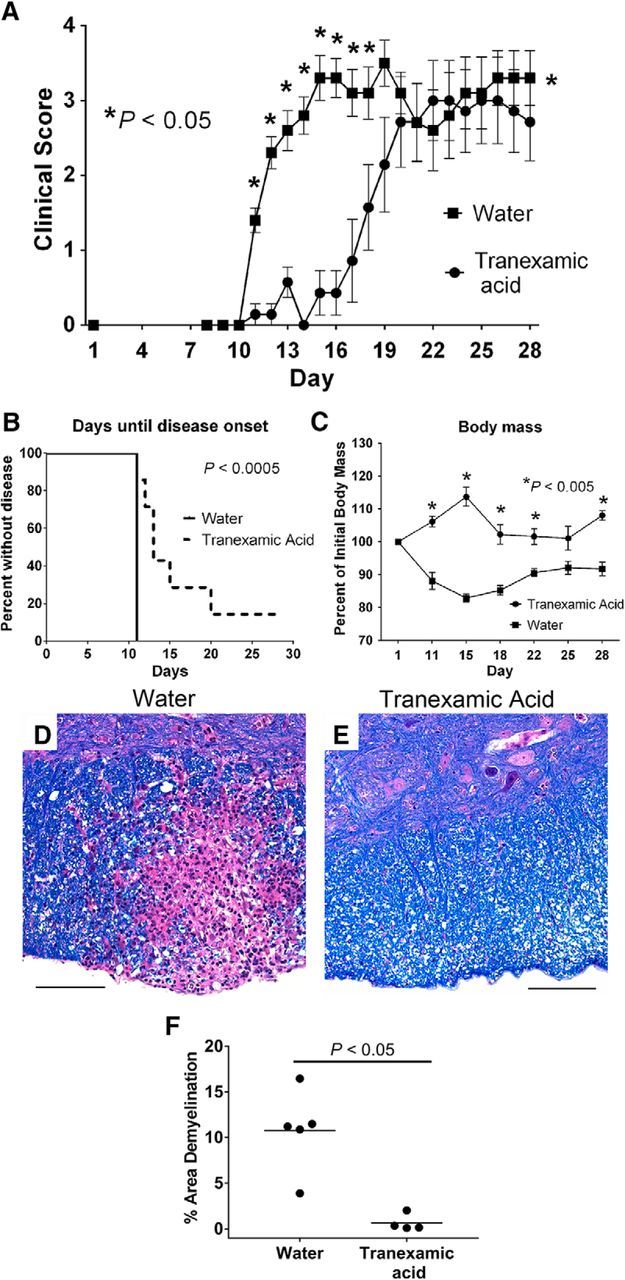
Treatment of mice with TXA is protective against disease severity associated with EAE. A, Cohorts of wild-type mice were treated with either TXA (50 mg/ml) in the drinking water or drinking water alone. Significant protection from disease was seen in the TXA-treated animals, especially early in the course of disease. *p < 0.05; ∧p < 0.01; §p < 0.005; #p < 0.001. B, Kaplan-Meier analysis of time until disease onset reveals significant prolongation of time until TXA-treated mice develop signs of disease (e.g., clinical score ≥1). C, Mice treated with TXA show protection from the body mass loss that is classically associated with development of autoinflammatory demyelinating disease. D, E, Evaluation of spinal cords harvested at the peak of disease from either mice on drinking water only (D; 20×) or TXA (E; 20×) shows decreased areas of demyelination and inflammation in mice treated with TXA. F, Quantification of the area of demyelination demonstrates a significant diminution of demyelination at the peak of disease in TXA-treated animals. Scale bars, 100 μm.
Histological analysis of spinal cords revealed prominent areas of demyelination throughout the cords of control animals that received water alone (Fig. 2D, representative image). In contrast, the spinal cords of the TXA-treated cohort had substantially fewer and smaller areas of demyelination and inflammation (Fig. 2E, representative image). Quantification of the area of demyelination also revealed a significant decrease in the area of demyelination at the peak of disease (Fig. 2F).
Regulation of peripheral T-cell responses is not a major function of plasminogen in EAE
To determine the mechanisms linking plasminogen to EAE disease onset and progression, we first analyzed whether diminished pathologies noted in Plg− mice were linked to a decreased autoreactive T-cell response following MOG peptide immunization. First, we examined the differential composition of the splenocytes of the Plg+ and Plg− mice at the peak of disease (day 14). There was no significant difference in CD3-, CD3/CD25-, CD19-, or F4/80-expressing cells between the two genotypes (Fig. 3A–D). Next, cultured splenocytes from the same cohorts of both Plg+ and Plg− animals were compared for specific proliferative response by measuring cellular [3H]-thymidine incorporation following stimulation with MOG35–55. Consistent with prior findings, there was a significant separation of clinical scores in Plg+ and Plg− mice at the time of splenocyte isolation. Nevertheless, no significant difference was noted in MOG peptide-induced T-cell proliferation in splenocytes isolated from Plg+ or Plg− mice in any of the treatment groups (Fig. 3E). We next assessed whether plasminogen contributes to T-cell polarization in EAE induction. Splenocytes of EAE-challenged Plg+ and Plg− mice were stimulated with media alone, MOG, or a CD-3 antibody. The supernatant from these cultures was harvested after 48 h and assessed for cytokines associated with T helper type 1 (Th-1), Th-2, or Th-17 cell response. There was no difference between Plg+ or Plg− splenocyte production of any cytokine among those that were incubated with media only. A modest, but statistically significant, decrease in IFN-γ production was noted in the MOG-stimulated Plg− splenocytes, compared with the Plg+ splenocytes (Fig. 3H). Otherwise, no significant differences were found among the MOG-stimulated splenocytes (Fig. 3G,I–L). Among those treated with the anti-CD3 antibody, a modest, statistically significant increase in IL-2 production was noted in the Plg− splenocytes (Fig. 3G).
Figure 3.

Response to MOG immunization. A–D, Splenocytes were harvested at the peak of disease (day 14), labeled, and analyzed via FACS. No differences among CD3, CD3/CD25, CD19, or F4/80 were observed between genotypes. E, Splenocytes from the same cohort of animals were used for a proliferation assay. Splenocytes were stimulated in vitro with either media alone, MOG35–55, or a stimulatory anti-CD3 antibody. No difference between genotypes was observed in any stimulatory group. Supernatants from the stimulated splenocytes were harvested and assayed to assess T-cell polarization. F, I–L, No significant differences between stimulated splenocytes from Plg+ or Plg− animals were noted for secretion of IL-6, IL-1β, IL-12p70, IL-4, or IL-17α. G, A significant increase in IL-2 secretion was seen in anti-CD3-stimulated splenocytes from Plg− animals. H, A small, but significant decrease in IFN-γ secretion was noted in MOG-stimulated splenocytes from Plg− animals.
Furthermore, we tested the effects of plasminogen in adoptive transfer EAE. As the PLP-induced model of EAE is better suited for passive transfer of disease, we immunized wild-type SJL mice with PLP and transferred PLP139–151-primed lymphocytes and splenocytes to wild-type mice treated with TXA or vehicle. Similar to active EAE (Fig. 2A), passive EAE mice treated with TXA had a delayed onset of EAE disease in contrast to control mice (Fig. 4). Overall, these data suggest that the regulation of the primary T-cell response is not a primary function of plasminogen in EAE.
Figure 4.

Passive transfer of EAE disease in mice treated with TXA shows a similar pattern to active disease induction. Wild-type SJL mice were immunized with PLP, and spleens and draining lymph nodes were harvested. These cells were stimulated with PLP in culture and transferred to mice treated with either water or TXA in the drinking water. A, Similar to TXA treatment of mice undergoing active disease induction with MOG, passive EAE disease severity was delayed in mice treated with TXA. B, There was also a statistically significant delay of disease onset in mice treated with TXA.
Decreased demyelination and inflammation in spinal cords of Plg− mice
To assess the degree of pathology within the CNS of EAE challenged mice at the peak of disease, spinal cords were harvested from cohorts of Plg− and Plg+ animals (17 d after immunization). As expected, inflammatory infiltrates correlated with areas of demyelination throughout the cords of Plg+ animals (Fig. 5A,C). In contrast, Plg− animals had dramatically decreased demyelination and inflammation compared with the Plg+ animals (Fig. 5B,D). Quantification showed that Plg− animals had over a fourfold decreased area of demyelination in the spinal cords in contrast to Plg+ animals (Fig. 5E).
Figure 5.
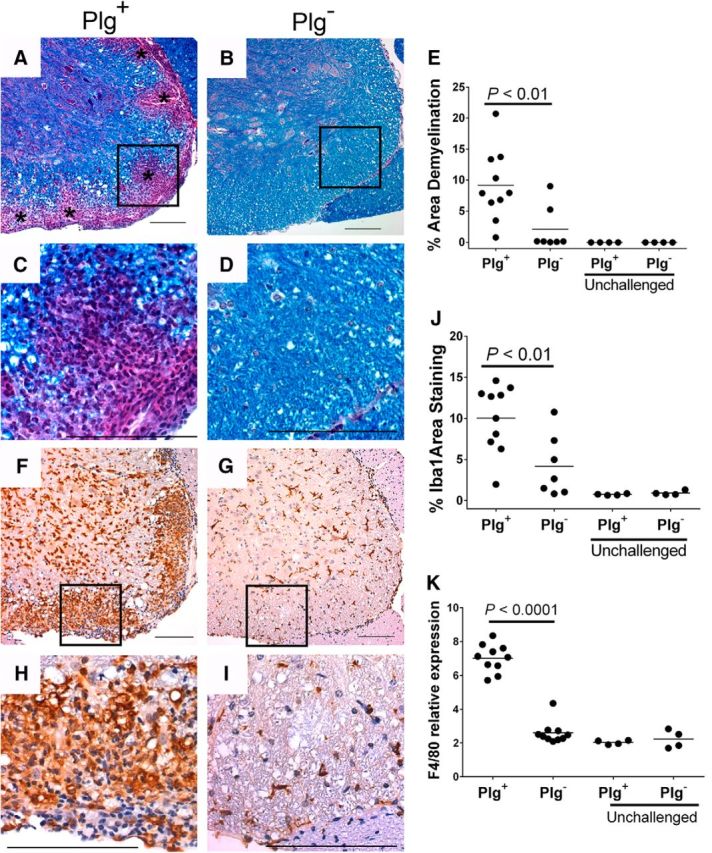
Diminished demyelination, inflammation, and microglial/macrophage accumulation in spinal cords of Plg− animals. A, Spinal cords were harvested at the peak of disease from both Plg+ and Plg− mice and stained with Luxol fast blue to delineate areas of demyelination. Plg+ animals exhibited prominent areas of demyelination throughout the spinal cord (indicated by asterisks; 20×). C, Higher-power view of an area of demyelination reveals a profuse inflammatory cell infiltrate (40×). B, D, However, Plg− animals had significantly less demyelination throughout the spinal cords. E, The area of demyelination was quantified using ImageJ, and a statistically significant decrease in demyelination was present in the Plg− animals compared with Plg+ mice. F, G, Significant macrophage accumulation was found in the Plg+ (F) animals while the Plg− (G) animals had substantially less macrophage accumulation. H, I, At high power (H), the macrophage predominance in the areas affected is well appreciated in the Plg+ animals; however, similar accumulation is not seen in the Plg− mice (I). J, The area of Iba1 staining was quantified using ImageJ. A significant diminution of area stained was present within the spinal cords of EAE-challenged Plg− animals compared with challenged Plg+ mice. F, F4/80 expression was determined within the spinal cords of EAE-challenged and EAE-unchallenged mice. Corresponding to the immunohistochemical analysis, a significant diminution of microglial/macrophage accumulation in challenged Plg− mice compared with Plg+ animals was noted in this assay. Scale bars, 100 μm.
Macrophage/microglial cell accumulation in the spinal cords was examined in tissue sections using the specific marker for microglial cells and macrophages Iba1. Iba1+ cells were present at low, baseline levels in both unchallenged Plg+ and Plg− spinal cords. While abundant Iba1+ cells were readily apparent throughout the spinal cords of control EAE-challenged Plg+ animals (Fig. 5F,H), Plg− animals exhibited fewer Iba1+ cells in the spinal cords (Fig. 5G,I). Indeed, Iba1+ cells in EAE-challenged Plg− CNS tissue sections were only modestly increased relative to unchallenged animals. Quantitative image analyses established that the mean area of Iba1 staining in EAE-challenged Plg− mice was less than half that observed in EAE-challenged Plg+ animals (Fig. 5J). As an additional objective measure of macrophage accumulation into the spinal cords of study group mice, total RNA was purified from spinal cords and used for qPCR of a second known monocyte/macrophage marker, F4/80. Consistent with the macrophage/microglial immunohistochemical studies, relative to EAE-challenged Plg+ animals, EAE-challenged Plg− mice exhibited 2.7-fold less F4/80 RNA in spinal cord tissues (Fig. 5K). As anticipated, the F4/80 RNA levels in spinal cord tissues collected from EAE-challenged Plg+ animals were an average of 3.4-fold higher than those observed in unchallenged Plg+ controls. In contrast, F4/80 RNA levels in spinal cord tissues from EAE-challenged Plg− animals exhibited a small increase (∼1.1-fold) over unchallenged Plg− mice.
To better understand the signals underlying the macrophage response, we looked at fibrinogen deposition, and CD4 T-cell and B-cell infiltration into the spinal cords of EAE-challenged mice. As previously reported (Adams et al., 2007), abundant fibrinogen was identified within the spinal cords of Plg+ animals at the peak of disease (Fig. 6A). Consistent with the difference in disease severity, Plg− mice demonstrated little fibrinogen deposition within the spinal cords at the peak of disease (Fig. 6B). CD4+ T cells were prominent at sites of demyelination within the spinal cords of Plg+ animals, while few CD4+ cells were found in the spinal cords from Plg− animals (Fig. 6C,D). Occasional CD20+ B cells were present within the inflammatory plaques of Plg+ animals; however, few B cells were observed in the spinal cords of Plg− animals (Fig. 6E,F). Thus, Plg− animals appear to have decreased numbers of CD4 cells in the spinal cord and diminished BBB leakage, fibrinogen deposition, and macrophage infiltration.
Figure 6.
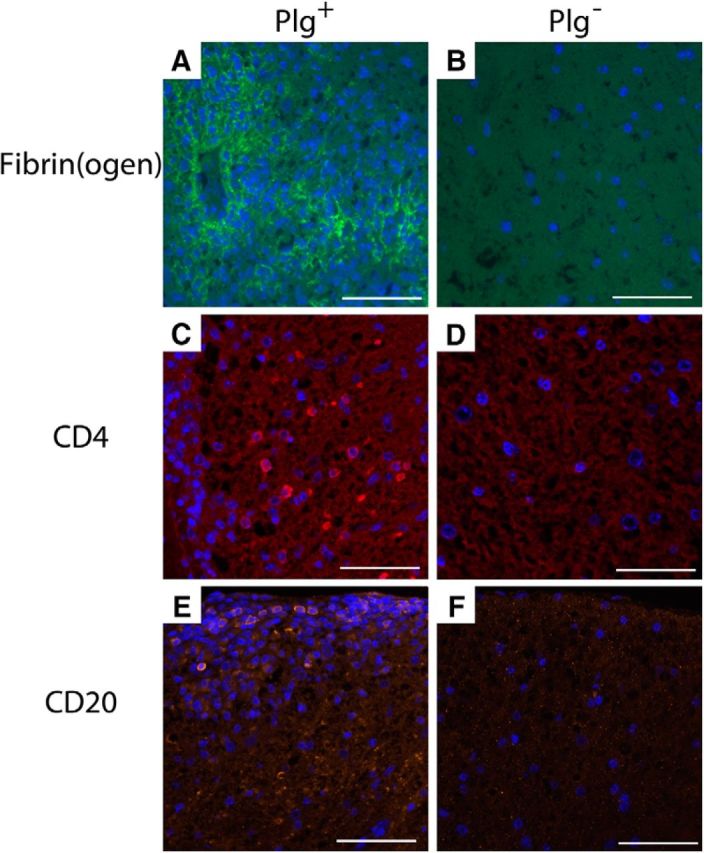
Fibrinogen deposition and CD4 and CD20 infiltration into spinal cords during disease. Spinal cords, harvested at the peak of disease, were examined for fibrinogen deposition. A, Plg+ cords were found to have abundant fibrinogen throughout the areas of demyelination. B, However, minimal fibrinogen was identified in the spinal cords of Plg− animals, corresponding with decreased levels of disease and diminished BBB leak. C, CD4 cells were readily identified in the spinal cords from Plg+ mice, typically associated with areas of demyelination and inflammation. D, In spinal cords of Plg− animals, few CD4 cells were identified. E, F, CD20 cells were identified in areas of demyelination in cords from Plg+ animals (E); however, very few CD20 cells were identified in the Plg− spinal cords (F).
Potential genotype-dependent differences in inflammatory signals critical to the pathogenesis of neuroinflammatory disease were also evaluated. Here, spinal cords from Plg− and Plg+ mice were harvested at the peak of neuroinflammatory disease (day 14), and total RNA preparations were analyzed for cytokine gene expression levels by qPCR. Given that the Th-17 response is crucial for the development of human MS and murine EAE (Komiyama et al., 2006), we assayed for IL-17a. EAE-challenged plasminogen-deficient mice had a significant threefold reduction in IL-17a levels compared with wild-type mice (Fig. 7A). Th-1 responses are also central to the development of EAE and MS (Merrill et al., 1992). Consistent with the reduced clinical signs and diminution in histological evidence of neuroinflammatory disease in EAE-challenged Plg− mice, the expression of IFN-γ, IL-6, and IL-1β were all significantly decreased in Plg− spinal cord tissue compared with Plg+ spinal cord preparations (Fig. 7B–D). Furthermore, RNA levels of proinflammatory chemokines CXCL1 and CCL5, which are known to support leukocyte chemotaxis in EAE (Godiska et al., 1995; Carlson et al., 2008), were similarly diminished in the Plg− mice compared with control animals (Fig. 7E–F).
Figure 7.

Reduction of proinflammatory cytokine and chemokine expression in Plg− spinal cords. Spinal cords from cohorts of Plg− and Plg+ animals were harvested at the peak of disease (day 14), and total RNA preps were prepared and used for quantitative PCR. A, Expression of IL-17a, the defining cytokine of the Th-17 response, was significantly decreased in the spinal cords of Plg− animals in contrast to Plg+ animals. B–D, The Th-1 response, also required for the development of neuroinflammatory disease, was assessed by determination of expression of IFN-γ, IL-6, and IL-1β. Significantly decreased expression of each of these crucial cytokines was found in the Plg− mice compared with Plg+ controls. E, F, Additionally, evaluation of the proinflammatory chemokines CXCL1 and CCL5 revealed significantly decreased expression in the Plg− mice in contrast to the control animals.
Plasminogen regulates the activation of matrix metalloproteinases in EAE
Previous reports have pointed to a mechanistic relationship between plasmin and MMP9 in inflammation and the recruitment of macrophages at sites of inflammation (Liu et al., 2005; Gong et al., 2008). To determine whether plasminogen deficiency directly and/or indirectly altered MMP9 activation within CNS tissues of EAE-challenged mice, spinal cord extracts were prepared from cohorts of plasminogen-sufficient and plasminogen-deficient mice at peak EAE disease (16 d post-MOG induction) and MMP9 activity was assessed by gelatin zymography. EAE-challenged Plg+ mice uniformly exhibited prominent proteolytic activity corresponding to MMP9 based on molecular weight (Fig. 8A). In contrast, spinal cord extracts from EAE-challenged Plg− animals were found in most cases to have essentially no detectable MMP9 activity (Fig. 8A). Interestingly, no MMP2 activity was detected. MMP9 activity in the spinal cords of Plg+ mice was induced in response to MOG peptide immunization, as little to no MMP9 activity was observed in unchallenged mice regardless of genotype (Fig. 8B). Densitometry of gel images revealed a statistically significant diminution in MMP9 activity for spinal cords harvested from EAE-challenged Plg− mice relative to Plg+ mice (Fig. 8C). MMP9 activity did not appear to be a function only of disease score, as Plg− mice with similar scores to Plg+ animals did not have similar levels of MMP9 activity. To determine whether the altered MMP9 activity was linked to plasminogen-dependent changes in overall MMP9 expression or MMP9 zymogen activation in spinal cords, qPCR-based MMP9 RNA analyses were performed. At the peak of disease in control mice (day 14), no significant difference in MMP9 mRNA levels was observed between the Plg+ and Plg− mice (Fig. 8D).
Figure 8.

Reduction of MMP9 activity in Plg− spinal cords. A, Zymography was performed on protein samples from spinal cords harvested at the peak of disease (day 16) from animals challenged with EAE. No significant MMP2 activity was noted. However, while all EAE-challenged Plg+ animals showed readily appreciable MMP9 bands, the majority of Plg− mice showed no significant bands (the lone Plg− animal with a notable band is shown here). B, Samples from spinal cords of unchallenged mice from either genotype demonstrated little to no evidence of MMP9 activity. C, Images of the gels were captured, and, using ImageJ, quantitative densitometry was performed. A significant decrease in relative activity was noted for the Plg− animals compared with the Plg+ animals. Further, there was no significant difference between the EAE-challenged Plg− mice and the unchallenged Plg− mice. D, Quantitative PCR was performed to assess MMP9 expression in spinal cords of mice at the peak of EAE disease (day 14). No significant difference in MMP9 expression between EAE-challenged Plg− and Plg+ animals was found. Further, no significant difference between EAE-challenged and EAE-unchallenged mice was observed.
Delay in EAE onset in plasminogen deficiency is dependent on fibrinogen degradation
The premier proteolytic target of plasmin is fibrin (Bugge et al., 1996). Fib− mice or mice treated with fibrinogen-depleting anticoagulants have decreased severity of neuroinflammation (Akassoglou et al., 2004; Adams et al., 2007). With this in mind, we sought to determine whether plasminogen was mechanistically linked to fibrinogen in the modification of neuroinflammatory disease. We interbred Plg− with Fib− animals and challenged cohorts of Plg+/Fib+, Plg−/Fib+, Plg+/Fib−, and Plg−/Fib− mice with EAE. Plg+/Fib− mice showed reduced disease severity compared with Plg+/Fib+ mice (Fig. 9A), similar to pharmacologic depletion of fibrinogen and the prevention of thrombin-mediated fibrinogen polymerization (Inoue et al., 1996; Akassoglou et al., 2004; Han et al., 2008). Among all experimental groups, only Plg−/Fib+ mice had a significantly prolonged time to the onset of paralysis and exhibited significant early protection from neurologic signs of EAE (Fig. 9A). Plg−/Fib+ mice had a significantly prolonged time to initial disease onset compared with Plg−/Fib− mice (median time, 15 vs. 12 d, respectively; p < 0.05). More impressively, the Plg−/Fib+ animals had a significantly prolonged time to onset of paralysis compared with the Plg−/Fib− mice. Plg+/Fib+, Plg+/Fib−, and Plg−/Fib− mice had essentially an identical time to the onset of paralysis. However, there was a median of 7 d difference between these groups and the Plg−/Fib+ animals (Fig. 9B). These data suggest that plasminogen and fibrinogen are mechanistically coupled to regulate disease onset in EAE.
Figure 9.

Early protection in plasminogen deficiency is dependent upon fibrinogen. A, In comparative studies of Plg+/Fib+, Plg+/Fib−, Plg−/Fib+, and Plg−/Fib− animals, we found that the Plg−/Fib+ animals (n = 8) had significant protection from early disease compared with Plg−/Fib− (n = 9), Plg+/Fib+ (n = 14), and Plg+/Fib− mice (n = 9). Plg−/Fib− and Plg+/Fib− mice had similar disease onset but decreased severity compared with Plg+/Fib+ mice. *p < 0.05, ∧p < 0.01, #p < 0.005. B, Impressively, Plg−/Fib+ animals had a significantly prolonged time to paralysis compared with all other genotypes, including Plg−/Fib− mice.
Discussion
Previous studies of neuroinflammatory disease in mice have established that fibrinogen and plasminogen activators are positive and negative determinants, respectively, of disease severity (Lu et al., 2002; Akassoglou et al., 2004; East et al., 2005; Adams et al., 2007; Davalos et al., 2012; Gur-Wahnon et al., 2013). A strong, but untested, inference of these findings was that a loss of plasmin-mediated fibrinolysis would worsen EAE. However, plasminogen has been linked to processes beyond fibrinolysis, including monocyte/macrophage cell migration, via mechanisms linked to other proteolytic targets, such as MMP9 (Gong et al., 2008). Therefore, we sought to determine the contribution of plasminogen to the initiation and progression of neuroinflammatory disease in EAE-challenged mice. Here, we report that mice with genetically imposed plasminogen deficiency developed significantly delayed onset of EAE and reduced paralysis. While we have not found an impairment in T-cell response in plasminogen-deficient animals, future studies to include additional early time points to study peripheral T-cell infiltration would be beneficial. Consistent with our genetics-based findings, pharmacologic suppression of plasminogen activation using TXA, a widely used pharmacological agent to suppress bleeding symptoms, also significantly delays the onset of EAE disease symptoms.
Fibrin is the premier, albeit not sole, proteolytic target of plasmin. Extravascular fibrin deposits are a conspicuous feature of sites of vascular leak and tissue injury. Accordingly, fibrin has been documented in demyelinating plaques of both MS patients and experimental animals with EAE (Sobel and Mitchell, 1989). In many disease settings, including neuroinflammation (Adams et al., 2007; Davalos et al., 2012), inopportune or exuberant fibrin promotes disease development and/or severity. Fibrin-rich matrices are an important local cue for leukocyte activation, mediated via interaction with the integrin receptor αMβ2 (Flick et al., 2004). Elimination of the αMβ2 binding motif on fibrinogen, while retaining clotting function, significantly protects mice in the settings of inflammatory joint disease, colitis, Duchene's muscular dystrophy, and, most notably, EAE (Adams et al., 2007; Flick et al., 2007; Steinbrecher et al., 2010; Vidal et al., 2012). In this latter context, fibrin matrices appear to drive microglial/macrophage migration and local activation events, leading to demyelination and loss of motor function in EAE (Adams et al., 2007; Davalos et al., 2012; Ryu et al., 2015). Based on prior findings in mice with altered fibrinogen and individual deficits in plasminogen activators, our studies were initiated with the working hypothesis that the loss of plasminogen and plasmin-mediated fibrinolysis would accelerate disease onset and/or exacerbate disease. However, alternatively, loss of plasmin-mediated proteolysis may impede macrophage migration, leading to diminished neuroinflammation. Indeed, this second hypothesis proved correct, as plasminogen deficiency led to a profound protection from the onset and progression of neuroinflammatory disease.
Contrary to our findings, loss of plasminogen frequently promotes inflammatory disease through impaired fibrinolysis. Specifically, Plg− mice develop exacerbated TNF-α-driven arthritis (Raghu et al., 2014), glomerulonephritis (Kitching et al., 1997), and inflammatory osteoporosis (Cole et al., 2014). Confirming dependence on fibrinolysis, genetically superimposed loss of fibrinogen effectively uncoupled plasminogen deficiency from worsened disease outcome in arthritis, glomerulonephritis, and osteoporosis. The role of plasminogen in neuroinflammatory disease appears to also depend, at least partially, on fibrinogen. Using genetic deficiencies of plasminogen and fibrinogen in combination and parallel, we determined that fibrinogen deficiency abrogates the early protection conferred by plasminogen deficiency alone. This suggests that the delay of EAE disease onset in plasminogen deficiency is mechanistically linked to an inability to clear fibrinogen. This early protection appears to be specific for the Plg− animals, since mice deficient only for fibrinogen show decreased EAE severity; however, without the significant delay of disease onset. Further, plasmin degradation of fibrin leads to the release of fibrin degradation products. These products are known to be increased in inflammatory conditions; however, direct contribution to leukocyte activation and biology have not been well characterized.
Plasminogen deficiency also leads to less inflammation in other contexts of neuroinflammation, such as LPS-induced neuroinflammation, despite the presence of spontaneous fibrinogen deposits (Hultman et al., 2014). The dual observations that fibrin promotes neuroinflammatory pathologies in EAE and that the loss of the premier fibrin clearance protease, plasmin, delays disease onset in the very same experimental setting, may point to fibrin-independent plasmin targets in neuroinflammation. Nevertheless, the absence of plasmin-mediated fibrin clearance appears to be at least one mechanism underlying the diminished EAE in Plg− mice. Beyond fibrin, other plasmin substrates could influence EAE pathogenesis. Since BBB breakdown is a prerequisite for disease progression (Floris et al., 2004), it is of particular interest that plasmin is reported to degrade components (e.g., laminin, fibronectin, collagen) critical to the architecture of the BBB (Liotta et al., 1981; Mackay et al., 1990). Plasmin is capable of proteolytically remodeling matrix components both directly and indirectly via matrix metalloproteinase activation (e.g., MMP1, MMP3, and MMP9; He et al., 1989; Mackay et al., 1990). MMP9 also plays a role in inflammatory diseases, including EAE; young MMP9-deficient animals have decreased disease severity (Dubois et al., 1999). Macrophage recruitment following thioglycollate-induced peritonitis is both plasminogen dependent (Ploplis et al., 1998) and MMP9 dependent (Gong et al., 2008). Consistent with this are our observations that MMP9 activity is significantly diminished in the spinal cords of EAE-challenged Plg− mice relative to challenged wild-type mice, and macrophage infiltrates within the CNS are reduced in Plg− mice compared with wild-type animals. While provocative, the direct linkage among plasminogen, MMP9 activity, leukocyte trafficking, and BBB leakage in disease outcome remains to be formally established. Future studies will dissect the effects of different plasmin substrates during the course of EAE.
Mice lacking singular plasminogen activator deficiencies do not phenocopy Plg− mice regarding the amelioration of EAE pathologies (Lu et al., 2002; East et al., 2005; Gur-Wahnon et al., 2013). The precise impact of tissue plasminogen activator (tPA) deficiency on EAE remains controversial; some studies report early protection followed by worsened late-stage disease and other studies report worsened disease throughout the disease course (Lu et al., 2002; East et al., 2005). Urokinase-type plasminogen activator (uPA)-deficient animals exhibit exacerbated disease following EAE challenge (Gur-Wahnon et al., 2013), whereas urokinase plasminogen activator receptor (uPAR)-deficient animals exhibit either early disease protection or worsened disease throughout (East et al., 2005; Gur-Wahnon et al., 2013). One model that may reconcile these seemingly disparate findings is that plasmin-mediated proteolysis of nonfibrin and fibrin substrates present temporal liabilities and benefits in the progression of neuroinflammatory disease. In this paradigm, the differential targeting of plasmin substrates resulting from the loss of individual plasminogen activators exacerbates EAE, whereas the loss of all plasmin-mediated proteolysis delays EAE onset. Absence of plasmin-mediated cleavage of matrix proteins that support BBB function could be key advantages not gained in tPA-, uPA-, or uPAR-deficient mice.
Evidence that plasminogen is a determinant of macrophage recruitment in inflammatory settings has led to studies demonstrating that multiple cell surface plasminogen binding proteins [e.g., histone H2B (Herren et al., 2006), α-enolase (Miles et al., 1991), S100A10 (MacLeod et al., 2003), and Plg-RKT (Andronicos et al., 2010)] also are central to macrophage migration (Das et al., 2007; Wygrecka et al., 2009; O'Connell et al., 2010; Lighvani et al., 2011). No direct analyses of cell surface plasminogen-binding proteins and EAE outcome have been reported, but their importance as a group is implied by our studies showing that pharmacological intervention with TXA ameliorates EAE pathologies. One mechanism of action of TXA is to suppress plasminogen binding to cell surface receptors and thereby suppress cell-associated plasminogen activation and pericellular proteolysis. Similarly, ε-aminocaproic acid, also a lysine analog, has been reported to limit EAE progression (Smith and Amaducci, 1982; Koh and Paterson, 1987). These drugs are used clinically as safe and effective agents to limit bleeding in surgical patients and to treat bleeding disorders (Tengborn et al., 2015). Future studies will determine whether there is any translational potential of these agents in early stages of neuroinflammation.
Footnotes
This work was funded by the Race to Erase MS Young Investigator Award (J.K.R.), National Institute of Neurological Disorders and Stroke Grant R35-NS-097976 (K.A.), and National Heart, Lung, and Blood Institute Grant 1K08-HL-105672 (E.S.M.). We thank Joseph Palumbo, Tanya Mullins, and Russell Ware for their insightful comments. We also thank Keith Kombrinck, David Sweet, Whitney Miller, Malinda Frederick, Leah Rosenfeldt, Pamela Rios Coronado, and Carolina Cruz for their excellent technical assistance.
E.S.M. has served on advisory boards for Baxalta, Shire, Bayer, and US WorldMeds on matters unrelated to this research. The authors declare no other competing financial interests.
References
- Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K (2007) The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med 204:571–582. 10.1084/jem.20061931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H, Probert L, Strickland S (2004) Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci U S A 101:6698–6703. 10.1073/pnas.0303859101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronicos NM, Chen EI, Baik N, Bai H, Parmer CM, Kiosses WB, Kamps MP, Yates JR 3rd, Parmer RJ, Miles LA (2010) Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood 115:1319–1330. 10.1182/blood-2008-11-188938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel D, Schindler S, Zipfel PF (2012) Plasminogen is a complement inhibitor. J Biol Chem 287:18831–18842. 10.1074/jbc.M111.323287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berri F, Rimmelzwaan GF, Hanss M, Albina E, Foucault-Grunenwald ML, Lê VB, Vogelzang-van Trierum SE, Gil P, Camerer E, Martinez D, Lina B, Lijnen R, Carmeliet P, Riteau B (2013) Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog 9:e1003229. 10.1371/journal.ppat.1003229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno NE, Yano Y, Takei Y, Gabazza EC, Qin L, Nagashima M, Morser J, D'Alessandro-Gabazza CN, Taguchi O, Sumida Y (2008) Protective role of thrombin activatable fibrinolysis inhibitor in obstructive nephropathy-associated tubulointerstitial fibrosis. J Thromb Haemost 6:139–146. 10.1111/j.1538-7836.2007.02826.x [DOI] [PubMed] [Google Scholar]
- Bugge TH, Flick MJ, Daugherty CC, Degen JL (1995) Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev 9:794–807. 10.1101/gad.9.7.794 [DOI] [PubMed] [Google Scholar]
- Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL (1996) Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 87:709–719. 10.1016/S0092-8674(00)81390-2 [DOI] [PubMed] [Google Scholar]
- Campbell PG, Andress DL (1997) Plasmin degradation of insulin-like growth factor-binding protein-5 (IGFBP-5): regulation by IGFBP-5-(201–218). Am J Physiol 273:E996–E1004. [DOI] [PubMed] [Google Scholar]
- Carlson T, Kroenke M, Rao P, Lane TE, Segal B (2008) The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med 205:811–823. 10.1084/jem.20072404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain D, Kreizman T, Shapira H, Shaltiel S (1991) Plasmin cleavage of vitronectin Identification of the site and consequent attenuation in binding plasminogen activator inhibitor-1. FEBS Lett 285:251–256. 10.1016/0014-5793(91)80810-P [DOI] [PubMed] [Google Scholar]
- Cole HA, Ohba T, Nyman JS, Hirotaka H, Cates JM, Flick MJ, Degen JL, Schoenecker JG (2014) Fibrin accumulation secondary to loss of plasmin-mediated fibrinolysis drives inflammatory osteoporosis in mice. Arthritis Rheumatol 66:2222–2233. 10.1002/art.38639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AD, De Nardo CM, Braine EL, Turner AL, Vlahos R, Way KJ, Beckman SK, Lenzo JC, Hamilton JA (2010) Urokinase-type plasminogen activator and arthritis progression: role in systemic disease with immune complex involvement. Arthritis Res Ther 12:R37. 10.1186/ar2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Burke T, Plow EF (2007) Histone H2B as a functionally important plasminogen receptor on macrophages. Blood 110:3763–3772. 10.1182/blood-2007-03-079392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3:1227. 10.1038/ncomms2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degen JL, Bugge TH, Goguen JD (2007) Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost 5 [Suppl 1]:24–31. 10.1111/j.1538-7836.2007.02519.x [DOI] [PubMed] [Google Scholar]
- De Nardo CM, Lenzo JC, Pobjoy J, Hamilton JA, Cook AD (2010) Urokinase-type plasminogen activator and arthritis progression: contrasting roles in systemic and monoarticular arthritis models. Arthritis Res Ther 12:R199. 10.1186/ar3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew AF, Tucker HL, Liu H, Witte DP, Degen JL, Tipping PG (2001) Crescentic glomerulonephritis is diminished in fibrinogen-deficient mice. Am J Physiol 281:F1157–F1163. [DOI] [PubMed] [Google Scholar]
- Dubois B, Masure S, Hurtenbach U, Paemen L, Heremans H, van den Oord J, Sciot R, Meinhardt T, Hämmerling G, Opdenakker G, Arnold B (1999) Resistance of young gelatinase B-deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J Clin Invest 104:1507–1515. 10.1172/JCI6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- East E, Baker D, Pryce G, Lijnen HR, Cuzner ML, Gveric D (2005) A role for the plasminogen activator system in inflammation and neurodegeneration in the central nervous system during experimental allergic encephalomyelitis. J Pathol 167:545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis S, Nuenke JM, Grindeland RE (1968) Identity Between the growth hormone degrading activity of the pituitary gland and plasmin. Endocrinology 83:1029–1042. 10.1210/endo-83-5-1029 [DOI] [PubMed] [Google Scholar]
- Flick MJ, Du X, Degen JL (2004) Fibrin(ogen)-alpha M beta 2 interactions regulate leukocyte function and innate immunity in vivo. Exp Biol Med 229:1105–1110. [DOI] [PubMed] [Google Scholar]
- Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, Thornton S, Degen JL (2007) Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest 117:3224–3235. 10.1172/JCI30134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris S, Blezer EL, Schreibelt G, Döpp E, van der Pol SM, Schadee-Eestermans IL, Nicolay K, Dijkstra CD, de Vries HE (2004) Blood-brain barrier permeability and monocyte infiltration in experimental allergic encephalomyelitis: a quantitative MRI study. Brain 127:616–627. [DOI] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS (2006) Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med 354:942–955. 10.1056/NEJMra052130 [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Gold R, Linington C, Lannes-Vieira J, Wekerle H, Kreutzberg GW (1993) Microglial involvement in experimental autoimmune inflammation of the central and peripheral nervous system. Glia 7:50–59. 10.1002/glia.440070110 [DOI] [PubMed] [Google Scholar]
- Godiska R, Chantry D, Dietsch GN, Gray PW (1995) Chemokine expression in murine experimental allergic encephalomyelitis. J Neuroimmunol 58:167–176. 10.1016/0165-5728(95)00008-P [DOI] [PubMed] [Google Scholar]
- Gong Y, Hart E, Shchurin A, Hoover-Plow J (2008) Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest 118:3012–3024. 10.1172/JCI32750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li J, Hagström E, Ny T (2011) Beneficial and detrimental effects of plasmin(ogen) during infection and sepsis in mice. PLoS One 6:e24774. 10.1371/journal.pone.0024774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur-Wahnon D, Mizrachi T, Maaravi-Pinto FY, Lourbopoulos A, Grigoriadis N, Higazi AA, Brenner T (2013) The plasminogen activator system: involvement in central nervous system inflammation and a potential site for therapeutic intervention. J Neuroinflammation 10:124. 10.1186/1742-2094-10-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L (2008) Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451:1076–1081. 10.1038/nature06559 [DOI] [PubMed] [Google Scholar]
- Hattori N, Degen JL, Sisson TH, Liu H, Moore BB, Pandrangi RG, Simon RH, Drew AF (2000) Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J Clin Invest 106:1341–1350. 10.1172/JCI10531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CS, Wilhelm SM, Pentland AP, Marmer BL, Grant GA, Eisen AZ, Goldberg GI (1989) Tissue cooperation in a proteolytic cascade activating human interstitial collagenase. Proc Natl Acad Sci U S A 86:2632–2636. 10.1073/pnas.86.8.2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, Waisman A, Rülicke T, Prinz M, Priller J, Becher B, Aguzzi A (2005) Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 11:146–152. 10.1038/nm1177 [DOI] [PubMed] [Google Scholar]
- Herren T, Burke TA, Das R, Plow EF (2006) Identification of histone H2B as a regulated plasminogen receptor. Biochemistry 45:9463–9474. 10.1021/bi060756w [DOI] [PubMed] [Google Scholar]
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N (1992) Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem 267:26031–26037. [PubMed] [Google Scholar]
- Hultman K, Cortes-Canteli M, Bounoutas A, Richards AT, Strickland S, Norris EH (2014) Plasmin deficiency leads to fibrin accumulation and a compromised inflammatory response in the mouse brain. J Thromb Haemost 12:701–712. 10.1111/jth.12553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Koh CS, Shimada K, Yanagisawa N, Yoshimura K (1996) Suppression of cell-transferred experimental autoimmune encephalomyelitis in defibrinated Lewis rats. J Neuroimmunol 71:131–137. 10.1016/S0165-5728(96)00150-6 [DOI] [PubMed] [Google Scholar]
- Kitching AR, Holdsworth SR, Ploplis VA, Plow EF, Collen D, Carmeliet P, Tipping PG (1997) Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J Exp Med 185:963–968. 10.1084/jem.185.5.963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CS, Paterson PY (1987) Suppression of clinical signs of cell-transferred experimental allergic encephalomyelitis and altered cerebrovascular permeability in Lewis rats treated with a plasminogen activator inhibitor. Cell Immunol 107:52–63. 10.1016/0008-8749(87)90265-6 [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y (2006) IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177:566–573. 10.4049/jimmunol.177.1.566 [DOI] [PubMed] [Google Scholar]
- Lassmann H, Schmied M, Vass K, Hickey WF (1993) Bone marrow derived elements and resident microglia in brain inflammation. Glia 7:19–24. 10.1002/glia.440070106 [DOI] [PubMed] [Google Scholar]
- Lighvani S, Baik N, Diggs JE, Khaldoyanidi S, Parmer RJ, Miles LA (2011) Regulation of macrophage migration by a novel plasminogen receptor Plg-RKT. Blood 118:5622–5630. 10.1182/blood-2011-03-344242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta LA, Goldfarb RH, Brundage R, Siegal GP, Terranova V, Garbisa S (1981) Effect of plasminogen activator (urokinase), plasmin, and thrombin on glycoprotein and collagenous components of basement membrane. Cancer Res 41:4629–4636. [PubMed] [Google Scholar]
- Liu Z, Li N, Diaz LA, Shipley M, Senior RM, Werb Z (2005) Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest 115:879–887. 10.1172/JCI23977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Bhasin M, Tsirka SE (2002) Involvement of tissue plasminogen activator in onset and effector phases of experimental allergic encephalomyelitis. J Neurosci 22:10781–10789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Thornton S, Duwel LE, Boivin GP, Giannini EH, Leiden JM, Bluestone JA, Hirsch R (1998) Inhibition of collagen-induced arthritis in mice by viral IL-10 gene transfer. J Immunol 161:1516–1524. [PubMed] [Google Scholar]
- Mackay AR, Corbitt RH, Hartzler JL, Thorgeirsson UP (1990) Basement membrane type IV collagen degradation: evidence for the involvement of a proteolytic cascade independent of metalloproteinases. Cancer Res 50:5997–6001. [PubMed] [Google Scholar]
- MacLeod TJ, Kwon M, Filipenko NR, Waisman DM (2003) Phospholipid-associated annexin A2–S100A10 heterotetramer and its subunits: characterization of the interaction with tissue plasminogen activator, plasminogen, and plasmin. J Biol Chem 278:25577–25584. 10.1074/jbc.M301017200 [DOI] [PubMed] [Google Scholar]
- Makowski GS, Ramsby ML (1998) Binding of latent matrix metalloproteinase 9 to fibrin: activation via a plasmin-dependent pathway. Inflammation 22:287–305. 10.1023/A:1022300216202 [DOI] [PubMed] [Google Scholar]
- Merrill JE, Kono DH, Clayton J, Ando DG, Hinton DR, Hofman FM (1992) Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice. Proc Natl Acad Sci U S A 89:574–578. 10.1073/pnas.89.2.574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF (1991) Role of cell-surface lysines in plasminogen binding to cells: identification of alpha-enolase as a candidate plasminogen receptor. Biochemistry 30:1682–1691. 10.1021/bi00220a034 [DOI] [PubMed] [Google Scholar]
- Munks MW, McKee AS, Macleod MK, Powell RL, Degen JL, Reisdorph NA, Kappler JW, Marrack P (2010) Aluminum adjuvants elicit fibrin-dependent extracellular traps in vivo. Blood 116:5191–5199. 10.1182/blood-2010-03-275529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell PA, Surette AP, Liwski RS, Svenningsson P, Waisman DM (2010) S100A10 regulates plasminogen-dependent macrophage invasion. Blood 116:1136–1146. 10.1182/blood-2010-01-264754 [DOI] [PubMed] [Google Scholar]
- Pepinsky RB, Sinclair LK, Chow EP, Mattaliano RJ, Manganaro TF, Donahoe PK, Cate RL (1988) Proteolytic processing of mullerian inhibiting substance produces a transforming growth factor-beta-like fragment. J Biol Chem 263:18961–18964. [PubMed] [Google Scholar]
- Pfaffl MW. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF (1998) Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood 91:2005–2009. [PubMed] [Google Scholar]
- Raghu H, Jone A, Cruz C, Rewerts CL, Frederick MD, Thornton S, Degen JL, Flick MJ (2014) Plasminogen is a joint-specific positive or negative determinant of arthritis pathogenesis in mice. Arthritis Rheumatol 66:1504–1516. 10.1002/art.38402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K (1996) Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med 2:287–292. 10.1038/nm0396-287 [DOI] [PubMed] [Google Scholar]
- Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP, Vagena E, Bedard C, Machado MR, Rios Coronado PE, Prod'homme T, Charo IF, Lassmann H, Degen JL, Zamvil SS, Akassoglou K (2015) Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun 6:8164. 10.1038/ncomms9164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmukhappa K, Matte U, Degen JL, Bezerra JA (2009) Plasmin-mediated proteolysis is required for hepatocyte growth factor activation during liver repair. J Biol Chem 284:12917–12923. 10.1074/jbc.M807313200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ME, Amaducci LA (1982) Observations on the effects of protease inhibitors on the suppression of experimental allergic encephalomyelitis. Neurochem Res 7:541–554. 10.1007/BF00965121 [DOI] [PubMed] [Google Scholar]
- Sobel RA, Mitchell ME (1989) Fibronectin in multiple sclerosis lesions. J Pathol 135:161–168. [PMC free article] [PubMed] [Google Scholar]
- Steinbrecher KA, Horowitz NA, Blevins EA, Barney KA, Shaw MA, Harmel-Laws E, Finkelman FD, Flick MJ, Pinkerton MD, Talmage KE, Kombrinck KW, Witte DP, Palumbo JS (2010) Colitis-associated cancer is dependent on the interplay between the hemostatic and inflammatory systems and supported by integrin alpha(M)beta(2) engagement of fibrinogen. Cancer Res 70:2634–2643. 10.1158/0008-5472.CAN-09-3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh TT, Holmbäck K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL (1995) Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev 9:2020–2033. 10.1101/gad.9.16.2020 [DOI] [PubMed] [Google Scholar]
- Swaisgood CM, Aronica MA, Swaidani S, Plow EF (2007) Plasminogen is an important regulator in the pathogenesis of a murine model of asthma. Am J Respir Crit Care Med 176:333–342. 10.1164/rccm.200609-1345OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengborn L, Blombäck M, Berntorp E (2015) Tranexamic acid—an old drug still going strong and making a revival. Thromb Res 135:231–242. 10.1016/j.thromres.2014.11.012 [DOI] [PubMed] [Google Scholar]
- Vidal B, Ardite E, Suelves M, Ruiz-Bonilla V, Janué A, Flick MJ, Degen JL, Serrano AL, Muñoz-Cánoves P (2012) Amelioration of Duchenne muscular dystrophy in mdx mice by elimination of matrix-associated fibrin-driven inflammation coupled to the alphaMbeta2 leukocyte integrin receptor. Hum Mol Genet 21:1989–2004. 10.1093/hmg/dds012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wygrecka M, Marsh LM, Morty RE, Henneke I, Guenther A, Lohmeyer J, Markart P, Preissner KT (2009) Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood 113:5588–5598. 10.1182/blood-2008-08-170837 [DOI] [PubMed] [Google Scholar]