

Abstract
In a subset of children experiencing prolonged febrile seizures (FSs), the most common type of childhood seizures, cognitive outcomes are compromised. However, the underlying mechanisms are unknown. Here we identified significant, enduring spatial memory problems in male rats following experimental prolonged FS (febrile status epilepticus; eFSE). Remarkably, these deficits were abolished by transient, post hoc interference with the chromatin binding of the transcriptional repressor neuron restrictive silencing factor (NRSF or REST). This transcriptional regulator is known to contribute to neuronal differentiation during development and to programmed gene expression in mature neurons. The mechanisms of the eFSE-provoked memory problems involved complex disruption of memory-related hippocampal oscillations recorded from CA1, likely resulting in part from impairments of dendritic filtering of cortical inputs as well as abnormal synaptic function. Accordingly, eFSE provoked region-specific dendritic loss in the hippocampus, and aberrant generation of excitatory synapses in dentate gyrus granule cells. Blocking NRSF transiently after eFSE prevented granule cell dysmaturation, restored a functional balance of γ-band network oscillations, and allowed treated eFSE rats to encode and retrieve spatial memories. Together, these studies provide novel insights into developing networks that underlie memory, the mechanisms by which early-life seizures influence them, and the means to abrogate the ensuing cognitive problems.
SIGNIFICANCE STATEMENT Whereas seizures have been the central focus of epilepsy research, they are commonly accompanied by cognitive problems, including memory impairments that contribute to poor quality of life. These deficits often arise before the onset of spontaneous seizures, or independent from them, yet the mechanisms involved are unclear. Here, using a rodent model of common developmental seizures that provoke epilepsy in a subset of individuals, we identify serious consequent memory problems. We uncover molecular, cellular, and circuit-level mechanisms that underlie these deficits and successfully abolish them by targeted therapeutic interventions. These findings may be important for understanding and preventing cognitive problems in individuals suffering long febrile seizures.
Keywords: epigenetics, febrile seizures, hippocampus, memory, NRSF, seizures
Introduction
Epilepsy, the third most common chronic brain disorder, is defined by spontaneous seizures, and these have been the central focus of research to date. Yet, epilepsy, especially temporal lobe epilepsy, a common type involving the hippocampal-limbic circuit, often involves cognitive deficits, including memory and decision-making problems, which contribute to poor quality of life (Hermann et al., 1997; Helmstaedter et al., 2003; Elger et al., 2004; Berg, 2011). The underlying mechanisms remain unclear (Mueller et al., 2012).
Many individuals with temporal lobe epilepsy have a history of long childhood febrile seizures (FSs). While FS are generally common and benign, FSs longer than 30 min, febrile status epilepticus (FSE), may cause hyperexcitability and epilepsy after an extended “silent period” (French et al., 1993; Berg and Shinnar, 1996; Dubé et al., 2007). A rodent model of experimental FSE (eFSE) has been characterized and used extensively to elucidate the mechanisms promoting epileptogenesis after eFSE (Baram et al., 1997; Dubé et al., 2010). eFSE also provokes enduring memory deficits (Dubé et al., 2009; Barry et al., 2015, 2016a), as observed in some children after FSE (Martinos et al., 2013).
Cognitive deficits can arise after an epilepsy-inducing insult before spontaneous seizures or independently from them (Elger et al., 2004; Hoppe et al., 2007; Bender et al., 2012). This suggests that epilepsy and memory problems following FSE may arise in the same limbic/hippocampal networks via independent or partially overlapping processes.
We have recently identified some of the mechanisms underlying functional changes in hippocampal networks that promote epilepsy after eFSE (Brewster et al., 2002; Dubé et al., 2007, 2010; McClelland et al., 2011, 2014; Patterson et al., 2015). Specifically, we uncovered coordinated transcriptionally regulated changes in expression of multiple genes that govern neuronal behavior. These changes, including repression of the ion channel HCN1 (Brewster et al., 2002; Dubé et al., 2007; McClelland et al., 2011), were common to several insults provoking temporal lobe epilepsy in several laboratories (Brewster et al., 2002; Jung et al., 2007, 2011; Huang et al., 2009; McClelland et al., 2011). Importantly, this large-scale, proepileptogenic transcriptional program involved the transcription factor Neuron Restrictive Silencing Factor (NRSF) (Huang et al., 2009; McClelland et al., 2014; Brennan et al., 2016). NRSF is canonically expressed in non-neuronal tissues where it suppresses neuron-specific genes (Schoenherr and Anderson, 1995; Z. F. Chen et al., 1998; Ballas and Mandel, 2005; Mandel et al., 2011). More recently, NRSF expression in mature neurons has been described where the factor may be crucial for normal function (Ballas and Mandel, 2005; Gao et al., 2011). Indeed, aberrant NRSF function might contribute to Huntington disease (Conforti et al., 2013) and aging/dementias (Lu et al., 2014). In hippocampus, epilepsy-inducing insults increase NRSF levels and activity in neurons (Palm et al., 1998; Roopra et al., 2001; Garriga-Canut et al., 2006; McClelland et al., 2011, 2014; Rodenas-Ruano et al., 2012; Brennan et al., 2016), likely via activation of sirtuins and repression of microRNA124 (Brennan et al., 2016). Blocking NRSF chromatin-binding after SE attenuated the ensuing epilepsy (McClelland et al., 2011), highlighting the importance of NRSF in the underlying hippocampal network transformation.
NRSF also contributes to regulating gene expression programs during neuronal maturation (Schoenherr and Anderson, 1995; Z. F. Chen et al., 1998; Gao et al., 2011). During the developmental stage of FSE (infancy/childhood in human, P10-P11 in rat), hippocampal dentate gyrus (DG) granule cells (GCs), are still differentiating and would be particularly susceptible to aberrantly increased NRSF levels. Indeed, abnormal maturation of GCs following SE contributes to formation of aberrant hippocampal networks (Thind et al., 2008; Murphy et al., 2012; Goldberg and Coulter, 2013). The complex roles of NRSF in immature and mature neurons render it a promising candidate for mediating structural and functional hippocampal defects following eFSE, promoting cognitive impairments. We tested this hypothesis by transiently blocking NRSF function after eFSE, then testing for memory. We examined hippocampal oscillations underlying memory processes and assessed structural integrity of the hippocampal network, including the effects of eFSE and NRSF blockade.
Materials and Methods
All experiments were conducted with approval of the institutional animal care committee and conformed to National Institutes of Health guidelines. Experimental design, performance, and analysis aimed to adopt guidelines for rigor and reproducibility in science. Group sizes were determined a priori, and animals were randomly assigned to experimental groups. All analyses were performed without knowledge of treatment group.
Induction of eFSE.
eFSE has been described (Baram et al., 1997; Dubé et al., 2010; Patterson et al., 2015). P10-P11 male rats (20–25 g) were used. Potential hyperthermia-related injury was prevented by adhering the tail to a thin piece of wood using a gauze and glue, and applying a generous amount of a glycerin-based hydrating ointment to the tail. The gauze was then wrapped 3× around the tail, and the bandage was taped. Ears and paws were coated with the glycerin-based hydrating ointment. Rats were placed in pairs inside a 3 L flask and subjected to a continuous stream of warm air. The beginning of the seizure was determined by the onset of freezing followed by chewing automatisms. Once seizures began, core temperatures were assessed and repeated every 2 min to ensure body temperature of 39.5C°-41°C during eFSE. Hyperthermia was maintained for 60 min. At the end of eFSE, animals were briefly immersed in room temperature water and returned to home cages. There was no mortality.
ICV infusions of oligodeoxynucleotides.
An intact NRSE or a scrambled (SCR) NRSE sequence oligodeoxynucleotide (ODN) was infused ICV 3 h after the end of eFSE (Sigma-Aldrich). Four experimental groups were used in these experiments. The first was a non-eFSE control animal that received a scrambled sequence of NRSE (CTL-SCR). The second was a non-eFSE control who received the NRSE sequenced ODN (CTL-NRSE). The third were eFSE animals infused with scrambled ODN (eFSE-SCR). Finally, there was a group of eFSE animals infused with NRSE ODNs (eFSE-NRSE). These groups will be identified by their respective acronyms. A sharp-tipped cannula was created using a 30 gauge hypodermic needle with the depth delineated for the ventricles of a P10 rat pup (3 mm) by a small amount of solder. The cannula was attached to microtubing and the tubing was attached to a 10 μl Hamilton syringe. The syringe cannula was then loaded with either NRSE or SCR ODNs and placed into a syringe pump (KD Scientific). ICV infusions were done free-hand by using bregma demarcations, which are visible through the skin in a P10 rat pup. The needle was inserted through the skin and skull and into the brain up until the stopper was reached. 2.5 nmol of either NRSE or SCR ODN were infused bilaterally into the ventricles at a rate of 0.5 μl/min and a volume of 2.5 μl/hemispheres. ICV infusions were repeated exactly as described again 24 h after eFSE to ensure complete abatement of NRSF during noted times of augmentation after seizures.
Surgery.
At age 3–5 months, rats underwent implantation of a bilateral array of recording electrodes. The rats were anesthetized with inhaled isoflurane and placed in a stereotaxic frame. The skull was exposed and four screws inserted, two anterior to the left and right ends of bregma and two left and right over the cerebellum. Grounding was achieved via the right cerebellar screw. Rats were chronically implanted with a custom implant that allowed for local field potential (LFP) recordings from a 2 × 8 array of 50 μm stainless steel electrodes (California Fine Wire) divided between the left and right hippocampus (−3.8 anteroposterior, ±3.8 mediolateral). The electrode tips ranged from 2.5–3.5 mm in length from the skull surface. All implants were fixed to the skull via the skull screws (FHC) and Grip Cement (Dentsply). The wound was sutured and topical antibiotic applied. The interval between surgery and the beginning of electrophysiological recording was 1 week.
Active avoidance.
Animals underwent testing in the active place avoidance task (Biosignal). In this task, animals learn to associate an unmarked region of space with a mild shock on a constantly rotating arena. The rats must attend to their ever-changing position in the room frame lest they be rotated into a predetermined shock zone where they receive a mildly painful electrical shock.
Rats were lightly anesthetized and implanted with a stainless-steel swivel in the skin between the shoulders. This allowed attachment of a cable with an light emitting diode (LED) at the end that allows for automated tracking and also the delivery of shock. Experimenters were blind as to which animals were control and FSE.
The arena consists of a steel disc 82 cm in diameter and is lighted from both above and below. The arena is centered in a room where it is ∼50 cm from black curtains on the S and E sides and 50 cm from white walls on the N and W sides. The N and W walls have an 11 cm gray power strip that forms a continuous line 50 cm above the floor of the arena. Two rectangular spatial cues (30 cm high × 43 cm wide) depicting a red star (centered at W position) and a black circle (centered at N position), both on a white background, were placed 18 cm above the arena floor. An additional rectangular polarizing cue (53 cm high × 84 cm wide) made of white paper (Color-Aid 2.5 gray) with five 2.5 cm wide diagonal black stripes (Color-Aid 9.5 gray) was centered at the N position, 5 cm above the gray power strip.
On day 1 of training, the animal was connected to the shock cable and introduced to the rotating arena for a 10 min habituation without shock. On all subsequent sessions on training days 1 and 2, rats received a 0.4 mA shock in an unmarked 876 cm wedge-shaped sector covering a 60° arc in the NE sector of the arena. The shock zone was stable in the room frame while the arena rotated. The entrance latency of the shock was 1 ms, the shock duration was 0.5 s, and the intershock latency was 2 s. Mixed groups of eFSE and control rats were trained in eight 10-min sessions per day for 2 d (16 sessions). Performance measures were recorded and analyzed using custom software (Biosignal).
Electrophysiology and recording protocols.
Rats were tethered to a recording cable while exploring the stable arena or during the active avoidance task. EEG signals were preamplified × 1 at the headstage and channeled through the tether cable to the signal amplifiers and computer interface. LFPs were sampled at 30 kHz and filtered at 1–9000 Hz (Neuralynx). All signals were referenced against a tetrode wire above the cerebellum. The rat's location in the arena was sampled at 60 Hz (Biosignal) using a digital camera that detected an LED placed near the animal's head. Position and electrophysiology timestamps were synchronized offline using custom software. An additional LED fixed to the rotating arena, in register with the animal LED, allowed for accurate calculation of speed when the arena was moving.
Recording sessions during exploration of the stable platform were used to assess baseline hippocampal EEG properties in the absence of cognitive demand. Two consecutive recording sessions were then made during performance of the active avoidance task. Rats were connected to the shock tether via a pin on the animal's neck. Two minutes later, the arena began to rotate and the recording session in the active avoidance context lasted 10 min. The alternation from stable arena exploration to avoidance was typically separated by 5–10 min.
Signal processing of LFPs.
LFP signals were analyzed using the MATLAB Signal Processing Toolbox (The MathWorks) and the Chronux toolbox. Spectrograms (1 s window, 1 s overlap, tapers, 1–140 Hz) (Barry et al., 2016b) computed from fast Fourier transforms were performed over time course of the recording sessions. Only segments where the animal's speed was ≥5 cm/s were included in the analysis. The mean frequency and power (normalized by the sum of the spectrum) of the signal were calculated in θ (5–12 Hz) and slow γ (25–50 Hz) bands. These frequency bands were chosen because of their established roles in memory processes (Buzsáki and Wang, 2012; Colgin, 2016). We also analyzed the relationship between animal speed and θ frequency in a similar manner to previous work (Richard et al., 2013; Barry et al., 2016b). Only one channel from the left and right hippocampus was used for analysis. In the case of multiple suitable electrodes, the electrode signal with the largest mean θ/δ ratio was selected for analysis.
ChIP.
The ChIP procedure was modified from McClelland et al. (2011) and Singh-Taylor et al. (2017). Briefly, 3 h after ODN infusion, pups were rapidly decapitated and their hippocampi were removed and stored at −80°C. Hippocampi were thawed, cross-linked with methanol-free formaldehyde for 10 min at room temperature in PBS. Neutralization of cross-linking was achieved with the addition of glycine. Tissue was added to homogenization buffer (50 mm HEPES, pH 8.0, 140 mm NaCl, 1 mm EDTA, 0.4% Igepel CA-630, 0.2% Triton X-100, and a mixture of protease inhibitors) and centrifugation aided in nuclei collection. Nuclei were sonicated for 30 min using a Diagenode sonicator to an average size of 500 bp. After cellular debris was removed, lysate was precleared overnight with Protein-A/G (Santa Cruz Biotechnology) at 4°C and then incubated with 10 μg of either control nonimmune serum (IgG) (Cell Signaling Technology) or anti-NRSF (Santa Cruz Biotechnology), overnight at 4°C in buffer containing 20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, and protease inhibitors. Protein A/G beads precleared with salmon sperm DNA (400 μg/ml) were added to lysate for 2 h. The beads were washed several times and then eluted using a buffer containing 2% SDS and 0.2 m sodium bicarbonate. After reversal of cross-linking at 65°C overnight, the bound DNA was purified and eluted using the QiaQuick MinElute PCR purification kit (QIAGEN). Quantitative PCR (qPCR) amplification was done using SYBR Green chemistry (Roche) on a Lightcycler 96 (Roche) with primers specific for Hcn1.
Golgi impregnation and analyses.
We have previously validated findings generated by Golgi in our hands with results obtained from the use of GFP-expressing mice (e.g., Y. Chen et al., 2016). Rats were deeply anesthetized with pentobarbital and transcardiacly perfused with aldehyde fixative (sliceGolgi Kit, Bioenno Tech 003760). After perfusion, brains were immediately removed and drop-fixed in aldehyde for 48 h. The sliceGolgi Kit protocol was used to impregnate neurons for structural analyses. Brains were sectioned into 200 μm coronal slices on a vibratome, and slices were immersed in impregnation solution for 7 d at room temperature in the dark. Sections were rinsed with 0.01 m PBS-T and transferred into a staining solution for 8 min followed by a 1 min 0.01 m PBS-T wash. Slices were treated with poststaining solution for 4 min followed by 4 × 5 min PBS washes. Slices were mounted onto gelatin-coated slides, dried overnight, dehydrated with 100% EtOH and xylene, and coverslipped with Permount (Fisher).
Golgi analyses were conducted without knowledge of treatment groups at the same level in dorsal hippocampus for all animals and groups. DG GCs were selected based on their structure (small soma with fan-like apical processes and few basal processes) and their location. GCs were counted in both the upper and lower blades of the hilus over ∼11,500 μm of dorsal hippocampus per animal. Hilar basal dendrites were categorized as a process pointing into the hilus that is ∼5-fold thicker than an axon. Sholl analysis of dorsal hippocampal CA1 cells was completed on fully impregnated cells. Only long-shaft CA1 neurons were selected, and 93 neurons (average 10/animal) were analyzed. Sholl of DG neurons was completed on 263 neurons with an average of 65 neurons per group as described elsewhere.
Hippocampal organotypic slice culture and in vitro “seizures.”
To test the direct effects seizure-like events on the structure of developing GCs, an in vitro system was used. Heterozygous Thy1-YFP male mice were bred to C57BL/6 female mice to produce heterozygote litters. These pups all expressed yellow fluorescent protein (YFP) driven by the Thy1 promoter, which allowed for easy visualization of GCs with fluorescent microscopy. At P7, pups were rapidly decapitated and their brains were removed under sterile conditions into ice-cold prep medium: MEM (Invitrogen), l-glutamine (Invitrogen), HEPES buffer (Fisher Scientific), magnesium sulfate (Sigma), and cell culture-grade water (GE Healthcare). Brains were bisected, and horizontal hippocampi slices 200 μm in thickness were obtained using a McIllwain Tissue Chopper. Dorsal sections of hippocampi were isolated from the rest of the slice and placed four at a time on a 0.4 μm, 30-mm-diameter cell culture insert (Merck Millipore). Inserts were then placed in 6-well plates and into growth medium: MEM, HBSS (Invitrogen), l-glutamine, magnesium sulfate, sodium bicarbonate (Invitrogen), HEPES, heat-inactivated horse serum (Invitrogen), ascorbic acid (Sigma), and cell culture-grade water. Six-well plates were moved to and maintained in a CO2-enriched incubator (Thermo). On DIV 7, seizure-like activity was induced by transferring inserts into culture medium containing 6 μm kainic acid (KA; Abcam) for 24 h (Richichi et al., 2008). Seizure-like activity was terminated by transferring cultures to fresh media. Structural abnormalities resulting from seizure-like activity were examined by fixing cultures with ice-cold PFA at 3 weeks after KA exposure. Hippocampi were then removed from inserts and mounted on gelatin-coated slides. Endogenous YFP in the processes of DGC was readily visible allowing for appropriate analyses.
To assess the role of NRSF in GC dysmaturation provoked by KA-induced seizures like events, we applied, following the exposure to KA, either the NRSF blocking oligodeoxynucleotide (NRSE-ODN) or a scrambled ODN (1 μm for 72 h) to subsets of both control and “seizure”-experiencing cultures. Cultures were maintained for 3 weeks and then harvested (at DIV 28) to examine GC maturation.
Hippocampal neuron culture.
Hippocampal neuron cultures were derived from postnatal day 0 rats. Hippocampi were quickly removed and incubated for 30 min in buffered salt solution containing 10 units/ml papain (Worthington). Cells were then mechanically triturated and plated at a density of 400–460 cells/mm2 on 12 mm coverslips that were precoated with poly-d-lysine (Sigma). Cultures began incubation with Neurobasal Medium with B-27 supplement (Invitrogen) at 36°C and 5% CO2. At 3 h later, half of the culture medium was replaced with a Neurobasal Medium/B-27-based medium that was preconditioned with 1- to 2-week old non-neuronal cell culture prepared from P3-P4 rat cortices. Cultures were refreshed every ∼72 h with conditioned medium. On DIV 7, cultures were exposed to 100 nm BODIPY linked ODNS (NRSE or SCR). BODIPY is a small, fluorescent molecule that allows for visualization of the ODN without structurally interfering with its transportation across membranes. New ODNs were added to cultures each time they were refreshed. Cultures were collected on DIV 28 by fixing them with fresh ice-cold 4% PFA (Fisher) and affixing coverslips to precoated microscope slices with mounting media (Southern Biotechnology).
Statistical considerations.
Rats were assigned to groups randomly, and group sizes were determined a priori based on expected effect size and variance. All analyses were performed without knowledge of treatment group. We used several a priori determined comparisons and aimed to apply the appropriate statistical tests. For example, because the behavioral and place cell datasets contained data from multiple sessions in single animals and the LFP dataset contained data from up to 3 recording sessions from each animal, the observations for each of these measures within single animals were likely to be correlated, and these data should be represented as a cluster. In this case, the existence of a relationship between each measure of interest within an individual animal may be assumed. In this study, we used generalized estimating equations (SPSS), a class of regression marginal model, for exploring multivariable relationships between clustered response data in control and FSE animals in the active avoidance task. At the behavioral level, we tested for differences in the number of shocks, the number of entrances into the shock zone, and the amount of time spent opposite the shock zone. We used one-way ANOVA and post hoc Bonferroni's tests to compare multiple groups, and two-way ANOVA for the Sholl analyses, involving treatment and distance from soma. Paired t tests were used to compare performance of an individual rat at two time points. Unless noted otherwise, Prism 6 was used (GraphPad Software) and data are presented as group means with SEs.
Results
NRSF nuclear levels and binding to the chromatin are augmented after eFSE and can be abrogated post hoc using a decoy strategy
The levels of NRSF (Fig. 1A) in nuclear extracts of hippocampi from eFSE rats were augmented 2 d after eFSE compared with controls (Fig. 1B,C) where interindividual variability was noted. This increase persisted at 19 and 60 d after the eFSE (Fig. 1C).
Figure 1.
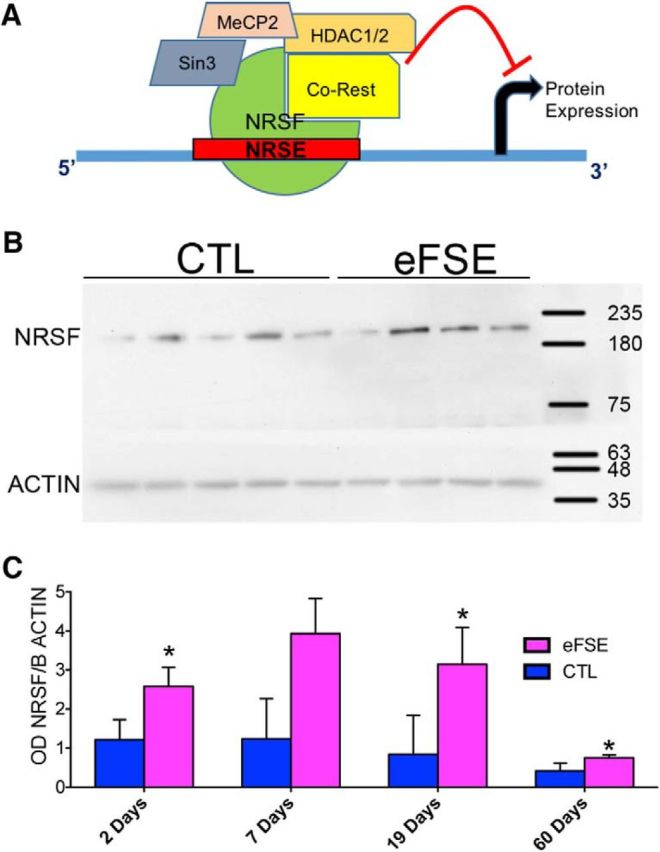
NRSF nuclear levels and binding to the chromatin are augmented after eFSE. A, Schematic of NRSF structure and cofactors. B, Western blot of representative nuclear extract from hippocampi of control or eFSE rats obtained 7 d after the seizures. C, The quantitative analyses of NRSF protein levels in nuclear extracts of hippocampi from eFSE rats were significantly augmented 2, 19, and 60 d after eFSE compared with controls. Data are mean ± SEM. Levels were lower in the older groups. *p < 0.05.
To assess the functional binding of NRSF to target-gene chromatin, we chose as a marker gene for NRSF chromatin-binding, hcn1, a gene that encodes for hyperpolarization-activated cyclic nucleotide-gate channel 1 (Fig. 2A): HCN1 is expressed within neurons in the hippocampal formation and is reduced after eFSE (Brewster et al., 2002). NRSF is expressed in principal cells in the hippocampal formation (Fig. 2B) and has previously been shown to bind and repress HCN1 within pyramidal cells after status epilepticus (McClelland et al., 2011, 2014). We examined the specificity of NRSF binding to NRSE by measuring the binding of the repressor to regions of the gene that lack an NRSE (schematic, Fig. 2A,E). To interfere with NRSF chromatin binding after eFSE, we used a decoy strategy using ODNs. Specifically, we designed synthetic, protected ODNs with a sequence of the NRSE of hcn1 (Fig. 2A), and used an SCR as a control (McClelland et al., 2011, 2014). NRSE-ODNs bind NRSF, sequestering the protein and preventing it from binding to chromatin and repressing gene expression. We assessed the ODNs' ability to enter neurons and their nuclei by the use of ODNs tagged with the fluorescent dye BODIPY and determined that ODNs entered hippocampal neurons grown in primary cultures, and accumulated inside their nuclei, the location of chromatin (Fig. 2C). We then compared NRSF function (chromatin binding) in four groups of rats: controls that received SCR-ODNs, controls that received NRSE-ODNs, and eFSE-experiencing rats that received either SCR-ODN or NRSE-ODNs.
Figure 2.
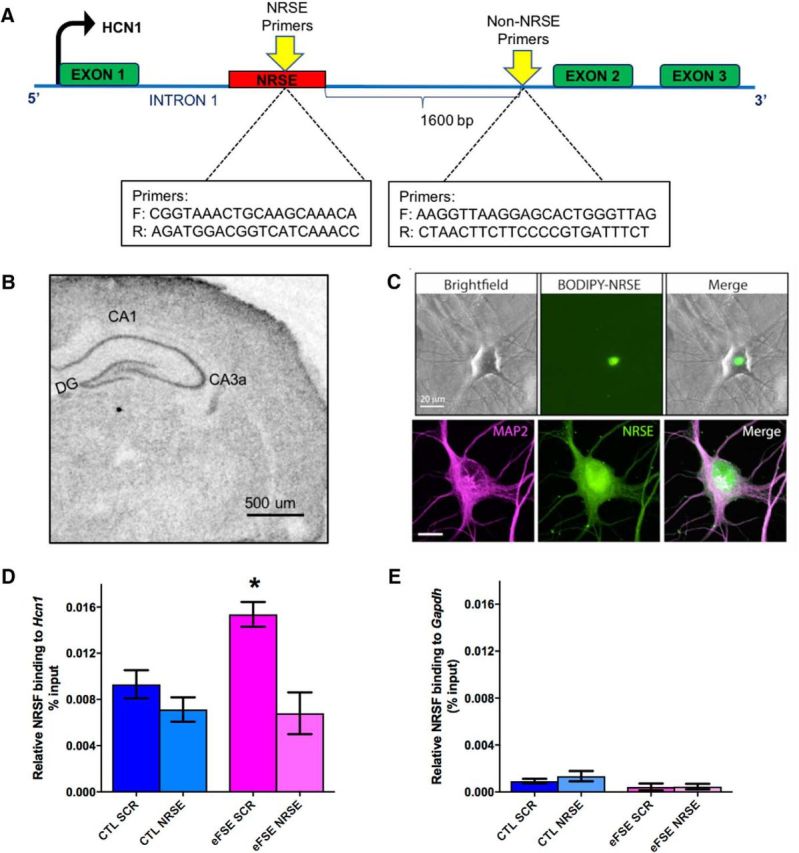
Augmented binding of NRSF to a target marker gene is specific, selective, increased after eFSE, and abrogated by a decoy strategy. A, Schematic showing the gene-structure and binding sites for NRSF (NRSF recognition element [NRSE]) within a target-gene, hcn1, and of a control region lacking an NRSE. B, In situ hybridization performed on a P11 rat brain section, demonstrating endogenous expression of NRSF in the hippocampus. A classical neuronal distribution is apparent. Scale bar, 500 μm. C, To interfere with NRSF chromatin binding after eFSE, we used a decoy strategy using ODNs with a sequence of the NRSE of hcn1. The ODNs enter hippocampal neurons grown in culture and their nuclei, as apparent by the use of ODNs tagged with the green fluorescent dye BODIPY. Top, Unstained neurons shown in bright-field, under green fluorescence setting (for visualizing the BODIPY) and as a combined image. Bottom, Neurons were subjected to immunohistochemistry for the dendritic marker MAP2, then imaged with both red and green fluorescence filters (left and middle), as well as combined. D, The use of ChIP demonstrated that NRSF occupancy at the hcn1 gene was not different between control rats that were given a scrambled ODN (CTL-SCR) and controls administered NRSE-ODN (CTL-NRSE; t(20) = 1.04; p > 0.05; Bonferroni's multiple-comparisons test). However, NRSF binding to HCN1 was significantly augmented in hippocampi of eFSE-SCR rats 3 h after eFSE (t(20) = 3.12, p < 0.05). Administration of NRSE-ODN after eFSE significantly reduced NRSF occupancy at the gene hcn1 in the eFSE-NRSE group compared with eFSE-SCR (t(20) = 4.07, p < 0.05), and this binding was not significantly different from controls. E, The specificity of NRSF binding to the NRSE is apparent from the minimal binding of the repressor to a gene (Gapdh) that lacks an NRSE. *p < 0.05.
The use of ChIP demonstrated significant group differences in the binding of NRSF to chromatin (one-way ANOVA, F = 6.57, p = 0.003; Fig. 2D). NRSF occupancy at the hcn1 gene was not different in CTL-SCR and CTL-NRSE rats (t(20) = 1.04; p > 0.05; Bonferroni's multiple-comparisons test). However, NRSF binding to hcn1 was significantly augmented in hippocampi of eFSE-SCR rats 3 h after insult (t(20) = 3.12, p < 0.05). Administration of the NRSE ODN after eFSE significantly reduced NRSF binding in the eFSE-NRSE group compared with eFSE-SCR (t(20) = 4.07, p < 0.05), and this binding was not significantly different from controls. Both the binding of NRSF and the interference by decoy NRSEs were selective: NRSF binding to a non–NRSE-containing gene (GAPDH) was minimal and not significantly different from binding when nonimmune IgG rather than anti-NRSF were used as a precipitant (Fig. 2E). This was also the case when binding of NRSF to non–NRSE-containing regions of the hcn1 gene (Fig. 2A) was assessed (data not shown). Together, these findings indicated that NRSF bound selectively to NRSE-containing gene loci and this binding was abolished by “flooding” neurons with decoy NRSEs at the doses and administration regimens used here.
In line with the repression of HCN1 in hippocampal principal cells after eFSE, observed with the use of in situ hybridization (Brewster et al., 2002), eFSE led to a nonsignificant 17% reduction of HCN1-mRNA in whole-hippocampus homogenates (including diverse neuronal types), measured using RT-PCR. Administration of the NRSF-blocking oligodeoxynucleotide abolished this repression (eFSE-SCR vs eFSE-NRSE, t(13) = 2.33; p = 0.038).
Blocking NRSF function after eFSE prevents cognitive deficits months later
Approximately 3 months after the eFSE and the interventions, rats were subjected to the active avoidance spatial task (Fig. 3). During this behavioral test, rats are exposed to a rotating arena containing a shock zone quadrant. Rats are scored on their ability to encode and remember the location of the shock zone using unique visual cues placed around the arena. Behavior in this task was scored over 2 ds with 8 trials per day. We compared performance in this task of four experimental groups: control (CTL) rats that received either SCR or NRSE ODNs, and eFSE rats that received either SCR or NRSE-ODNs post hoc.
Figure 3.

eFSE provokes serious and enduring impairments of spatial memory, and these are abrogated by blocking NRSF function (chromatin binding) after the eFSE. A, As adults, rats experiencing eFSE and treated with a random ODN (eFSE-SCR) received significantly more shocks than both control groups (CTL-SCR, CTL-NRSE), as well as from eFSE rats treated post hoc with a blocker of NRSF (eFSE-NRSE). This indicated that they could not use spatial cues to remember the location of the shock zone (for the second day of training on the active avoidance test, one-way ANOVA; F = 11.11; p < 0.0001; CTL-SCR vs eFSE-SCR, t(148) = 4.65, p < 0.05; CTL-SCR vs eFSE-NRSE, t(148) = 0.16, p > 0.05; eFSE-SCR vs eFSE-NRSE, t(148) = 5.02, p < 0.05). eFSE-SCR rats also entered into the shock zone more than any other experimental group (group effect: F = 10.21, p < 0.0001; post hoc significance exists between CTL-SCR vs eFSE-SCR, and eFSE-SCR vs eFSE-NRSE). Similar results are obtained when assessing the duration of time spent in the opposite quadrant to the shock zone (F = 9.441, p < 0.0001; eFSE-SCR significantly different from CTL-SCR and eFSE-NRSE). B, Assessing the ability of the four groups to remember between training sessions, eFSE-SCR rats learned less well, as evident by equal number of entries into the shock zone on days 1 and 2 of the active avoidance test. The other experimental groups entered fewer times on day 2 compared with day 1 (paired t tests, day 1 vs day 2: CTL-SCR, p < 0.0001; CTL-NRSE, p < 0.0001; eFSE-SCR, p = 0.076; eFSE-NRSE, p < 0.0001). A similar result was obtained when time spent opposite the shock zone was compared between days 1 and 2 (paired t tests, day 1 vs day 2: CTL-SCR, p = 0.0006; CTL-NRSE, p = 0.001; eFSE-SCR, p = 0.69; eFSE-NRSE, p = 0.01). C, eFSE-SCR animals did worse on the three outcome measures compared with other groups when retested in the active avoidance test 1 month after initial testing (one-way ANOVA, F = 3.49, p = 0.04). D, Representative traces of rats from each of the four experimental groups, depicting a rat's movement around the active avoidance arena. Data are mean ± SEM. CTL-SCR, n = 4; CTL-NRSE, n = 4; eFSE-SCR, n = 6; eFSE-NRSE, n = 6. *p < 0.05.
On the first day, eFSE-SCR rats received significantly more shocks (15.48 ± 2.68) than each of the other three groups (CTL-SCR, 9.03 ± 2.29; CTL-NRSE, 7.718 ± 1.83; eFSE-NRSE, 5.71 ± 0.95), suggesting that they were not able to learn the location of the shock quadrant (F = 4.82, p = 0.003; CTL-SCR vs eFSE-SCR, p < 0.05). A second measure of spatial learning in this task is the duration of time a rat spends in the quadrant opposite the shock zone, indicating the presence of a strategy to avoid shocks (Fig. 3D). eFSE-SCR rats spent significantly less time in the opposite quadrant (214.7 ± 11.64 s) compared with the other three groups, suggesting that these animals used an alternative, nonspatial strategy to avoid shocks: that is, by running out of the shock zone immediately after a shock (CTL-SCR, 261.0 ± 17.81; CTL-NRSE, 287.6 ± 16.70; eFSE-NRSE, 282.5 ± 16.77; F3 = 5.39; p = 0.0014; CTL-SCR vs eFSE-SCR, p < 0.05). By the second day, the differences among groups were larger (Fig. 3A): eFSE-SCR rats received more shocks, entered the shock zone quadrant more times, and spent significantly less time in the opposite quadrant than any group: for example, for shock numbers, effects of treatment group (F = 11.1; p < 0.0001; CTL-SCR, 0.93 ± 0.2; eFSE-SCR, 6.5 ± 1.27). Strikingly, eFSE-NRSE rats were indistinguishable from either control group on any measure used to assess spatial memory (CTL-SCR vs eFSE-NRSE, p > 0.05 for number of shocks and time spent in opposite quadrant), suggesting that transiently blocking the function of NRSF after eFSE rescues from cognitive deficits provoked by eFSE (Fig. 3A).
Over the 2 d of testing, CTL-SCR animals improved significantly in this task (Fig. 3B), with less shocks on day 2 compared with day 1 (paired t test; t(31) = 3.67, p = 0.0009), less entrances into the shock zone (p < 0.00001), and more time spent in the opposite quadrant (p = 0.0006). CTL-NRSE rats were indistinguishable from the CTL-SCR group in improved performance in the three memory parameters (number of shocks, p = 0.0002; number of entrances, p < 0.00001; time spent in the opposite quadrant, p = 0.001). In contrast, eFSE-SCR rats did not improve over the 2 d. While they received fewer shocks on the second day, they entered the shock zone equally on both days (p = 0.08) and spent the same amount of time opposite the shock zone (p = 0.7), suggesting that they merely improved in leaving the shock zone after receiving a shock. Notably, treatment with the NRSE-ODN enabled the eFSE-NRSE group to perform as well as controls on the three measures of the active avoidance test. They received less shocks on day 2 compared with day 1 (p < 0.0001), entered the shock zone less on day 2 (p < 0.0001), and spent more time opposite the shock zone (p = 0.01; Fig. 3B).
Finally, retesting the rats a month later to assess the longevity of this memory demonstrated the enduring memory of the CTL and eFSE-NRSE groups (one-way ANOVA, F = 3.45, p = 0.02; Fig. 3C). In contrast, the eFSE-SCR group failed to remember the location of the shock zone, as evident from large numbers of shock zone entries: CTL-SCR versus eFSE-SCR, p < 0.05, shocks: CTL-SCR versus eFSE-SCR, p < 0.05, and reduced time in the opposite quadrant (p < 0.05). Blocking the action of NRSF after eFSE enabled rats to retain memories from the active avoidance test, rendering them indistinguishable from controls. This was evident in the number of shocks they received, entrances into the shock zone and time spent in the opposite quadrant (CTL-SCR vs eFSE-NRSE, p > 0.05 for all; Fig. 3C).
Together, these data indicate that eFSE greatly reduced a rat's ability to create spatial memories. Importantly, blocking NRSF activity transiently after the eFSE allowed NRSE-ODNs rats to learn and perform the active avoidance task. However, the mechanisms for both of these striking observations remained unclear.
Brain oscillations underlying spatial memory are altered after eFSE and further modulated by the administration of NRSE ODNs
To probe the functional mechanisms of the cognitive deficits caused by eFSE, and their reversal by post hoc blocking of NRSF function, rats were implanted with a bilateral array of recording electrodes in hippocampal region CA1. We focused on oscillations well associated with spatial memory (Colgin et al., 2009; Colgin, 2016; Buzsáki and Wang, 2012; Richard et al., 2013; Vaidya and Johnston, 2013; Barry et al., 2016b), such as θ and slow and fast γ oscillations. These rhythms have distinct origins and roles: slow γ is often considered to involve CA3-CA1 pathways and contribute to memory retrieval, whereas fast γ may reflect information flow in the entorhinal cortex-CA1 node of the hippocampal network (Colgin et al., 2009). We investigated these oscillations during exploration in the stable arena to obtain measures of the baseline network available for learning. We then recorded in the rotating arena during active avoidance, when the rats have a need to remember the location of the shock zone.
During exploration in the stable arena, we detected a significant effect of group on rats' mean movement speed (p = 0.001). Specifically, mean speed of eFSE-SCR (14.87 ± 0.668 cm/s) was significantly faster than that of CTL-SCR (11.52 ± 0.68, p < 0.001), CTL-NRSE (11.13 ± 1.04, p = 0.002), and eFSE-NRSE (12.17 ± 0.64, p = 0.003). Importantly, the speed of the eFSE-experiencing group treated transiently with NRSE-ODNs was indistinguishable from those of either control groups. This finding is notable because increased mean speed should correlate with an increased speed/θ relationship (Richard et al., 2013), yet animals in the eFSE-SCR group tended to have the lowest speed/θ correlations, implying circuit dysfunction (Barry et al., 2016b). Indeed, whereas θ frequency itself was not altered by either eFSE or the NRSE-ODN, the linear relationship between motor speed and CA1 θ frequency was influenced (Fig. 4A,B): Speed/θ correlation coefficients were robust in the CTL-SCR group (0.25 ± 0.041), reduced drastically by eFSE (to 0.07 ± 0.06 in the eFSE-SCR group), and partially restored in the eFSE-NRSE group (0.14 ± 0.02; Fig. 4C).
Figure 4.

Early-life eFSE modulates hippocampal rhythms of adult rats. A, Relationship of rat movement-speed in the active avoidance arena and amplitude and frequency of θ oscillations recorded from hippocampal electrodes. A single 10 min active avoidance session of a control (CTL-SCR) rat is shown. The spectrogram shows signal amplitude in relation to signal frequency (Top) and rat speed (Bottom). B, Speed-filtered spectrogram of A, indicating that the power and frequency of θ increase with speed. C, Relationship between speed of movement and θ frequency (blue dots) throughout active avoidance sessions of a representative rat from each of the four experimental groups. The line of best fit (black line) and the corresponding correlation coefficient between θ frequency and speed indicates a significant linear relationship. Significant linear relationship between speed and frequency occurred in all groups, except the eFSE-SCR rats. D, Fast γ-band frequency, recorded from CA1 was increased in eFSE-SCR rats and restored to control levels in eFSE-NRSE rats. E, F, eFSE led to reduction of the amplitude of slow γ-band oscillations both in the stable (E) and active-avoidance (F) arenas. Slow γ amplitude was similar to control levels when NRSF activity was transiently blocked after eFSE. Thus, eFSE disrupted the balance of fast and slow γ band oscillations, and this disruption was prevented by an intervention blocking NRSF function. *p < 0.05.
Fast γ mean frequency during exploration was significantly accelerated by eFSE (FSE-SCR rats: 81.64 ± 3.02 Hz; CTRL-SCR: 77.62 ± 1.88; CTL-NRSE: 78.57 ± 0.88; Fig. 4D). Remarkably, blocking NRSF function after eFSE abolished this acceleration (eFSE-NRSE: 74.09 ± 0.53). There was also a significant group effect (p = 0.003) on the normalized mean amplitude of fast γ oscillations. Compared with the CTL-SCR group (0.502 ± 0.003), mean amplitudes were slightly higher after eFSE (eFSE-SCR: 0.505 ± 0.003) and reverted to control levels with blocking NRSF (eFSE-NRSE: 0.4966 ± 0.0014). Thus, eFSE influenced baseline measures of both mean amplitude and mean frequency of fast γ oscillations in a manner that was congruent with the ability of the rat to remember the location of the shock zone during the active avoidance test. Faster and larger CA1 fast γ oscillations were associated with worse performance. The inability to filter fast γ oscillations (i.e., information flow between hippocampus and neocortex) could interfere with the acquisition of spatial information or its recall, a process thought to be associated with slow γ rhythms and information flow in the CA3-CA1 node of the hippocampal network (Colgin, 2016). Therefore, we examined the effects of eFSE and of blocking of NRSF function on CA1 slow γ rhythms.
During exploration of the stable arena, normalized mean amplitudes of slow γ were lowest in the eFSE-SCR group (0.187 ± 0.0015) and significantly higher (p < 0.001) in eFSE rats that were treated with NRSE-ODN (0.192 ± 0.001). Amplitude in this group was actually higher than in controls (CTL-NRSE: 0.188 ± 0.001; CTL-SCR: 0.190 ± 0.001; Fig. 4E). For frequencies, compared with CTL-SCR (32.56 ± 0.327) and CTL-NRSE (32.25 ± 0.059), slow γ frequency in eFSE rats was lower (31.69 ± 0.40), but this was no longer observed in the eFSE-NRSE group (32.44 ± 0.07). During the active avoidance test, when a demand for encoding and retrieval of spatial memory was great, similar and enhanced effects of eFSE on slow γ oscillation properties were noted. Frequencies were lowest in rats experiencing eFSE (eFSE-SCR: 31.37 ± 0.40) and restored by the early administration of NRSE-ODN to eFSE rats (eFSE-NRSE: 33.37 ± 0.555), yielding frequencies similar to those of both control groups (CTL-NRSE: 32.03 ± 0.5; CTL-SCR: 32.10 ± 0.66). As found in the stable arena, normalized mean amplitudes of slow γ were lowest in the eFSE-SCR group (0.1878 ± 0.0006) and significantly higher in eFSE rats that were treated with NRSE-ODN (0.1913 ± 0.0008; Fig. 4F).
Together, these findings point at potential mechanisms for both the deleterious effects of eFSE on spatial memory and for the ameliorating actions of blocking NRSF function transiently. The faster and higher-amplitude fast γ oscillations are consistent with failure of filtering of this cortical input within CA1 place cells, which is reported to depend on gradients of ion channels along apical dendrites (Buzsáki and Wang, 2012; Vaidya and Johnston, 2013). These filtering gradients ensure that each synaptic input carries equal weight during subthreshold integration at the soma preventing summation (Migliore et al., 2004; Noam et al., 2011; Buzsáki and Wang, 2012). In addition, eFSE led to attenuation of amplitudes and frequencies of slow γ oscillations, and these may derive from abnormal function of inputs to CA3 and CA1. Transient treatment with NRSE-ODN enabled the FSE-NRSE group to form spatial memories, associated with modulation of γ oscillations.
Blocking NRSF after eFSE ameliorates sources of aberrant excitatory synaptogenesis in DG GCs
The results above suggested that eFSE influences several properties of brain oscillations that underlie encoding and retrieval of spatial information. These included the correlation of θ rhythms recorded from CA1 neurons with animal speed, as well as amplitudes and frequencies of slow and fast γ oscillations, which reflect the routing of neural information throughout the hippocampal formation. Interestingly, blockade of NRSF function in the eFSE-NRSE rats enabled them to encode and retain spatial memories, associated with modulation of both fast γ and slow γ amplitude and frequency. Because it is thought that these two types of oscillations involve distinct microcircuitries, we proceeded to examine separately the effects of both eFSE and NRSF on the structural integrity of principal neurons in different compartments of the hippocampal formation.
We first examined the structure of apical and basal dendrites that carry excitatory synapses within the DG, which gates input onto CA3 and CA1 (Goldberg and Coulter, 2013). Seizures can influence the numbers of excitatory synapses onto GC via two mechanisms. First, seizures may promote retention of hilar basal dendrites (HBDs) that are normally lost with GC maturation (Kron et al., 2010; Koyama et al., 2012; Hester and Danzer, 2014). These basal dendrites are targets of both autaptic excitatory synapses as well as those from neighboring GCs (Thind et al., 2008; Kron et al., 2010; Murphy et al., 2012). These aberrant loops alter excitability within the DG, and might disrupt information flow within the DG “gate” to CA3 and CA1 (Thind et al., 2008; Goldberg and Coulter, 2013). Therefore, we examined whether eFSE promotes HBD retention in GCs and whether there was an effect of blocking NRSF after the insult.
GCs were identified in dorsal hippocampal sections obtained postmortem from all four groups of rats, based on the classic structural phenotype of this cell type (Schlessinger et al., 1975; Thind et al., 2008; Kron et al., 2010; Murphy et al., 2012) (Fig. 5A–D). The density of GCs per se did not vary among the groups (F = 1.78, p = 0.16; Fig. 5F). By contrast, the number of GCs with the dysmature retention of HBD varied by treatment group (F = 10.41, p < 0.0001). The number of HBDs on GCs was not different between the CTL-SCR (1.04 ± 0.44) or CTL-NRSE (0.65 ± 0.5) groups. However, the number of HBDs on GCs from eFSE-SCR rats were significantly increased (3.14 ± 1.22; p < 0.05). Blocking NRSF activity after eFSE abolished the difference in the number of HBD-possessing GCs (eFSE-NRSE: 1.21 ± 0.18; CTL-SCR vs eFSE-NRSE, p > 0.05; Fig. 5G). The results persisted when data were analyzed as the percentage of GCs with retained HBDs: CTL-SCR (1.38 ± 0.22%), CTL-NRSE (0.79 ± 0.59%), and eFSE-NRSE (1.68 ± 0.56%) did not differ (p = 0.15), whereas the percentage of dysmature GCs with persistent HBDs was significantly higher in the eFSE-SCR group (4.57 ± 0.79%) (Fig. 5E).
Figure 5.

eFSE disrupts dendritic maturation of DG GCs, promoting excessive excitatory synapses, and this is prevented by blocking NRSF function. A–D, Representative photomicrographs of the DG from each experimental group. Insets, Magnified Golgi-Cox-filled GCs. Red arrows indicate persistent HBDs. A′–D′, Representative traces of GCs from each experimental group, as used for analysis. E, A significantly higher percentage of GCs retain HBDs in eFSE-SCR rat, compared with other groups; this is not observed in eFSE-NRSE rats (group effect: one-way ANOVA, F = 25.56, p = 0.0002; Bonferroni's multiple-comparison tests, eFSE-SCR: p < 0.05 vs CTL-SCR, CTL-NRSE, and eFSE-NRSE). F, There was no group difference in the total number of GCs (p = 0.16). G, The total number of GCs with HBDs was increased in eFSE-SCR animals compared with both controls and eFSE-NRSE. H, Apical dendrites were exuberant in eFSE-SCR rats, with augmented dendritic arborization mainly in the GC layer of the DG, and this effect was not found in eFSE-NRSE rats (two-way ANOVA; p < 0.0001 for distance from cell body, treatment, and interaction). I, Total dendritic length was augmented in eFSE-SCR rats compared with all others, including the eFSE-NRSE group. Data are mean ± SEM. CTL-SCR: n = 3 (rats), n = 89 (neurons); CTL-NRSE: n = 3 (rats), n = 47 (neurons); eFSE-SCR: n = 4 (rats), n = 58 (neurons); eFSE-NRSE: n = 4 (rats), n = 65 (neurons). *p < 0.05.
The presence of dysmature basal dendrites provides a source for excitatory synapses that disrupt circuit function in the DG. A second possible source for aberrant synapses involves exuberant branching of apical GC dendrites, as recently reported after eFSE (Raijmakers et al., 2016). Therefore, we examined the structure of GC apical dendrites. Sholl analyses revealed a group effect (two-way ANOVA; interaction, F = 12.49 p < 0.00001; distance from soma, F = 627.3 p < 0.00001; experimental condition, F = 209.3, p < 0.00001). Branching in eFSE-SCR GCs was higher compared with those from CTL-SCR rats (p < 0.05), with augmented arborization primarily in the GC layer rather than the outer molecular layer (Fig. 5H). eFSE-NRSE GCs were indistinguishable from both control groups. For dendritic length, a group difference was found (F = 10.4, p = 0.00028), deriving from a significant increase in the eFSE-SCR group (519.5 ± 67.05 μm) versus all others (CTL-SCR: 415 ± 10.80; CTL-NRSE: 387.4 ± 19.71; eFSE-NRSE: 323.5 ± 44.1; Fig. 5I).
Together, these data indicate that eFSE increased sources of excitatory synapses of GCs via two mechanisms: dysmaturation, apparent as retention of HBDs and their synapses, as well as hyperbranching of apical dendrites, which promotes formation of excitatory synapses that contact other GCs and cell types within the DG, altering information flow.
eFSE-induced dendritic defects in CA1 pyramidal cells are independent of NRSF
eFSE generated impairments of spatial memory, associated with abnormal rhythms recorded from CA1 pyramidal cells. The higher amplitude and frequencies of fast γ oscillations suggested an impaired capacity of CA1 pyramidal cell apical dendrites to dampen this cortical input. Therefore, we examined the structural integrity of these dendrites, as well as the consequences of blocking NRSF function after FSE, a treatment that effectively restored spatial memory function.
We studied CA1 neurons from the same animals used for the cognitive studies, and used Sholl analyses of Golgi-impregnated neurons taken without knowledge of group (Fig. 6A–D). One-way ANOVA revealed a group effect (F = 13.74, p < 0.0001). Notably, no differences in arborization or in total dendritic length were found between the two control groups (CTL-SCR: 838.9 ± 66.04 μm; CTL-NRSE: 986.7 ± 47.60). Dendritic length was lower in the eFSE-SCR group (666.3 ± 38.36), and this was not ameliorated by transiently interfering with the function of NRSF (eFSE-NRSE: 590.6 ± 42.92; Fig. 6E). Together, these data indicated that eFSE reduced overall apical dendritic integrity in CA1 pyramidal cells by an NRSF-independent mechanism. Importantly, the data suggested that the improved cognitive function afforded by early NRSE-ODN treatment did not involve prevention of structural dendritic defects in CA1. Rather, blocking NRSF may have prevented repression of crucial functional components of CA1 apical dendrites, such as the ion channel HCN1, involved in regulating dendritic summation of synaptic input.
Figure 6.

eFSE-induced dendritic defects in CA1 pyramidal cells are independent of NRSF. A–D, Representative photomicrographs of area CA1 of dorsal hippocampus following with Golgi-Cox impregnation. A′–D′, Representative masks generated from CA1 pyramidal cells for analysis. E, Quantification of total dendritic length demonstrates impoverished dendritic arborization after eFSE, and no effect of blocking NRSF. Data are mean ± SEM. CTL-SCR: n = 3 (rats), n = 19 (neurons); CTL-NRSRE: n = 3 (rats), n = 21 (neurons); eFSE-SCR: n = 4 (rats), n = 20 (neurons); eFSE-NRSE: n = 4 (rats), n = 34 (neurons). *p < 0.05.
NRSF acts within the DG to mediate the disruptive effects of eFSE on GC maturation and connectivity
The data shown up to now suggested that eFSE promotes aberrant retention of excitatory synapses on basal dendrites of GCs, as well as the formation of supernumerary apical dendritic branching, and that these effects might be abrogated by blocking NRSF function during a critical period that follow eFSE. However, in vivo, we infused the NRSE-ODN throughout the whole brain. Therefore, it was possible that the effects of the ODNs influenced gene expression and neuronal structure in areas connecting to the DG rather than acting to prevent the effects of NRSF within the DG of eFSE rats.
To study the direct effects of NRSF on the structure of developing DG GCs, we used an in vitro organotypic slice culture system. Hippocampi cultured from P7 Thy1-YFP-expressing mice enabled direct visualization of GCs and their development (Fig. 7A,B). Notably, in vitro, organotypic cultures faithfully follow patterns of in vivo development (Bender et al., 2007) (Fig. 7A,B). Hippocampi were exposed to seizure-like activity at an age parallel to eFSE in vivo (DIV 7), using administration of KA at doses shown to provoke epileptiform discharges but not cell death (Richichi et al., 2008). Following the exposure to KA, we treated subsets of both control and “seizure”-experiencing cultures with either the NRSF blocking oligodeoxynucleotide (NRSE-ODN) or a scrambled ODN. Cultures were harvested at several time points to examine GC maturation. The seizure-like events promoted a robust increase in NRSF protein in DG nuclear extracts 24 h after the end of KA exposure (control DG: 1.047 ± 0.026 OD units/actin; KA-DG: 7.00 ± 1.33 OD units/actin; p < 0.01; Fig. 7C). Over the 3 weeks following KA exposure, the numbers of Thy1-expressing GCs varied by group (ANOVA, p = 0.0004). It was similar in control and KA groups a day after the seizure-like events (14.17 ± 4.8 and 13.40 ± 4.2 in controls and KA, respectively), was higher 1 week later (CTL: 18.8 ± 9.1; KA: 93.6 ± 17.7 p < 0.05, Bonferroni's post hoc test), but was no longer significantly higher at 2 weeks (36.9 ± 6.3; and 59.8 ± 4.8) and 3 weeks (18.4 ± 13.9 and 43.3 ± 9.1) after KA, in line with the in vivo data (Fig. 5F). In both control and KA cultures, most GCs appeared structurally mature with a typical tuft of apical dendrites radiating into the DG molecular layer (Fig. 7A,B, insets). However, some immature-looking GCs with a retained HBDs were found as well, and their numbers were influenced by group (F = 7.47; p = 0.027). To better assess the proportion of immature-appearing GCs, we examined the ratio of GCs that appeared dysmature by retaining the hilar basal dendrite to the total number of GCs. In addition, as exposure to a scrambled ODN did not influence the action of KA, we combined the KA and KA/SCR-ODN groups. We found significant differences among groups in both the total numbers of HBD-carrying GCs (F2 = 10.94; p = 0.001; Fig. 7D) and in the ratio of these immature cells to the total GC numbers (F2 = 5.98. p = 0.013; Fig. 7E). The ratio of immature to total GCs was significantly increased by exposure to seizure-like events (CTL, 1.587 ± 1.59% and KA/KA-SCR, 8.94 ± 1.63%; post hoc, p < 0.05). Treatment with an NRSE-ODN after the termination of KA-induced seizure-like events prevented the retention of HBDs in hippocampi exposed to KA (post hoc: KA/KA-SCR vs KA-NRSE, p < 0.05). Indeed, the percentage of immature/ dysmature GC in the KA-NRSE group was not different from in controls (2.54 ± 1.05%; post hoc, p > 0.05; Fig. 7E).
Figure 7.

NRSF acts directly on DG GCs to mediate the effects of seizure-like activity on their maturation and dendritic branching. A, Representative low-magnification photomicrograph of the DG after 14 d in culture (left panel) with high-magnification photomicrographs of dentate GCs after 14 d (right, top) and 28 d (right, bottom) in culture. Slice cultures were generated from Thy1-YFP mice. Scale bar, 50 μm. B, Representative low-magnification photomicrograph of the DG in KA-treated cultures at 14 d in culture (left) with high-magnification images of dentate GCs with atypical HBDs after 14 d (right, top) and 28 d (right, bottom) in culture. Red arrows indicate HBDs. C, Representative Western blot from nuclear extracts of control DG cultures (C1–C3) and those subjected to seizure-like activity using KA (K1–K4). NRSF and actin bands demonstrate augmented NRSF protein expression after seizure-like activity (unpaired t test; p = 0.009; CTL = 3, KA = 4). D, Seizure-like activity in vitro (Richichi et al., 2008) influences the number of GCs with dysmature HBD. Total numbers of HBD-carrying GCs were influenced by group (F2 = 10.94; p = 0.001; exposure to a scrambled ODN did not influence the effects of KA, so the KA and KA/SCR-ODN groups were combined). E, The percentage of HBD-carrying cells within the total GC numbers was also group-dependent (F2 = 5.98, p = 0.013). The percentage of immature GCs was significantly increased by exposure to seizure-like events (CTL, 1.587 ± 1.59; KA/KA-SCR, 8.94 ± 1.63; post hoc, p < 0.05). Treatment with an NRSE-ODN after the termination of KA exposure prevented the retention of HBDs in hippocampi exposed to KA (post hoc: KA/KA-SCR vs KA-NRSE, p < 0.05): the percentage of dysmature GC in the KA-NRSE group was not different from that in controls (2.54 ± 1.05; post hoc, p > 0.05). *p < 0.05; **p < 0.01.
Together, these data indicate that GC maturation within DG is perturbed by early-life seizures and that NRSF contributes crucially and directly to these disturbances to the normal formation and connectivity of the DG network.
Discussion
The principal findings of the current experiments are as follows: long, early-life seizures in an FSE model promote enduring memory impairments. Remarkably, these are abrogated by transient, post hoc interference with the transcriptional repressor NRSF, known to contribute to neuronal differentiation and programmed gene expression. The mechanisms of the memory problems involve abnormal hippocampal rhythms, likely resulting from impaired dendritic and synaptic function via regionally specific dendritic loss in CA1 and aberrant generation of excitatory synapses in DG. Together, these studies provide novel information about the development of memory processing and the mechanisms by which disease may influence them. They suggest approaches to prevent cognitive problems after early-life seizures.
Spatial memory deficits after eFSE arise from profound disturbances of the hippocampal network
eFSE rats had difficulties locating the static shock zone on a rotating arena, evident from numerous entries to the zone and increased shock numbers. The problems involved both short-term memory (avoiding additional shocks in the same session), and long-term memory (poor improvement on the second day and a month later). These findings are in line with reports of memory difficulties after rodent developmental seizures (Lugo et al., 2014; Barry et al., 2016a). Interestingly, the longer-duration eFSE worsened cognitive outcomes compared with our previous work with shorter eFS (Dubé et al., 2009; Barry et al., 2015), supporting that repeated/long seizures in children might disrupt memory (Bender et al., 2012; Visser et al., 2012; Martinos et al., 2013; Tsai et al., 2015).
Probing the structural and functional underpinnings of memory deficits, we surveyed the hippocampal network using in vivo electrophysiology and neuroanatomical tools. We identified perturbations of hippocampal oscillations, including enhanced amplitude and frequency of fast γ oscillations, reduced parameters of slow γ rhythms, and disrupted θ/speed relationships. These defects might arise via numerous mechanisms, including loss of rhythm-generating pyramidal cells or of interneurons that comprise the microcircuitry responsible for temporal organization of network activity (Colgin et al., 2009; Buzsáki and Wang, 2012). Neuronal loss is minimal in our model (Toth et al., 1998; Dubé et al., 2007, 2009; Baram et al., 2011).Therefore, we investigated potential structural or functional changes in surviving neurons. We found significant structural defects potentially explaining the physiological deficits: the observed paucity of apical dendritic branches should influence filtering properties of CA1 apical dendrites, augmenting oscillations involving input from cortical regions (i.e., fast γ). New, aberrant excitatory synapses in DG might disrupt its gating function onto CA3, influencing amplitude and frequency of slow γ, as observed.
Convergent studies of hippocampal oscillations and neuronal structure inform mechanisms of spatial memory problems, and rescue by NRSF blockers
How do we explain the profound effects of eFSE and the remarkable improvement induced by short treatment with NRSF blockers? These questions are particularly intriguing because both eFSE-SCR and eFSE-NRSE groups had significant perturbation of CA1 apical dendritic trees, crucial for place-cell and hippocampal network function.
The electrophysiological findings in both stable arena and during active avoidance are informative. During baseline conditions, eFSE-SCR rats had larger and faster fast γ oscillations in CA1 than controls. Fast γ rhythms may reflect input to CA1 from cortical regions, including entorhinal cortex via the temporoammonic pathway (Colgin, 2016). Increased amplitude and frequency of these oscillations might indicate poor filtering of this input and might repress intrinsic hippocampal oscillations such as slow γ. Indeed, during active avoidance, when memory demands were greatest, eFSE rats had low frequency and amplitude of slow γ, considered to reflect CA3-CA1 input influenced by DG gating (Colgin et al., 2009; Colgin, 2016; Goldberg and Coulter, 2013; Vaidya and Johnston, 2013). Together, the balance of routed neural information to CA1 from EC (fast γ) and CA3 (slow γ) appeared disrupted by eFSE, interfering with acquisition of spatial information associated with entorhinal inputs, and recall of spatial information typically associated with CA3 inputs.
We previously found that eFS provoked aberrant firing fields and poor stability of CA1 place cells, associated with cognitive impairment (Dube et al., 2009; Barry et al., 2015). Additionally, we described problems with θ rhythms: phase preference at peak θ in control CA1 place cells shifted during active avoidance to that observed in CA3. However, cognitively impaired eFS rats did not exhibit phase preference in foraging or avoidance accompanied by poor CA1-CA3 cross θ interaction (Barry et al., 2016a). Thus, disrupted dynamic temporal coordination of neuronal activity by θ-phase may explain cognitive problems following eFSE.
Here we identify novel disruption of γ rhythms: Slow γ organization of newly described θ sequences (Zheng et al., 2016) reflects planning of future movements from stored spatial memories, whereas fast γ sequences reflect sensory driven details of current location (Zheng et al., 2016). Here, eFSE altered the balance of slow and fast γs. Combined with our prior finding, we propose that the principal spatial problem in eFSE animals is reduced network efficacy in integrating current spatial information on the rotating arena with stored representation of the environment that allows animals to plan escape from the shock zone.
Blocking NRSF had remarkable effects: in the active avoidance task, eFSE-NRSE rats had larger and faster slow γ oscillations than all other groups, and the amplitude of these oscillations was significantly higher than of controls. Restoration of slow γ properties, considered crucial for memory retrieval, in eFSE-NRSE rats suggested that NRSF blockade might have ameliorated the effects of eFSE on DG-CA3 pathways, and this hypothesis was tested.
NRSF uses several mechanisms to mediate seizure-induced disruption of hippocampal processes underlying memory
Seizures including eFSE increase the expression and chromatin-binding of NRSF. This transient augmentation of NRSF function seems to disturb hippocampal networks enduringly via several region-specific mechanisms.
In mature hippocampal CA1, there are ∼400 potential targets for NRSF: genes containing the recognition site NRSE (Johnson et al., 2007; McClelland et al., 2011, 2014). However, only ∼40 of these genes are repressed by augmented NRSF levels following SEs (McClelland et al., 2011, 2014). Notably, these genes comprise crucial components of neuronal function, including glutamate receptors, calcium-dependent signaling, and ion channels.
Focusing on the filtering properties of CA1 apical dendrites, NRSF directly represses the expression of HCN1, the principal contributor to dendritic Ih in hippocampus (McClelland et al., 2011). Ih counteracts dendritic summation (i.e., location-dependent temporal difference of dendritic inputs at the soma) (Migliore et al., 2004; George et al., 2009; Noam et al., 2011; Vaidya and Johnston, 2013) and contributes to γ oscillations (Buzsáki and Wang, 2012; Neymotin et al., 2013). Reduced Ih after SE in adults was reported (Jung et al., 2007, 2011; Huang et al., 2009; Noam et al., 2011), and the current was restored by blocking NRSF (McClelland et al., 2011). HCN1 is reduced also after early-life seizures, including eFSE (Brewster et al., 2002). Here, blocking NRSF function shortly after eFSE reversed increases in fast γ properties without reversing structural defects in CA1 dendrites, consistent with NRSF-mediated changes in HCN1 expression. In support, blocking NRSF function in controls influenced the amplitude of fast γ without modifying dendritic trees. Thus, eFSE-induced changes of NRSF function might influence hippocampal rhythms via disruption of the carefully regulated levels of HCN1 and coregulated ion channels (George et al., 2009).
There is relatively little Ih in DGCs in developing rats (Bender et al., 2007), suggesting alternative mechanisms for the major effects of upregulated NRSF in DG. Both in vivo and in cultures, early life seizures provoked anomalous sources of excitatory synapses, which were reversed by NRSF blockade. Specifically, dysmaturation of GCs, retention of HBDs, which form excitatory synapses on neighboring GCs, was observed after eFSE and prevented by NRSF. Persistent HBDs are found on cells that are immature during seizures (Parent et al., 1997; Kron et al., 2010; Murphy et al., 2012; Singh et al., 2013; Hester and Danzer, 2014), a population strongly influenced by NRSF (Gao et al., 2011). NRSF regulates numerous genes involved in structural and synaptic neuronal maturation (Schoenherr and Anderson, 1995; Z. F. Chen et al., 1998; Gao et al., 2011; McClelland et al., 2014), and immature GCs are primarily subject to maladaptive plasticity following insults (Parent et al., 1997; Toth et al., 1998; Kron et al., 2010; Murphy et al., 2012). Thus, it is reasonable to consider NRSF as a potent mediator of seizure-induced defects in the DG network, which is crucial for gating information flow to the hippocampus proper. Indeed, our in vitro studies using organotypic cultures demonstrate that NRSF levels are augmented within DG and result in dysmaturation of GCs apparent as retention of HBDs and exuberant growth of apical dendrites. Further, both anomalies are prevented by blocking NRSF function during a critical developmental period in GC life cycles. Remarkably, preventing dysregulation of DG maturation sufficed to influence hippocampal oscillations and allowed memory encoding, processing, and retrieval in treated eFSE rats.
Thus, long experimental febrile seizures provoke profound, enduring spatial memory problems. The mechanisms of these problems involve complex disruption of memory-related oscillations within hippocampal network, likely deriving from impaired dendritic filtering and synaptic function in CA1 and DG, respectively. Importantly, these deficits are abrogated by transient postinsult interference with chromatin binding of the transcriptional repressor NRSF, an approach with significant translational potential.
Footnotes
This work was supported by National Institutes of Health Grants NS78279 (to G.L.H. and T.Z.B.), NS35439 (to T.Z.B.), T32 NS045540 (to T.Z.B., K.P.P., and M.M.C.), T32 GM08620 (to M.M.C.), and an AES/EFA Predoctoral Award (to K.P.P.). We thank Jennifer Daglian for excellent technical assistance.
The authors declare no competing financial interests.
References
- Ballas N, Mandel G (2005) The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol 15:500–506. 10.1016/j.conb.2005.08.015 [DOI] [PubMed] [Google Scholar]
- Baram TZ, Gerth A, Schultz L (1997) Febrile seizures: an appropriate-aged model suitable for long-term studies. Dev Brain Res 98:265–270. 10.1016/S0165-3806(96)00190-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Jensen FE, Brooks-Kayal A (2011) Does acquired epileptogenesis in the immature brain require neuronal death. Epilepsy Curr 11:21–26. 10.5698/1535-7511-11.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry JM, Choy M, Dube C, Robbins A, Obenaus A, Lenck-Santini PP, Scott RC, Baram TZ, Holmes GL (2015) T2 relaxation time post febrile status epilepticus predicts cognitive outcome. Exp Neurol 269:242–252. 10.1016/j.expneurol.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry JM, Tian C, Spinella A, Page M, Holmes GL (2016a) Spatial cognition following early-life seizures in rats: performance deficits are dependent on task demands. Epilepsy Behav 60:1–6. 10.1016/j.yebeh.2016.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry JM, Sakkaki S, Barriere SJ, Patterson KP, Lenck-Santini PP, Scott RC, Baram TZ, Holmes GL (2016b) Temporal coordination of hippocampal neurons reflects cognitive outcome post-febrile status epilepticus. EBioMedicine 7:175–190. 10.1016/j.ebiom.2016.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AC, Morse RP, Scott RC, Holmes GL, Lenck-Santini PP (2012) SCN1A mutations in Dravet syndrome: impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav 23:177–186. 10.1016/j.yebeh.2011.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Kirschstein T, Kretz O, Brewster AL, Richichi C, Rüschenschmidt C, Shigemoto R, Beck H, Frotscher M, Baram TZ (2007) Localization of HCN1 channels to presynaptic compartments: novel plasticity that may contribute to hippocampal maturation. J Neurosci 27:4697–4706. 10.1523/JNEUROSCI.4699-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT. (2011) Epilepsy, cognition, and behavior: the clinical picture. Epilepsia 52:7–12. 10.1111/j.1528-1167.2010.02905.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, Shinnar S (1996) Complex febrile seizures. Epilepsia 37:126–133. 10.1111/j.1528-1157.1996.tb00003.x [DOI] [PubMed] [Google Scholar]
- Brennan GP, Dey D, Chen Y, Patterson KP, Magnetta EJ, Hall AM, Dube CM, Mei YT, Baram TZ (2016) Dual and opposing roles of microRNA-124 in epilepsy are mediated through inflammatory and NRSF-dependent gene networks. Cell Rep 14:2402–2412. 10.1016/j.celrep.2016.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster A, Bender RA, Chen Y, Dube C, Eghbal-Ahmadi M, Baram TZ (2002) Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci 2:4591–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Wang XJ (2012) Mechanisms of γ oscillations. Annu Rev Neurosci 35:203–225. 10.1146/annurev-neuro-062111-150444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Molet J, Lauterborn JC, Trieu BH, Bolton JL, Patterson KP, Gall CM, Lynch G, Baram TZ (2016) Converging, synergistic actions of multiple stress hormones mediate enduring memory impairments after acute simultaneous stresses. J Neurosci 36:11295–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZF, Paquette AJ, Anderson DJ (1998) NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat Genet 20:136–142. 10.1038/2431 [DOI] [PubMed] [Google Scholar]
- Colgin LL. (2016) Rhythms of the hippocampal network. Nat Rev Neurosci 17:239–249. 10.1038/nrn.2016.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL, Denninger T, Fyhn M, Hafting T, Bonnevie T, Jensen O, Moser MB, Moser EI (2009) Frequency of γ oscillations routes flow of information in the hippocampus. Nature 19:353–357. 10.1038/nature08573 [DOI] [PubMed] [Google Scholar]
- Conforti P, Zuccato C, Gaudenzi G, Ieraci A, Camnasio S, Buckley NJ, Mutti C, Cotelli F, Contini A, Cattaneo E (2013) Binding of the repressor complex REST-mSIN3b by small molecules restores neuronal gene transcription in Huntington's disease models. J Neurochem 127:22–35. 10.1111/jnc.12348 [DOI] [PubMed] [Google Scholar]
- Dubé CM, Brewster AL, Richichi C, Zha Q, Baram TZ (2007) Fever, febrile seizures and epilepsy. Trends Neurosci 30:490–496. 10.1016/j.tins.2007.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé CM, Zhou JL, Hamamura M, Zhao Q, Ring A, Abrahams J, McIntyre K, Nalcioglu O, Shatskih T, Baram TZ, Holmes GL (2009) Cognitive dysfunction after experimental febrile seizures. Exp Neurol 215:167–177. 10.1016/j.expneurol.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé CM, Ravizza T, Hamamura M, Zha Q, Keebaugh A, Fok K, Andres AL, Nalcioglu O, Obenaus A, Vezzani A, Baram TZ (2010) Epileptogenesis provoked by prolonged experimental febrile seizures: mechanisms and biomarkers. J Neurosci 30:7484–7494. 10.1523/JNEUROSCI.0551-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elger CE, Helmstaedter C, Kurthen M (2004) Chronic epilepsy and cognition. Lancet Neurol 3:663–672. 10.1016/S1474-4422(04)00906-8 [DOI] [PubMed] [Google Scholar]
- French JA, Williamson PD, Thadani VM, Darcey TM, Mattson RH, Spencer SS, Spencer DD (1993) Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol 34:774–780. 10.1002/ana.410340604 [DOI] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ding P, Nashaat M, Yuan L, Ma J, Hammer RE, Hsieh J (2011) The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J Neurosci 31:9772–9786. 10.1523/JNEUROSCI.1604-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, Morrison JF, Ockuly J, Stafstrom C, Sutula T, Roopra A (2006) 2-Deoxy-d-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci 9:1382–1387. 10.1038/nn1791 [DOI] [PubMed] [Google Scholar]
- George MS, Abbott LF, Siegelbaum SA (2009) HCN hyperpolarization-activated cation channel inhibit EPSPs by interactions with M-type K(+) channel. Nat Neurosci 5:577–584. 10.1038/nn.2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EM, Coulter DA (2013) Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci 14:337–349. 10.1038/nrn3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter C, Kurthen M, Lux S, Reuber M, Elger CE (2003) Chronic epilepsy and cognition: a longitudinal study in temporal lobe epilepsy. Ann Neurol 54:425–432. 10.1002/ana.10692 [DOI] [PubMed] [Google Scholar]
- Hermann BP, Seidenberg M, Schoenfeld J, Davies K (1997) Neuropsychological characteristics of the syndrome of mesial temporal lobe epilepsy. Arch Neurol 54:369–376. 10.1001/archneur.1997.00550160019010 [DOI] [PubMed] [Google Scholar]
- Hester MS, Danzer SC (2014) Hippocampal granule cell pathology in epilepsy: a possible structural basis for comorbidities of epilepsy? Epilepsy Behav 38:105–116. 10.1016/j.yebeh.2013.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe C, Elger CE, Helmstaedter C (2007) Long-term memory impairment in patients with focal epilepsy. Epilepsia 9:26–29. 10.1111/j.1528-1167.2007.01397.x [DOI] [PubMed] [Google Scholar]
- Huang Z, Walker MC, Shah MM (2009) Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci 29:10979–10988. 10.1523/JNEUROSCI.1531-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316:1497–1502. 10.1126/science.1141319 [DOI] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo JN Jr, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP (2007) Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci 27:13012–13021. 10.1523/JNEUROSCI.3605-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Warner LN, Pitsch J, Becker AJ, Poolos NP (2011) Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J Neurosci 31:14291–14295. 10.1523/JNEUROSCI.1148-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama R, Tao K, Sasaki T, Ichikawa J, Miyamoto D, Muramatsu R, Matsuki N, Ikegaya Y (2012) GABAergic excitation after febrile seizures induces ectopic granule cells and adult epilepsy. Nat Med 18:1271–1278. 10.1038/nm.2850 [DOI] [PubMed] [Google Scholar]
- Kron MM, Zhang H, Parent JM (2010) The developmental stage of dentate granule cells dictates their contribution to seizure-induced plasticity. J Neurosci 6:2051–2059. 10.1523/JNEUROSCI.5655-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, Bennett DA, Colaiácovo MP, Yankner BA (2014) REST and stress resistance in ageing and Alzheimer's disease. Nature 507:448–454. 10.1038/nature13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo JN, Swann JW, Anderson AE (2014) Early-life seizures result in deficits in social behavior and learning. Exp Neurol 256:74–80. 10.1016/j.expneurol.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel G, Fiondella CG, Covey MV, Lu DD, Loturco JJ, Ballas N (2011) Repressor element 1 silencing transcription factor (REST) controls radial migration and temporal neuronal specification during neocortical development. Proc Natl Acad Sci U S A 108:16789–16794. 10.1073/pnas.1113486108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinos MM, Yoong M, Patil S, Chong WK, Mardari R, Chin RF, Neville BG, de Haan M, Scott RC (2013) Early developmental outcomes in children following convulsive status epilepticus: a longitudinal study. Epilepsia 54:1012–1019. 10.1111/epi.12136 [DOI] [PubMed] [Google Scholar]
- McClelland S, Flynn C, Dubé C, Richichi C, Zha Q, Ghestem A, Esclapez M, Bernard C, Baram TZ (2011) Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann Neurol 70:454–464. 10.1002/ana.22479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland S, Brennan GP, Dubé C, Rajpara S, Iyer S, Richichi C, Bernard C, Baram TZ (2014) The transcription factor NRSF contributes to epileptogenesis by selective repression of a subset of target genes. eLife 3:e01267. 10.7554/eLife.01267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore M, Messineo L, Ferrante M (2004) Dendritic Ih selectively blocks temporal summation of unsynchronized distal inputs in CA1 pyramidal neurons. J Comput Neurosci 16:5–13. 10.1023/B:JCNS.0000004837.81595.b0 [DOI] [PubMed] [Google Scholar]
- Mueller SG, Laxer KD, Scanlon C, Garcia P, McMullen WJ, Loring DW, Meador KJ, Weiner MW (2012) Different structural correlates for verbal memory impairment in temporal lobe epilepsy with and without mesial temporal lobe sclerosis. Hum Brain Mapp 33:489–499. 10.1002/hbm.21226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BL, Hofacer RD, Faulkner CN, Loepke AW, Danzer SC (2012) Abnormalities of granule cell dendritic structure are a prominent feature of the intrahippocampal kainic acid model of epilepsy despite reduced postinjury neurogenesis. Epilepsia 53:908–921. 10.1111/j.1528-1167.2012.03463.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neymotin SA, Hilscher MM, Moulin TC, Skolnick Y, Lazarewicz MT, Lytton WW (2013) Ih tunes θ/γ oscillations and cross-frequency coupling in an in silico CA3 model. PLoS One 8:e76285. 10.1371/journal.pone.0076285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noam Y, Bernard C, Baram TZ (2011) Towards an integrated view of HCN channel role in epilepsy. Curr Opin Neurobiol 21:873–879. 10.1016/j.conb.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm K, Belluardo N, Metsis M, Timmusk T (1998) Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci 18:1280–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH (1997) Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 10:3727–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KP, Brennan GP, Curran M, Kinney-Lang E, Dubé C, Rashid F, Ly C, Obenaus A, Baram TZ (2015) Rapid, coordinate inflammatory responses after experimental febrile status epilepticus: Implications for epileptogenesis. eNeuro 2:ENEURO.0034–15.2015. 10.1523/ENEURO.0034-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raijmakers M, Clynen E, Smisdom N, Nelissen S, Brône B, Rigo JM, Hoogland G, Swijsen A (2016) Experimental febrile seizures increase dendritic complexity of newborn dentate granule cells. Epilepsia 5:717–726. 10.1111/epi.13357 [DOI] [PubMed] [Google Scholar]
- Richard GR, Titiz A, Tyler A, Holmes GL, Scott RC, Lenck-Santini PP (2013) Speed modulation of hippocampal θ frequency correlates with spatial memory performance. Hippocampus 12:1269–1279. 10.1002/hipo.22164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richichi C, Brewster AL, Bender RA, Simeone TA, Zha Q, Yin HZ, Weiss JH, Baram TZ (2008) Mechanisms of seizure-induced ‘transcriptional chanelopathy’ of hyperpolarization-activated cyclic nucleotide gated (HCN) channels. Neurobiol Dis 29:297–305. 10.1016/j.nbd.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenas-Ruano A, Chávez AE, Cossio MJ, Castillo PE, Zukin RS (2012) REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat Neurosci 15:1382–1390. 10.1038/nn.3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopra A, Huang Y, Dingledine R (2001) Neurological disease: listening to gene silencers. Mol Interv 1:219–228. [PubMed] [Google Scholar]
- Schlessinger AR, Cowan WM, Gottlieb DI (1975) An autoradiographic study of the time of origin and the pattern of granule cell migration in the dentate gyrus of the rat. J Comp Neurol 159:149–175. 10.1002/cne.901590202 [DOI] [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ (1995) The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science 267:1360–1363. 10.1126/science.7871435 [DOI] [PubMed] [Google Scholar]
- Singh SP, He X, McNamara JO, Danzer SC (2013) Morphological changes among hippocampal dentate granule cells exposed to early kindling-epileptogenesis. Hippocampus 23:1309–1320. 10.1002/hipo.22169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Taylor A, Molet J, Jiang S, Korosi A, Bolton JL, Noam Y, Simeone K, Cope J, Chen Y, Mortazavi A, Baram TZ (2017) NRSF-dependent epigenetic mechanisms contribute to programming of stress-sensitive neurons by neonatal experience, promoting resilience. Mol Psychiatry, in press. 10.1038/mp.2016.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thind KK, Ribak CE, Buckmaster PS (2008) Synaptic input to dentate granule cell basal dendrites in a rat model of temporal lobe epilepsy. J Comp Neurol 509:190–202. 10.1002/cne.21745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ (1998) Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci 18:4285–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai ML, Hung KL, Tsan YY, Tung WT (2015) Long-term neurocognitive outcome and auditory event-related potentials after complex febrile seizures in children. Epilepsy Behav 47:55–60. 10.1016/j.yebeh.2015.04.067 [DOI] [PubMed] [Google Scholar]
- Vaidya SP, Johnston D (2013) Temporal synchrony and γ-to θ power conversion in the dendrites of CA1 pyramidal neurons. Nat Neurosci 16:1812–1820. 10.1038/nn.3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser AM, Jaddoe VW, Ghassabian A, Schenk JJ, Verhulst FC, Hofman A, Tiemeier H, Moll HA, Arts WF (2012) Febrile seizures and behavioral and cognitive outcomes in preschool children: the Generation R study. Dev Med Child Neurol 11:1006–1011. 10.1111/j.1469-8749.2012.04405.x [DOI] [PubMed] [Google Scholar]
- Zheng C, Bieri KW, Hsiao YT, and Colgin LL (2016) Spatial sequence coding differs during slow and fast gamma rhythms in the hippocampus. Neuron 89:398–408. 10.1016/j.neuron.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]