

Abstract
Axonal degeneration is a pathophysiological mechanism common to several neurodegenerative diseases. The slow Wallerian degeneration (WldS) mutation, which results in reduced axonal degeneration in the central and peripheral nervous systems, has provided insight into a redox-dependent mechanism by which axons undergo self-destruction. We studied early molecular events in axonal degeneration with single-axon laser axotomy and time-lapse imaging, monitoring the initial changes in transected axons of purified retinal ganglion cells (RGCs) from wild-type and WldS rat retinas using a polarity-sensitive annexin-based biosensor (annexin B12-Cys101,Cys260-N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) ethylenediamine). Transected axons demonstrated a rapid and progressive change in membrane phospholipid polarity, manifested as phosphatidylserine externalization, which was significantly delayed and propagated more slowly in axotomized WldS RGCs compared with wild-type axons. Delivery of bis(3-propionic acid methyl ester)phenylphosphine borane complex, a cell-permeable intracellular disulfide-reducing drug, slowed the onset and velocity of phosphatidylserine externalization in wild-type axons significantly, replicating the WldS phenotype, whereas extracellular redox modulation reversed the WldS phenotype. These findings are consistent with an intra-axonal redox mechanism for axonal degeneration associated with the initiation and propagation of phosphatidylserine externalization after axotomy.
SIGNIFICANCE STATEMENT Axonal degeneration is a neuronal process independent of somal apoptosis, the propagation of which is unclear. We combined single-cell laser axotomy with time-lapse imaging to study the dynamics of phosphatidylserine externalization immediately after axonal injury in purified retinal ganglion cells. The extension of phosphatidylserine externalization was slowed and delayed in Wallerian degeneration slow (WldS) axons and this phenotype could be reproduced by intra-axonal disulfide reduction in wild-type axons and reversed by extra-axonal reduction in WldS axons. These results are consistent with a redox mechanism for propagation of membrane polarity asymmetry in axonal degeneration.
Keywords: axon membrane, axonal degeneration, phosphatidylserine, redox, Wallerian degeneration slow
Introduction
Axonal degeneration is a common feature of many traumatic and neurodegenerative diseases (Lingor et al., 2012). It takes place in two directions, one toward the cell body (retrograde degeneration) and the other toward the distal axon terminal (Wallerian degeneration) (Waller, 1850; Luo and O'Leary, 2005). Morphologically, degenerative changes of axons in Wallerian degeneration are well characterized, starting with axonal swelling, followed by membrane beading, and finally fragmentation of the axon (Coleman and Freeman, 2010).
Acute axonal damage leads to the loss of the integrity of the membrane and axonal degeneration (Kerschensteiner et al., 2005; Wang et al., 2012). Asymmetry of the axonal membrane is an integral property of a healthy neuron, which is maintained by restriction of phosphatidylserine and phosphatidylethanolamine to the cytosolic (inner) leaflet of the plasma membrane, whereas phosphatidylcholine, sphingomyelin, and glycosphingolipids are limited to the noncytosolic (outer) leaflet (Verkleij et al., 1973; Rothman and Lenard, 1977; Maiese and Vincent, 2000; Fadeel and Xue, 2009; Leventis and Grinstein, 2010). Externalization of phosphatidylserine after axonal or soma injury is an early apoptotic marker and acts as a signal enabling microglia/macrophages to recognize and actively remove damaged somas and axons (Martin et al., 1995; Rimon et al., 1997; Fadok et al., 2001).
Although the processes that maintain asymmetric distribution of axonal lipids is well studied (van Meer et al., 2008), the mechanism by which axonal injury signals phosphatidylserine externalization is not well understood. Using real-time confocal microscopy in living animals, we showed previously that optic nerve transection causes a burst of superoxide in retinal ganglion cells (RGCs) that precedes apoptosis (Kanamori et al., 2010). RGC axons form the optic nerve, which conducts visual information from the retina to the brain. RGC axonal injury leads to axonal degeneration, which can be ameliorated by redox-dependent disulfide reduction (Almasieh et al., 2011). Elevated levels of superoxide anion could result in oxidative modification of sulfhydryl groups on critical lipids and proteins in the axon. One such target is phosphatidylserine, which undergoes oxidation during apoptosis, resulting in the production of oxidized phosphatidylserine (Matsura, 2014). It has been reported that oxidation of phosphatidylserine results in a significant increase in phosphatidylserine externalization (Tyurina et al., 2004). Therefore, in this study, we investigated the hypothesis that the loss of axonal membrane asymmetry after damage is related to axonal redox status.
On average, a transected wild-type mouse axon survives for up to 2 d. Remarkably, transected axons of delayed Wallerian degeneration (WldS) mutant mice survive for up to 3 weeks while maintaining the capacity to conduct action potentials (Perry et al., 1990; Tsao et al., 1999). Discovery of WldS mice provided support for the idea that a molecularly distinct self-destruct program is responsible for the axonal degeneration (Lunn et al., 1989; Perry et al., 1991; Raff et al., 2002). The WldS mutation results from the tandem triplication of nicotinamide mononucleotide adenylyltransferase 1 (NMNAT-1) and the N-terminal 70 aa of ubiquitination factor e4b (Lyon et al., 1993; Coleman et al., 1998). NMNAT-1 is a central enzyme in nicotinamide adenine dinucleotide (NAD+) biosynthesis (Mack et al., 2001), which plays an important role in cellular energy metabolism and redox reactions, acting as an oxidizing and reducing (NADH) agent in the respiratory chain and subsequently the production of ATP and reactive oxygen species (ROS) (Orrenius, 2007; Ying, 2008).
We hypothesized that the extended survival of WldS axons after axonal injury is a reflection of their ability to maintain the axonal redox status and by extension the membrane asymmetry of phosphatidylserine. To test this hypothesis, axons of purified RGCs from wild-type and WldS rat retinas were transected individually with an ultrafast laser. Using a polarity-sensitive annexin-based biosensor with switchable fluorescence states (Kim et al., 2010a), the spatial and temporal pattern of axonal phosphatidylserine externalization in transected wild-type RGC axons was assessed and compared with WldS axons. Cell-permeable and cell-impermeable disulfide-reducing agents were used to demonstrate that phosphatidylserine externalization after axonal injury is redox dependent.
Materials and Methods
Animals.
All procedures were performed in accordance with the guidelines of Association for Research in Vision and Ophthalmology and the Canadian Council on Animal Care for the use of animals in research and were approved by the Maisonneuve-Rosemont Hospital Research Centre animal care committee. The WldS transgenic rats were kindly provided to us by Dr. Michael Coleman (Babraham Institute, Cambridge, UK). To generate the WldS rats, Ube4b/Nmnat cDNA was cloned downstream of the β-actin promoter in pHbetaAPr-1 plasmid and injected into embryonic pronuclei of Sprague Dawley rats (Mack et al., 2001; Adalbert et al., 2005). WldS pups were generated in an in-house breeding program. The WldS strain is on a Sprague Dawley background and is homozygous, precluding the use of littermate controls. Wild-type pregnant Sprague Dawley rats were obtained from Charles River Laboratories and acclimated in the animal facility until litters were born.
Materials.
Glass-bottom dishes for imaging were from MatTek. Goat anti-rabbit IgG (H+L) and goat anti-mouse IgM (μ-chain specific) were from Jackson ImmunoResearch (catalog #111–005-003, RRID: AB_2337913 and catalog #115–005-020, AB_2338450, respectively). Rabbit anti-rat macrophage sera was from Cedarlane (catalog #CLAD51240, RRID: AB_10059912). Thy-1 IgM was produced from the T11D7e2 hybridoma line (American Type Culture Collection; catalog #TIB-103, RRID: CVCL_F769). Poly d-lysine, DNase I, insulin, Forskolin, BSA, trypsin, trypsin inhibitor, human apo-transferrin, progesterone, sodium selenite, putrescine, thyroxine (T3), N-acetyl-l-cysteine (NAC), 1,4-dithiothreitol (DTT), and tris (2-carboxyethyl) phosphine hydrochloride (TCEP) were from Sigma-Aldrich. Cultrex mouse laminin I was from Trevigen. Dulbecco's PBS (DPBS) and fetal bovine serum (FBS) were from Invitrogen. Papain was from Worthington Biochemical. L-cysteine was from Wisent. Neurobasal-A, penicillin/streptomycin, sodium pyruvate, and l-glutamine were from Invitrogen. hBDNF and CNTF were from Alomone Laboratories. Recombinant human FGF-basic (154 aa) was from PeproTech. Defined serum supplement GS-21 was from MTI-GlobalStem. Annexin B12-Cys101,Cys260-N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) ethylenediamine (annexin B12-IANBD) was from Novus Biologicals. Bis(3-propionic acid methyl ester) phenylphosphine-borane complex (PB1) was synthesized according to our previously published methods (Schlieve et al., 2006) at the Keck–University of Wisconsin Comprehensive Cancer Center Small Molecule Screening Facility (Madison, WI). The purity of PB1 was >99% by HPLC.
RGC purification and culture.
Purified RGC cultures were produced from dissociated Sprague Dawley or WldS P2-P7 pup retinas of either sex immunopurified with sequential anti-macrophage and anti-Thy-1 panning, using modified version of a protocol first developed by Barres et al. (1988) and modified by Hu et al. (2010). Briefly, freshly dissected retinas were placed in a filtered DPBS solution containing 16.5 U/ml papain, 0.2 mg/ml L-cysteine, 124 U/ml DNase I, and 0.1% of a 1 n sodium hydroxide solution. The retinas were digested for 30 min at 37°C, gently swirling the solution at 15 min intervals. After digestion, the papain solution was gently aspirated, retaining the partially digested retinas. Ovomucoid (0.6 mg/ml) and BSA (0.6 mg/ml) were added to halt digestion, followed by anti-rat macrophage antibody. The retinas were gently triturated to produce a single cell suspension. After 10 min to allow antibody binding, the cells were pelleted by centrifugation and resuspended in DPBS containing 10 mg/ml ovomucoid and BSA. Cells were pelleted again and resuspended in DBPS containing 0.2 mg/ml BSA and 5 μg/ml insulin.
The cell suspension was added to a 150 mm Petri dish that had been incubated overnight with goat anti-rabbit IgG in Tris-HCl and incubated for 20 min at room temperature to allow macrophages to adhere to the plate, agitating after 10 min. Nonadherent cells were transferred to a second identical 150 mm dish and incubated for 45 min (agitating at 15 min) to further deplete macrophages. The binding of macrophages was confirmed on a microscope. Nonadherent cells were then gently shaken loose and seeded onto a 100 mm Petri dish that had been previously coated with goat anti-mouse IgM in Tris-HCl, followed by supernatant from the T11D7e2 hybridoma line containing anti-Thy-1 IgM. Cells were incubated 50 min at room temperature to allow binding of RGCs, agitating every 10 min. Nonadherent cells were then aspirated and the plate was very gently rinsed four to six times with DPBS until only adherent cells remained, as assessed by microscopy.
The RGCs adherent to the anti-Thy-1 dish were removed with a trypsin incubation for 4 min at 37°C, followed by inactivation with 30% heat-inactivated FBS and dislodging by pipetting. Cells were collected in a 20 ml plastic tube, pelleted, and resuspended in 500 μl of 30% FBS for counting on a hemocytometer, at which point the quality of isolation was estimated by uniformity, shape, and size of isolated cells.
The purified RGCs were plated on MatTek glass-bottom culture dishes that had been previously coated with poly-d-lysine (10 μg/ml) for at least 30 min, followed by laminin in Neurobasal A (2 μg/ml). The RGC culture medium consisted of Neurobasal A supplemented with 2% GS-21 (a defined serum supplement), penicillin (100 U/ml), streptomycin (100 μg/ml), insulin (5 μg/ml), sodium pyruvate (1 mm), l-glutamine (1 mm), transferrin (100 μg/ml), BSA (100 μg/ml), progesterone (0.2 μm), putrescine (16 μg/ml), sodium selenite (40 ng/ml), T3 (40 ng/ml), N-acetyl cysteine (5 μg/ml), forskolin (5 μm), BDNF (50 ng/ml), CNTF (10 μg/ml), and bFGF (10 μg/ml). Cells were kept in a humidified 10% CO2 incubator at 37°C for at least 3 d to allow time for the ganglion cells to adhere and extend processes and fed every 3 d by removal of half the medium and replacement with fresh medium.
Single-axon transection.
We developed a platform for producing submicron ablations in cultured RGC axons using ultrafast lasers. A Spectra-Physics Mai Tai fully automated laser producing 3 W at 800 nm with 100 fs pulses at 80 MHz was coupled to an Olympus IX71 inverted fluorescence microscope with a custom-made Thorlabs scanning laser module. This permits alignment of a high-power beam directly through the aperture of a 1.2 numerical aperture water-immersion 60× objective coated for infrared wavelengths, which was used to focus the laser beam on axons to produce injury.
The motorized microscope stage and laser trigger were controlled by our custom-made LabVIEW program (National Instruments Corporation, TX). The program moves the stage in the desired direction while simultaneously triggering the laser, guiding the axon through the path of the laser and thereby enabling axonal transection. At the beginning of each set of experiments, the following calibration protocol was followed to ensure consistency of experiments. The alignment of the laser beam was checked using a target so that the beam was centered on the axis of the lens. After centering the beam, the microscope was switched to the 60× objective and the laser power output at the focal point was adjusted to 65 mW using a power meter interface (PM100; Thorlabs) and sensor (LM-10 HTD; Coherent). A MatTek glass-bottom dish clad with a thin gold film was placed in the micro-incubator (positioned on the microscope stage) at the same spot that RGC dish would sit. Then using the LabVIEW program, 3 lines (40 μm length, 1 μm width) were engraved into the gold film at 0°, 45°, and 90° to provide reference points for the laser transection start and end points (Fig. 1A). Axotomy was performed only on RGCs with clearly identifiable axons. After the RGC was selected and before axotomy, the RGC soma and the full extent of its axon was captured in a series of bright-field images (60× objective), enabling calculation of the length of the axon and the distance of transection site to the soma (Fig. 1B). The axon was then positioned so that the intended transection site would be in the path of the laser beam and images with 488 nm excitation and bright-field illumination were obtained, the former as a baseline for subsequent fluorescence detection and the latter to determine the diameter of axon at the transection site and as a baseline for subsequent laser-induced morphological changes at the site of axotomy. To perform the axotomy, all filters were removed from the optical path and the LabVIEW program was used to trigger the laser and move the stage simultaneously, resulting in axonal transection. Immediately after axotomy, a bright-field image was obtained and then fluorescence imaging immediately commenced by restoring the FITC filter set to the optical path. The bright-field image was used for measuring the lesion size (Fig. 2).
Figure 1.

Example of the axonal extent and soma of a rat retinal ganglion cell in culture. Retinal ganglion cells were isolated from a cell suspension of P2–P7 rat pup retinas by immunopanning and placed on PDL/laminin-coated glass bottom dishes in neurotrophin-enriched medium. A, Laser beam was used to engrave 3 lines into a gold film, each starting from point 0 in three directions (indicated by 1, 2, and 3) to provide reference points for positioning the axon before transection. B, Wild-type retinal ganglion cell 3 d in culture with a full-length axon captured in series of bright-field images (60× water-immersion objective). The arrow shows the RGC soma at the lower right corner of the frame. The axon extends with few branches. The axon terminal is indicated by an asterisk. Scale bars, 40 μm.
Figure 2.

In vitro axotomy of cultured retinal ganglion cells. A pulse laser at 800 nm with 100 fs pulses at 80 MHz was directed through the scanning laser module of an Olympus IX71 inverted fluorescence microscope to provide a high-power beam directly through the aperture of a 1.2 numerical aperture water-immersion 60× objective. Axon transection was achieved by photodisruption, a phenomenon in which electrons within the tissue absorb the incident photons and their energy exceeds the ionization potential; the resultant plasma creates a cavitation bubble and shock wave that cleaves the tissue at the focal point of the laser. A, Bright-field image of cultured wild-type RGC axon before axonal transection. The white arrow shows the future site of axotomy, which is magnified in the insert. The diameter of the axon at the site axotomy is 1.16 μm. B, Same axon 6 min after laser axotomy. The transection site is indicated by the white arrow and magnified in the insert. The width of the incision is 0.76 μm. Scale bars, 40 μm (5 μm in insert).
Detection of phosphatidylserine externalization after axotomy.
To detect the initiation and spread of phosphatidylserine externalization, annexin B12-IANBD was added to the medium before axotomy. Annexin B12 belongs to the family of highly preserved cytosolic proteins that in the presence of Ca2+ bind exclusively to the negatively charged membrane phospholipids in particular to the phosphatidylserine (Gerke et al., 2005). Similar to the other members of annexin family, the annexin B12 structure has a main core consisting of four domains. Arrangement of these domains gives the molecule a 3D shape of a bent disc, with convex and concave surfaces (Patel et al., 2005). Each domain is 70 aa long and contains five α-helices; four of them (helices A, B, D, E) are arranged in a cylindrical formation and perpendicularly positioned in the disc and the fifth α-helix (C) covers the formation on the concave surface (Isas et al., 2004). On the convex surface of annexin molecule, there are amino acid loops that connect the helices (A to B and D to E) of each domain; these loops contain the regions that provide the Ca2+- and membrane-binding sites (Isas et al., 2005). Kim et al. (2010a) attached the thiol-reactive fluorophore IANBD to the cysteine residing in Ca2+ and membrane-binding sites of the loop in annexin B12 to create an environment-sensitive fluorescent biosensor. IANBD amide only emits fluorescence when the molecule is in a nonpolar environment (e.g., the membrane-bound state of annexin) and the fluorescence is negligible (“off”) when in solution. Binding of annexin B12- IANBD to membranes places the label in a nonpolar lipid enclosure and switches “on” the fluorescence signal (Kim et al., 2010b). The use of annexin B12-IANBD in our study enabled extremely rapid detection of early membrane changes after axon transection. This provides a significant advantage over the use of conventional fluorescent-labeled annexin V (Sievers et al., 2003), which requires several washes to reduce the fluorescent background. These washes delay the detection of the signal and cause cell stress, which could confound the observations.
Quantification of axonal degeneration after axotomy.
The fluorescent probe annexin B12-IANBD (4 μg/ml) was added to the medium of MatTek glass-bottom dishes to visualize externalization of phosphatidylserine along the axon. After laser axotomy, time-lapse imaging was used to record the progress of fluorescent signal along the axon length, corresponding to a wave of membrane lipid asymmetry along the length of axon (Fig. 3A).
Figure 3.
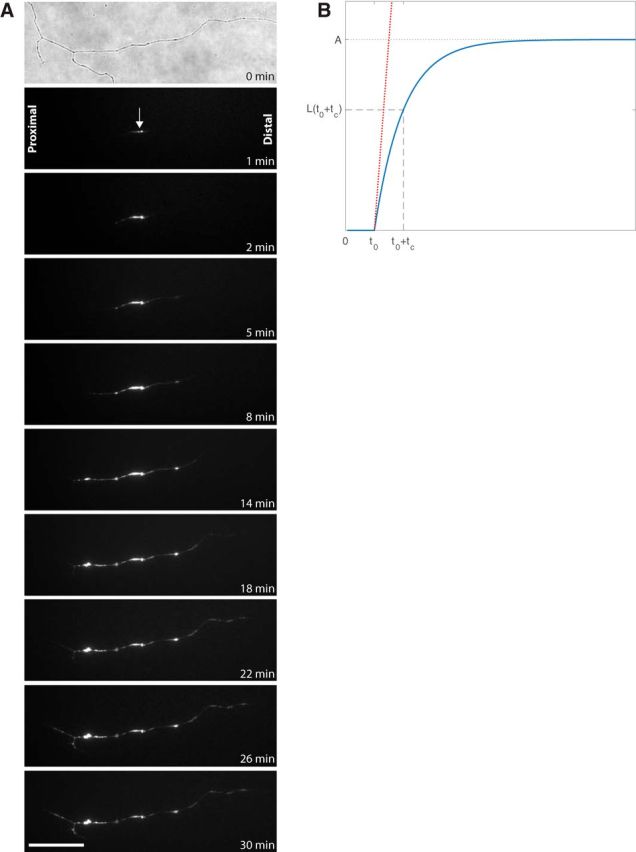
Phosphatidylserine externalization in a transected wild-type axon. A, First frame shows a bright-field image of the site of axotomy taken immediately before axonal transection of cultured wild-type RGC, followed by a selection of fluorescent time-lapse images showing progression of phosphatidylserine externalization visualized by the polarity-sensitive annexin-based biosensor, annexin B12-IANBD. The arrow shows the site of axotomy, with proximal indicating the propagation of signal toward the soma and distal indicating propagation toward the axon terminal. Scale bar, 40 μm. B, Derivation of the velocity (slope) and t0 (delay) of propagation from the time course of annexin B12-IANBD fluorescence.
Transection was followed immediately by real-time fluorescence imaging with the same 60× water-immersion objective using a sensitive Retiga 2000R video-rate camera. Initial images were captured every 10 s until fluorescence was visible and then every 60 s for a total of 30 min. Images were used to measure the delay, initiation, and velocity of the fluorescent signal of annexin B12-IANBD along the proximal and distal segments of the transected axon.
The length of the fluorescent segment of an axon as a function of time l(t) was fitted using an empiric mathematical model as follows:
 |
where A is a constant corresponding to the asymptotic value of the length of the fluorescent section of the axon (Fig. 3B). The time interval between axon transection and the first observation of fluorescence is defined as t0. The time constant tc characterizes the propagation of the fluorescent signal along the axon. H(t) is the Heaviside step function, defined as follows:
 |
where δ is the Dirac delta function. The three parameters (A, t0, and tc) were obtained with a linear least-square fit implemented in MATLAB. Axons that could not be fitted (based on the calculated A being longer than the axon itself or tc <1 min, both of which were physically impossible) were not analyzed. The excluded axons (eight of a total of 154 axons) were evenly distributed (by χ2 and t test analysis) across species, distal versus proximal axons, long versus short axons, distant versus near transection sites, age in culture, and age of animals.
The initial velocity was calculated as the right derivative of the length, evaluated at t0 as follows:
 |
Morphological evidence of axonal degeneration was based on the development of swelling and beading propagating away from the transection site along the axon (Fig. 4).
Figure 4.
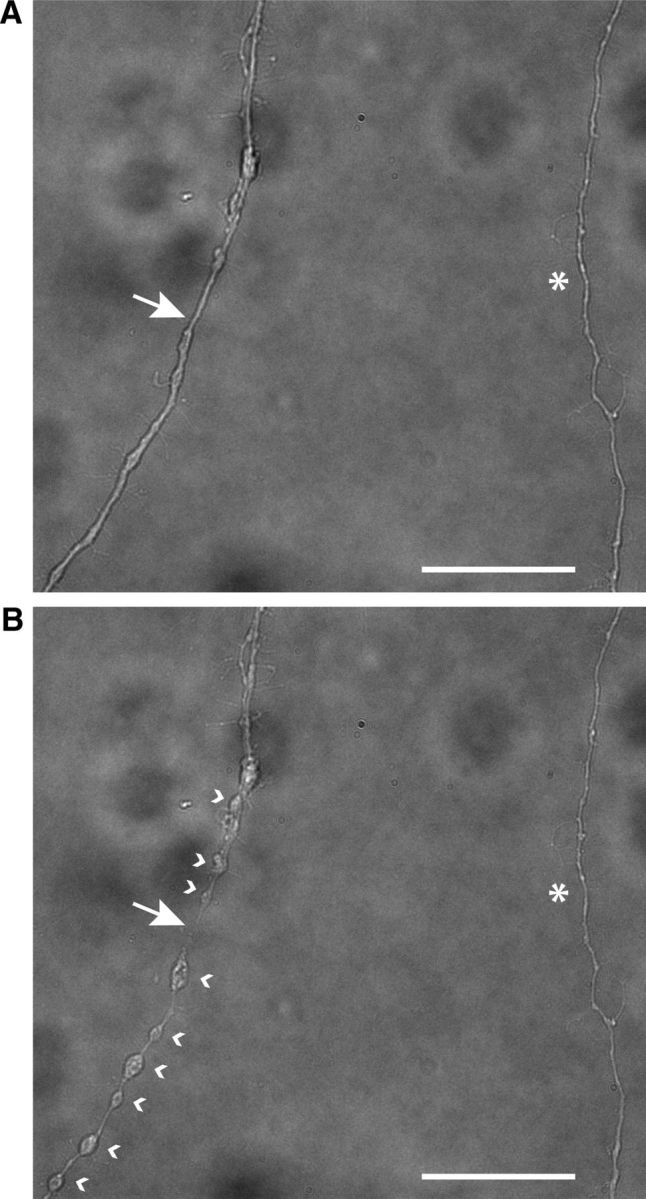
Morphological changes of axon after axotomy. A, Bright-field image of cultured wild-type RGC axons before axotomy. The axon at left was subject to axotomy, with the site of axotomy indicated by a white arrow. The axon at right, indicated by an asterisk, belong to a different RGC and was not transected. B, Forty-five minutes after the axotomy the beading and swelling of the transected axon are clearly visible. The white arrow shows the site of axotomy, the white arrowheads at the right side of transected axon show the beading of distal segment, and the arrowheads at the left side show the beading of proximal segment of transected axon. There were no morphological changes in a control nontransected (asterisk) axon. Scale bar, 40 μm.
Statistical analysis.
Because the velocity and delay distributions were not normally distributed, analyses were with nonparametric methods. Comparison of two groups was performed with the Wilcoxon test. Comparison of more than two groups was with the Kruskal–Wallis test, followed by Dunn nonparametric post hoc testing with joint ranking for comparison of more than two groups against a control. More complex models were analyzed using fit model for mixed models. All analyses were with JMP (SAS Institute; RRID: SCR_014242).
Results
Annexin B12-IANBD can be used to measure the velocity and delay until initiation of phosphatidylserine externalization in transected retinal ganglion cell axons
Within seconds after laser axotomy, the fluorescent signal of annexin B12-IANBD, reflected binding to externalized phosphatidylserine, was detected at the site of axotomy and to adjacent proximal and distal axonal segments (Fig. 3A). This reflects the externalization of phosphatidylserine to the outer leaflet of the axonal membrane. The length of the fluorescent segment for each axon was fitted as a function of time to obtain the velocity values (in micrometers per minute) for each segment. In the first 30 min after axotomy, the annexin B12-IANBD signal progressed at similar rates in the proximal and distal segments, with a mean velocity of 12.68 ± 1.57 μm/min (n = 22) proximally compared with 10.49 ± 1.56 μm/min (n = 19) distally (p = 0.346).
The velocity of phosphatidylserine externalization along the axon is one measure of axonal degeneration, but we also observed differences in the delay from the time of axonal transection to the first detection of phosphatidylserine externalization. Assuming that this delay reflects intracellular processes to initiate phosphatidylserine externalization, whereas velocity reflects the transmission of the degeneration signal along the axon, it is likely that different mechanisms are being probed by measuring each process.
The delay until initiation of phosphatidylserine externalization in the wild-type axons was 3.18 ± 0.64 min (n = 42). Together with the similar velocities of phosphatidylserine externalization in the proximal and distal segments, this indicates that the dynamics of retrograde and Wallerian degeneration with respect to membrane polarization are similar.
WldS axons have significantly slower velocity of phosphatidylserine externalization after axotomy compared with wild-type axons
The slow degeneration of the distal axon after transection in WldS mutant animals is well documented (Mack et al., 2001; Adalbert et al., 2005). We hypothesized that slowed degeneration in WldS axons is mediated in part by differences in axonal degeneration and, in particular, would correlate with the polarity of phosphatidylserine in the axonal membrane after axotomy. The use of the fluorescent reporter annexin B12-IANBD allows direct imaging of the flipping of phosphatidylserine from the inner to outer leaflets (Fig. 5A).
Figure 5.
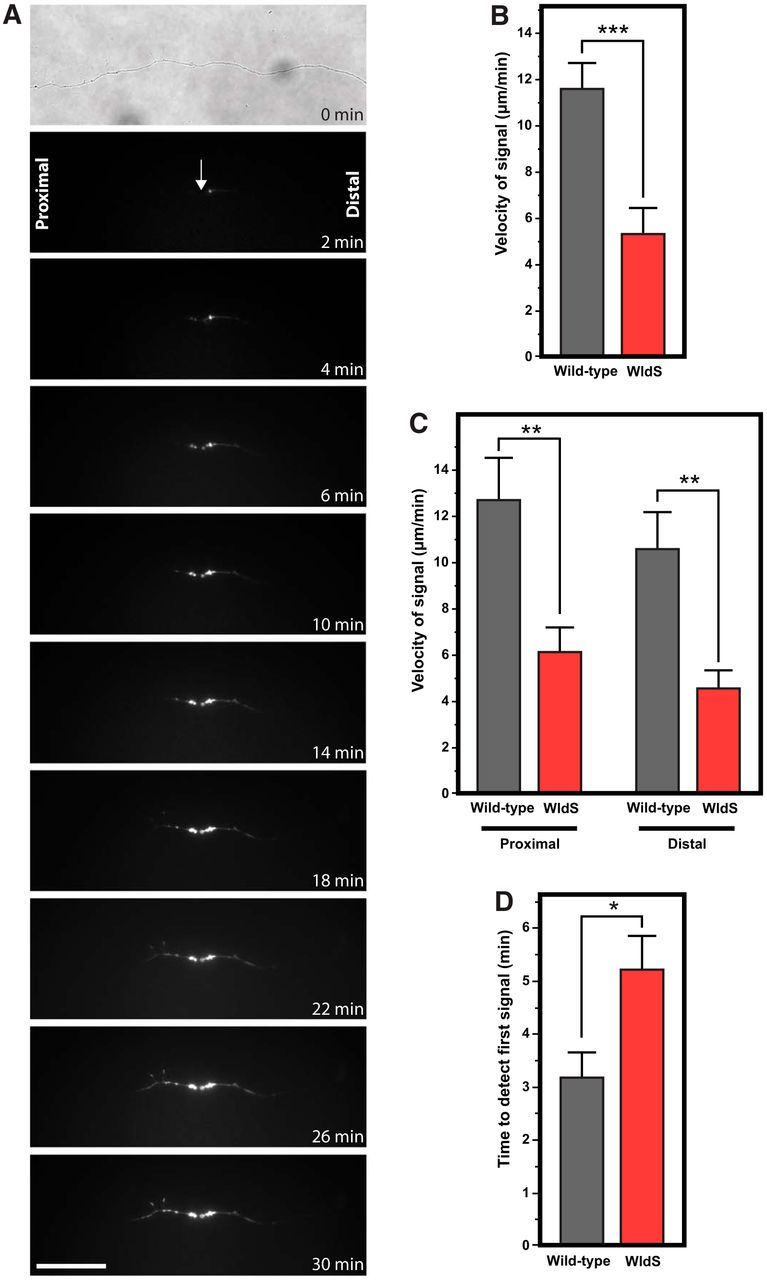
Phosphatidylserine externalization propagates more slowly along transected WldS axons compared with wild-type axons. A, Time-lapse series of images from a transected WldS axon show significantly slower propagation of phosphatidylserine externalization. B, WldS-transected axons had significantly slower phosphatidylserine externalization compared with wild-type axons. C, Slowed velocity of phosphatidylserine externalization was seen in both the proximal and distal axon segments. D, Compared with wild-type axons, there was a significantly longer delay before the onset of phosphatidylserine externalization signal in WldS axons. *p < 0.05; **p < 0.01; ***p < 0.001. Scale bar, 40 μm.
We found that transected WldS axons had a significantly slower velocity of phosphatidylserine externalization along the axon compared with wild-type axons (Fig. 5B). In WldS axons, the mean velocity was 5.24 ± 1.29 μm/min (n = 30) compared with 11.67 ± 1.11 μm/min (n = 41) in wild-type axons (p = 0.0001). To determine whether this difference was specific to retrograde versus anterograde signaling, the data for the proximal and distal segments were analyzed separately. The mean velocity of phosphatidylserine externalization along the proximal segment of transected WldS axons was 6.14 ± 1.90 μm/min (n = 15) compared with 12.68 ± 1.57 μm/min (n = 22) for wild-type axons (p = 0.011). In the distal axon, phosphatidylserine externalization spread with a mean velocity of 4.35 ± 1.76 μm/min (n = 15) for WldS axons compared with 10.49 ± 1.56 μm/min (n = 19) for wild-type axons (p = 0.002). The velocity of phosphatidylserine externalization in WldS axons was 48% slower for the proximal segment and 41% slower in the distal segment compared with wild-type axons (Fig. 5C). Finally, there was no difference in velocity of signaling between proximal and distal segments of WldS axons (p = 0.229), similar to what was observed in wild-type axons.
The initial morphological alterations resulting from laser axotomy were limited to a 2–3 μm area surrounding the focal point of the laser (Fig. 2), which became visible within minutes after axotomy. With the progress of time, morphological changes associated with axonal degeneration were detected along the axon (Fig. 4). To further understand the difference in velocity of phosphatidylserine externalization between WldS and wild-type axons, we compared it with the morphological changes associated with axonal degeneration, detected as swelling and beading along axon. The mean velocity of axonal degeneration was 0.90 ± 0.15 μm/min in WldS axons and 1.51 ± 0.14 μm/min in wild-type axons (p = 0.01).
We then measured the delay between the transection of the axon and the first detection of the annexin B12-IANBD signal in wild-type and WldS axons, which represents the time until the phosphatidylserine polarity first flips from internal facing to external facing. In WldS axons, the mean delay was significantly longer than in wild-type axons (5.17 ± 0.71 min, n = 34 vs 3.18 ± 0.64 min, n = 42; p = 0.042). Therefore, in WldS axons, the velocity of spread of phosphatidylserine externalization was significantly slower, whereas the delay from transection until the first evidence of externalization was significantly longer (Fig. 5D).
Redox modulation significantly slows the velocity of phosphatidylserine externalization in transected wild-type axons
The mechanism by which phosphatidylserine externalization is slowed after axotomy in WldS axons could be related to the mutation's effect on redox signaling (O'Donnell et al., 2013). To explore this hypothesis, we used a cell-permeable intracellular disulfide reducing agent, PB1 (Fig. 6A) (Schlieve et al., 2006; Niemuth et al., 2016), which we had shown previously to slow axonal degeneration (Almasieh et al., 2011). Preincubation of primary RGCs for 30 min before axotomy in medium containing PB1 (100 μm) affected the dynamics of phosphatidylserine externalization. The mean velocity after transection of axons in medium with PB1 was significantly slower (6.73 ± 2.14 μm/min, n = 15) compared with control medium (11.66 ± 1.29 μm/min, n = 41; p = 0.004; Fig. 6B). In comparison, the mean velocity of morphological changes associated with axonal degeneration (swelling and beading) for PB1-treated wild-type axons was 0.81 ± 0.16 μm/min compared with 1.51 ± 0.14 μm/min in untreated wild-type axons (p = 0.002) and similar to the 0.90 ± 0.15 μm/min observed in untreated WldS axons (p = 0.169).
Figure 6.
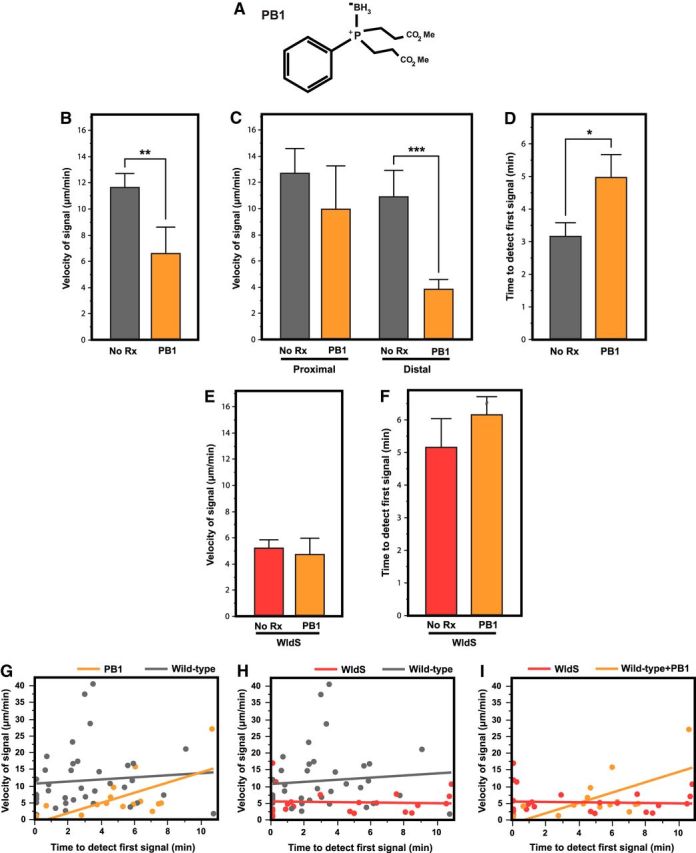
A, Structure of the cell-permeable redox-modulating compound bis(3-propionic acid methyl ester) phenylphosphine-borane complex (PB1), used for intra-axonal disulfide reduction. B, Incubation of wild-type RGCs in 100 μm PB1 significantly slowed the propagation of phosphatidylserine externalization compared with untreated cultures. C, Analysis of the proximal and distal segments showed that the velocity of phosphatidylserine externalization was significantly slowed by PB1 along the distal segments of transected wild-type axons. D, PB1 treatment significantly delayed the initiation of phosphatidylserine externalization. E, F, Preincubation of WldS cultures with PB1 did not affect the delay or velocity of phosphatidylserine externalization. G–I, Mixed-model analysis of the interaction between the velocity/delay relationship and WldS or PB1 treatment. *p < 0.05; **p < 0.01; ***p < 0.001.
To better understand these results, we analyzed the proximal and distal segments separately. The velocity of phosphatidylserine externalization was less slowed by PB1 along the proximal segment of transected wild-type axons (9.91 ± 3.36 μm/min, n = 7 vs 12.68 ± 1.89 μm/min, n = 22; p = 0.47) than along the distal segment (3.91 ± 2.86 μm/min, n = 7 vs 10.97 ± 1.79 μm/min, n = 18; p = 0.001; Fig. 6C). There was a significant difference in delay until initiation of phosphatidylserine externalization between PB1-treated wild-type axons and untreated wild-type axons (4.89 ± 0.69 min, n = 16 vs 3.18 ± 0.42, n = 42; p = 0.028; Fig. 6D). These results demonstrate that the redox-dependent mechanism for initiation of phosphatidylserine externalization is similar to that for its propagation.
To test whether the signaling of redox modulation on phosphatidylserine externalization was occurring in the intracellular or extracellular compartments, the effects of PB1 was compared with two non-cell-permeable disulfide reducing agents, DTT and TCEP. In contrast to the effects of PB1 in slowing velocity of phosphatidylserine externalization, preincubation of primary RGCs with DTT (100 μm) or TCEP (100 μm) for 30 min did not affect the velocity (8.93 ± 3.42 μm/min, n = 6, p > 0.9 for DTT and 13.41 ± 2.41 μm/min, n = 12, p > 0.9 for TCEP). Unlike PB1, which increased the delay until phosphatidylserine externalization after axonal transection, the delay was significantly decreased with DTT (0.24 ± 1.17 min, n = 6, p = 0.013 vs no treatment) and TCEP (1.08 ± 0.82 min, n = 12 p = 0.042 vs no treatment). Together, these findings are consistent with intracellular redox signaling of the spread of axonal degeneration after axotomy and for extracellular redox signaling in its initiation.
In contrast to the effects of PB1 on wild-type RGCs after axotomy, PB1 treatment of WldS RGCs had no effect on the velocity (4.74 ± 1.26 μm/min, n = 8 vs 5.24 ± 0.65 μm/min, n = 30, p > 0.9; Fig. 6E) or delay (6.26 ± 2.05 min, n = 8 vs 5.17 ± 0.99 min, n = 34, p = 0.665) until phosphatidylserine externalization after axotomy (Fig. 6F). Conversely, preincubation of WldS RGCs with DTT and TCEP, which, unlike PB1, affects the extracellular redox state, reversed the WldS phenotype. Specifically, they significantly increased velocity (16.18 ± 2.06 μm/min, n = 16 and 17.17 ± 2.38 μm/min, n = 12, p = 0.002 and 0.0001 vs no treatment, respectively) and decreased the delay (1.44 ± 1.17 min, n = 16 and 0.16 ± 1.35 min, n = 12, p = 0.040 and p = 0.0001 vs no treatment, respectively).
To study the interaction between the delay and the velocity of phosphatidylserine externalization in wild-type, WldS, and PB1-treated wild-type axons, we analyzed the two variables together using a mixed-model approach. Delayed initiation of phosphatidylserine externalization was not correlated with the velocity of the signal in wild-type axons (r = 0.09; p = 0.58) or WldS axons (r = 0.08; p = 0.66), but was seen in PB1-treated wild-type axons (r = 0.62; p = 0.014). The mixed-model analysis demonstrated an interaction between velocity/delay and whether an axon was WldS or PB1-treated wild-type (p = 0.0011; Fig. 6G–I), suggesting that intra-axonal disulfide reduction recapitulates most but not all features of the WldS phenotype. These findings together suggest a common mechanism to the WldS mutation and intracellular redox signaling in slowing the signaling of axonal degeneration, with extracellular reduction having opposite effects.
Axon length significantly slows the velocity and increases the delay until phosphatidylserine externalization in axotomized RGCs
Neurons with long axons have more cell surface and time for axonal transport to carry retrograde and anterograde survival signals and therefore may be more susceptible to axonal injury, as seen in some peripheral neuropathies with respect to energy metabolism along the axon. Recent evidence indicates a reduced mobility of mitochondria in the distal segment of axons as they mature (Lewis et al., 2016) and similar slowed mitochondrial movement has been shown to contribute to the initiation of neurodegenerative disease (Takihara et al., 2015). We therefore investigated whether axonal length affects the velocity or delay of phosphatidylserine externalization after axotomy. The median of all axon lengths was calculated (717 μm) and RGCs with axons shorter than the median were placed in the short axon group and RGCs with axons longer than the median were placed in the long axon group. The short axons had a mean length of 524.5 ± 16.6 μm compared with long axons with mean length of 1004.0 ± 48.5 μm. The minimum length of an axon included in the short group was 182.6 μm and the maximum length was 714.3 μm. The minimum length of an axon included in the long group was 720 μm and the maximum length was 2677 μm. The mean diameter of short axons was slightly greater than the mean diameter of long axons (1.03 ± 0.02 vs 0.94 ± 0.03; p = 0.01), although the diameter did not correlate with axonal length (r = 0.0625; p = 0.63).
Short wild-type axons had a significantly faster velocity of phosphatidylserine externalization compared with their longer counterparts (13.00 ± 1.66 μm/min, n = 28 vs 8.27 ± 2.66 μm/min, n = 11; p = 0.04; Fig. 7A). However, short wild-type axons were significantly slower to initiate externalization compared with long axons (3.51 ± 0.47 min, n = 28 vs 1.56 ± 0.76 min, n = 11; p = 0.021; Fig. 7B).
Figure 7.
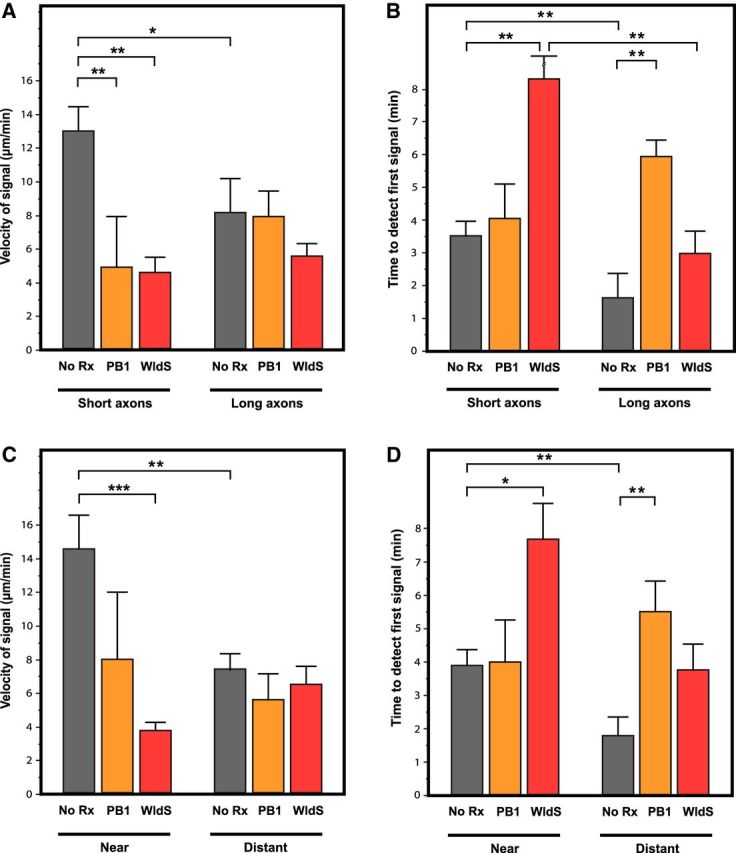
Axon length and distance between the axotomy site and the soma affects the velocity and delay until onset of phosphatidylserine externalization. A, Short, wild-type axons had a significantly faster velocity of propagation of phosphatidylserine externalization after transection compared with longer axons, to short PB1-treated axons, and to short WldS axons. The effect of PB1 treatment and the WldS mutation versus wild-type were significantly less in longer axons. B, Short, wild-type axons had a longer delay until initiation of phosphatidylserine externalization compared with long wild-type axons. This pattern was also seen in WldS axons, but not PB1-treated axons. C, Wild-type axons with the axotomy site near the soma had a significantly faster velocity of phosphatidylserine externalization compared with where the axotomy site was distant from the soma. The velocity was also significantly slower for WldS axons transected near the soma compared with wild-type. When axotomy was distant from the soma, neither PB1 treatment nor the WldS mutation affected velocity compared with wild-type, untreated axons. D, Axotomy near the soma in wild-type axons resulted in a longer delay until initiation of phosphatidylserine externalization compared with distant transections. This pattern was also seen in WldS axons, but not PB1-treated axons. *p < 0.05; **p < 0.01; ***p < 0.001.
To study the redox-dependency of the effect of axon length on velocity and delay, RGCs were treated with PB1 and then axotomized. Preincubation of RGCs with PB1 significantly slowed the velocity of phosphatidylserine externalization in short axons, from 13.00 ± 1.78 μm/min (n = 28) in control media to 4.89 ± 3.86 μm/min (n = 6) with PB1 treatment (p = 0.01) (Fig. 7A), but had no effect in long axons (8.27 ± 1.75 μm/min, n = 11 to 7.94 ± 1.93 μm/min, n = 9; p = 0.196). Conversely, PB1 significantly delayed the onset of phosphatidylserine externalization in long axons by increasing the delay from 1.56 ± 0.76 min (n = 11) in control axons to 5.90 ± 0.67 min (n = 10, p = 0.001; Fig. 7B), but had no effect in short axons (3.51 ± 0.53 min, n = 11 in control axons vs 4.02 ± 1.14 μm/min, n = 6 with PB1 treatment; p = 0.984). Together, these findings imply that the difference in the redox mechanisms underlying phosphatidylserine externalization initiation and propagation extends to differences between short and long axons.
Given that PB1 slowed velocity in short, but not long, wild-type axons, we compared phosphatidylserine externalization in short and long WldS axons to wild-type axons. Short WldS axons had a significantly slower velocity of phosphatidylserine externalization than short wild-type axons (4.58 ± 2.42 μm/min, n = 12 vs 13.00 ± 1.6 μm/min, n = 28; p = 0.001), but this difference was not as pronounced between long WldS axons and long wild-type axons (5.68 ± 0.98 μm/min, n = 18 vs 8.27 ± 1.26 μm/min, n = 11; p = 0.069; Fig. 7A). The greater effect of the mutation in short versus long axons paralleled what had been seen with PB1 treatment. We also found that short WldS axons had a significantly longer delay until the onset of phosphatidylserine externalization compared with their wild-type counterparts (8.60 ± 1.27 min, n = 12 vs 3.51 ± 0.83 min, n = 28; p = 0.002). The delay in the onset of signal was not significantly different between long WldS and wild-type axons (3.06 ± 0.69 min, n = 18 vs 1.56 ± 0.88 min, n = 11; p = 0.227; Fig. 7B). Finally, we compared short and long WldS axons, and although the velocity of phosphatidylserine externalization was not significantly different between short and long WldS axons (4.58 ± 1.04 μm/min, n = 12 vs 5.68 ± 0.85 μm/min, n = 18; p = 0.421), there was a longer delay in its initiation in short compared with long WldS axons (8.60 ± 1.40 min, n = 12 vs 3.06 ± 1.20 min, n = 18; p = 0.01; Fig. 7A). This suggests that there are differences in the nature of redox-dependent phosphatidylserine externalization that are related to axon length.
Distance from transection site to soma affects the dynamics of phosphatidylserine externalization along the axon
Previous studies demonstrated that the distance from the site of axonal injury to the soma affects the rate of neuronal death, based on studies of RGCs after intraorbital versus intracranial axonal injury (Villegas-Pérez et al., 1993). The magnitude of depolarization recorded at the soma after axotomy is also dependent on the distance of the axotomy site from the soma (Berdan et al., 1993). The neuronal soma contains most of the metabolic machinery to support the axon and is the recipient of retrograde positive and negative signals. We hypothesized that the distance from soma to site of axonal injury would affect the dynamics of axonal degeneration signaling. To test whether the relationship of the axotomy site to the cell body affects the velocity and delay of phosphatidylserine externalization, we performed laser axotomy at different distances from the RGC soma and studied the time course of annexin B12-IANBD binding. The closest distance from axotomy site to soma was 95 μm and the furthest was 2272 μm from the soma. Analysis was performed by dividing all axons into “near” or “distant” based on whether the distance from soma to axotomy was less than or greater than the median distance of 343 μm determined from all axons that were studied. The mean distance for the near group was 230.9 ± 7.9 μm and for the distant group was 568.2 ± 42.5 μm.
The distance of the axotomy from the soma significantly affected the velocity of phosphatidylserine externalization in wild-type RGC axons, with axotomy near the soma producing a significant increase in velocity compared with distant locations (14.46 ± 1.64 μm/min, n = 24 vs 8.61 ± 1.93 μm/min, n = 14; p = 0.029; Fig. 7C). Axotomy near the soma also resulted in a significantly delay until first detection of phosphatidylserine externalization compared with axotomy distant from the soma (3.93 ± 0.54 min, n = 22 vs 1.78 ± 0.65 min, n = 15; p = 0.013; Fig. 7D).
To determine whether the association of velocity and distance from the soma was redox dependent, RGCs were preincubated with PB1 (100 μm) for 30 min and then axotomized. PB1 did not interact with the effect of distance from axotomy to soma on the velocity of phosphatidylserine externalization (p = 0.304; Fig. 7C). However, PB1 treatment significantly prolonged the delay until detection of the annexin signal for transections distant from the soma (5.42 ± 0.93 min, n = 10 vs 1.78 ± 0.65 min, n = 15; p = 0.010), but not near the soma (4.00 ± 1.34 min, n = 6 vs 3.93 ± 0.54 min, n = 22; p = 0.846; Fig. 7D).
Given the above results and the likelihood that the WldS phenotype causes slowed axonal degeneration via a redox-dependent mechanism, we studied the velocity of phosphatidylserine externalization in WldS axons that were axotomized near and distant to the soma. WldS axons transected near the soma had a significantly slower velocity of phosphatidylserine externalization compared with wild-type axons (3.88 ± 2.14 μm/min, n = 14 vs 14.46 ± 1.64 μm/min, n = 24; p = 0.0001; Fig. 7C). When axons were transected distant from the soma, there were minimal differences in the velocity of phosphatidylserine externalization between WldS and wild-type (6.43 ± 1.09 μm/min, n = 16 vs 7.46 ± 1.06 μm/min, n = 17; p = 0.377; Fig. 7C). The same pattern of WldS affecting axons axotomized near the soma was observed with respect to the delay until the first detection of phosphatidylserine externalization. There was no significant difference between WldS and wild-type axons in delay when axons were transected near (7.64 ± 1.25 min, n = 14 vs 3.93 ± 0.54 min, n = 22; p = 0.197) or distant from the soma (3.75 ± 0.83, n = 18 vs 1.78 ± 0.65 min, n = 15; p = 0.261; Fig. 7D). Finally, preincubation of WldS RGCs with PB1 had no effect on the interaction between transection site distance from soma and velocity, suggesting that pharmacological modulation of redox signaling overlaps with the intracellular mechanism(s) affected by the WldS mutation.
Effect of the age and time in culture on the velocity of phosphatidylserine externalization after axotomy
For purified RGC cultures we harvested retinas from postnatal day 4 (P4) pups and older (up to P6) because younger RGCs have a higher rate of survival in culture because of ease in enzymatically dissociating the retina. However, the process of removing the retina from the eye severs the RGC axons in the retinal nerve fiber layer. Early postnatal RGCs are not fully developed neurons and maintain the capacity to regenerate a new axon in culture in the presence of trophic factors (Goldberg et al., 2002). This axonal regeneration in culture is the basis of studying axonal degeneration in vitro.
However, a potential confounding factor is that developmental RGC target-dependent death occurs from soon after birth in rats. The majority of RGC death occurs between P0 and P5, and although RGC death continues to P10, the numbers are negligible (Potts et al., 1982; Galli-Resta and Ensini, 1996). It was therefore critical to assess whether the velocity of the axonal degeneration was dependent on age of the animal and, specifically, whether there was a difference if RGCs were derived from pups before versus during the time of development-associated degeneration. To do this, the measurements of phosphatidylserine externalization were analyzed with respect to age and time in culture.
There was no significant age-dependent difference in the velocity of phosphatidylserine externalization after transection among RGCs harvested from pups ranging from P2–P7 (p = 0.286; Fig. 8A). The length of axons obtained from young (P2–P5) pups was ∼25% less than those from older (P6–P7) pups (652 ± 48 vs 813 ± 40; p = 0.047). Age-dependent increases in cellular neurotrophic levels are thought to promote higher survival rate for neurons in postnatal injury models (Pollin et al., 1991; Kuzis et al., 1999). However, the present data do not support a difference in the rate at which axonal degeneration spreads in vitro in these neurons.
Figure 8.
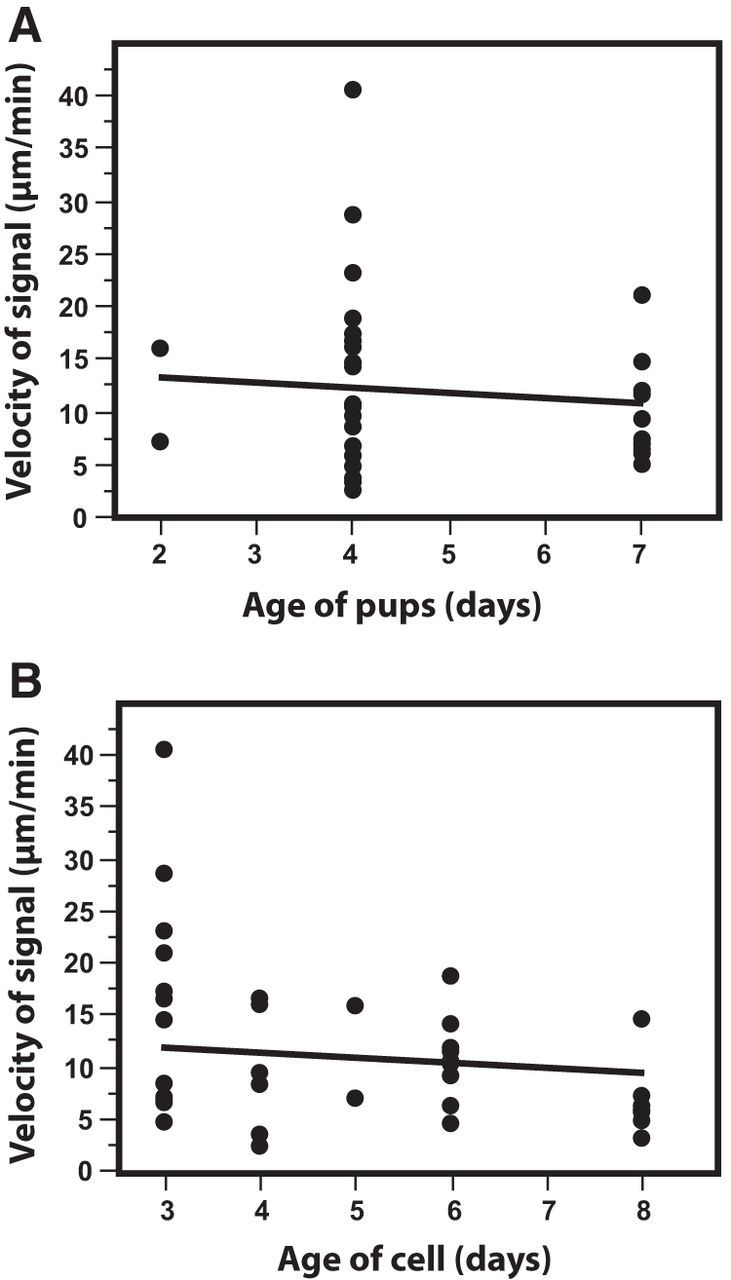
Age of rat pups used for RGC cultures and length of time in culture did not affect the velocity of phosphatidylserine externalization. A, There was no significant difference in the velocity of annexin B12-IANBD signal among RGCs harvested from pups ranging from P2 to P7. B, Length of time in culture for RGCs used in axotomy experiments ranged between 3 and 8 d. There was no significant effect on the velocity of phosphatidylserine externalization.
Laser axotomy was performed on cultured RGCs 3 d after plating to allow the growth of identifiable axons. The length of time that RGCs were in culture before cutting the axon (r = 0.37; p = 0.0228) correlated with the velocity of phosphatidylserine externalization after axotomy, but with lower velocity in older cells (Fig. 8B). In contrast, in a study of axotomized dorsal root ganglion (DRG) axons, the time in culture had a significant effect on the velocity of the Wallerian degeneration, with greater velocity when there was greater time in culture (Buckmaster et al., 1995; Sievers et al., 2003). The differences between the results seen in DRG axons and the velocity of phosphatidylserine externalization after RGC axotomy could be related to differences in peripheral and CNS axon responses to injury or other factors.
Discussion
RGC axotomy in vivo induces intracellular superoxide generation followed by phosphatidylserine externalization, with both events occurring over days (Kanamori et al., 2010). In the present study, the combination of single-cell laser axotomy with time-lapse imaging enabled the study of the dynamics of phosphatidylserine externalization beginning within minutes after the onset of axonal injury. These findings help improve our understanding of the mechanisms that maintain the asymmetry of lipids in the axonal membrane and its dysregulation after axonal injury.
In this study, we demonstrated an immediate change in axonal membrane lipid asymmetry of cultured RGCs after axotomy, manifested as phosphatidylserine externalization into the outer leaflet of the axonal membrane, which was redox sensitive and was ∼8× faster than the rate of axonal degeneration. Movement of lipids between the inner and outer leaflets of axonal membrane is mediated by two categories of transmembrane lipid transporters. The first category are ATP-dependent carriers transporting lipids in a single direction. Depending on the direction of transport, these are referred to as flippases (from the outer leaflet to the inner leaflet) or floppases (from the inner leaflet to the outer leaflet; Pomorski and Menon, 2006). In addition to ATP, the activity of these transporters is regulated by caspases 3, 6, and 7 (Segawa et al., 2014). The second category, scramblases, transport lipids bidirectionally without requiring energy. Scramblases are either constitutively active (Goren et al., 2014) or regulated by intracellular signals such as Ca2+ (Suzuki et al., 2010; Williamson, 2015), making Ca2+ entry into the axon both an initiator of axonal degeneration (George et al., 1995; Knöferle et al., 2010; Vargas et al., 2015) and a major factor in loss of asymmetry of membrane lipids because its influx triggers externalization of phosphatidylserine by scramblases (Bevers and Williamson, 2010).
It is unclear what regulates the velocity by which phosphatidylserine externalization propagates down the injured axon, which in wild-type RGC axons is ∼12 μm/min and in WldS axons 5 μm/min. Sievers et al. (2003) demonstrated spread of annexin-A5 binding at 5–6 μm/min in DRG explants where the axons were transected with a microscalpel, similar to the present findings. This rapid propagation of phosphatidylserine externalization is unlikely to be related to axonal conduction, which for unmyelinated axon occurs at 0.5–10 m/s, ∼8 orders of magnitude faster. Nor does correlate with the velocity by which the transient calcium wave propagates down an axon after axotomy which is ∼2.7–16 μm/s (160–960 μm/min) (Ziv and Spira, 1993; Rishal and Fainzilber, 2014; Vargas et al., 2015). The calcium increase lasts ∼30 s and returns to normal levels within a minute after the onset of axonal injury (Knöferle et al., 2010). After this initial transient calcium wave, there was a gradual increase in intra-axonal calcium 4–6 h later, which remained elevated and led to axonal fragmentation via activation of calpains (LoPachin et al., 1990; Vargas et al., 2015). The tempo for this secondary calcium elevation also does not correlate with the velocity of phosphatidylserine externalization down the axon.
The mechanisms responsible for the decreased axonal degeneration in WldS animals are not well understood. One theory suggests that the WldS mutation maintains low axoplasmic Ca2+ levels after axonal damage. Immediately after axotomy, a transient calcium elevation occurs in both wild-type and WldS axons, but WldS axons do not show a second elevation in calcium levels (Adalbert et al., 2012). This theory could explain the small difference between wild-type and WldS axons in the delay until initial detection of phosphatidylserine externalization. The absence of the second axonal calcium wave could be due to the greater buffering capacity of WldS mitochondria (Avery et al., 2012). Conversely, a comparable degree of mitochondrial membrane potential loss occurs in WldS and wild-type mitochondria incubated with calcium, suggesting that WldS mitochondria do not buffer calcium differently from wild-type (Barrientos et al., 2011). Axotomy of wild-type neurons in vivo also affects the redox state of mitochondria, resulting in a rapid increase of ROS production that can be prevented by transgenic expression of WldS (O'Donnell et al., 2013).
We had shown previously a redox-dependent role for RGC apoptosis after axonal injury (Kanamori et al., 2010; Almasieh et al., 2011). We hypothesized there may be a redox component to the axonal degeneration for which WldS may be protective. The present studied demonstrated that RGC cultures incubated in the redox-modulating compound PB1 resulted in significantly slower velocity of phosphatidylserine externalization and a longer delay until the detection of the first signal. These data are consistent with a mechanism whereby an intra-axonal oxidative reaction, possibly related to mitochondrial depolarization (Sievers et al., 2003), initiates and propagates phosphatidylserine externalization after axotomy. This is supported by the data of Fadeel et al. (1999), who elicited robust phosphatidylserine externalization when Raji, HL-60, and other non-neuronal cell lines were incubated with the oxidants N-ethylmaleimide (NEM) and diamide (Fadeel et al., 1999).
Evidence for a redox-dependent mechanism for phosphatidylserine externalization is provided by studies of flippases in oxidative and apoptotic conditions. As mentioned previously, constant activity of flippases is required to move phosphatidylserine from the outer leaflet to the inner leaflet to maintain its asymmetric distribution (Pomorski and Menon, 2006). Segawa et al. (2014) demonstrated that the cleavage of ATP11C (a flippase) by caspases 3, 6, and 7 during apoptosis blocked its activity and led to exposure of phosphatidylserine in the outer leaflet. The inhibition of caspases by the pan-caspase inhibitor z-VAD-fmk did not prevent phosphatidylserine exposure induced by the oxidizing agents N-ethylmaleimide or pyridyldithioethylamine (PDA) (Balasubramanian et al., 2007), suggesting that oxidizing agents trigger phosphatidylserine exposure in an apoptosis-independent manner. It is thought that PDA oxidizes a sensitive site in flippases and blocks the association of transporter and phosphatidylserine, resulting in the accumulation of phosphatidylserine in the outer leaflet of membrane (Connor and Schroit, 1990, 1991). Conversely, incubation of cultures with a high concentration of the disulfide-reducing dithiol dithioerythritol, an epimer of DTT, resulted in inhibition of calcium-activated transfer of phosphatidylserine from the inner to outer leaflet, that is, flopping, whereas incubation with the oxidizing agent NEM increased externalization (Kamp et al., 2001).
DTT and TCEP are poorly membrane permeable and, when added to the culture medium, act as extra-axonal disulfide-reducing agents. This differs from the effect of PB1, which is only active when its methyl esters are hydrolyzed by intra-axonal esterases and therefore has intra-axonal disulfide-reducing activity. We found that the delay to initiation of phosphatidylserine externalization was longer with PB1 and shorter with DTT and TCEP. The effects of PB1 on wild-type axons were similar to that of the WldS mutation with respect to velocity of phosphatidylserine externalization, delay until onset, and velocity of propagation of morphological evidence of axonal degeneration. A model that explains these results is depicted in Figure 9.
Figure 9.
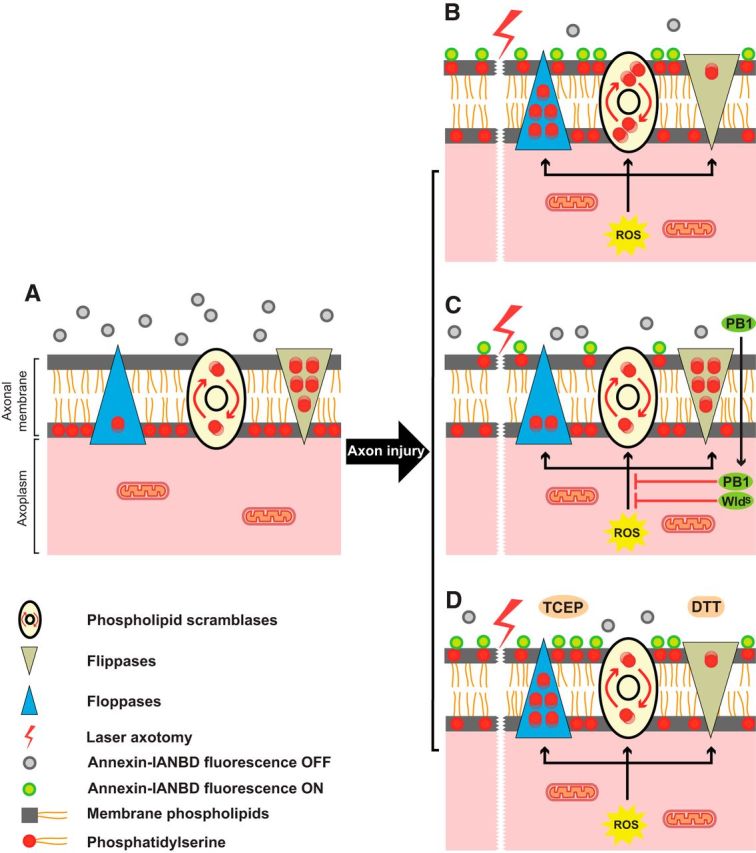
Diagram illustrating a putative mechanism by which intra-axonal disulfide reduction with PB1 and the WldS mutation promote axonal protection. A, In the intact axon, the distribution of phosphatidylserine (red circles) is actively restricted to the cytosolic (inner) leaflet of the plasma membrane. Scramblases (ovals) randomly shuffle phosphatidylserine between the inner and outer leaflet of membrane. Floppases (blue triangles) move the phosphatidylserine from the inner leaflet to the outer leaflet of the membrane, whereas flippases (beige triangles) actively remove phosphatidylserine from the outer leaflet and insert them in the inner leaflet. The net effect is to maintain phosphatidylserine asymmetry in the inner leaflet. B, After axotomy, the generation of intracytoplasmic ROS affects these transporters and the balance shifts, with floppases moving more phosphatidylserine to the outer leaflet while the rate of inward transport of phosphatidylserine by flippases slows. Scramblases possibly increase their rate of shuffling. The result is a net accumulation of phosphatidylserine in the outer leaflet of membrane. The exposure of phosphatidylserine to the outer leaflet and its interaction with annexin B12-IANBD (gray circles) switches “on” the fluorescence signal (green circles). C, Either the WldS mutation or intra-axonal disulfide reduction with membrane-permeable PB1 (which is inactive outside the axon) can inhibit the effects of ROS on flippases, floppases, and scramblases and are thereby able to maintain the asymmetry of phosphatidylserine across the membrane. D, Poorly permeable disulfide-reducing agents 1,4-dithiothreitol (DTT) and tris (2-carboxyethyl) phosphine hydrochloride (TCEP) reduce extra-axonal disulfides and inhibit transporter activity, with the effect on flippases maintaining phosphatidylserine at the outer leaflet, thereby reversing the WldS phenotype.
In addition to the effects of reduction on the oxidation status of the transporters slowing externalization (Castegna et al., 2004; de Jong and Kuypers, 2006), the oxidation status of phosphatidylserine itself also has a significant effect on the rate of its externalization (Kagan et al., 2000; Tyurina et al., 2004). The observation that PB1, a membrane-permeable disulfide-reducing agent, was effective in slowing the rate of phosphatidylserine externalization is consistent with the target of redox modification inside the damaged axon being phosphatidylserine itself.
The faster velocity of phosphatidylserine externalization in shorter axons could reflect the state of axonal maturity. Axon maturation involves several changes that stabilize the membrane, including formation of protein–lipid complexes (Ledesma et al., 1998), upregulation of sphingomyelin synthesis (Tiveron et al., 1994; Ledesma et al., 1999), reduced expression of membrane-trafficking proteins such as Sar1 (Aridor and Fish, 2009) and distribution of proteins like Thy-1 along the membrane (Xue et al., 1991; Rege and Hagood, 2006; Smrz et al., 2007). Given that both the WldS mutation and PB1 slowed phosphatidylserine exposure in short axons more than long axons, it is possible that a less mature axonal membrane or actively growing axon amplifies the effect of redox modulation. Another possibility is that short axons had slightly greater diameters than long axons and even slight differences in the surface/volume ratio could interact with redox processes. Finally, proximity to the soma is associated with improved axonal regeneration because of soma-derived signals (Benfey and Aguayo, 1982; Plunet et al., 2002; Conta Steencken et al., 2011). The effect of WldS on slowing velocity of phosphatidylserine externalization was mostly seen in transections near the soma. This could either be from a difference in redox signaling that depends on distance to the soma or the effect of a soma-derived factor that counteracts a redox injury process.
The propagation of phosphatidylserine externalization in transected RGC axons in the present study (11.7 μm/min) was several times faster than the spread of morphological axonal degeneration (1.5 μm/min). Similar results were seen in vivo by Knöferle et al. (2010) in an optic nerve crush model in which the velocity of morphological changes of RGC axonal degeneration in vivo was 1.2 μm/min, similar to the 1.5 μm/min observed in vitro in the present study. Phosphatidylserine externalization was eight times faster than the morphological changes associated with axonal degeneration, raising the question of how phosphatidylserine externalization relates to consequent axonal degeneration. The exposure of negatively charged lipids such as phosphatidylserine into outer leaflet of the membrane increases ion influx through Ca2+-activated potassium channels (Tillman and Cascio, 2003), whereas at inner leaflet, phosphatidylserine facilitates cellular functions such as exocytosis, endocytosis, membrane scaffolding, and G-protein signaling (Swairjo and Seaton, 1994). It is likely that prolonged loss of asymmetric distribution of phosphatidylserine across the inner and outer leaflets of the axonal membrane could initiate degeneration of the damaged axon.
The present study used an in vitro model of axotomized purified RGCs to study the role of membrane lipid asymmetry and redox state in axonal degeneration. However, those factors can also be affected by elements missing from this system such as astrocytes, oligodendrocytes, microglia, and vascular cells. For example, phosphatidylserine externalization triggers phagocytic removal of injured axons by activated microglia and astrocytes (Nguyen et al., 2011; Brown and Neher, 2014). Neuron-glial coculture improves the survival of RGCs by releasing antioxidants (Skytt et al., 2016; Vecino et al., 2016). Use of in vitro myelination models such as oligodendrocyte–neuron cocultures could provide helpful information about degenerative events in myelinated axons (Callizot et al., 2011; Pang et al., 2012). However, detailed study of axonal membrane polarity in disease is difficult to reproduce in vitro, yet imaging techniques do not currently provide single-axon resolution to allow study in vivo.
In summary, the initiation and propagation of phosphatidylserine externalization after axotomy is associated with an intra-axonal redox mechanism for axonal degeneration. Redox-active drugs, which could decrease the oxidation of phosphatidylserine and its transfer to the outer leaflet, may be candidates for axonal protection in diseases characterized by axonal degeneration, such as glaucoma, multiple sclerosis, peripheral neuropathies, and others.
Footnotes
This work was supported in part by the Canadian Foundation for Innovation (L.A.L. and S.C.), the Canada Research Chair Program (L.A.L.), the National Institutes of Health (Grants R21 EY025074 and P30EY016665 to L.A.L.), Research to Prevent Blindness (unrestricted departmental grant, University of Wisconsin), the Natural Sciences and Engineering Research Council of Canada (S.C.), Fonds de Recherche du Quebec–Santé (salary awards to M.A. and S.C.), and the Fonds de Recherche en Ophtalmologie de l'Universite de Montreal (L.B.). We thank Professor Michael Coleman for the kind gift of WldS rats and Isabelle Prévost and Javier Mazzaferri for technical support with laser axotomy.
L.A.L. is an inventor for patents on PB1 and related phosphine-borane complexes. The patents have been assigned to the Wisconsin Alumni Research Foundation. The remaining authors declare no competing financial interests.
References
- Adalbert R, Gillingwater TH, Haley JE, Bridge K, Beirowski B, Berek L, Wagner D, Grumme D, Thomson D, Celik A, Addicks K, Ribchester RR, Coleman MP (2005) A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur J Neurosci 21:271–277. 10.1111/j.1460-9568.2004.03833.x [DOI] [PubMed] [Google Scholar]
- Adalbert R, Morreale G, Paizs M, Conforti L, Walker SA, Roderick HL, Bootman MD, Siklós L, Coleman MP (2012) Intra-axonal calcium changes after axotomy in wild-type and slow Wallerian degeneration axons. Neuroscience 225:44–54. 10.1016/j.neuroscience.2012.08.056 [DOI] [PubMed] [Google Scholar]
- Almasieh M, Lieven CJ, Levin LA, Di Polo A (2011) A cell-permeable phosphine-borane complex delays retinal ganglion cell death after axonal injury through activation of the pro-survival extracellular signal-regulated kinases 1/2 pathway. J Neurochem 118:1075–1086. 10.1111/j.1471-4159.2011.07382.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Fish KN (2009) Selective targeting of ER exit sites supports axon development. Traffic 10:1669–1684. 10.1111/j.1600-0854.2009.00974.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, Freeman MR (2012) WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol 22:596–600. 10.1016/j.cub.2012.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian K, Mirnikjoo B, Schroit AJ (2007) Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem 282:18357–18364. 10.1074/jbc.M700202200 [DOI] [PubMed] [Google Scholar]
- Barres BA, Silverstein BE, Corey DP, Chun LL (1988) Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron 1:791–803. 10.1016/0896-6273(88)90127-4 [DOI] [PubMed] [Google Scholar]
- Barrientos SA, Martinez NW, Yoo S, Jara JS, Zamorano S, Hetz C, Twiss JL, Alvarez J, Court FA (2011) Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci 31:966–978. 10.1523/JNEUROSCI.4065-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfey M, Aguayo AJ (1982) Extensive elongation of axons from rat brain into peripheral nerve grafts. Nature 296:150–152. 10.1038/296150a0 [DOI] [PubMed] [Google Scholar]
- Berdan RC, Easaw JC, Wang R (1993) Alterations in membrane potential after axotomy at different distances from the soma of an identified neuron and the effect of depolarization on neurite outgrowth and calcium channel expression. J Neurophysiol 69:151–164. [DOI] [PubMed] [Google Scholar]
- Bevers EM, Williamson PL (2010) Phospholipid scramblase: an update. FEBS Lett 584:2724–2730. 10.1016/j.febslet.2010.03.020 [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ (2014) Microglial phagocytosis of live neurons. Nat Rev Neurosci 15:209–216. 10.1038/nrn3710 [DOI] [PubMed] [Google Scholar]
- Buckmaster EA, Perry VH, Brown MC (1995) The rate of Wallerian degeneration in cultured neurons from wild type and C57BL/WldS mice depends on time in culture and may be extended in the presence of elevated K+ levels. Eur J Neurosci 7:1596–1602. 10.1111/j.1460-9568.1995.tb01155.x [DOI] [PubMed] [Google Scholar]
- Callizot N, Combes M, Steinschneider R, Poindron P (2011) A new long term in vitro model of myelination. Exp Cell Res 317:2374–2383. 10.1016/j.yexcr.2011.07.002 [DOI] [PubMed] [Google Scholar]
- Castegna A, Lauderback CM, Mohmmad-Abdul H, Butterfield DA (2004) Modulation of phospholipid asymmetry in synaptosomal membranes by the lipid peroxidation products, 4-hydroxynonenal and acrolein: implications for Alzheimer's disease. Brain Res 1004:193–197. 10.1016/j.brainres.2004.01.036 [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR (2010) Wallerian degeneration, Wld(S), and Nmnat. Annu Rev Neurosci 33:245–267. 10.1146/annurev-neuro-060909-153248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH (1998) An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A 95:9985–9990. 10.1073/pnas.95.17.9985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor J, Schroit AJ (1990) Aminophospholipid translocation in erythrocytes: evidence for the involvement of a specific transporter and an endofacial protein. Biochemistry 29:37–43. 10.1021/bi00453a005 [DOI] [PubMed] [Google Scholar]
- Connor J, Schroit AJ (1991) Transbilayer movement of phosphatidylserine in erythrocytes. Inhibitors of aminophospholipid transport block the association of photolabeled lipid to its transporter. Biochim Biophys Acta 1066:37–42. 10.1016/0005-2736(91)90247-6 [DOI] [PubMed] [Google Scholar]
- Conta Steencken AC, Smirnov I, Stelzner DJ (2011) Cell survival or cell death: differential vulnerability of long descending and thoracic propriospinal neurons to low thoracic axotomy in the adult rat. Neuroscience 194:359–371. 10.1016/j.neuroscience.2011.05.052 [DOI] [PubMed] [Google Scholar]
- de Jong K, Kuypers FA (2006) Sulphydryl modifications alter scramblase activity in murine sickle cell disease. Br J Haematol 133:427–432. 10.1111/j.1365-2141.2006.06045.x [DOI] [PubMed] [Google Scholar]
- Fadeel B, Xue D (2009) The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Crit Rev Biochem Mol Biol 44:264–277. 10.1080/10409230903193307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadeel B, Gleiss B, Högstrand K, Chandra J, Wiedmer T, Sims PJ, Henter JI, Orrenius S, Samali A (1999) Phosphatidylserine exposure during apoptosis is a cell-type-specific event and does not correlate with plasma membrane phospholipid scramblase expression. Biochem Biophys Res Commun 266:504–511. 10.1006/bbrc.1999.1820 [DOI] [PubMed] [Google Scholar]
- Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL (2001) Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem 276:1071–1077. 10.1074/jbc.M003649200 [DOI] [PubMed] [Google Scholar]
- Galli-Resta L, Ensini M (1996) An intrinsic time limit between genesis and death of individual neurons in the developing retinal ganglion cell layer. J Neurosci 16:2318–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EB, Glass JD, Griffin JW (1995) Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci 15:6445–6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerke V, Creutz CE, Moss SE (2005) Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol 6:449–461. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Espinosa JS, Xu Y, Davidson N, Kovacs GT, Barres BA (2002) Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron 33:689–702. 10.1016/S0896-6273(02)00602-5 [DOI] [PubMed] [Google Scholar]
- Goren MA, Morizumi T, Menon I, Joseph JS, Dittman JS, Cherezov V, Stevens RC, Ernst OP, Menon AK (2014) Constitutive phospholipid scramblase activity of a G protein-coupled receptor. Nat Commun 5:5115. 10.1038/ncomms6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Cho S, Goldberg JL (2010) Neurotrophic effect of a novel TrkB agonist on retinal ganglion cells. Invest Ophthalmol Vis Sci 51:1747–1754. 10.1167/iovs.09-4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isas JM, Langen R, Hubbell WL, Haigler HT (2004) Structure and dynamics of a helical hairpin that mediates calcium-dependent membrane binding of annexin B12. J Biol Chem 279:32492–32498. 10.1074/jbc.M402568200 [DOI] [PubMed] [Google Scholar]
- Isas JM, Kim YE, Jao CC, Hegde PB, Haigler HT, Langen R (2005) Calcium- and membrane-induced changes in the structure and dynamics of three helical hairpins in annexin B12. Biochemistry 44:16435–16444. 10.1021/bi051751m [DOI] [PubMed] [Google Scholar]
- Kagan VE, Fabisiak JP, Shvedova AA, Tyurina YY, Tyurin VA, Schor NF, Kawai K (2000) Oxidative signaling pathway for externalization of plasma membrane phosphatidylserine during apoptosis. FEBS Lett 477:1–7. 10.1016/S0014-5793(00)01707-5 [DOI] [PubMed] [Google Scholar]
- Kamp D, Sieberg T, Haest CW (2001) Inhibition and stimulation of phospholipid scrambling activity: consequences for lipid asymmetry, echinocytosis, and microvesiculation of erythrocytes. Biochemistry 40:9438–9446. 10.1021/bi0107492 [DOI] [PubMed] [Google Scholar]
- Kanamori A, Catrinescu MM, Kanamori N, Mears KA, Beaubien R, Levin LA (2010) Superoxide is an associated signal for apoptosis in axonal injury. Brain 133:2612–2625. 10.1093/brain/awq105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T (2005) In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med 11:572–577. 10.1038/nm1229 [DOI] [PubMed] [Google Scholar]
- Kim YE, Chen J, Chan JR, Langen R (2010a) Engineering a polarity-sensitive biosensor for time-lapse imaging of apoptotic processes and degeneration. Nat Methods 7:67–73. 10.1038/nmeth.1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Chen J, Langen R, Chan JR (2010b) Monitoring apoptosis and neuronal degeneration by real-time detection of phosphatidylserine externalization using a polarity-sensitive indicator of viability and apoptosis. Nat Protoc 5:1396–1405. 10.1038/nprot.2010.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, Tönges L, Stadelmann C, Brück W, Bähr M, Lingor P (2010) Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A 107:6064–6069. 10.1073/pnas.0909794107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzis K, Coffin JD, Eckenstein FP (1999) Time course and age dependence of motor neuron death following facial nerve crush injury: role of fibroblast growth factor. Exp Neurol 157:77–87. 10.1006/exnr.1999.7014 [DOI] [PubMed] [Google Scholar]
- Ledesma MD, Simons K, Dotti CG (1998) Neuronal polarity: essential role of protein-lipid complexes in axonal sorting. Proc Natl Acad Sci U S A 95:3966–3971. 10.1073/pnas.95.7.3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma MD, Brügger B, Bünning C, Wieland FT, Dotti CG (1999) Maturation of the axonal plasma membrane requires upregulation of sphingomyelin synthesis and formation of protein-lipid complexes. EMBO J 18:1761–1771. 10.1093/emboj/18.7.1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventis PA, Grinstein S (2010) The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys 39:407–427. 10.1146/annurev.biophys.093008.131234 [DOI] [PubMed] [Google Scholar]
- Lewis TL Jr, Turi GF, Kwon SK, Losonczy A, Polleux F (2016) Progressive decrease of mitochondrial motility during maturation of cortical axons in vitro and in vivo. Curr Biol 26:2602–2608. 10.1016/j.cub.2016.07.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingor P, Koch JC, Tönges L, Bähr M (2012) Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res 349:289–311. 10.1007/s00441-012-1362-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM Jr, LoPachin VR, Saubermann AJ (1990) Effects of axotomy on distribution and concentration of elements in rat sciatic nerve. J Neurochem 54:320–332. 10.1111/j.1471-4159.1990.tb13317.x [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S (1989) Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci 1:27–33. 10.1111/j.1460-9568.1989.tb00771.x [DOI] [PubMed] [Google Scholar]
- Luo L, O'Leary DD (2005) Axon retraction and degeneration in development and disease. Annu Rev Neurosci 28:127–156. 10.1146/annurev.neuro.28.061604.135632 [DOI] [PubMed] [Google Scholar]
- Lyon MF, Ogunkolade BW, Brown MC, Atherton DJ, Perry VH (1993) A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4. Proc Natl Acad Sci U S A 90:9717–9720. 10.1073/pnas.90.20.9717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack TGA, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP (2001) Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci 4:1199–1206. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent AM (2000) Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J Neurosci Res 59:568–580. 10.1002/(SICI)1097-4547(20000215)59:4%3C568::AID-JNR13%3E3.0.CO%3B2-R [DOI] [PubMed] [Google Scholar]
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR (1995) Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med 182:1545–1556. 10.1084/jem.182.5.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsura T. (2014) Oxidized phosphatidylserine: production and bioactivities. Yonago Acta Med 57:119–127. [PMC free article] [PubMed] [Google Scholar]
- Nguyen JV, Soto I, Kim KY, Bushong EA, Oglesby E, Valiente-Soriano FJ, Yang Z, Davis CH, Bedont JL, Son JL, Wei JO, Buchman VL, Zack DJ, Vidal-Sanz M, Ellisman MH, Marsh-Armstrong N (2011) Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci U S A 108:1176–1181. 10.1073/pnas.1013965108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemuth NJ, Thompson AF, Crowe ME, Lieven CJ, Levin LA (2016) Intracellular disulfide reduction by phosphine-borane complexes: Mechanism of action for neuroprotection. Neurochem Int 99:24–32. 10.1016/j.neuint.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KC, Vargas ME, Sagasti A (2013) WldS and PGC-1alpha regulate mitochondrial transport and oxidation state after axonal injury. J Neurosci 33:14778–14790. 10.1523/JNEUROSCI.1331-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S. (2007) Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev 39:443–455. 10.1080/03602530701468516 [DOI] [PubMed] [Google Scholar]
- Pang Y, Zheng B, Kimberly SL, Cai Z, Rhodes PG, Lin RC (2012) Neuron-oligodendrocyte myelination co-culture derived from embryonic rat spinal cord and cerebral cortex. Brain Behav 2:53–67. 10.1002/brb3.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DR, Isas JM, Ladokhin AS, Jao CC, Kim YE, Kirsch T, Langen R, Haigler HT (2005) The conserved core domains of annexins A1, A2, A5, and B12 can be divided into two groups with different Ca2+-dependent membrane-binding properties. Biochemistry 44:2833–2844. 10.1021/bi047642+ [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER, Tree P, Gordon S (1990) Evidence that very slow Wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur J Neurosci 2:802–808. 10.1111/j.1460-9568.1990.tb00472.x [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER (1991) Very slow retrograde and Wallerian degeneration in the CNS of C57BL/Ola mice. Eur J Neurosci 3:102–105. 10.1111/j.1460-9568.1991.tb00815.x [DOI] [PubMed] [Google Scholar]
- Plunet W, Kwon BK, Tetzlaff W (2002) Promoting axonal regeneration in the central nervous system by enhancing the cell body response to axotomy. J Neurosci Res 68:1–6. 10.1002/jnr.10176 [DOI] [PubMed] [Google Scholar]
- Pollin MM, McHanwell S, Slater CR (1991) The effect of age on motor neurone death following axotomy in the mouse. Development 112:83–89. [DOI] [PubMed] [Google Scholar]
- Pomorski T, Menon AK (2006) Lipid flippases and their biological functions. Cell Mol Life Sci 63:2908–2921. 10.1007/s00018-006-6167-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts RA, Dreher B, Bennett MR (1982) The loss of ganglion cells in the developing retina of the rat. Brain Res 255:481–486. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT (2002) Axonal self-destruction and neurodegeneration. Science 296:868–871. 10.1126/science.1068613 [DOI] [PubMed] [Google Scholar]
- Rege TA, Hagood JS (2006) Thy-1 as a regulator of cell-cell and cell-matrix interactions in axon regeneration, apoptosis, adhesion, migration, cancer, and fibrosis. FASEB J 20:1045–1054. 10.1096/fj.05-5460rev [DOI] [PubMed] [Google Scholar]
- Rimon G, Bazenet CE, Philpott KL, Rubin LL (1997) Increased surface phosphatidylserine is an early marker of neuronal apoptosis. J Neurosci Res 48:563–570. 10.1002/(SICI)1097-4547(19970615)48:6%3C563::AID-JNR9%3E3.0.CO%3B2-8 [DOI] [PubMed] [Google Scholar]
- Rishal I, Fainzilber M (2014) Axon-soma communication in neuronal injury. Nat Rev Neurosci 15:32–42. 10.1038/nrn3609 [DOI] [PubMed] [Google Scholar]
- Rothman JE, Lenard J (1977) Membrane asymmetry. Science 195:743–753. 10.1126/science.402030 [DOI] [PubMed] [Google Scholar]
- Schlieve CR, Tam A, Nilsson BL, Lieven CJ, Raines RT, Levin LA (2006) Synthesis and characterization of a novel class of reducing agents that are highly neuroprotective for retinal ganglion cells. Exp Eye Res 83:1252–1259. 10.1016/j.exer.2006.07.002 [DOI] [PubMed] [Google Scholar]
- Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S (2014) Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344:1164–1168. 10.1126/science.1252809 [DOI] [PubMed] [Google Scholar]
- Sievers C, Platt N, Perry VH, Coleman MP, Conforti L (2003) Neurites undergoing Wallerian degeneration show an apoptotic-like process with Annexin V positive staining and loss of mitochondrial membrane potential. Neurosci Res 46:161–169. 10.1016/S0168-0102(03)00039-7 [DOI] [PubMed] [Google Scholar]
- Skytt DM, Toft-Kehler AK, Brændstrup CT, Cejvanovic S, Gurubaran IS, Bergersen LH, Kolko M (2016) Glia-neuron interactions in the retina can be studied in cocultures of Muller cells and retinal ganglion cells. Biomed Res Int 2016:1087647. 10.1155/2016/1087647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrz D, Dráberová L, Dráber P (2007) Non-apoptotic phosphatidylserine externalization induced by engagement of glycosylphosphatidylinositol-anchored proteins. J Biol Chem 282:10487–10497. 10.1074/jbc.M611090200 [DOI] [PubMed] [Google Scholar]
- Suzuki J, Umeda M, Sims PJ, Nagata S (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468:834–838. 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- Swairjo MA, Seaton BA (1994) Annexin structure and membrane interactions: a molecular perspective. Annu Rev Biophys Biomol Struct 23:193–213. 10.1146/annurev.bb.23.060194.001205 [DOI] [PubMed] [Google Scholar]
- Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, Takamura Y, Sakamoto H, Satoh K, Kondo M, Sakamoto T, Goldberg JL, Nabekura J, Tanihara H (2015) In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc Natl Acad Sci U S A 112:10515–10520. 10.1073/pnas.1509879112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillman TS, Cascio M (2003) Effects of membrane lipids on ion channel structure and function. Cell Biochem Biophys 38:161–190. 10.1385/CBB:38:2:161 [DOI] [PubMed] [Google Scholar]
- Tiveron MC, Nosten-Bertrand M, Jani H, Garnett D, Hirst EM, Grosveld F, Morris RJ (1994) The mode of anchorage to the cell surface determines both the function and the membrane location of Thy-1 glycoprotein. J Cell Sci 107:1783–1796. [DOI] [PubMed] [Google Scholar]
- Tsao JW, George EB, Griffin JW (1999) Temperature modulation reveals three distinct stages of Wallerian degeneration. J Neurosci 19:4718–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurina YY, Tyurin VA, Zhao Q, Djukic M, Quinn PJ, Pitt BR, Kagan VE (2004) Oxidation of phosphatidylserine: a mechanism for plasma membrane phospholipid scrambling during apoptosis? Biochem Biophys Res Commun 324:1059–1064. 10.1016/j.bbrc.2004.09.102 [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124. 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas ME, Yamagishi Y, Tessier-Lavigne M, Sagasti A (2015) Live imaging of calcium dynamics during axon degeneration reveals two functionally distinct phases of calcium influx. J Neurosci 35:15026–15038. 10.1523/JNEUROSCI.2484-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC (2016) Glia-neuron interactions in the mammalian retina. Prog Retin Eye Res 51:1–40. 10.1016/j.preteyeres.2015.06.003 [DOI] [PubMed] [Google Scholar]
- Verkleij AJ, Zwaal RF, Roelofsen B, Comfurius P, Kastelijn D, van Deenen LL (1973) The asymmetric distribution of phospholipids in the human red cell membrane: a combined study using phospholipases and freeze-etch electron microscopy. Biochim Biophys Acta 323:178–193. 10.1016/0005-2736(73)90143-0 [DOI] [PubMed] [Google Scholar]
- Villegas-Pérez MP, Vidal-Sanz M, Rasminsky M, Bray GM, Aguayo AJ (1993) Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol 24:23–36. 10.1002/neu.480240103 [DOI] [PubMed] [Google Scholar]
- Waller AV. (1850) Experiments on the section of glossopharyngeal and hypoglossal nerves of the frog, and observations on the alterations produced thereby in the structure of their of their primitive fibers. Philos Trans R Soc Lond B Biol Sci 140:423–429. [Google Scholar]
- Wang JT, Medress ZA, Barres BA (2012) Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol 196:7–18. 10.1083/jcb.201108111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson P. (2015) Phospholipid Scramblases. Lipid Insights 8:41–44. 10.4137/LPI.S31785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue GP, Rivero BP, Morris RJ (1991) The surface glycoprotein Thy-1 is excluded from growing axons during development: a study of the expression of Thy-1 during axogenesis in hippocampus and hindbrain. Development 112:161–176. [DOI] [PubMed] [Google Scholar]
- Ying W. (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10:179–206. 10.1089/ars.2007.1672 [DOI] [PubMed] [Google Scholar]
- Ziv NE, Spira ME (1993) Spatiotemporal distribution of Ca2+ following axotomy and throughout the recovery process of cultured Aplysia neurons. Eur J Neurosci 5:657–668. 10.1111/j.1460-9568.1993.tb00531.x [DOI] [PubMed] [Google Scholar]