

Abstract
Studies comparing neuronal activity at the dorsal and ventral poles of the hippocampus have shown that the scale of spatial information increases and the precision with which space is represented declines from the dorsal to ventral end. These dorsoventral differences in neuronal output and spatial representation could arise due to differences in computations performed by dorsal and ventral CA1 neurons. In this study, we tested this hypothesis by quantifying the differences in dendritic integration and synaptic plasticity between dorsal and ventral CA1 pyramidal neurons of rat hippocampus. Using a combination of somatic and dendritic patch-clamp recordings, we show that the threshold for LTP induction is higher in dorsal CA1 neurons and that a G-protein-coupled inward-rectifying potassium channel mediated regulation of dendritic plateau potentials and dendritic excitability underlies this gating. By contrast, similar regulation of LTP is absent in ventral CA1 neurons. Additionally, we show that generation of plateau potentials and LTP induction in dorsal CA1 neurons depends on the coincident activation of Schaffer collateral and temporoammonic inputs at the distal apical dendrites. The ventral CA1 dendrites, however, can generate plateau potentials in response to temporally dispersed excitatory inputs. Overall, our results highlight the dorsoventral differences in dendritic computation that could account for the dorsoventral differences in spatial representation.
SIGNIFICANCE STATEMENT The dorsal and ventral parts of the hippocampus encode spatial information at very different scales. Whereas the place-specific firing fields are small and precise at the dorsal end of the hippocampus, neurons at the ventral end have comparatively larger place fields. Here, we show that the dorsal CA1 neurons have a higher threshold for LTP induction and require coincident timing of excitatory synaptic inputs for the generation of dendritic plateau potentials. By contrast, ventral CA1 neurons can integrate temporally dispersed inputs and have a lower threshold for LTP. Together, these dorsoventral differences in the threshold for LTP induction could account for the differences in scale of spatial representation at the dorsal and ventral ends of the hippocampus.
Keywords: CA1 pyramidal neurons, dorsoventral axis, G-protein-coupled inward-rectifying potassium channels, long-term potentiation, membrane potential, plateau potentials
Introduction
The laminar connectivity of the excitatory pathways repeats itself across the entire dorsoventral (DV) axis of the hippocampus. At all DV levels, information from the entorhinal cortex reaches CA1 pyramidal neurons via two topographically segregated inputs: distal regions of CA1 apical dendrites are innervated by temporoammonic (TA) inputs from layer 3 of the entorhinal cortex; proximal regions of the apical and basal dendrites are innervated by Schaffer collateral (SC) inputs from hippocampal CA3 neurons (Steward, 1976; Amaral and Witter, 1989; Kajiwara et al., 2008). While this intrinsic pattern of connectivity is conserved along the DV axis, the scale of spatial information increases and the precision with which space is represented declines from the dorsal-to-ventral end (Moser and Moser, 1998; Strange et al., 2014). Specifically, the ventral CA1 neurons have larger place fields compared with dorsal CA1 neurons (Jung et al., 1994; Maurer et al., 2005; Kjelstrup et al., 2008). What are the underlying mechanisms for this functional segregation in neuronal output along the DV axis? This DV difference in place field size could arise due to the differences in spatial and nonspatial inputs reaching different DV levels of the hippocampus (Brun et al., 2008b; Lyttle et al., 2013; Keinath et al., 2014). According to this postulate, dorsal and ventral CA1 neurons act as linear integrators and perform similar computation and the differences in neural firing arise primarily due to the variation in the inputs.
Theoretical and experimental evidence suggests that LTP at the excitatory inputs to CA1 neurons underlies the formation of place fields (Blum and Abbott, 1996; Wilson and Tonegawa, 1997; Mehta and Wilson, 2000; Nakazawa et al., 2004). Nonlinear integration of excitatory inputs mediated by dendritic spikes and plateau potentials is critical for robust LTP and more recently has been shown to be important for place field formation (Golding et al., 2002; Takahashi and Magee, 2009; Bittner et al., 2015). Additionally, in vivo manipulation of channels that regulate dendritic excitability in CA1 neurons affects LTP and the size of place fields (Hussaini et al., 2011). Interestingly, we and others have shown that ventral CA1 neurons are intrinsically more excitable compared with dorsal neurons (Dougherty et al., 2012; Malik et al., 2016). A differential expression of voltage-gated ion channels and differences in morphology underlie this DV difference in excitability. Specifically, the dorsal neurons have a higher resting conductance of G-protein coupled inward-rectifying K+ (GIRK) channels and higher numbers of A-type (KA) and M-type K+ channels (Marcelin et al., 2012; Hönigsperger et al., 2015; Kim and Johnston, 2015). Because GIRK and KA channels are expressed in CA1 dendrites and are known to regulate the integration of excitatory inputs (Drake et al., 1997; Chen and Johnston, 2005), it is likely that a higher expression of these channels in dorsal dendrites would inhibit active dendritic mechanisms and limit the plasticity at the excitatory inputs to dorsal neurons.
In this study, we asked whether the DV differences in place field size could arise due to differences in the threshold for LTP induction. We hypothesized that the small place fields in dorsal hippocampus could arise from a higher threshold for LTP induction such that only strong and/or coincident inputs would be potentiated in dorsal neurons. By contrast, a lower threshold of LTP induction in ventral neurons would cause potentiation of even weak and temporally dispersed excitatory inputs leading to formation of large or multiple place fields (Rich et al., 2014). In this study, we tested these hypotheses by quantifying the DV differences in gating of LTP. We found that the dorsal neurons have a higher threshold for LTP induction. Additionally, a higher resting GIRK conductance in dorsal dendrites inhibits the generation of plateau potentials and constricts the window for temporal summation of SC and TA inputs. Similar regulation of LTP was absent in ventral dendrites. Together, our results provide a potential cellular mechanism for the dorsoventral differences in place field size.
Materials and Methods
Animal use.
Adult male 1.5- to 2-month-old Sprague Dawley rats (RRID: RGD_5508397) were used in accordance to the rules and regulations of the University of Texas at Austin Institutional Animal Care and Use Committee. Rats were group housed (2 or 3 per cage) and had ad libitum access to water and food.
Acute hippocampal slice preparation.
Rats were anesthetized with an intraperitonial injection of a mixture of ketamine and xylazine and transcardially perfused with an ice-cold cutting solution containing the following (in mm): 210 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, and 7 dextrose. Vibrating blade microtome (VT1000A, Leica Microsystems) was used to obtain 350-μm-thick dorsal and ventral hippocampus slices using slicing protocols described previously (Dougherty et al., 2012; Malik et al., 2016). Specifically, three slices from the ventral end (∼1 mm) and 4 slices from the dorsal end (∼1.2 mm) were used for all experiments (Malik et al., 2016). From a given rat, both dorsal and ventral slices were obtained from opposite hemispheres. Slices were allowed to recover at 34°C for 30 min followed by 30 min recovery at room temperature in a holding solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, 12.5 dextrose, 1.3 ascorbic acid, and 3 sodium pyruvate.
Whole-cell patch-clamp recordings.
Whole-cell current-clamp recordings were obtained from submerged slices perfused in a heated (32°C–34°C) recording solution (bubbled with 95% O2/5% CO2, pH ∼7.4) containing the following (in mm): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 12.5 dextrose. In all experiments, GABAA- and GABAB-mediated inhibition was blocked by adding 2 μm gabazine and 5 μm CGP 55845 hydrochloride to the recording solution. Area CA3 was removed before placing the slice in the recording chamber to prevent spontaneous epileptiform activity. Dendrites and somas of pyramidal neurons were visualized using infrared differential interference contrast optics (Zeiss Axioskop) fitted with a 60× water-immersion objective. Pyramidal neurons located in the middle of the proximodistal/transverse (CA3-subiculum) axis of CA1 region, close to stratum radiatum, were targeted for recordings. Somatic patch electrodes (4–6 mΩ) and dendritic patch electrodes (6–8 mΩ) were pulled from borosilicate capillary glass of external diameter 1.65 mm (World Precision Instruments) using a Flaming/Brown micropipette puller (model P-97, Sutter Instruments). Electrodes were filled with an internal solution containing the following (in mm): 120 K-gluconate, 20 KCl, 10 HEPES, 4 NaCl, 7 K2-phosphocreatine, 0.3 Na-GTP, and 4 Mg-ATP (pH ∼7.3 adjusted with KOH), Neurobiotin (Vector Laboratories) was included (0.1%–0.2%) for subsequent histological processing, 15 μm AlexaFluor-594 (Invitrogen) was also added to the internal solution. In some experiments, K2-phosphocreatine was replaced by Tris-phosphocreatine. In these cases, concentration of K-gluconate was increased to 134 mm to keep the K+ reversal potential consistent across experiments. Electrophysiology data were recorded using a Dagan BVC-700 amplifier with custom-written acquisition software in the IgorPro environment (Wavemetrics, RRID: SCR_000325). Code is available upon request. Signals were low-pass filtered at 3–10 kHz and sampled at 10–40 kHz using an ITC-18 computer interface (InstruTech). Voltages have not been corrected for measured liquid junction potential (∼8 mV). Upon successful transition to the whole-cell configuration, the neuron was given at least 5 min to stabilize before data were collected. Access resistance and pipette capacitance were appropriately compensated before each recording. Experiments were terminated if access resistances exceeded 25 mΩ for somatic recordings and 30 mΩ for dendritic recordings or when access resistance varied by >20% during the experiment.
LTP induction protocol.
Synaptic EPSPs were evoked by orthodromic stimulation through a bipolar tungsten electrode (0.1 ms, 0.01–0.2 μA) located 120–400 μm from the soma and 15–25 μm lateral to the visually identified apical dendrite. To ensure that we were not directly stimulating dendrites with our stimulating electrodes, separate control experiments were performed in which 25 μm AP5 and 20 μm DNQX were added to the bath, and a complete block of the stimulation-induced depolarization was observed (data not shown). Back-propagating action potentials (bAPs) were evoked by antidromic stimulation (0.1 ms), 5–20 μm from the initial segment of the axon of the same neuron. The protocol for θ-burst pairing (TBP) to induce LTP consisted of five EPSPs evoked in bursts of 100 Hz, repeated in 10 trains of 5 Hz (θ-frequency). These trains were repeated three times at 0.1 Hz. For somatic TBP-LTP recordings, the EPSPs were paired with single action potentials elicited by short current injections to the soma (2 ms, 1–1.5 nA). During dendritic TBP-LTP recordings, EPSPs were paired with bAPs. Two forms of TBP protocols were used in this study: strong TBP protocol, in which a single bAP was paired with each EPSP (5 bAPs per burst); and weak TBP protocol, in which a single bAP was paired with the last two EPSPs in the burst (1–2 bAPs per burst). In all induction conditions, the bAP was timed to occur close to the peak of the EPSP. Experiments were excluded if the bAP appeared to be evoked by basal synaptic stimulation instead of antidromic stimulation (i.e., if the bAP was riding on top of an EPSP and/or it had a latency >3 ms). The two-pathway pairing protocol consisted of pairing five SC and five TA EPSPs in bursts of 100 Hz, repeated in 10 trains of 5 Hz. These trains were repeated three times at 0.1 Hz. Independence of the two pathways was tested by paired-pulse stimulation (at 50 ms interval) before the start of the LTP experiments. The membrane potential of the neurons was maintained at their initial resting membrane potential (RMP) during the course of LTP recordings. For certain experiments, membrane potential during TBP induction was depolarized or hyperpolarized. In cases where the membrane potential was depolarized, either small steps of depolarizing current or long duration depolarizing current steps were used.
Blockers used.
All drugs were made from stock solutions in water or DMSO. NMDA receptors were blocked by adding 25 μm d-AP5 and L-type voltage-gated calcium channels (L-VGCCs) were blocked by adding 10 μm nimodipine to the recording solution. GIRK channels were blocked by addition of 25 μm barium chloride or 0.3 μm Tertiapin-Q (TerQ).
Data analysis.
All data analyses were performed using custom routines written in Igor Pro. Code is available upon request. All statistical comparisons were done using MATLAB (RRID: SCR_001622). All plots were generated using Igor Pro. LTP was quantified as the change in initial EPSP slope. Initial EPSP slope was estimated by fitting a straight line to a fixed time window (1 ms duration, 1–3 ms after EPSP onset) to all the EPSPs in an LTP experiment. The slopes of the EPSPs measured during the 5 min (baseline) duration preceding the LTP stimulus were averaged. This baseline average was considered to be 100%, and the slopes of each of the individual responses for that slice were expressed as a percentage of this baseline average (normalized slopes). The depolarization area during TBP train was estimated by measuring the time integral of the voltage trace. Plateau duration was estimated as the width of the half-maximal voltage of the after depolarization at the end of the TBP burst. Plateau potentials for all the bursts of the first TBP train were averaged. Input resistance (Rin) was calculated as the slope of the linear fit of the voltage-current plot generated from a family of hyperpolarizing and depolarizing current injections (−50 to 50 pA, steps of 10 pA). Sag ratio was calculated as the ratio of the peak voltage to the steady-state voltage in response to a −50 pA current injection. Resonance frequency (fR) was measured using a sinusoidal current injection of constant amplitude and linearly spanning 0–15 Hz in 15 s (CHIRP15). The impedance amplitude profile (ZAP) was determined by taking the ratio of the fast Fourier transform of the voltage response to the fast Fourier transform of the CHIRP current. fR was defined as the peak of the ZAP.
Histological processing.
Slices were fixed with 3% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, and stored at 4°C for at least 48 h. Slices were processed using an avidin-HRP system activated by DAB (Vector Laboratories). DAB-processed slices were mounted in glycerol. Anatomical reconstructions were performed using Neurolucida 6.0 imaging software (MBF Bioscience) (RRID: SCR_001775).
Statistics.
Equality of variance for grouped electrophysiology data was tested using the Bartlett test. Normality of distribution was tested using the Kolmogorov–Smirnov test and Lilliefors test. Statistical significance was assessed using a two-tailed Student's t test or a one-way ANOVA followed by a post hoc Tukey HSD test. Statistical significance for datasets where the groups were not normally distributed was assessed using Kruskal–Wallis ANOVA followed by a Dunn's multiple-comparison test. Sample sizes refer to the number of neurons. Group data are presented as mean ± SEM.
Results
Robust LTP in ventral but not dorsal CA1 neurons
SC axons originating from area CA3 pyramidal neurons provide the numerically largest excitatory synaptic input received by the area CA1 pyramidal neurons. Plasticity at these inputs is thought to underlie place field formation (Rotenberg et al., 1996; Wilson and Tonegawa, 1997; Nakazawa et al., 2004). To investigate the DV differences in plasticity at SC inputs, we used TBP to induce LTP (Fig. 1A,B). Similar-sized subthreshold EPSPs (dorsal: 4.1 ± 0.2 mV; ventral: 3.6 ± 0.3 mV; unpaired t test, t(18) = 0.29, p = 0.1) were recorded from somatic regions of the dorsal and ventral neurons (Fig. 1C), and TBP-LTP was induced after obtaining a stable baseline. The majority of the dorsal neurons showed very little or no LTP after TBP (150 ± 24.6%; n = 10) (Fig. 1D,F). Interestingly, all ventral CA1 neurons exhibited robust LTP (370.4 ± 32.5%, n = 10) as indicated by a large increase in EPSP slope after TBP (Fig. 1E,F). At 30 min after TBP, the normalized change in EPSP slope was significantly larger (unpaired t test, t(18) = 5.4, p = 4e−05) in ventral neurons compared with dorsal neurons (Fig. 1G).
Figure 1.

Larger TBP-LTP in ventral CA1 neurons compared with dorsal neurons. A, Schematic representing the experimental configuration. Voltage responses were measured at the soma. EPSPs were evoked using a stimulating electrode placed at the SC inputs (∼120–150 μm from soma). B, Top, Schematic showing the pairing of SC EPSPs with somatic current injections (Iinj.) during TBP protocol. Bottom, Example voltage traces recorded during a TBP train from a dorsal and a ventral neuron. C, Typical EPSPs recorded during baseline and 30 min after TBP in dorsal and ventral neurons. D, E, Time course plots representing the change in normalized EPSP slope, Rin, and RMP during a typical LTP experiment recorded from a dorsal neuron (D) and a ventral neuron (E). Gray dashed lines indicate the average baseline values. F, Summary graph showing the difference in TBP-LTP between dorsal (n = 10) and ventral neurons (n = 10). G, Scatter plot illustrating the amount of LTP in dorsal and ventral neurons. Individual data points indicate average EPSP slopes (percentage baseline) measured 25–30 min after LTP induction (unpaired t test, t(18) = 5.4, p = 4e−05). Open circles represent individual cells. Closed circles represent mean ± SEM. **p < 0.01.
A difference in summation of EPSPs and APs during TBP trains might underlie this DV difference in LTP. Thus, we tested whether the number of APs fired during TBP and also the depolarization area (which might be correlated with the amount of calcium influx) were different between the dorsal and ventral neurons. Contrary to our prediction, no DV differences were observed in the number of APs fired per burst (dorsal: 4.9 ± 0.11 APs; ventral: 4.7 ± 0.08 APs; unpaired t test, t(18) = 1.68, p = 0.11), or in the depolarization area during TBP trains (dorsal: 18.2 ± 2.1 mV × s; ventral: 15.1 ± 2.26 mV × s; unpaired t test, t(18) = 1.24, p = 0.23).
It has been reported previously that, along with synaptic plasticity, TBP protocol induces intrinsic plasticity in CA1 pyramidal neurons (Fan et al., 2005; Narayanan and Johnston, 2007). This intrinsic plasticity is an important regulator of neuronal excitability and is caused by a change in the HCN (hyperpolarization-activated cyclic nucleotide-gated) channel mediated h-current. This TBP-induced intrinsic plasticity can be quantified in current-clamp recordings by measuring the changes in h-current sensitive properties: Rin, sag ratio, and fR. Although the role of intrinsic plasticity in place cell formation is not known, several studies have shown that the perturbation of HCN channel gradients in CA1 neurons can affect LTP and the size of place fields (Nolan et al., 2004; Hussaini et al., 2011). Because the amount of LTP induced by TBP was different between dorsal and ventral neurons, we tested whether the TBP-induced intrinsic plasticity was also different. Input resistance (paired t test, t(9) = 6.15, p = 1.6e−4; Fig. 2A,D), sag ratio (paired t test, t(9) = 3.1, p = 0.006; Fig. 2A,F), and fR (paired t test, t(9) = 5, p = 7.3e−4; Fig. 2B,H) of ventral neurons measured 30 min after TBP were significantly different from baseline. Similar TBP-induced changes in Rin (paired t test, t(9) = 7.3, p = 4.2e−5; Fig. 2A,C), sag ratio (paired t test, t(9) = 2.7, p = 0.002; Fig. 2A,E), and fR (paired t test, t(9) = 7.66, p = 3.1e−5; Fig. 2A,G) were observed in dorsal neurons. It is important to note that these changes in Rin (paired t test, dorsal: t(6) = 0.65, p = 0.53; ventral: t(5) = 0.7, p = 0.51; Figure 2C,D), sag ratio (paired t test, dorsal: t(6) = 1, p = 0.34; ventral: t(5) = 0.63, p = 0.55), and fR (paired t test, dorsal: t(6) = 1.2, p = 0.37; ventral: t(5) = 0.45, p = 0.67) were specific to the LTP induction and were not observed in control recordings. Contrary to our prediction, TBP-induced percentage changes in Rin (unpaired t test, t(18) = 0.7, p = 0.48; Fig. 2I), sag ratio (unpaired t test, t(18) = 1.8, p = 0.09; Fig. 2J), and fR (unpaired t test, t(18) = 1.48, p = 0.16; Fig. 2K) after TBP were not different between dorsal and ventral neurons.
Figure 2.

No dorsoventral differences in TBP-induced intrinsic plasticity. A, Representative voltage responses to 800 ms long depolarizing and hyperpolarizing current injections (−50 to 50 pA, steps of 10 pA) during baseline and after TBP in dorsal (black) and ventral (red) neurons. B, Representative voltage traces illustrating the membrane resonance in response to a CHIRP stimulus (0–15 Hz, 15 s) during baseline and after TBP recorded from example dorsal and ventral CA1 neurons. Left, Traces were recorded during baseline. Right, Traces were recorded 30 min after TBP. C, D, Significant reduction in the Rin of dorsal (C) (paired t test, t(9) = 7.3, p = 4.2e−5; n = 10) and ventral neurons (D) (paired t test, t(9) = 6.15, p = 1.6e−4; n = 10) after TBP. No change in Rin was observed in dorsal (C) (gray; paired t test, t(6) = 0.65, p = 0.53; n = 7) and ventral neurons (D) (orange; paired t test, t(5) = 0.7, p = 0.51; n = 6) in control recordings (with no TBP induction). E, F, Significant increase in the sag ratio after TBP in dorsal (E) (paired t test, t(9) = 2.7, p = 0.002; n = 10) and ventral (F) (paired t test, t(9) = 3.1, p = 0.006; n = 10) CA1 neurons. G, H, Significant increase in the fR after TBP in dorsal neurons (G) (paired t test, t(9) = 7.66, p = 3.1e−5) and ventral neurons (H) (paired t test, t(9) = 5, p = 7.3e−4). I, TBP-induced percentage decrease in Rin was similar for dorsal and ventral CA1 neurons (unpaired t test, t(18) = 0.7, p = 0.486). J, TBP-induced percentage increase in the sag ratio was similar for dorsal and ventral neurons (unpaired t test, t(18) = 1.8, p = 0.09). K, Percentage increase in fR after TBP in dorsal and ventral neurons was similar (unpaired t test, t(18) = 1.48, p = 0.15). Open circles represent individual cells. Closed circles represent mean ± SEM. **p < 0.01.
TBP-LTP is dependent on NMDARs and VGCCs in ventral CA1 neurons
Next, we tested whether the LTP in dorsal and ventral neurons is dependent on activation of NMDARs and L-VGCCs (Magee and Johnston, 1997; Clemens and Johnston, 2014). In these experiments, slices were incubated in d-AP5 (25 μm) (Fig. 3A,B), nimodipine (10 μm) (Fig. 3C,D), or a mixture of d-AP5 and nimodipine (Fig. 3E,F). Following the baseline measurements, TBP-LTP was induced and the amount of LTP obtained in the presence of different pharmacological blockers was compared. In dorsal neurons, the TBP-induced LTP in control conditions was not significantly different from baseline; thus, the addition of d-AP5 or nimodipine had little effect (AP5: 127.2 ± 10.1%, n = 5; nimodipine: 140.9 ± 19.5%, n = 5; AP5 and nimodipine: 88.8 ± 19%, n = 5; one-way ANOVA, F(3,21) = 1.63, p = 0.21; Fig. 3G). The TBP-LTP in ventral neurons, however, was significantly reduced by blocking NMDARs alone and was further reduced when both NMDARs and L-VGCCs were blocked (AP5: 163 ± 37.8%, n = 6; nimodipine: 236.5 ± 30.3%, n = 5; AP5 and nimodipine: 141.9 ± 15.8%, n = 5; one-way ANOVA, F(3,22) = 12.9, p = 6.5e−05, Tukey HSD post hoc test, p < 0.05; Fig. 3H).
Figure 3.
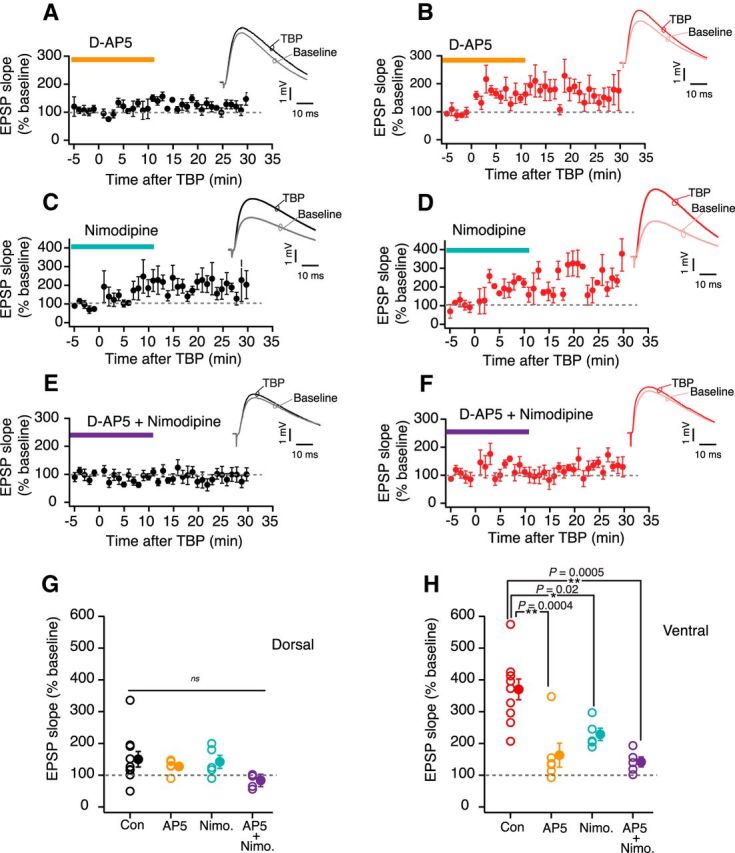
TBP-LTP is dependent on NMDARs and VGCCs. A, B, Time course plots of averaged normalized EPSP slope during LTP recordings in the presence of d-AP5 (25 μm) in dorsal neurons (black, n = 5) (A) and ventral neurons (red, n = 6) (B). Top right, Example EPSP traces recorded from dorsal and ventral neurons during baseline and 30 min after TBP-LTP induction. C, D, Time course plots of averaged normalized EPSP slope during LTP recordings in the presence of nimodipine (10 μm) in dorsal neurons (black, n = 5) (C) and ventral neurons (red, n = 5) (D). Top right, Example EPSP traces recorded from dorsal and ventral neurons during baseline and 30 min after LTP induction. E, F, Time course plots of averaged normalized EPSP slope during LTP recordings in the presence of both d-AP5 (25 μm) and nimodipine (10 μm) in dorsal neurons (black, n = 5) (E) and ventral neurons (red, n = 5) (F). Top right, Example EPSP traces recorded from dorsal and ventral neurons during baseline and 30 min after LTP induction. G, Summary plot representing the normalized change in EPSP slope in control condition compared with d-AP5 alone, nimodipine (Nimo.) alone, and d-AP5 + nimodipine (AP5 + Nimo.) for dorsal neurons (one-way ANOVA, F(3,21) = 1.63, p = 0.21). H, Summary plot representing the normalized change in EPSP slope in control condition compared with d-AP5 alone, nimodipine (Nimo.) alone, and d-AP5 + nimodipine (AP5 + Nimo.) for ventral neurons (one-way ANOVA, F(3,22) = 12.9, p = 6.5e−05, Tukey HSD post hoc test). *p < 0.05. **p < 0.01. Open circles represent individual cells. Closed circles represent mean ± SEM.
Induction threshold for TBP-LTP is lower in ventral neurons
A lower threshold for LTP induction in ventral neurons could underlie the DV differences in TBP-LTP. To test this prediction, we modulated the strength of the induction paradigm. First, we tested whether a weak TBP protocol (5 EPSPs were paired with 1–2 APs) would be sufficient to induce LTP in ventral but not dorsal CA1 neurons (Fig. 4A). Similar to the strong induction paradigm, the weak TBP protocol induced a robust increase in EPSP slope in ventral neurons (180 ± 13.1%, n = 6). The majority of the dorsal neurons (5 of 6 neurons) failed to show any potentiation (110.3 ± 14.2%; Fig. 4B,C) in response to the weak induction protocol. Next, we tested whether a larger initial EPSP in dorsal neurons might provide the induction strength to produce ventral-like LTP. In this experiment, large somatic EPSPs (8–10 mV) were recorded during baseline and LTP was induced using the strong TBP protocol. Interestingly, even the strong TBP induction with large initial EPSPs was insufficient to produce ventral-like robust LTP in dorsal neurons (Fig. 4D).
Figure 4.
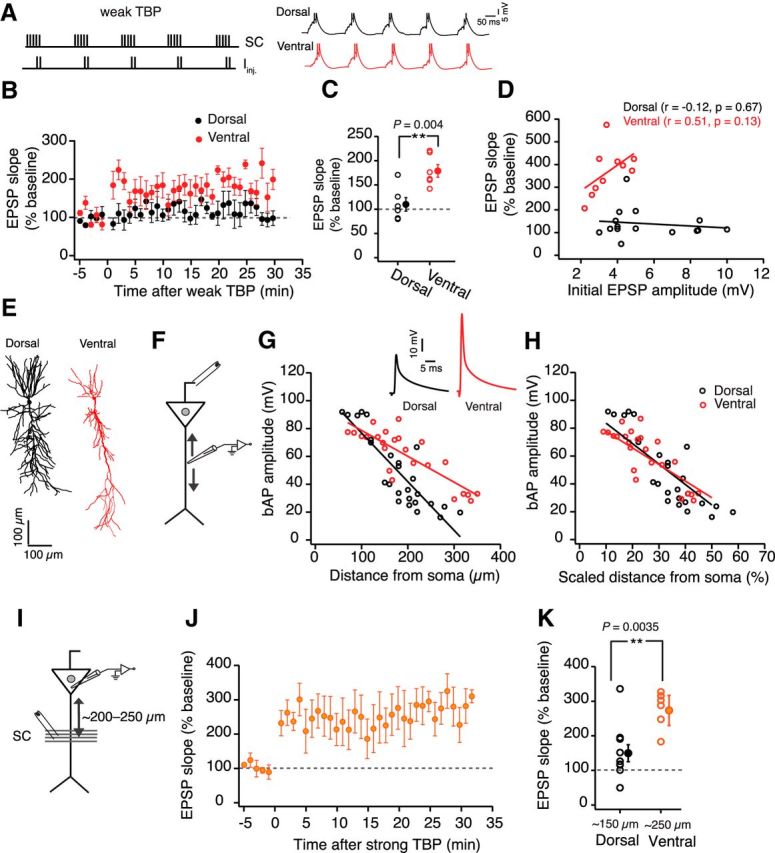
Higher threshold for LTP induction in dorsal neurons is not due to smaller bAPs. A, Top, Schematic showing the pairing of five SC EPSPs with two somatic current injections (Iinj.) during a weak TBP protocol. Bottom, Example voltage traces recorded during a weak TBP train from a dorsal (black) and ventral (red) neuron. B, Time course of change in averaged normalized EPSP slope after weak TBP induction in dorsal (black, n = 6) and ventral neurons (red, n = 6). C, Scatter plot comparing weak TBP-induced LTP in dorsal and ventral neurons (unpaired t test, t(10) = 3.62, p = 0.0047). D, Scatter plot illustrating the relationship between the initial EPSP amplitude and the percentage LTP observed in dorsal (black) and ventral (red) CA1 neurons. Lines indicate linear fits. Correlation was not significant for dorsal and ventral neurons (tested using Pearson correlation coefficient). Dorsal neurons did not show large LTP even with large initial EPSPs. E, Representative morphological reconstructions of neurobiotin-filled dorsal (black) and ventral (red) CA1 neurons. F, Schematic of the experimental configuration. Voltage responses were measured at varying dendritic locations. Antidromic stimulating electrode was used to elicit bAPs. G, Scatter plot illustrating the distance-dependent decrease in the bAP amplitude in dorsal (black, n = 26) and ventral (red, n = 24) dendrites. Lines indicate the linear fits (one-way ANCOVA, F(1,48) = 15.9, p = 0.0002). Inset, Example bAPs recorded from dorsal and ventral dendrites at ∼200 μm distance from soma. H, The amplitudes of bAPs from G are plotted against the scaled dendritic distance (calculated as the ratio of dendritic recording location to the radial dendritic length). Lines indicate the linear fits (one-way ANCOVA, F(1,48) = 3.9, p = 0.06). I, Schematic of the experimental configuration. Voltage responses were measured at the soma. EPSPs were evoked using a stimulating electrode placed at a scaled location (200–250 μm). J, Time course plot representing the averaged normalized EPSP slope in ventral neurons (n = 6) when LTP was measured at scaled location. K, Summary plot comparing the LTP in dorsal neurons at ∼150 μm with LTP in ventral neurons at ∼200 μm (unpaired t test, t(14) = 3.5, p = 0.0035). Open circles represent individual cells. Closed circles represent mean ± SEM. **p < 0.01.
Why is the threshold for LTP induction lower in ventral CA1 neurons? Previous studies have shown that a lower expression of KA channels and larger bAPs in CA1 neurons can decrease the threshold for TBP-LTP (Golding et al., 2002; Takahashi and Magee, 2009). One possible explanation for the DV difference in the threshold for LTP induction could be larger bAPs in the ventral dendrites. Recently, it was reported that the bAP amplitude at distal dendritic locations (>300 μm away from soma) in ventral neurons is larger compared with the dorsal neurons (Marcelin et al., 2012). The same study reported a lower expression of KA channels in the ventral neurons. It is, however, not known whether the amplitude of bAPs at proximal dendrites (∼150 μm) is also different between dorsal and ventral neurons. To explore the role of bAPs in TBP-LTP induction, we first compared the distance-dependent attenuation of bAPs in dorsal and ventral dendrites.
Whole-cell current-clamp recordings were obtained from varying dendritic locations, and the bAPs were elicited using an antidromic stimulating electrode (Fig. 4F). The attenuation of the bAP amplitude along the apical dendrite was significantly larger (one-way ANCOVA, F(1,48) = 15.9, p = 0.0002) in dorsal dendrites (n = 26) compared with the ventral dendrites (n = 24) (Fig. 4G). This DV difference in bAP amplitude was, however, small at proximal dendrites (50–150 μm) but more pronounced at distal dendritic locations (150–350 μm). It has been shown previously that the radial length of the apical dendrite is significantly longer in ventral neurons (Dougherty et al., 2012; Malik et al., 2016) (Fig. 4E). We wondered whether the decreased attenuation of bAPs in ventral dendrites might be a compensatory mechanism for the longer apical dendritic length of these neurons. To test this, we normalized the dendritic recording location in dorsal and ventral neurons to their respective average apical dendritic lengths (dorsal: 540 μm, ventral: 760 μm). The attenuation of the bAP amplitude along the scaled apical dendritic distance was now similar in dorsal and ventral neurons (one-way ANCOVA, F(1,48) = 3.9, p = 0.06; Fig. 4H). If the larger bAP amplitude in ventral dendrites underlies larger LTP in ventral neurons, then LTP at scaled distal dendritic location (200–250 μm) in ventral neurons should be similar to LTP at proximal locations (120–150 μm) in dorsal neurons (Fig. 4I). Contrary to this prediction, however, LTP at distal locations in ventral neurons (280 ± 75.3%; n = 6) was still significantly larger (unpaired t test, t(14) = 3.5, p = 0.0035) than LTP at proximal locations in dorsal neurons (Fig. 4J,K). It is important to note that the stimulating electrodes placed at the distal location can activate SC axons that form synapses close to the soma. We have, however, not controlled for this caveat in our experiments. Overall, these results illustrate that the differences in bAP amplitude between dorsal and ventral neurons might contribute to but cannot fully explain the DV differences in the threshold for LTP induction. Additionally, it is likely that bAPs are not always necessary for LTP induction in dorsal and ventral CA1 neurons. We have, however, not tested this in the present study.
What is happening at the dendrites during LTP induction?
Along with the bAPs, local dendritic events, such as dendritic spikes and plateau potentials, are known to have a significant impact on the summation and potentiation of synaptic inputs (Golding et al., 2002; Takahashi and Magee, 2009). To explore whether there are differences in dendritic summation during the strong TBP protocol, we obtained current-clamp recordings from dendrites of dorsal and ventral neurons during LTP experiments (Fig. 5A). Similar to the somatic recordings, the strong TBP protocol induced a robust LTP in ventral dendrites (341.9 ± 45.2%, n = 7), which was significantly larger (unpaired t test, t(13) = 4.65, p = 0.0004) than the LTP observed in the dorsal dendrites (140.4 ± 9.4%; n = 8; Fig. 5D).
Figure 5.
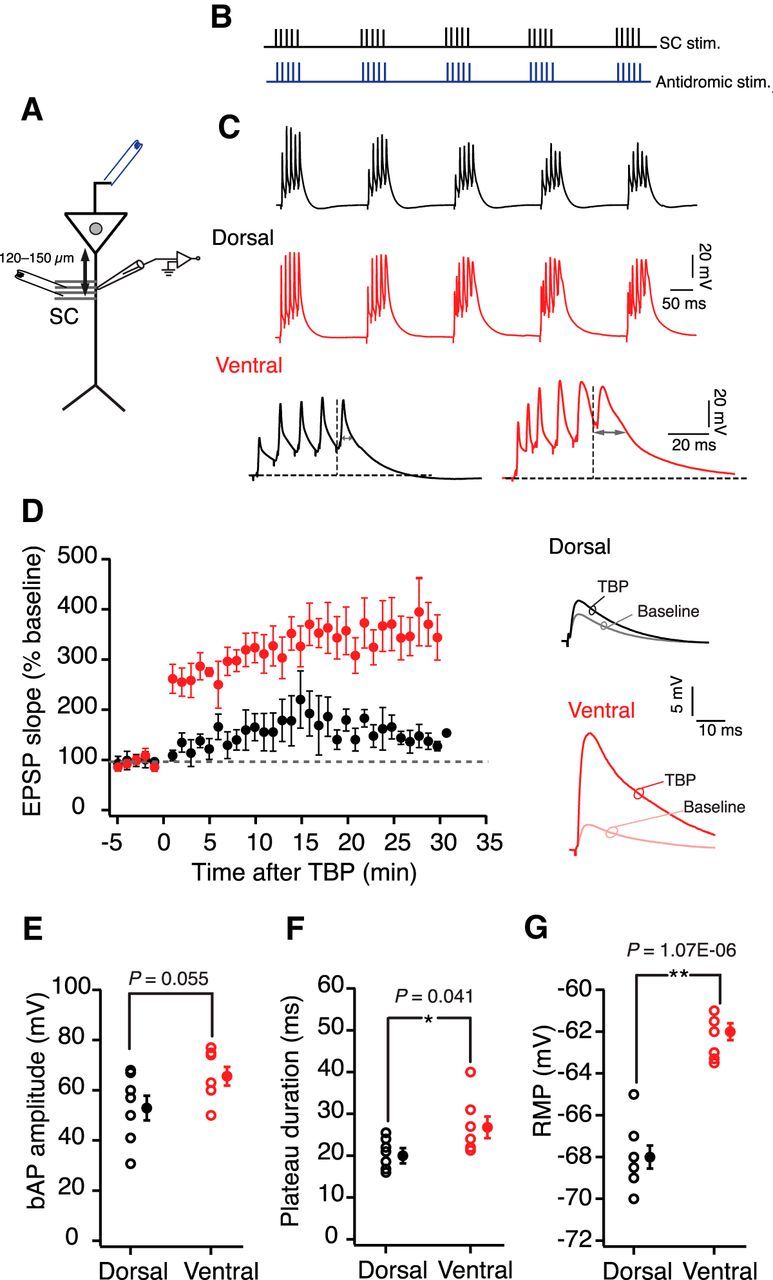
Recording at the apical dendrite during TBP-LTP in dorsal and ventral neurons. A, Schematic of the experimental configuration. Voltage responses were measured at the dendrite (120–150 μm). EPSPs were evoked using a stimulating electrode placed at the SC inputs. Antidromic stimulating electrode (blue) was used to elicit bAPs. B, Schematic showing the pairing of SC EPSPs with bAPs during TBP. C, Top, Example voltage traces recorded during five bursts of TBP trains in dorsal (black) and ventral (red) dendrites. Bottom, The last burst in the TBP train is expanded. Two-headed arrows indicate measurement of plateau potential duration. D, Summary graph showing the difference in time course of TBP-LTP between dorsal (n = 8) and ventral neurons (n = 7) (unpaired t test, t(13) = 4.65, p = 0.0004). Right, Traces represent example EPSPs recorded from dorsal and ventral dendrites during baseline and after TBP. E, Backpropagating spike amplitude at the dendritic recording location before LTP induction for dorsal and ventral neurons (unpaired t test, t(13) = 2.1, p = 0.055). F, Duration of plateau potentials during TBP trains recorded from dorsal and ventral dendrites (unpaired t test, t(13) = 2.1, p = 0.055). G, Initial RMP of dorsal and ventral dendrites (unpaired t test, t(13) = 8.5, p = 1.07e−06). E–G, Open circles represent individual cells. Closed circles represent mean ± SEM. *p < 0.05. **p < 0.01.
To determine whether nonlinear dendritic events, such as plateau potentials, underlie the lower threshold for LTP induction in ventral dendrites, we compared the individual bursts during TBP trains in the dorsal and ventral dendrites. We used previously published methods (Takahashi and Magee, 2009; Bittner et al., 2015) to measure the duration of plateau potentials at the end of each burst of TBP train (Fig. 5C). Interestingly, the duration of plateau potentials during LTP induction was significantly longer (unpaired t test, t(13) = 2.25, p = 0.0004) in ventral dendrites (26.3 ± 3.2 ms) compared with the dorsal dendrites (20.1 ± 1.2 ms; Fig. 5F).
Depolarization during induction is critical for LTP in dorsal dendrites but not the ventral dendrites
It has been shown previously that depolarizing the dendritic membrane during LTP induction can increase the duration of plateau potentials (Takahashi and Magee, 2009). Interestingly, the RMP of ventral dendrites is depolarized (−62.7 ± 0.33 mV) compared with the RMP of dorsal dendrites (−68 ± 0.66 mV) (unpaired t test, t(13) = 8.5, p = 1.07e−06; Fig. 5G). To explore the role of the membrane potential (Vm) in regulating the plateau potentials and LTP in dorsal dendrites, we depolarized the dorsal dendrites during LTP induction. The Vm of the dendrites was maintained at the initial RMP during the baseline and post-TBP recordings. During the TBP protocol dendritic Vm was depolarized by ∼8–10 mV. Interestingly, whereas the induction of TBP-LTP at RMP caused only a small change in EPSP slope (Fig. 1), TBP protocol applied at depolarized Vm led to a large increase in EPSP slope in dorsal dendrites (300 ± 34.2%, n = 10; Fig. 6C,D). Additionally, large dendritic plateau potentials (31.73 ± 3 ms) were observed in dorsal dendrites during the depolarized TBP trains (Fig. 6B,E).
Figure 6.
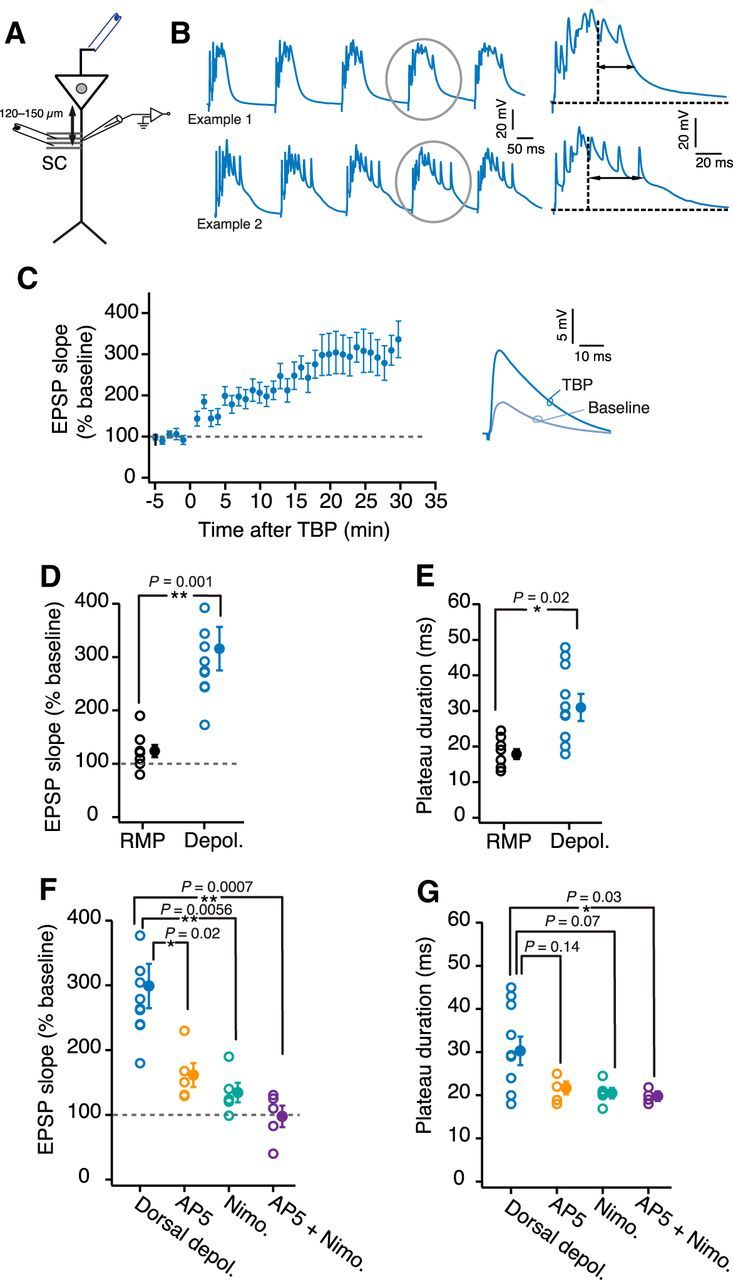
Depolarizing the dorsal dendrites increases TBP-LTP. A, Schematic of the experimental configuration. Voltage responses were measured at the dendrite (120–150 μm). EPSPs were evoked using a stimulating electrode placed at the SC inputs. Antidromic stimulating electrode (blue) was used to elicit bAPs. B, Left, Example voltage traces recorded from two dorsal dendrites during five bursts of TBP train. The membrane potential was adjusted to depolarize the neurons by ∼8–10 mV. The bursts highlighted by gray circles are expanded on the right to illustrate the long duration of plateau potentials (two-headed arrows). C, Time course plot of averaged normalized EPSP slope in dorsal dendrites (n = 10) when TBP-LTP was induced at depolarized potential. Right, Example EPSP traces recorded from dorsal dendrites during baseline and after TBP. D, E, Scatter plot comparing the amount of LTP (unpaired t test, t(16) = 3.88, p = 0.0013) (D) and plateau potential duration (unpaired t test, t(16) = 2.35, p = 0.035) (E) between dorsal LTP induction at RMP (n = 8) and dorsal LTP induction at depolarized potential (n = 10). F, G, Summary plot comparing the normalized EPSP slope (F) and plateau potential duration (G) between dorsal depolarized TBP-LTP recordings without blockers (n = 10; blue), with d-AP5 (n = 5; orange), with nimodipine (n = 5; turquoise), and with d-AP5 + nimodipine (n = 5; purple) (percentage EPSP slope: F(3,21) = 9.47, p = 0.0004; plateau duration: F(3,21) = 4.28, p = 0.016; one-way ANOVA with Tukey HSD post hoc test). *p < 0.05. **p < 0.01. Open circles represent individual cells. Closed circles represent mean ± SEM.
This LTP induced by the depolarized TBP protocol in dorsal dendrites was partially blocked by the addition of either d-AP5 or Nimodipine (Fig. 6F). Complete block of LTP was observed when the blockers for both NMDARs and L-VGCCs were present during LTP induction (94.6 ± 21.1%; one-way ANOVA with Tukey HSD post hoc test, F(3,21) = 9.47, p < 0.05; Fig. 6F). Further, the block of TBP-LTP by d-AP5 and nimodipine was correlated with a significant reduction in the duration of plateau potentials (20 ± 1.4 ms; one-way ANOVA with Tukey HSD post hoc test, F(3,21) = 4.28, p < 0.05; Fig. 6G). These results suggest that the Vm and the plateau potentials play a significant role in mediating the LTP in dorsal dendrites.
Next, we checked whether similar regulation of TBP-LTP by Vm exists in ventral dendrites. Ventral dendrites rest at more depolarized potentials compared with dorsal dendrites and exhibit large LTP in response to the TBP protocol applied at RMP. We tested whether hyperpolarizing (∼8–10 mV) the ventral dendrites would block or reduce the amount of LTP in these neurons. Contrary to our prediction, LTP induced by TBP protocol at hyperpolarized Vm (271.6 ± 30.7%, n = 6) was not different from LTP induced at RMP (Fig. 7C,F). Additionally, the duration of plateau potentials was not reduced during the bursts of TBP at hyperpolarized voltages (21.5 ± 1.6 ms; Fig. 7B,G). This lack of change in ventral LTP and the duration of plateau potentials was in stark contrast to the dorsal recordings where depolarization-increased plateau duration was correlated with an increase in LTP. To explore this contrariety further, we asked whether depolarizing the ventral dendrites (∼8–10 mV) would affect the plateau potentials and the LTP. Interestingly, depolarizing the ventral dendrites during TBP also did not change the duration of plateau potentials (37 ± 8.5 ms, n = 5) or the amount of LTP (363.9 ± 66.4%) in ventral dendrites (Fig. 7D–G). Together, these results from altering the Vm during LTP induction suggest that the Vm plays a more important role in regulating LTP in dorsal than ventral dendrites.
Figure 7.
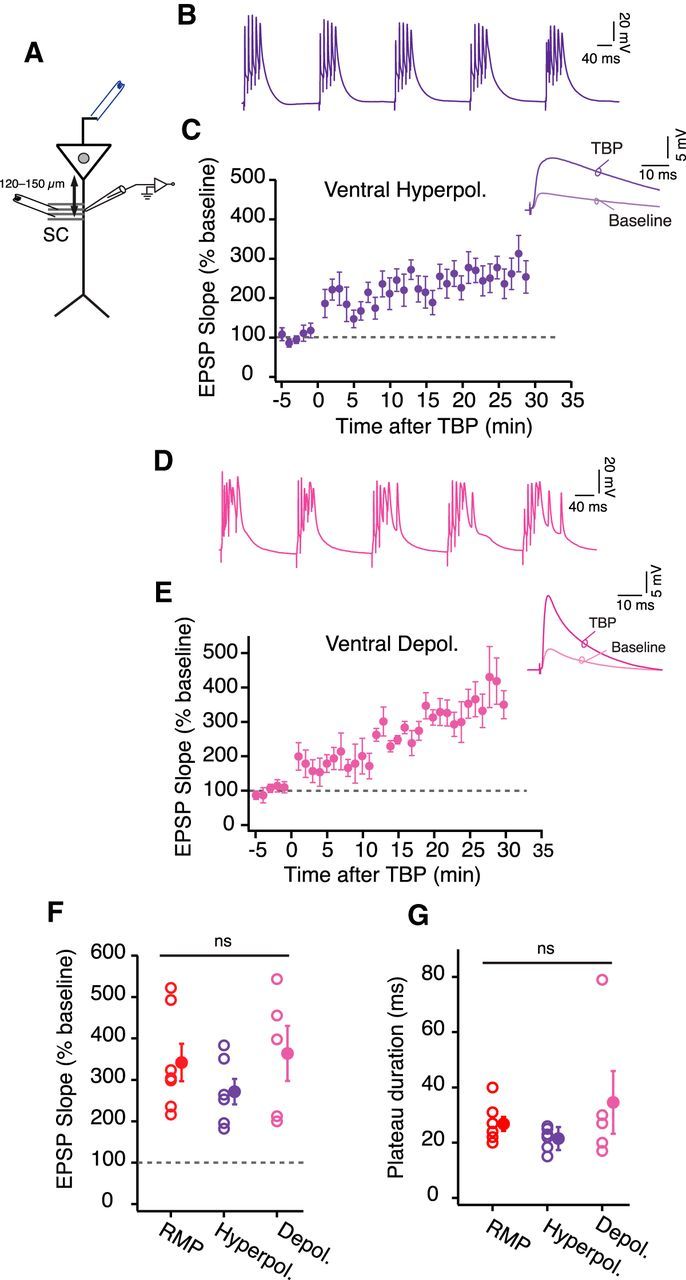
Dendritic membrane voltage during TBP has no effect on LTP in ventral neurons. A, Schematic of the experimental configuration. Voltage responses were measured at the dendrite (120–150 μm). EPSPs were evoked using a stimulating electrode placed at the SC inputs. Antidromic stimulating electrode (blue) was used to elicit bAPs. B, Example voltage trace recorded from ventral dendrites during five bursts of TBP train. The membrane potential was adjusted to hyperpolarize the dendrites by ∼8–10 mV. C, Time course plot of normalized EPSP slope (mean ± SEM) recorded in ventral dendrites when TBP-LTP was induced at hyperpolarized potential (n = 6). Inset, Example EPSP traces during baseline and after TBP. D, Example voltage trace recorded from ventral dendrites during five bursts of TBP train. The membrane potential was adjusted to depolarize the dendrites by ∼8–10 mV. E, Time course plot of normalized EPSP slope (mean ± SEM) recorded in ventral dendrites when TBP-LTP was induced at depolarized potential (n = 5). Inset, Example EPSP traces recorded from ventral dendrites during baseline and after TBP. F, G, Summary plot comparing the amount of LTP (F) (one-way ANOVA, F(2,15) = 0.97, p = 0.4) and duration of plateau potentials (G) (one-way ANOVA, F(2,15) = 1.19, p = 0.33) for three experimental conditions: LTP induction at RMP, LTP induction at hyperpolarized voltage, and LTP induction at depolarized voltage in ventral dendrites. Open circles represent individual cells. Closed circles represent mean ± SEM.
GIRK channels regulate the membrane potential and LTP of dorsal neurons
A recent study reported that the resting conductance from GIRK channels plays an important role in regulating the RMP and excitability of dorsal but not ventral neurons (Kim and Johnston, 2015). This study compared the resting GIRK conductance at the soma of dorsal and ventral neurons. It is known that the dendrites of CA1 neurons express a higher density of GIRK channels (Drake et al., 1997; Chen and Johnston, 2005). Thus, we speculated that dorsal dendrites would have a higher resting GIRK conductance and this higher GIRK conductance might underlie the higher threshold for LTP induction in dorsal neurons. GIRK channels have a high sensitivity to barium (Ba2+) and can be blocked by low concentration of barium chloride (BaCl2) (Chen and Johnston, 2005; Hibino et al., 2010; Makara and Magee, 2013; Kim and Johnston, 2015). We used 25 μm BaCl2 to block GIRKs in dorsal and ventral neurons. To test whether the resting GIRK conductance is higher in the dorsal dendrites, we first measured the changes in Vm and Rin after bath application of BaCl2. Similar to the previous study (Kim and Johnston, 2015), blocking GIRKs depolarized the dorsal neurons and increased the Rin. Furthermore, the Ba2+ induced change in Vm (dorsal: 4 ± 0.7 mV, n = 5; ventral: 1.3 ± 0.5 mV, n = 5; unpaired t test, t(8) = 2.3, p = 0.019) and Rin (dorsal: 31.3 ± 3.7%; ventral: 5.2 ± 2.25%; unpaired t test, t(8) = 6.47, p = 0.0003) was significantly larger in dorsal neurons (Fig. 8A,B). Next, we compared the effect of Ba2+ on the amount of LTP in dorsal and ventral neurons. To delineate the effects of Vm and the block of GIRKs, the Vm of dendrites was maintained at the initial RMP (measured before application of BaCl2) during the LTP experiment. Large changes in EPSP slope were observed in both dorsal and ventral neurons when TBP protocol was applied in the presence of BaCl2 (Fig. 9B–E). Interestingly, the amount of LTP induced in the presence of BaCl2 (284.4 ± 72.03%) was significantly larger (Kruskal–Wallis ANOVA, H = 10.32, p < 0.01) than LTP induced in control conditions only for dorsal dendrites. Similar increase in LTP with BaCl2 was not observed in ventral dendrites (409 ± 97.2%). Large plateau potentials were, however, observed in both dorsal and ventral dendrites during TBP in the presence of BaCl2 (dorsal: 76.2 ± 21.8 ms; ventral: 65.8 ± 22 ms) (Fig. 9B,D,K). A similar barium-induced enhancement of TBP-LTP in dorsal neurons was observed in somatic LTP recordings (Fig. 10D,F).
Figure 8.
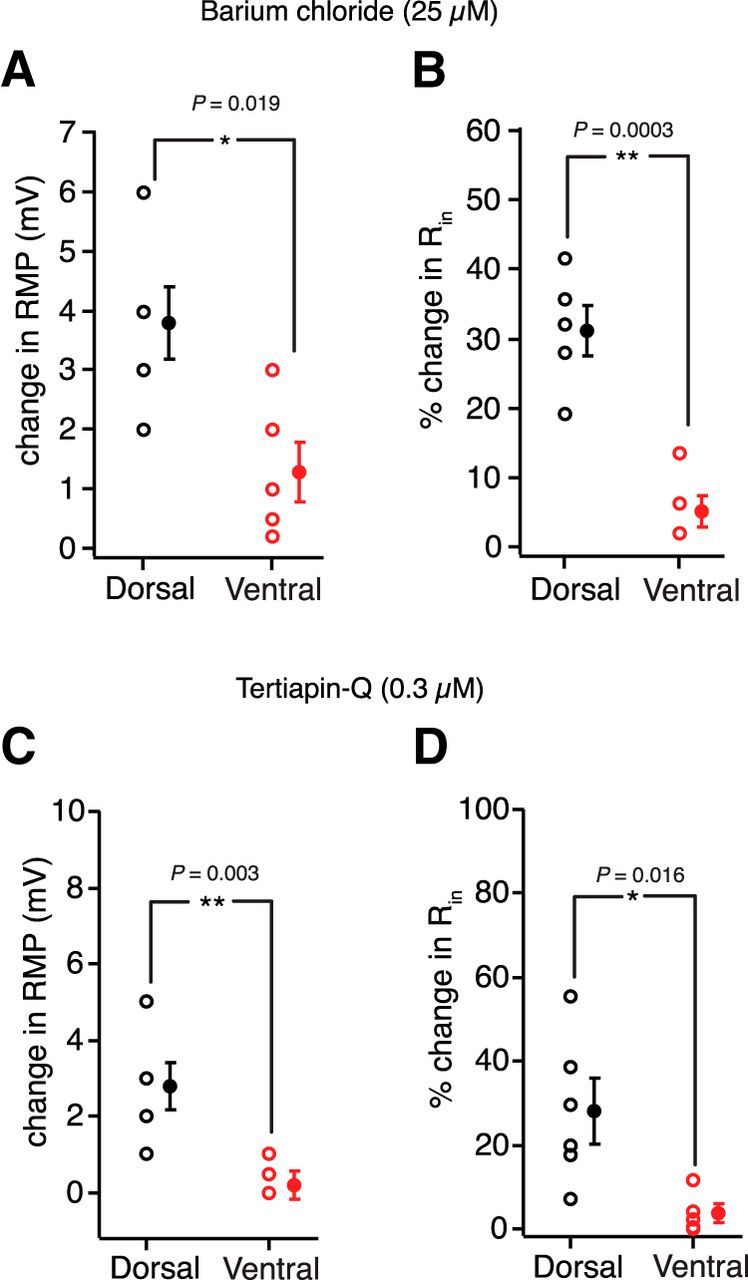
Differences in resting GIRK conductance between dorsal and ventral neurons. A, B, Change in RMP (A) (unpaired t test, t(8) = 2.3, p = 0.019) and percentage change in Rin (B) (unpaired t test, t(8) = 6.47, p = 0.0003) after addition of 25 μm BaCl2 during dorsal (n = 5) and ventral (n = 5) dendritic recordings. C, D, Change in RMP (C) (unpaired t test, t(8) = 3.67, p = 0.003) and percentage change in Rin (D) (unpaired t test, t(8) = 2.8, p = 0.016) after addition of 0.3 μm TerQ during dorsal (n = 5) and ventral (n = 5) dendritic recordings. Open circles represent individual cells. Closed circles represent mean ± SEM. *p < 0.05. **p < 0.01.
Figure 9.

Blocking resting GIRK conductance increases LTP in dorsal neurons. A, Schematic of the experimental configuration. Voltage responses were measured at the dendrite (120–150 μm). EPSPs were evoked using a stimulating electrode placed at the SC inputs. Antidromic stimulating electrode (blue) was used to elicit bAPs. B, D, Example voltage traces recorded from dorsal dendrites (B) and ventral dendrites (D) during five bursts of TBP train in the presence of BaCl2. C, E, Time course plot of normalized EPSP slope (mean ± SEM) in dorsal dendrites (n = 5) (C) and ventral dendrites (n = 5) (E) when TBP-LTP was induced in the presence of BaCl2. Inset, Example EPSP traces recorded from dorsal and ventral dendrites during baseline and after TBP. F, H, Example voltage traces recorded from dorsal dendrites (F) and ventral dendrites (H) during five bursts of TBP in the presence of TerQ. G, I, Time course plot of normalized EPSP slope (mean ± SEM) in dorsal dendrites (n = 5) (G) and ventral dendrites (n = 5) (I) when TBP-LTP was induced in the presence of TerQ. Inset, Example EPSP traces recorded from dorsal and ventral dendrites during baseline and after TBP. J, K, Comparison of the effect of BaCl2 and TerQ on the amount of TBP-LTP (J) (dorsal: H = 10.32, p = 0.0057; ventral: H = 0.89, p = 0.64; Kruskal–Wallis ANOVA followed by Dunn's multiple-comparison test). *p < 0.05. **p < 0.01. K, Plateau duration (dorsal: H = 12.68, p = 0.0018; ventral: H = 9.64, p = 0.0081; Kruskal–Wallis ANOVA followed by Dunn's multiple-comparison test) in dorsal and ventral dendrites. *p < 0.05. **p < 0.01. Open circles represent individual cells. Closed circles represent mean ± SEM.
Figure 10.
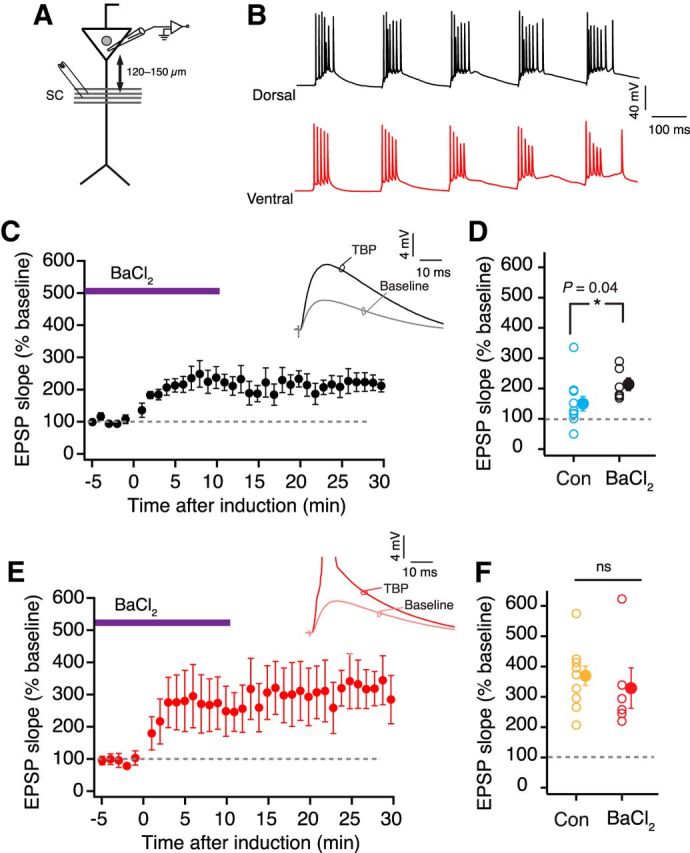
Somatic LTP recordings with resting GIRK conductance blocked by Ba2+. A, Schematic representing the experimental configuration. Voltage responses were measured at the soma. EPSPs were evoked using a stimulating electrode placed at the SC inputs (∼120–150 μm from soma). B, Example voltage traces recorded at the soma from dorsal (A) and ventral neurons (C) during five bursts of TBP train in the presence of BaCl2. C, Time course plot of normalized EPSP slope (mean ± SEM) in dorsal neurons (n = 6) when TBP-LTP was induced in the presence of BaCl2. Inset, Example EPSP traces recorded from dorsal somatic recordings during baseline and after TBP. D, Scatter plot comparing the amount of LTP in control recordings (blue, n = 10) versus LTP recordings in the presence of BaCl2 (black, n = 6) in dorsal CA1 neurons (unpaired t test, t(14) = 1.7, p = 0.04). E, Time course plot of normalized EPSP slope (mean ± SEM) in ventral neurons (n = 6) when TBP-LTP was induced in the presence of BaCl2. Inset, Example EPSP traces recorded from ventral neurons during baseline and after TBP. F, Scatter plot comparing the amount of LTP induced in control recordings (orange, n = 10) versus LTP recordings in the presence of BaCl2 (red, n = 6) in ventral CA1 neurons (unpaired t test, t(14) = 0.6, p = 0.26). Open circles represent individual cells. Closed circles represent mean ± SEM.
Because barium has affinity for GIRKs and other inward-rectifying K+ channels (IRKs), we repeated the above experiment with TerQ (Jin and Lu, 1999), which is a specific blocker for GIRK channels. TerQ also changed resting properties of dorsal neurons (Vm: 2.8 ± 0.66 mV; Rin: 28 ± 8.6%; n = 5) with only small effects on ventral dendrites (Vm: 0.2 ± 0.3 mV; Rin: 3.6 ± 2.3%; n = 5) (Fig. 8C,D). Similar to the effects of Ba2+, TerQ significantly increased (Kruskal–Wallis ANOVA, H = 10.32, p < 0.01) TBP-LTP in dorsal dendrites but did not affect the amount of LTP in ventral dendrites (dorsal: 280.2 ± 104.5%; ventral: 338.3 ± 134.5%) (Fig. 9G,I,J). Additionally, TerQ increased the duration of plateau potentials in both dorsal and ventral dendrites (dorsal: 62.8 ± 9.3 ms; ventral: 84.2 ± 18.1 ms; Kruskal–Wallis ANOVA, p < 0.01) (Fig. 9F,H,K). Together, these results suggest that a higher resting GIRK conductance in dorsal neurons underlies the higher threshold for LTP induction in these neurons.
Physiological role of regulating LTP in dorsal neurons
The results thus far indicate a resting GIRK conductance-mediated gating of LTP in dorsal neurons and a lack of a similar gating in ventral neurons. This gating of LTP in dorsal neurons can be overcome by either a pharmacological block of GIRK channels or by depolarization of dendritic membrane during LTP induction. Is there a physiological role for this gating? Apart from the large number of SC axons innervating the apical dendrite, CA1 neurons also receive TA inputs onto their distal dendrites (Steward, 1976; Amaral and Witter, 1989; Witter et al., 1989; Kajiwara et al., 2008). Recent studies have emphasized the importance of the summation and timing between the SC and TA inputs in regulating LTP and in the formation of place fields (Brun et al., 2008a; Bittner et al., 2015). We speculated that the higher threshold for LTP induction in dorsal neurons could enforce the requirement for coincident timing of SC and TA inputs. According to this postulate, dorsal neurons would exhibit robust LTP only when the two inputs arrive at the apical dendrite at the same time. To test this prediction, we obtained recordings from the dorsal dendrites at the border of stratum radiatum and stratum lacunosum-moleculare (Fig. 11A; Table 1). LTP was induced by activation of SC and TA inputs using a modified TBP protocol. During the burst of TBP, the interinput timing of the two inputs was set to be either 0 or 100 ms (Fig. 11B). Interestingly, long plateau potentials (42.6 ± 7.6 ms) and large LTP were observed at SC (313 ± 57.1%) and TA inputs (172.3 ± 18.7%) in dorsal dendrites when the two inputs were coincident during LTP induction (n = 8; Fig. 11F–H). By contrast, LTP induction with the two inputs separated by 100 ms led to a significantly smaller LTP in SC inputs (195 ± 14.2%) and failed to induce plasticity in TA inputs (103.1 ± 8.4%) (n = 6; Fig. 11G,H). Further, the decreased plasticity in the 100 ms induction protocol was accompanied by shorter duration of plateau potentials (33.9 ± 3.7 ms) in dorsal dendrites (Fig. 11F).
Figure 11.

Coincident SC and TA inputs boost LTP in dorsal neurons. A, Schematic represents the experimental configuration. Voltage responses were measured at the distal apical dendrite (300–400 μm). EPSPs were evoked using stimulating electrodes placed at the SC inputs and TA inputs. B, C, Top, Schematic showing the pairing of SC EPSPs with TA EPSPs during TBP protocol. Bottom, Example voltage traces recorded from dorsal dendrites (black) and ventral (red) dendrites during five bursts of TBP train; time interval between two inputs during LTP induction was 0 ms (B) and 100 ms (C). D, E, Example SC and TA EPSPs recorded during baseline and after TBP. Black represents dorsal traces. Red represents ventral traces. The time difference between two inputs during LTP induction was 0 ms (D) and 100 ms (E). F–H, Summary plots for plateau duration (F) (unpaired t test, dorsal: t(12) = 2.2, p = 0.04; ventral: t(14) = 0.87, p = 0.39), LTP at SC inputs (G) (unpaired t test; dorsal: t(12) = 2.47, p = 0.03; ventral: t(14) = 0.97, p = 0.34), and LTP at TA inputs (H) (unpaired t test; dorsal: t(12) = 3, p = 0.01; ventral: t(14) = 0.006, p = 0.99) for dorsal (black) and ventral (red) dendrites. Circles represent data points corresponding to 0 ms pairing protocol (dorsal, n = 8; ventral, n = 10). Triangles represent data points corresponding to 100 ms pairing protocol (dorsal, n = 6; ventral, n = 6). Open symbols represent individual cells. Closed symbols represent mean ± SEM. *p < 0 0.05.
Table 1.
Dendritic recordings during two-pathway pairing induced LTP in dorsal and ventral CA1 neuronsa
| Parameter | Dorsal | Ventral | p value | t value |
|---|---|---|---|---|
| Initial SC EPSP amplitude (mV) | 4.4 ± 0.46 | 4.4 ± 0.68 | 0.7 | 0.22 |
| Initial TA EPSP amplitude (mV) | 3.6 ± 0.4 | 4 ± 0.44 | 0.45 | 0.79 |
| 0 ms protocol depolarization area (mV × s) | 46.8 ± 6.8 | 75.6 ± 8.2 | 0.004** | 2.68 |
| 0 ms protocol plateau duration (ms) | 42.6 ± 7.6 | 91.8 ± 12.1 | 0.006** | 3.14 |
| 0 ms protocol SC pathway LTP (%) | 313 ± 57.1 | 331.4 ± 62 | 0.83 | 0.37 |
| 0 ms protocol TA pathway LTP (%) | 172.3 ± 18.7 | 183.5 ± 39.5 | 0.8 | 0.1 |
| 100 ms protocol depolarization area (mV × s) | 48.01 ± 4.5 | 80.23 ± 11.7 | 0.006** | 2.25 |
| 100 ms protocol plateau duration (ms) | 33.9 ± 3.7 | 78.5 ± 11.8 | 0.004** | 0.15 |
| 100 ms protocol SC pathway LTP (%) | 195 ± 14.2 | 440 ± 102.4 | 0.03* | 0.1 |
| 100 ms protocol TA pathway LTP (%) | 103.1 ± 8.4 | 183.1 ± 36.1 | 0.055 | 0.1 |
aThe amplitude of the initial EPSPs was similar for dorsal and ventral neurons. The depolarization areas and the plateau durations during TBP trains were significantly different for both 0 ms and 100 ms pairing protocols between dorsal and ventral neurons. LTP at the SC inputs to dorsal and ventral neurons was similar for 0 ms protocol but was different for the 100 ms protocol. LTP at the TA inputs to dorsal and ventral neurons was similar for both the 0 ms and 100 ms protocols.
*p < 0.05;
**p < 0.01; unpaired t test.
Next, we conducted the same two-pathway pairing experiments in ventral dendrites. In the ventral dendrites, similar LTP was obtained at the SC inputs (0 ms: 331.4 ± 62%; 100 ms: 440.6 ± 102.4%) and TA inputs (0 ms: 183.5 ± 39.5%; 100 ms: 183.1 ± 36.1%) when the interinput timing of the two inputs was either 0 or 100 ms (Fig. 11G,H; Table 1). Additionally, the duration of plateau potentials was also similar in the 0 ms (91.8 ± 12.1 ms, n = 10) and the 100 ms (78.5 ± 11.8 ms, n = 6) protocols (Fig. 11F). Together, these data suggest that the plasticity in dorsal neurons is boosted by activation of SC and TA inputs within a narrow time window. By contrast, the ventral neurons can generate plateau potentials and exhibit robust LTP in response to temporally dispersed inputs. These results suggest an important role of TA inputs in regulating plasticity in dorsal CA1 neurons. The spatial location of the TA inputs in dorsal neurons and the differences in the ion channel gradients at the distal apical dendrite might underlie this special role of TA inputs in plateau potential generation and LTP induction in the dorsal dendrites.
Discussion
Here we show that the dorsal CA1 neurons have a higher threshold for LTP induction and require coincident activation of SC and TA inputs for generation of plateau potentials. The ventral neurons, however, can integrate temporally dispersed excitatory inputs and have a lower threshold for LTP induction. We found that the higher threshold for LTP in the dorsal neurons is mediated by a higher resting conductance of GIRK channels. It should be noted that our results of larger LTP in ventral than dorsal neurons are in contrast to previous studies (Papatheodoropoulos and Kostopoulos, 2000; Maggio and Segal, 2007; Milior et al., 2016). The disparity in the results from our study and those of the previous studies could be due to differences in methodology. Specifically, these previous studies used extracellular field potential recordings from slices with intact inhibition, different bath solutions, and high-frequency stimulation to induce LTP.
We and others have previously reported that ventral neurons are more excitable compared with dorsal neurons and that this stems from differences in morphology and the expression of HCN, GIRKs, and A-type-K+ channels (Dougherty et al., 2012; Marcelin et al., 2012; Kim and Johnston, 2015; Malik et al., 2016). The results in the present study corroborate these previous findings obtained from somatic recordings and extend the comparison to the entire somatodendritic axis. Previously, it was shown that the resting GIRK conductance at the soma of dorsal neurons was higher compared with ventral neurons (Kim and Johnston, 2015); we provide evidence for a similar DV difference in resting GIRK conductance at the apical dendrites. This higher resting GIRK conductance in the dorsal dendrites lowers the excitability and also increases the threshold for LTP induction. In agreement with the previously reported higher expression of KA channels (Marcelin et al., 2012), the attenuation of bAPs was greater in dorsal dendrites compared with ventral dendrites. Interestingly, we found that the decreased attenuation of bAPs in ventral neurons was a compensatory mechanism for the longer radial length of their apical dendrites. Our results indicate that this DV difference in bAP amplitude does not underlie the DV differences in LTP, but we cannot rule out a contribution of KA channels in LTP regulation. It is becoming increasingly evident that conductances mediated by channels active at rest (e.g., HCN and GIRK) can interact with KA channels (John and Manchanda, 2011; Mishra and Narayanan, 2015) to regulate integration of excitatory inputs and generation of active dendritic events (Berger et al., 2003; Tsay et al., 2007). Although the current study does not address this interaction, it might play a role in regulation of LTP in dorsal neurons. Somewhat surprisingly, we did not find DV differences in TBP-induced intrinsic plasticity. Although dorsal and ventral neurons showed different amounts of LTP, the TBP-induced decrease in intrinsic excitability was similar in these neurons. It has been shown previously that the activation kinetics of HCN channels in dorsal and ventral neurons are different (Dougherty et al., 2013), but the effects of resting HCN conductance on subthreshold membrane properties of dorsal and ventral neurons are similar when recorded at their respective RMPs (Dougherty et al., 2013; Malik et al., 2016). In the present study, the DV differences in LTP induction were evident when the recordings were made at the respective RMP of the dorsal and ventral neurons. Thus, we speculate that the DV differences in the threshold for LTP induction are not due to differences in kinetics of HCN channels but due to the differences in resting GIRK conductance.
GIRK channels belong to the family of strong inwardly rectifying K+ channels, but despite this name, under physiological conditions, the net potassium flux through these channels is in the outward direction (Ehrengruber et al., 1997; Nichols and Lopatin, 1997; Yamada et al., 1998). Because of their higher expression at the dendrites and near the excitatory synapses (Drake et al., 1997; Chen and Johnston, 2005), GIRK channels can suppress the summation of EPSPs, reduce the availability of NMDARs, and influence the threshold and shape of active events in the dendrite. Multiple computational studies provide evidence for the above-mentioned roles of GIRK channels (Steephen and Manchanda, 2009; John and Manchanda, 2011; Sanders et al., 2013); our study provides the first experimental evidence for the role of GIRK channels in regulation of dendritic plateau potentials and LTP in CA1 neurons. Based on the subunit composition, GIRK channels can be constitutively active or can be activated/modulated by a variety of neurotransmitters (e.g., GABA, serotonin, adenosine, dopamine, and acetylcholine) in a G-protein-dependent manner. The levels of these neurotransmitters change during spatial navigation (Hasselmo et al., 1996; Stancampiano et al., 1999); thus, it is likely that the modulation of GIRKs could further narrow (or widen) the integration window for synaptic plasticity in dorsal neurons (Carr and Surmeier, 2007; Shen et al., 2007). It is important to note that the results in the present study were obtained from disinhibited slices. The influence of GIRK conductance might be higher under in vivo conditions because of the influence of inhibition on the CA1 microcircuit. There is evidence suggesting that the net inhibitory input to dorsal neurons is higher than ventral neurons (Papatheodoropoulos et al., 2002; Milior et al., 2016). This stronger inhibition coupled with a higher resting GIRK conductance would further increase the gating of LTP in dorsal neurons.
What is the functional role of distinct input integration schemes in dorsal and ventral neurons? Network models have speculated that the DV differences in place field size could arise due to the differences in spatial and nonspatial inputs across the DV axis (Moser and Moser, 1998). The findings in the present study provide the synaptic mechanisms that might underlie this DV difference in place field size. Specifically, a GIRK-mediated higher threshold for LTP induction in dorsal neurons enforces the requirement for coincident activation of the SC and TA inputs and inhibits the generation of NMDAR-dependent plateau potentials. This gating would ensure a preferential potentiation of the synchronized inputs arriving from area CA3 neurons and entorhinal cortex. This potentiation would tune the firing output of dorsal neurons to these inputs and thus would restrict the size of their place fields. A network modeling study showed that the pairing of GIRK channels with NMDARs is sufficient to produce bistable states (crucial for neurons involved in novelty detection) (Sanders et al., 2013). Because dorsal CA1 neurons are tasked to reliably encode novel spatial information and also to maintain a stable representation of previously stored information, GIRK channel-mediated suppression of NMDARs would keep the circuit under check by inhibiting LTP under resting conditions. Only when the incoming synaptic activity would sufficiently depolarize the neurons (to overcome the GIRK-mediated block of LTP), the neurons would be able to form new representations. Evidence in support of this prediction comes from two in vivo patch-clamp studies where many dorsal CA1 neurons were silent during spatial exploration (Epsztein et al., 2011; Lee et al., 2012; Bittner et al., 2015). Interestingly, depolarizing the neurons was sufficient to transform these silent cells to place cells. It is important to note that RMP values of the dorsal neurons recorded in our study are similar to the average RMP recorded in some (Lee et al., 2006; Harvey et al., 2009) but not all previous in vivo whole-cell patch-clamp studies (Epsztein et al., 2011; Bittner et al., 2015). This disparity might be caused due to differences in network activity states and the differences in liquid junction potential. The requirement for coincident activation of the two pathways or depolarization for plateau generation and plasticity in dorsal neurons reported in our study is, however, consistent with previous in vivo studies (Epsztein et al., 2011; Bittner et al., 2015). Because ventral neurons lack this GIRK-mediated regulation of plasticity and have an overall high excitability, we predict that, during spatial navigation, the majority of ventral CA1 neurons would be active in response to even weak and temporally dispersed synaptic inputs. This could lead to the formation of large or multiple place fields per ventral neuron (Rich et al., 2014). Based on these predictions, the spatial encoding in the ventral CA1 would be generalized to represent the entire context (Bannerman et al., 2004; Komorowski et al., 2013). This might be important to encode large environments or might play a role in associating crude contextual information with emotionally salient cues (Komorowski et al., 2013; Ciocchi et al., 2015).
The findings in this and previous studies strongly indicate the heterogeneity in the population of CA1 pyramidal neurons. They vary in gene expression, synaptic properties, intrinsic properties, connectivity and place field size, and properties, not only along the DV axis (Strange et al., 2014; Malik et al., 2016) but also along the transverse axis and by depth within stratum pyramidale (Cembrowski et al., 2016; Danielson et al., 2016). Overall, our study highlights the differences in regulation of synaptic plasticity between dorsal and ventral neurons, which could account for the DV differences in scale of spatial representation.
Footnotes
This work was supported by National Institutes of Health Grant 4R01NS084473 to D.J. We thank Komal H. Parikh and Mohammed Arrafi for histological processing and morphological reconstructions; Dr. Laura Colgin, Dr. Richard Gray, Dr. Rahul Rathour, and Elizabeth Arnold for helpful comments on the manuscript; and all members of D.J. laboratory for valuable discussions throughout the study.
The authors declare no competing financial interests.
References
- Amaral DG, Witter MP (1989) The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience 31:571–591. 10.1016/0306-4522(89)90424-7 [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Rawlins JN, McHugh SB, Deacon RM, Yee BK, Bast T, Zhang WN, Pothuizen HH, Feldon J (2004) Regional dissociations within the hippocampus: memory and anxiety. Neurosci Biobehav Rev 28:273–283. 10.1016/j.neubiorev.2004.03.004 [DOI] [PubMed] [Google Scholar]
- Berger T, Senn W, Lüscher HR (2003) Hyperpolarization-activated current Ih disconnects somatic and dendritic spike initiation zones in layer V pyramidal neurons. J Neurophysiol 90:2428–2437. 10.1152/jn.00377.2003 [DOI] [PubMed] [Google Scholar]
- Bittner KC, Grienberger C, Vaidya SP, Milstein AD, Macklin JJ, Suh J, Tonegawa S, Magee JC (2015) Conjunctive input processing drives feature selectivity in hippocampal CA1 neurons. Nat Neurosci 18:1133–1142. 10.1038/nn.4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum KI, Abbott LF (1996) A model of spatial map formation in the hippocampus of the rat. Neural Comput 8:85–93. 10.1162/neco.1996.8.1.85 [DOI] [PubMed] [Google Scholar]
- Brun VH, Leutgeb S, Wu HQ, Schwarcz R, Witter MP, Moser EI, Moser MB (2008a) Impaired spatial representation in CA1 after lesion of direct input from entorhinal cortex. Neuron 57:290–302. 10.1016/j.neuron.2007.11.034 [DOI] [PubMed] [Google Scholar]
- Brun VH, Solstad T, Kjelstrup KB, Fyhn M, Witter MP, Moser EI, Moser MB (2008b) Progressive increase in grid scale from dorsal to ventral medial entorhinal cortex. Hippocampus 18:1200–1212. 10.1002/hipo.20504 [DOI] [PubMed] [Google Scholar]
- Carr DB, Surmeier DJ (2007) M1 muscarinic receptor modulation of Kir2 channels enhances temporal summation of excitatory synaptic potentials in prefrontal cortex pyramidal neurons. J Neurophysiol 97:3432–3438. 10.1152/jn.00828.2006 [DOI] [PubMed] [Google Scholar]
- Cembrowski MS, Bachman JL, Wang L, Sugino K, Shields BC, Spruston N (2016) Spatial gene-expression gradients underlie prominent heterogeneity of CA1 pyramidal neurons. Neuron 89:351–368. 10.1016/j.neuron.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Chen X, Johnston D (2005) Constitutively active G-protein-gated inwardly rectifying K+ channels in dendrites of hippocampal CA1 pyramidal neurons. J Neurosci 25:3787–3792. 10.1523/JNEUROSCI.5312-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocchi S, Passecker J, Malagon-Vina H, Mikus N, Klausberger T (2015) Selective information routing by ventral hippocampal CA1 projection neurons. Science 348:560–563. 10.1126/science.aaa3245 [DOI] [PubMed] [Google Scholar]
- Clemens AM, Johnston D (2014) Age- and location-dependent differences in store depletion-induced h-channel plasticity in hippocampal pyramidal neurons. J Neurophysiol 111:1369–1382. 10.1152/jn.00839.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson NB, Zaremba JD, Kaifosh P, Bowler J, Ladow M, Losonczy A (2016) Sublayer-specific coding dynamics during spatial navigation and learning in hippocampal area CA1. Neuron 91:652–665. 10.1016/j.neuron.2016.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KA, Islam T, Johnston D (2012) Intrinsic excitability of CA1 pyramidal neurones from the rat dorsal and ventral hippocampus. J Physiol 590:5707–5722. 10.1113/jphysiol.2012.242693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KA, Nicholson DA, Diaz L, Buss EW, Neuman KM, Chetkovich DM, Johnston D (2013) Differential expression of HCN subunits alters voltage-dependent gating of h-channels in CA1 pyramidal neurons from dorsal and ventral hippocampus. J Neurophysiol 109:1940–1953. 10.1152/jn.00010.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Bausch SB, Milner TA, Chavkin C (1997) GIRK1 immunoreactivity is present predominantly in dendrites, dendritic spines, and somata in the CA1 region of the hippocampus. Proc Natl Acad Sci U S A 94:1007–1012. 10.1073/pnas.94.3.1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU, Doupnik CA, Xu Y, Garvey J, Jasek MC, Lester HA, Davidson N (1997) Activation of heteromeric G protein-gated inward rectifier K+ channels overexpressed by adenovirus gene transfer inhibits the excitability of hippocampal neurons. Proc Natl Acad Sci U S A 94:7070–7075. 10.1073/pnas.94.13.7070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztein J, Brecht M, Lee AK (2011) Intracellular determinants of hippocampal CA1 place and silent cell activity in a novel environment. Neuron 70:109–120. 10.1016/j.neuron.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D (2005) Activity-dependent decrease of excitability in rat hippocampal neurons through increases in Ih. Nat Neurosci 8:1542–1551. 10.1038/nn1568 [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N (2002) Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature 418:326–331. 10.1038/nature00854 [DOI] [PubMed] [Google Scholar]
- Harvey CD, Collman F, Dombeck DA, Tank DW (2009) Intracellular dynamics of hippocampal place cells during virtual navigation. Nature 461:941–946. 10.1038/nature08499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Wyble BP, Wallenstein GV (1996) Encoding and retrieval of episodic memories: role of cholinergic and GABAergic modulation in the hippocampus. Hippocampus 6:693–708. 10.1102/(SICI)1098-1063(1996)6:6%3C693::AID-HIPO12%3E3.0.CO.2-W [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366. 10.1152/physrev.00021.2009 [DOI] [PubMed] [Google Scholar]
- Hönigsperger C, Marosi M, Murphy R, Storm JF (2015) Dorsoventral differences in Kv7/M-current and its impact on resonance, temporal summation and excitability in rat hippocampal pyramidal cells. J Physiol 593:1551–1580. 10.1113/jphysiol.2014.280826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussaini SA, Kempadoo KA, Thuault SJ, Siegelbaum SA, Kandel ER (2011) Increased size and stability of CA1 and CA3 place fields in HCN1 knockout mice. Neuron 72:643–653. 10.1016/j.neuron.2011.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Lu Z (1999) Synthesis of a stable form of tertiapin: a high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry 38:14286–14293. 10.1021/bi991205r [DOI] [PubMed] [Google Scholar]
- John J, Manchanda R (2011) Modulation of synaptic potentials and cell excitability by dendritic KIR and KAs channels in nucleus accumbens medium spiny neurons: a computational study. J Biosci 36:309–328. 10.1007/s12038-011-9039-8 [DOI] [PubMed] [Google Scholar]
- Jung MW, Wiener SI, McNaughton BL (1994) Comparison of spatial firing characteristics of units in dorsal and ventral hippocampus of the rat. J Neurosci 14:7347–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara R, Wouterlood FG, Sah A, Boekel AJ, Baks-te Bulte LT, Witter MP (2008) Convergence of entorhinal and CA3 inputs onto pyramidal neurons and interneurons in hippocampal area CA1: an anatomical study in the rat. Hippocampus 18:266–280. 10.1002/hipo.20385 [DOI] [PubMed] [Google Scholar]
- Keinath AT, Wang ME, Wann EG, Yuan RK, Dudman JT, Muzzio IA (2014) Precise spatial coding is preserved along the longitudinal hippocampal axis. Hippocampus 24:1533–1548. 10.1002/hipo.22333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CS, Johnston D (2015) A1 adenosine receptor-mediated GIRK channels contribute to the resting conductance of CA1 neurons in the dorsal hippocampus. J Neurophysiol 113:2511–2523. 10.1152/jn.00951.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjelstrup KB, Solstad T, Brun VH, Hafting T, Leutgeb S, Witter MP, Moser EI, Moser MB (2008) Finite scale of spatial representation in the hippocampus. Science 321:140–143. 10.1126/science.1157086 [DOI] [PubMed] [Google Scholar]
- Komorowski RW, Garcia CG, Wilson A, Hattori S, Howard MW, Eichenbaum H (2013) Ventral hippocampal neurons are shaped by experience to represent behaviorally relevant contexts. J Neurosci 33:8079–8087. 10.1523/JNEUROSCI.5458-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AK, Manns ID, Sakmann B, Brecht M (2006) Whole-cell recordings in freely moving rats. Neuron 51:399–407. 10.1016/j.neuron.2006.07.004 [DOI] [PubMed] [Google Scholar]
- Lee D, Lin BJ, Lee AK (2012) Hippocampal place fields emerge upon single-cell manipulation of excitability during behavior. Science 337:849–853. 10.1126/science.1221489 [DOI] [PubMed] [Google Scholar]
- Lyttle D, Gereke B, Lin KK, Fellous JM (2013) Spatial scale and place field stability in a grid-to-place cell model of the dorsoventral axis of the hippocampus. Hippocampus 23:729–744. 10.1002/hipo.22132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D (1997) A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275:209–213. 10.1126/science.275.5297.209 [DOI] [PubMed] [Google Scholar]
- Maggio N, Segal M (2007) Unique regulation of long term potentiation in the rat ventral hippocampus. Hippocampus 17:10–25. 10.1002/hipo.20237 [DOI] [PubMed] [Google Scholar]
- Makara JK, Magee JC (2013) Variable dendritic integration in hippocampal CA3 pyramidal neurons. Neuron 80:1438–1450. 10.1016/j.neuron.2013.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Dougherty KA, Parikh K, Byrne C, Johnston D (2016) Mapping the electrophysiological and morphological properties of CA1 pyramidal neurons along the longitudinal hippocampal axis. Hippocampus 26:341–361. 10.1002/hipo.22526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin B, Lugo JN, Brewster AL, Liu Z, Lewis AS, McClelland S, Chetkovich DM, Baram TZ, Anderson AE, Becker A, Esclapez M, Bernard C (2012) Differential dorso-ventral distributions of Kv4.2 and HCN proteins confer distinct integrative properties to hippocampal CA1 pyramidal cell distal dendrites. J Biol Chem 287:17656–17661. 10.1074/jbc.C112.367110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer AP, Vanrhoads SR, Sutherland GR, Lipa P, McNaughton BL (2005) Self-motion and the origin of differential spatial scaling along the septo-temporal axis of the hippocampus. Hippocampus 15:841–852. 10.1002/hipo.20114 [DOI] [PubMed] [Google Scholar]
- Mehta MR, Wilson MA (2000) From hippocampus to V1: effect of LTP on spatio-temporal dynamics of receptive fields. Neurocomputing 32- 33:905–911. [Google Scholar]
- Milior G, Castro MA, Sciarria LP, Garofalo S, Branchi I, Ragozzino D, Limatola C, Maggi L (2016) Electrophysiological properties of CA1 pyramidal neurons along the longitudinal axis of the mouse hippocampus. Sci Rep 6:38242. 10.1038/srep38242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Narayanan R (2015) High-conductance states and A-type K+ channels are potential regulators of the conductance-current balance triggered by HCN channels. J Neurophysiol 113:23–43. 10.1152/jn.00601.2013 [DOI] [PubMed] [Google Scholar]
- Moser MB, Moser EI (1998) Functional differentiation in the hippocampus. Hippocampus 8:608–619. 10.1002/(SICI)1098-1063(1998)8:6%3C608::AID-HIPO3%3E3.0.CO%3B2-7 [DOI] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S (2004) NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci 5:361–372. 10.1038/nrn1385 [DOI] [PubMed] [Google Scholar]
- Narayanan R, Johnston D (2007) Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron 56:1061–1075. 10.1016/j.neuron.2007.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN (1997) Inward rectifying potassium channels. Annu Rev Physiol 59:171–191. 10.1146/annurev.physiol.59.1.171 [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A (2004) A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119:719–732. 10.1016/j.cell.2004.11.020 [DOI] [PubMed] [Google Scholar]
- Papatheodoropoulos C, Kostopoulos G (2000) Decreased ability of rat temporal hippocampal CA1 region to produce long-term potentiation. Neurosci Lett 279:177–180. 10.1016/S0304-3940(99)01002-2 [DOI] [PubMed] [Google Scholar]
- Papatheodoropoulos C, Asprodini E, Nikita I, Koutsona C, Kostopoulos G (2002) Weaker synaptic inhibition in CA1 region of ventral compared to dorsal rat hippocampal slices. Brain Res 948:117–121. 10.1016/S0006-8993(02)02958-X [DOI] [PubMed] [Google Scholar]
- Rich PD, Liaw HP, Lee AK (2014) Place cells: large environments reveal the statistical structure governing hippocampal representations. Science 345:814–817. 10.1126/science.1255635 [DOI] [PubMed] [Google Scholar]
- Rotenberg A, Mayford M, Hawkins RD, Kandel ER, Muller RU (1996) Mice expressing activated CaMKII lack low frequency LTP and do not form stable place cells in the CA1 region of the hippocampus. Cell 87:1351–1361. 10.1016/S0092-8674(00)81829-2 [DOI] [PubMed] [Google Scholar]
- Sanders H, Berends M, Major G, Goldman MS, Lisman JE (2013) NMDA and GABAB (KIR) conductances: the “perfect couple” for bistability. J Neurosci 33:424–429. 10.1523/JNEUROSCI.1854-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, Surmeier DJ (2007) Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci 10:1458–1466. 10.1038/nn1972 [DOI] [PubMed] [Google Scholar]
- Stancampiano R, Cocco S, Cugusi C, Sarais L, Fadda F (1999) Serotonin and acetylcholine release response in the rat hippocampus during a spatial memory task. Neuroscience 89:1135–1143. 10.1016/S0306-4522(98)00397-2 [DOI] [PubMed] [Google Scholar]
- Steephen JE, Manchanda R (2009) Differences in biophysical properties of nucleus accumbens medium spiny neurons emerging from inactivation of inward rectifying potassium currents. J Comput Neurosci 27:453–470. 10.1007/s10827-009-0161-7 [DOI] [PubMed] [Google Scholar]
- Steward O. (1976) Topographic organization of the projections from the entorhinal area to the hippocampal formation of the rat. J Comp Neurol 167:285–314. 10.1002/cne.901670303 [DOI] [PubMed] [Google Scholar]
- Strange BA, Witter MP, Lein ES, Moser EI (2014) Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 15:655–669. 10.1038/nrn3785 [DOI] [PubMed] [Google Scholar]
- Takahashi H, Magee JC (2009) Pathway interactions and synaptic plasticity in the dendritic tuft regions of CA1 pyramidal neurons. Neuron 62:102–111. 10.1016/j.neuron.2009.03.007 [DOI] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, Siegelbaum SA (2007) HCN1 channels constrain synaptically evoked Ca(2+) spikes in distal dendrites of CA1 pyramidal neurons. Neuron 56:1076–1089. 10.1016/j.neuron.2007.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Tonegawa S (1997) Synaptic plasticity, place cells and spatial memory: study with second generation knockouts. Trends Neurosci 20:102–106. 10.1016/S0166-2236(96)01023-5 [DOI] [PubMed] [Google Scholar]
- Witter MP, Groenewegen HJ, Lopes da Silva FH, Lohman AH (1989) Functional organization of the extrinsic and intrinsic circuitry of the parahippocampal region. Prog Neurobiol 33:161–253. 10.1016/0301-0082(89)90009-9 [DOI] [PubMed] [Google Scholar]
- Yamada M, Inanobe A, Kurachi Y (1998) G protein regulation of potassium ion channels. Pharmacol Rev 50:723–760. [PubMed] [Google Scholar]