

Studies of children with paroxysmal nocturnal hemoglobinuria (PNH) are scarce and include limited numbers of patients. We compared demographic and clinical characteristics of children and adults in the International PNH Registry. Our data show that, compared to adults with PNH, children had smaller PNH clones with more frequent severe cytopenia or previous marrow aplasia. In contrast, hemolysis was present in similar proportions, and both children and adults suffered from major adverse vascular events (MAVE), such as thrombotic events (TE), which seemed more prominent in adults.
PNH is an acquired clonal hematopoietic stem cell disorder characterized by complement-mediated hemolysis, thrombosis, and bone marrow failure.1–3 It is a very rare disease with a worldwide incidence estimated at 1.3 cases per million population.4 The onset of PNH is typically in adulthood, with pediatric cases accounting for only 5–10% of reported cases;1,5 studies of children with PNH have, therefore, been limited to case reports and small series.5–7 The largest PNH pediatric series included 26 patients and was published in 1991, when flow cytometry was not yet routinely available.1
The International PNH Registry is the largest PNH data repository to date, and contains a wide range of laboratory, clinical, and patient-reported information.8 The Registry is overseen by an independent committee of international medical PNH experts. Patients with detectable glycosylphosphatidylinositol (GPI)-deficient granulocytes or a “granulocyte PNH clone” (≥0.01%) are eligible for inclusion in the Registry, regardless of disease severity, comorbidities, or treatments.
This retrospective analysis included patients enrolled in or before June 2015. Because the objective of this analysis was to study the natural presentation of PNH, patients being treated with eculizumab at the time of enrollment were excluded. Children were <18 years of age at the time of disease start and time of enrollment, while adults were ≥18 years of age at disease start and enrollment. Patients who were <18 years at disease start but over 18 years at enrollment were excluded. The degree of cytopenia was classified using the following criteria:9,10 none (neutrophils ≥1.5×109/L and platelets ≥100×109/L), moderate (neutrophils <1.5×109/L or platelets <100×109/L), or severe (neutrophils <0.5×109/L or platelets <20×109/L). Hemolytic activity was defined as serum lactate dehydrogenase ≥1.5× the upper limit of normal and/or reticulocytes ≥60×109/L. Duration of disease was the time between disease start and enrollment into the Registry. MAVE included TE, as well as pulmonary embolus, amputation (non-traumatic; non-diabetic), myocardial infarction, cerebral arterial occlusion or transient ischemic attack, unstable angina, and gangrene (non-traumatic, non-diabetic). Adult and pediatric demographic and disease characteristics at enrollment were compared using the Wilcoxon-Mann-Whitney test for medians and the Pearson χ2 test for frequencies. Rates of TE and MAVE between disease start and enrollment were calculated per 100 person-years, and 95% confidence intervals (95% CI) were based on a Poisson distribution for event counts.
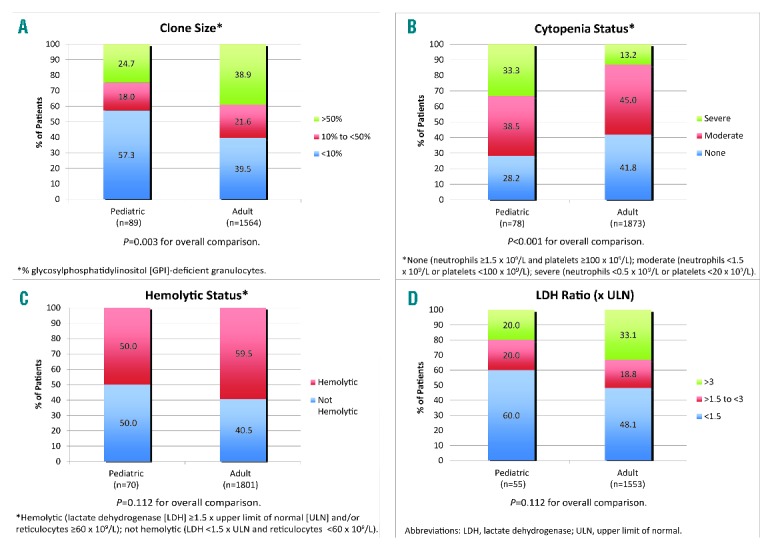
A total of 2,367 of 3,681 patients enrolled in the Registry fulfilled the above inclusion criteria for the analysis: 99 (4.2%) children and 2,268 (95.8%) adults (Online Supplementary Figure S1). Median (minimum, maximum) age at enrollment was 14 (1, 17) years for children and 45 (18, 99) years for adults; the median duration of disease at enrollment was shorter in children than in adults (0.7 years versus 2.1 years, P<0.001) (Table 1). Children had a shorter average time of observation by approximately 16.8 months compared to adults. While most patients in both age groups were White/Caucasian, there was a lower proportion of patients of Asian ancestry in the pediatric cohort than among the adults (7.1% versus 17.7%, P=0.017 for overall comparison of ethnicity). The percentage of GPI-deficient granulocytes differed between the two cohorts, with more children having smaller PNH clones at enrollment compared to adults (<10% clone: 57.3% versus 39.5%, 10–50% clone: 18.0% versus 21.6%, >50% clone: 24.7% versus 38.9%, respectively; P=0.003 for the overall comparison) (Figure 1A). More children were classified as having severe cytopenia at enrollment compared to adults (33.3% versus 13.2%, P<0.001 for overall comparison) (Figure 1B). Similar proportions were classified as hemolytic at the time of enrollment (50.0% versus 59.5%, P=0.112) (Figure 1C), although lactate dehydrogenase levels and reticulocyte counts were significantly higher in adults (Table 1). A history of aplastic or hypoplastic anemia at or before enrollment was more frequent in children than in adults (77.8% versus 54.4%, P<0.001). In contrast, a history of myelodysplastic syndrome was more frequent among adults than among children (10.2% versus 2.0%, P=0.007). A history of TE was less common among children than among adults (2.0% versus 8.6%, P=0.020), as was a history of MAVE (4.0% versus 14.4%, P=0.004). Using person-years from disease start to enrollment, rates of TE and MAVE were lower in children than in adults, although the 95% confidence intervals overlapped due to low numbers of events: [1.2 (95% CI: 0.2–4.4) and 2.3 (95% CI: 2.0–2.6), respectively, for TE; 2.5 (95% CI: 0.7–6.3) and 4.2 (95% CI: 3.9–4.6), respectively, for MAVE]. There were no differences in the frequency of impaired renal function or diagnosed pulmonary hypertension between the two cohorts. About twice as many children than adults had received antithymocyte globulin or immunosuppressive therapy within 12 months of enrollment (59.6% of children versus 28.5% of adults; P<0.001) (Table 2). The frequency of some symptoms was lower among children than adults, including dyspnea (20.2% versus 43.5%, respectively; P<0.001) and hemoglobinuria (19.3% versus 44.6%, respectively; P<0.001).
Table 1.
Demographics, medical history, and laboratory characteristics at enrollment in pediatric and adult patients.
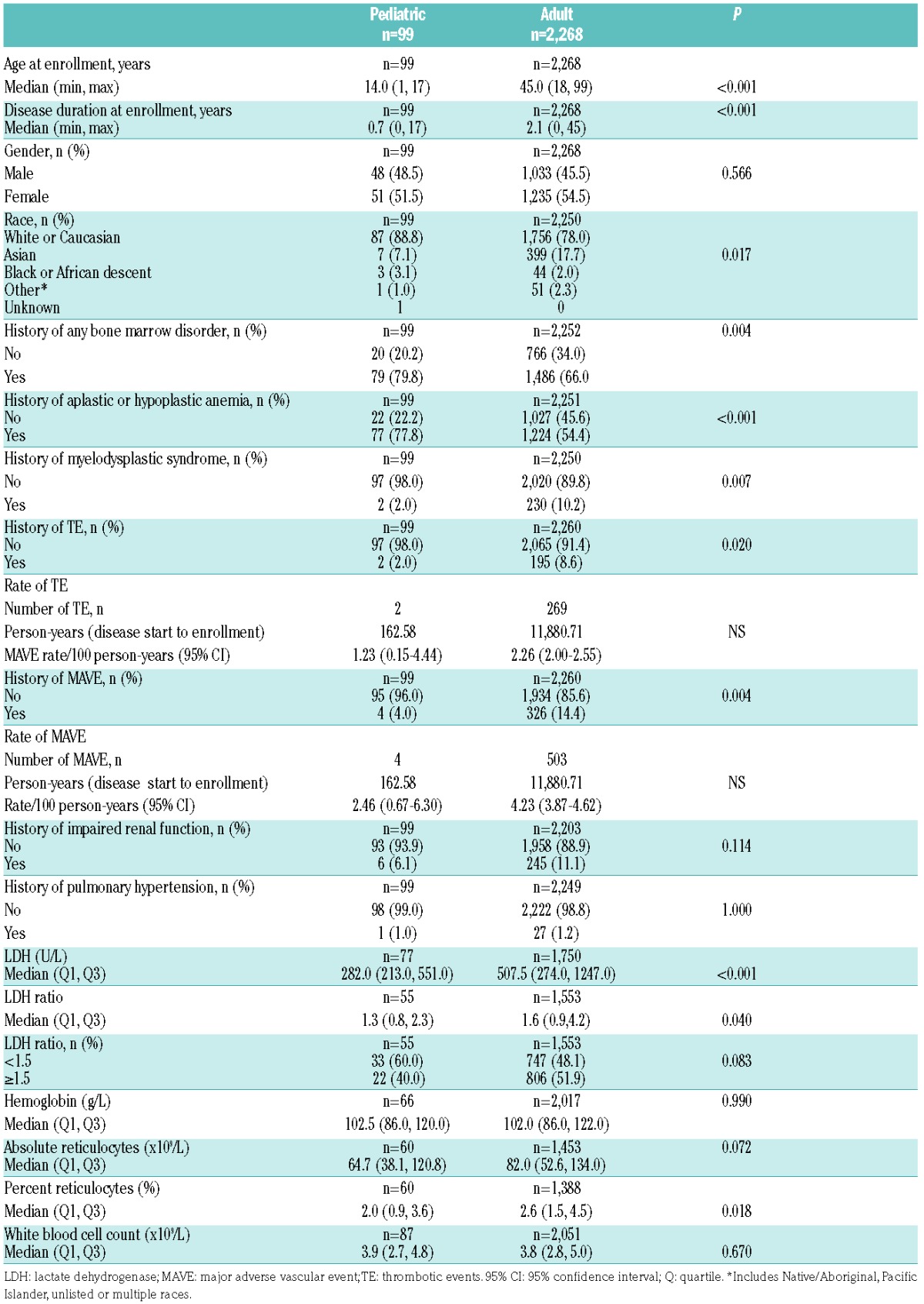
Figure 1.
Distribution of characteristics in pediatric and adult patients with paroxysmal noctural hemoglobinuria at enrollment. (A) Clone size, (B) cytopenia status, (C) hemolytic status, and (D) lactate dehydrogenase ratio.
Table 2.
Treatment received by pediatric and adult patients prior to and at enrollment.
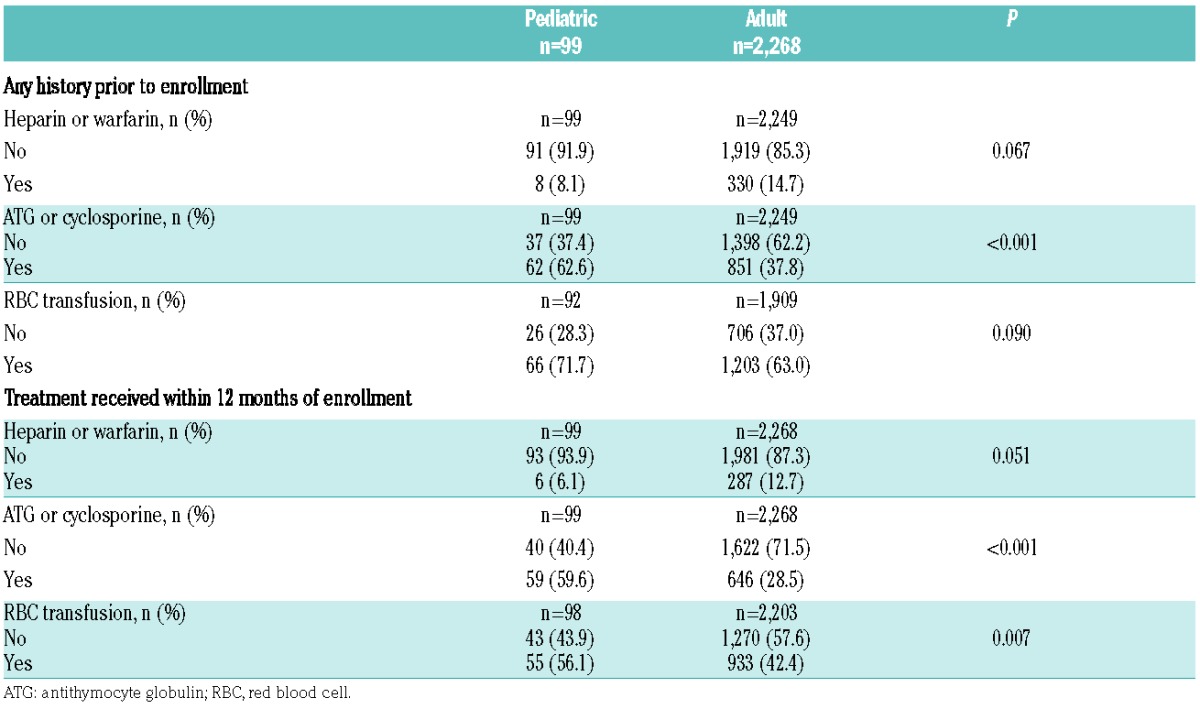
This summary of 99 patients with PNH younger than 18 years enrolled in the International PNH Registry represents more pediatric PNH patients than reported in the entire published literature to date. The main finding is that there are substantial differences between the clinical manifestations and hematologic laboratory values of these young patients and those of a group of 2,268 patients with PNH older than 18 years. The first observation was a significant difference in PNH clone size. The average GPI-deficient clone in children with PNH was much smaller than in adults, with more than half having a clone size <10%. A large GPI-deficient clone (>50%) is typically associated with classical hemolytic PNH, whereas a small GPI-deficient clone is more frequently observed in the aplastic anemia-PNH subtype.11 Our laboratory findings support this association: severe cytopenia was more frequently observed in children than in adults, as was a history of aplastic or hypoplastic anemia, and twice as many children had previously received antithymocyte globulin or cyclosporine before or at enrollment. In a retrospective analysis of 207 patients with severe aplastic anemia who were treated with horse antithymocyte globulin from 2000 to 2008, 14 pediatric patients had a PNH clone >1% at baseline.12 There was a statistically significant decrease in median clone size in these patients before and after immunosuppressive therapy over 12 months. Our findings support the concept that children with hypoplastic or aplastic anemia, even when successfully treated with immunosuppressive therapy, must be monitored serially for evidence of a GPI-deficient clone, since late clonal evolution from marrow failure to PNH can occur.11 Despite having smaller PNH clones and more often an aplastic component, children with PNH had evidence of hemolysis at a similar frequency as adults. We assume, therefore, that many children with PNH have coexistence of both aplastic and hemolytic components of PNH. Hemoglobinuria, lactate dehydrogenase levels, and reticulocyte counts in children were significantly lower than in adults, suggesting that hemolysis was less severe than in adults.
The size of the GPI-deficient clone has been associated with the risk of thrombosis. In a large British cohort of 163 unselected PNH patients,13 the incidence of thrombosis was significantly lower among patients with a small PNH clone, with a 50% threshold suggested for risk of clinical thrombosis and benefit of primary prophylaxis with warfarin in this group. Although thrombosis was less frequent in children, there were several severe cases and the authors are aware of fatal thrombosis in this setting. It is also well known that severe PNH-related thrombosis can occur during primary prophylactic anticoagulation.14 The true incidence of TE in pediatric PNH has been difficult to determine; previous pediatric series have reported values as low as 0–4% in Asian cohorts and as high as 50% among Caucasians.1,5–7,15,16 Our data confirm that thrombosis (TE and MAVE) is an important complication of PNH not only in adults (2.26 per 100 person-years), but also in children (1.23 per 100 person-years). However, myelodysplastic syndrome or myelodysplastic features previously reported in two smaller series5,7 were not confirmed in our registry cohort.
A limitation of our analyses is the descriptive nature of the data, which are subject to the inherent limitations of non-interventional, observational data collection, including missing or potentially confounding data. Additionally, the sample size of the pediatric cohort, as well as the differences in time of observation between the pediatric and adult cohorts precluded further subgroup analyses.
In conclusion, many children with PNH have smaller GPI-deficient clones than adults, as well as a higher incidence of bone marrow failure, less severe hemolysis, and probably a lower incidence of thrombosis. Future studies should examine longitudinal outcomes in pediatric and adult PNH patients, as well as the impact of conventional thrombosis prophylaxis and eculizumab or other anticomplement therapies.
Supplementary Material
Acknowledgments
Logistical and financial support for the International PNH Registry was provided by Alexion Pharmaceuticals. The International PNH Registry is overseen by an independent committee of international medical PNH experts: Wendell Rosse, MD (Duke University Medical Center, Durham, NC, USA); Robert Brodsky, MD (Johns Hopkins School of Medicine, MD, USA); Yuzuru Kanakura, MD (Osaka University School of Medicine, Japan); Peter Hillmen, MD, PhD (St. James’s University Hospital, United Kingdom); Jaroslaw Maciejewski, MD, PhD (Cleveland Clinic, OH, USA); Petra Muus, MD (Radboudumc, Nijmegen, the Netherlands); Hubert Schrezenmeier, MD (University of Ulm, Germany); Gérard Socié, MD (Paris Diderot University, France); Jeff Szer, MD (Royal Melbourne Hospital, Australia); Álvaro Urbano-Ispizua, MD (University of Barcelona, Spain). The authors would like to thank Dr. Tmirah Haselkorn of EpiMetrix, Inc. for editorial support.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Ware RE, Hall SE, Rosse WF. Paroxysmal nocturnal hemoglobinuria with onset in childhood and adolescence. N Engl J Med. 1991;325(14):991–996. [DOI] [PubMed] [Google Scholar]
- 2.Hillmen P, Lewis SM, Bessler M, et al. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253–1258. [DOI] [PubMed] [Google Scholar]
- 3.Socie G, Mary JY, de Gramont A, et al. Paroxysmal nocturnal hemoglobinuria: long-term follow-up and prognostic factors. French Society of Hematology. Lancet. 1996;348(9027):573–577. [DOI] [PubMed] [Google Scholar]
- 4.Gulbis B, Eleftheriou A, Angastiniotis M, et al. Epidemiology of rare anaemias in Europe. Adv Exp Med Biol. 2010;686:375–396. [DOI] [PubMed] [Google Scholar]
- 5.van den Heuvel-Eibrink MM, Bredius RG, te Winkel ML, et al. Childhood paroxysmal nocturnal hemoglobinuria (PNH), a report of 11 cases in the Netherlands. Br J Haematol. 2005;128(4):571–577. [DOI] [PubMed] [Google Scholar]
- 6.Naithani R, Mahapatra M, Dutta P, et al. Paroxysmal nocturnal hemoglobinuria in childhood and adolescence: a retrospective analysis of 18 cases. Ind J Pediatr. 2008;75(6):575–578. [DOI] [PubMed] [Google Scholar]
- 7.Curran KJ, Kernan NA, Prockop SE, et al. Paroxysmal nocturnal hemoglobinuria in pediatric patients. Pediatr Blood Cancer. 2012; 59(3):525–529. [DOI] [PubMed] [Google Scholar]
- 8.Schrezenmeier H, Muus P, Socie G, et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica. 2014;99(5):922–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camitta BM, Storb R, Thomas ED. Aplastic anemia (first of two parts): pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306(11):645–652. [DOI] [PubMed] [Google Scholar]
- 10.Camitta BM, Storb R, Thomas ED. Aplastic anemia (second of two parts): pathogenesis, diagnosis, treatment, and prognosis. N Engl J Med. 1982;306(12):712–718. [DOI] [PubMed] [Google Scholar]
- 11.Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheinberg P, Marte M, Nunez O, et al. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. 2010;95(7): 1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2003;102(10):3587–3591. [DOI] [PubMed] [Google Scholar]
- 14.Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(25):4985–4996. [DOI] [PubMed] [Google Scholar]
- 15.Ge M, Shi J, Li X, et al. Clinical features and survival of Asian pediatric patients with paroxysmal nocturnal hemoglobinuria: results from a single center in China. Acta Haematol. 2015;134(1):1–6. [DOI] [PubMed] [Google Scholar]
- 16.Sreedharanunni S, Sachdeva MU, Bose P, et al. Frequency of paroxysmal nocturnal hemoglobinuria clones by multiparametric flow cytometry in pediatric aplastic anemia patients of Indian ethnic origin. Pediatr Blood Cancer. 2016;63(1):93–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.