

Abstract
Deletions and mutations affecting lymphoid transcription factor IKZF1 (IKAROS) are associated with an increased relapse risk and poor outcome in B-cell precursor acute lymphoblastic leukemia. However, additional genetic events may either enhance or negate the effects of IKZF1 deletions on prognosis. In a large discovery cohort of 533 childhood B-cell precursor acute lymphoblastic leukemia patients, we observed that single-copy losses of BTG1 were significantly enriched in IKZF1-deleted B-cell precursor acute lymphoblastic leukemia (P=0.007). While BTG1 deletions alone had no impact on prognosis, the combined presence of BTG1 and IKZF1 deletions was associated with a significantly lower 5-year event-free survival (P=0.0003) and a higher 5-year cumulative incidence of relapse (P=0.005), when compared with IKZF1-deleted cases without BTG1 aberrations. In contrast, other copy number losses commonly observed in B-cell precursor acute lymphoblastic leukemia, such as CDKN2A/B, PAX5, EBF1 or RB1, did not affect the outcome of IKZF1-deleted acute lymphoblastic leukemia patients. To establish whether the combined loss of IKZF1 and BTG1 function cooperate in leukemogenesis, Btg1-deficient mice were crossed onto an Ikzf1 heterozygous background. We observed that loss of Btg1 increased the tumor incidence of Ikzf1+/− mice in a dose-dependent manner. Moreover, murine B cells deficient for Btg1 and Ikzf1+/− displayed increased resistance to glucocorticoids, but not to other chemotherapeutic drugs. Together, our results identify BTG1 as a tumor suppressor in leukemia that, when deleted, strongly enhances the risk of relapse in IKZF1-deleted B-cell precursor acute lymphoblastic leukemia, and augments the glucocorticoid resistance phenotype mediated by the loss of IKZF1 function.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common form of cancer in children and is characterized by recurrent genetic aberrations and chromosomal abnormalities, which represent distinct genetic subtypes that are used for risk stratification.1 In the past, we and others have demonstrated that genomic alterations affecting the lymphoid transcription factor gene IKZF1 represent a strong prognostic factor associated with relapse in childhood B-cell precursor acute lymphoblastic leukemia (BCP-ALL).2–5 Recently, we established that loss of IKZF1 affects glucocorticoid (GC)-mediated gene regulation and confers GC resistance in BCP-ALL.6 Deletion of IKZF1 is a hallmark of BCR-ABL1-positive BCP-ALL,7 and within this high-risk cytogenetic subtype, IKZF1 loss is associated with an even worse outcome.8 IKZF1 gene lesions are also frequently present in high-risk leukemias with a Philadelphia- or BCR-ABL1-like expression signature,2,5,9 which often carry activated tyrosine kinases (i.e., ABL1, PDGFRB, JAK2, FLT3) or mutations targeting the RAS pathway.10–12 Mouse studies have confirmed that two of these genomic alterations, namely BCR-ABL1 and mutant RAS, cooperate with loss of IKZF1 during leukemic transformation.13,14 On the other hand, ERG gene deletions constitute a specific subtype of BCP-ALL with favorable outcome, despite the frequent co-occurrence of IKZF1 deletions.15,16 Thus, specific genetic interactions may modulate the tumor suppressive functions of IKZF1 during leukemia development at initial diagnosis and at relapse after chemotherapy treatment.
Besides IKZF1, many other recurrent genetic aberrations have been observed in BCP-ALL, which include deletions affecting BTG1, CDKN2A/B, EBF1, ETV6, PAX5, RAG1, RB1 and TCF3.17–19 For some of these co-occurring genetic lesions, synergistic effects have been reported on leukemia development. For example, loss of Rag1 was shown to accelerate the onset of B-cell lymphoblastic leukemia in Cdkn2a/p19Arf-deficient mice,20 while increasing the incidence of T-cell lymphomas in Tcf3−/− mice.21 Furthermore, combined heterozygosity for Ebf1 and Pax5 results in a strong increase in the frequency of pro-B-cell leukemia in mice.22
Herein we describe the impact of deletions affecting the transcriptional coregulator B-cell translocation gene 1 (BTG1), in co-occurrence with IKZF1 loss on both leukemogenesis as well as outcome. We demonstrate that BTG1 deletions are enriched in IKZF1-deleted pediatric BCP-ALL cases, and correlate with increased relapse risk in this patient group. Using mouse knockout models, we further demonstrate that loss of Btg1 cooperates with Ikzf1+/− in the onset of ALL. Finally, our data indicate that both BTG1 and IKZF1 are important determinants of the GC therapy response, and that combined loss of these tumor suppressors enhances GC resistance.
Methods
Clinical samples
The discovery cohort comprised of 533 pediatric patients with newly diagnosed BCP-ALL from three consecutive Dutch Childhood Oncology Group trials (DCOG ALL-8, ALL-9 and ALL-10) and two German Cooperative ALL trials (COALL 06-97 and 07-03). The validation cohort consisted of 515 pediatric patients enrolled in the Australian and New Zealand Children’s Haematology and Oncology Group (ANZCHOG) ALL8 protocol. In accordance with the Declaration of Helsinki, written informed consent was obtained from parents or legal guardians, and institutional review boards approved the use of excess diagnostic material for research purposes. Details on the patient cohorts, treatment regimens and outcomes, were described previously.5,23,24
Statistics patient data
Cumulative incidence of relapse (CIR) was estimated using a competing risks model, equality of CIRs was tested with Gray’s test. Relapse and non-response to induction chemotherapy were considered as events, with death and secondary malignancy as competing events. Event-free survival (EFS) was calculated with non-response, relapse, secondary malignancy and death considered as events. EFS probabilities were estimated using the Kaplan-Meier method, and survival data between groups were compared using univariate and multivariate Cox regression analyses. The proportions of patients with IKZF1 and other B-cell development gene deletions as well as other categorical variables were compared using the Fisher’s exact test. All P-values are two-sided, and a significance level of 0.05 or less was considered to be significant. Analyses were performed in R 3.0.1 (2013-05-16), using the packages cmprsk version 2.2-7, mstate version 0.2.7, and survival version 2.37-4.25–27
Mice
Btg1 and Ikzf1 (IkNeo) knockout lines have been described previously,28,29 and were intercrossed on a C57Bl/6J background. Mice were maintained under specific pathogen-free conditions at our Central Animal Laboratory facility. Genotyping of the offspring was performed by polymerase chain reaction (PCR) (primer sequences are listed in the Online Supplementary Table S1). Animal experiments were approved by the Animal Experimental Committee of the Radboud university medical center and were performed in accordance with institutional and national guidelines.
Functional characterization of murine lymphocytes
Detailed information on functional characterization of normal and leukemic lymphocytes by flow cytometry, immunohistochemistry, immunoglobulin (IG)/T-cell receptor (TR) PCR and cell viability assays can be found in the Online Supplementary Methods section. To analyze the glucocorticoid response in B lymphocytes, mononuclear splenocytes were isolated from wild-type and the different knockout mice, and stimulated in vitro with 5 mg/mL lipopolysaccharide (LPS) for 48 hours. The obtained activated B lymphocytes (≥ 80% B220+) were isolated by ficoll gradient and cultured for another 48 hours in the absence or presence of the synthetic glucocorticoids prednisolone or dexamethasone. Thereafter, relative cell viability was assessed by MTS assay and AnnexinV/7-AAD staining.
Results
BTG1 deletions are enriched in IKZF1-deleted pediatric BCP-ALL
Gene deletion of the tumor suppressor IKZF1, creating either dominant-negative IKZF1 isoforms or haploinsufficiency, is an important predictor of poor outcome in BCP-ALL,2–5 but to what extent other additional common single gene deletions, such as CDKN2A/B, PAX5, BTG1, ETV6, EBF1 or RB1 impact the prognostic value of IKZF1 has not been clearly established. To address this question, we studied a previously described childhood BCP-ALL cohort of 533 cases enrolled in consecutive DCOG and COALL trials.5 The representation of the different BCP-ALL subtypes was similar between the DCOG and COALL cohorts, and comparable to that described in the literature (Online Supplementary Table S2). We identified 105 BCP-ALL patient samples containing an IKZF1 deletion. Within the IKZF1-deleted group, we observed a significant enrichment for BTG1 deletions (P=0.007), where 17 of the 105 IKZF1-deleted cases (16%) harbored BTG1 deletions as compared to 31 of the 428 IKZF1 wild-type cases (7%) (Table 1). These focal BTG1 deletions mainly covered the second exon of BTG1 and downstream adjacent sequences, as described previously.30 Similarly, deletions affecting PAX5 (P<0.0001), CDKN2A/B (P=0.0003) and RB1 (P=0.026) were present at higher frequencies in IKZF1-deleted cases, whilst this was not observed for EBF1 or ETV6 deletions (Table 1). To the contrary, in the BTG1-deleted group we observed significant enrichment for IKZF1 (P=0.007), EBF1 (P=0.0011), RB1 (P=0.042), and ETV6 (P=0.0046) deletions (Online Supplementary Table S3), which is in agreement with our previous findings.30 Previously, it has been shown that both IKZF1 and BTG1 deletions are strongly enriched in the cytogenetic BCR-ABL1 subtype.7,31 Consistent with this notion, we observed that of 24 BCR-ABL1-positive cases, 7 (29%) harbored deletions in both IKZF1 and BTG1 (Table 2), which represents 47% of IKZF1-deleted and 100% of the BTG1-deleted cases found in the BCR-ABL1-positive group (Table 2).
Table 1.
Co-occurrence of IKZF1 deletion with other common gene deletions in BCP-ALL.
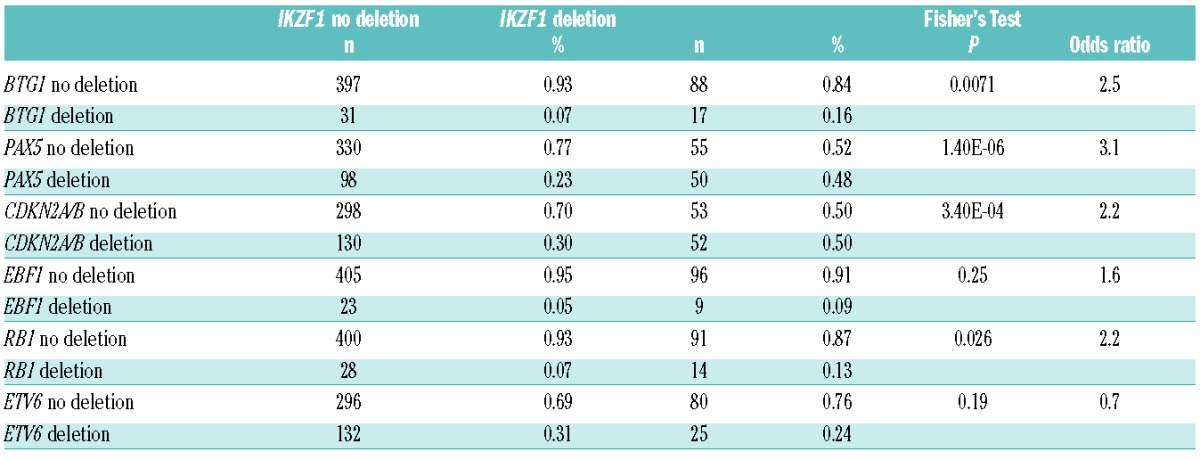
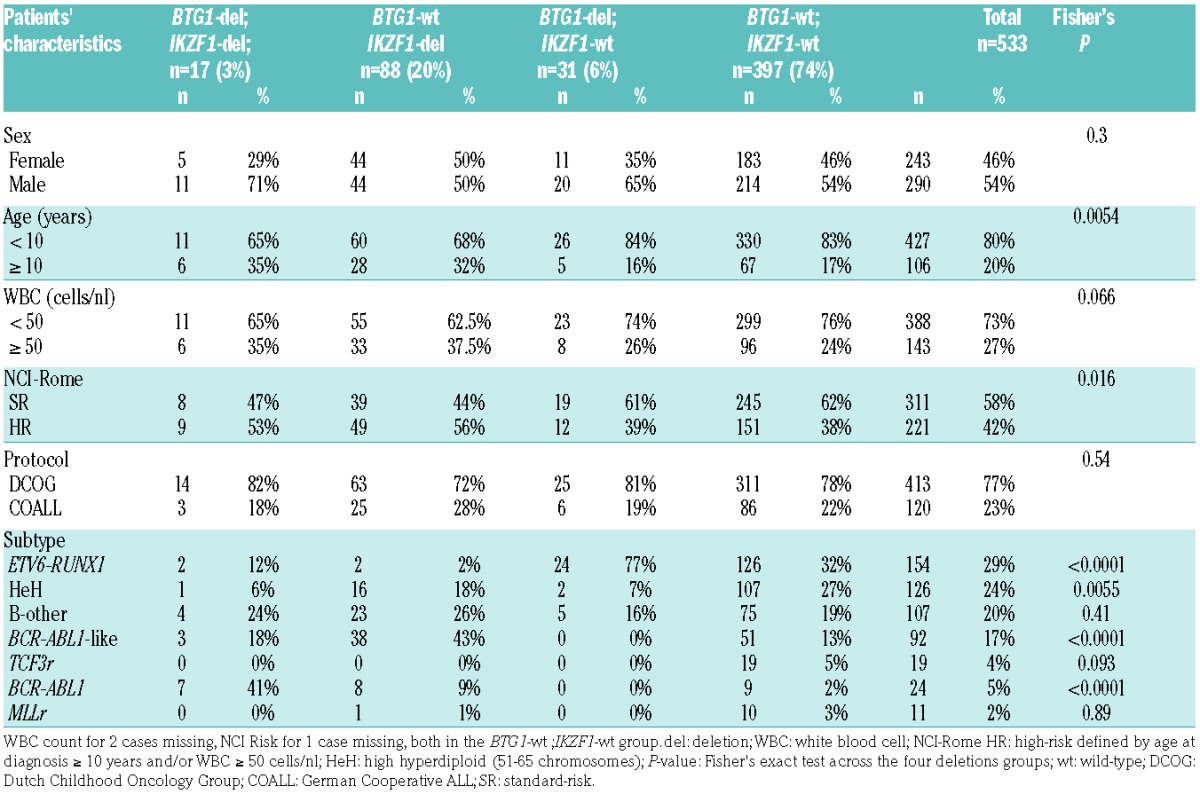
Table 2.
Pediatric BCP-ALL patient characteristics in the BTG1 and IKZF1 sole and BTG1;IKZF1 double-deleted groups.
Combined deletions of BTG1 and IKZF1 predict inferior outcome in BCP-ALL
We next compared clinical characteristics of BTG1;IKZF1 double-deleted cases, sole deletion of IKZF1 and sole deletion of BTG1 cases, and cases without IKZF1 or BTG1 deletion in our complete childhood BCP-ALL cohort. The characteristics of these four groups were similar to the total cohort with respect to gender and treatment protocol (Table 2). The double-deleted and sole deletion of IKZF1 groups contained more patients over 10 years of age and increased white blood cell counts, and hence more National Cancer Institute (NCI)-Rome criteria high-risk cases32 (Table 2). We compared the EFS and CIR between cases with both IKZF1 and BTG1 deletions (BTG1-del;IKZF1-del, n=17) with the cases of sole deletion of IKZF1 (BTG1-wt;IKZF1-del, n=88) (Figure 1A,B). The 5-year CIR in IKZF1- plus BTG1-deleted cases was 53% ± 13% compared with 28% ± 5% in the cases of sole deletion of IKZF1 (P=0.005; Table 3A). Similarly, the 5-year EFS was lower in double-deleted cases compared with the cases of sole deletion of IKZF1 (HR 3.5, P=0.0003; Table 3A). In contrast, the cases of sole deletion of BTG1 (BTG1-del;IKZF1-wt, n=31) showed a similar outcome (5-year CIR:10% ± 6%) to the reference cases without IKZF1 or BTG1 deletions (n=397; 5-year CIR:13% ± 2%). The synergistic effect of loss of BTG1 and IKZF1 on outcome remained after correction for subtype in the Cox model (Table 3A), and after leaving out the BCR-ABL1-positive cases (Figure 1C,D; Table 3B). For the BCR-ABL1-positive cases, BTG1 deletions did not further impact the poor treatment outcome as observed for the cases of sole deletion of IKZF1 (Figure 1E,F; Table 3C).
Figure 1.
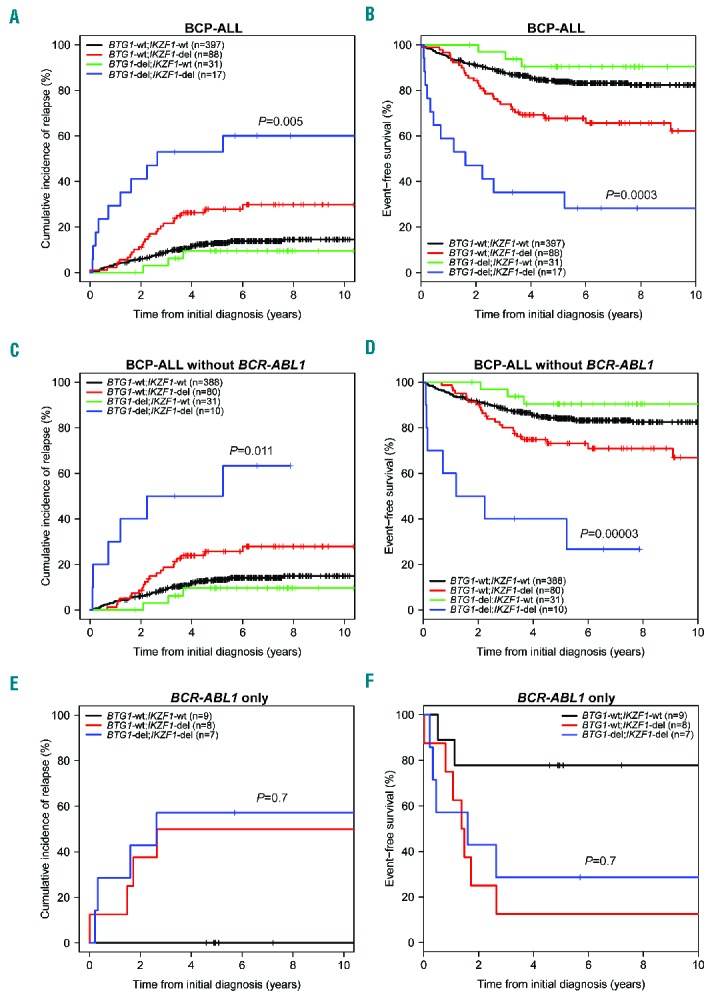
Cumulative incidence of relapse (CIR) and event-free survival (EFS) curves for pediatric BCP-ALL cases with or without IKZF1 and BTG1 deletions. (A) CIR and (B) EFS curves for total BCP-ALL cohort (n=533), IKZF1 plus BTG1 deletion, (C) CIR and (D) EFS curves for BCP-ALL without BCR-ABL1-positive cases (n=509), IKZF1 plus BTG1 deletion, (E) CIR (F) EFS curves for BCR-ABL1-positive cases (n=24), IKZF1 plus BTG1 deletion. Colors: black, IKZF1 and BTG1 wild-type; green, IKZF1 wild-type, BTG1-deleted; red, IKZF1-deleted, BTG1 wild-type; blue, both BTG1- and IKZF1-deleted. For CIR graphs (A,C,E) the Gray P-value and for the EFS graphs (B,D,F) the Cox P-value is indicated comparing BTG1-del;IKZF1-del with BTG1-wt;IKZF1-del. wt: wild-type; del: deletion; n: total number; BCP-ALL: B-cell precursor acute lymphoblastic leukemia.
Table 3.
CIR and EFS analysis of BTG1;IKZF1 double-deleted pediatric BCP-ALL cases compared with cases of sole deleted IKZF1.
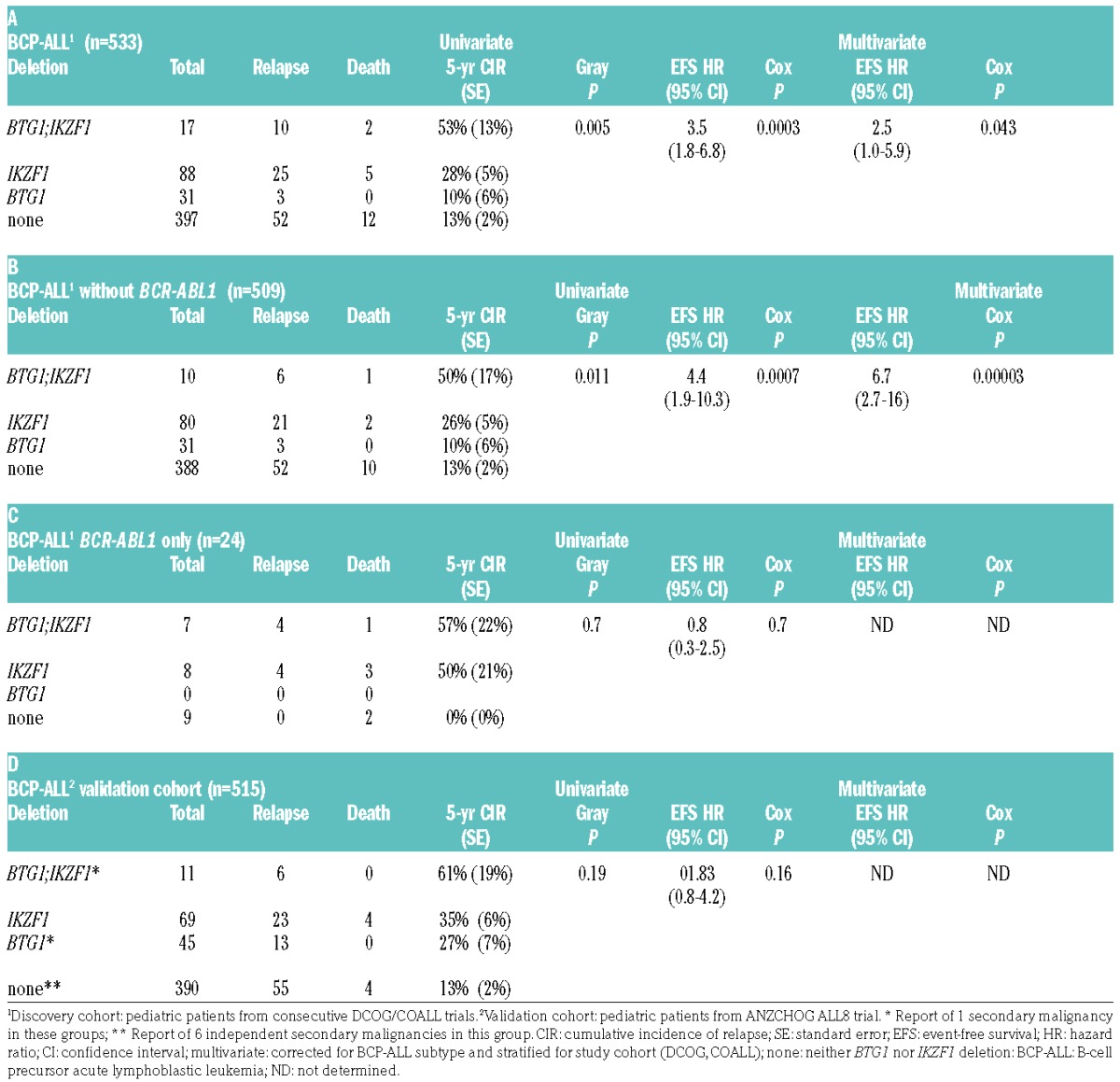
As deletions of PAX5, CDKN2A/B and RB1 were similarly enriched in IKZF1-deleted BCP-ALL, we examined the impact of these deletions on the outcome of IKZF1-deleted cases. In contrast to BTG1;IKZF1 double-deleted patients, the outcome of patients with co-occurring PAX5, CDKN2A/B or RB1 deletions did not differ from cases of sole deletion of IKZF1 (Figure 2; Online Supplementary Table S4). Similarly, co-occurrences of IKZF1 deletions with either EBF1 or ETV6 deletions did not affect outcome compared with the sole deletion of IKZF1. To validate our findings, we analyzed the Australian and New Zealand ANZCHOG ALL8 cohort (n=515)23,24 to assess the prognostic value of BTG1;IKZF1 double-deletions. In this cohort, 6 out of 11 BTG1-del;IKZF1-del patients developed a relapse (Online Supplementary Figure S1). The 5-year CIR in the BTG1;IKZF1 double-deleted group was 61% ± 19% versus 35% ± 6% in the group with the sole deletion of IKZF1 (P=0.19; Table 3D). Hence, the same trend was observed in this independent validation cohort, albeit statistically non-significant. Together, these data indicate that BTG1 deletions in an unselected leukemia population have no prognostic value, but BTG1 copy number losses specifically exacerbate the effects of IKZF1 deletion on inferior outcome in BCP-ALL.
Figure 2.
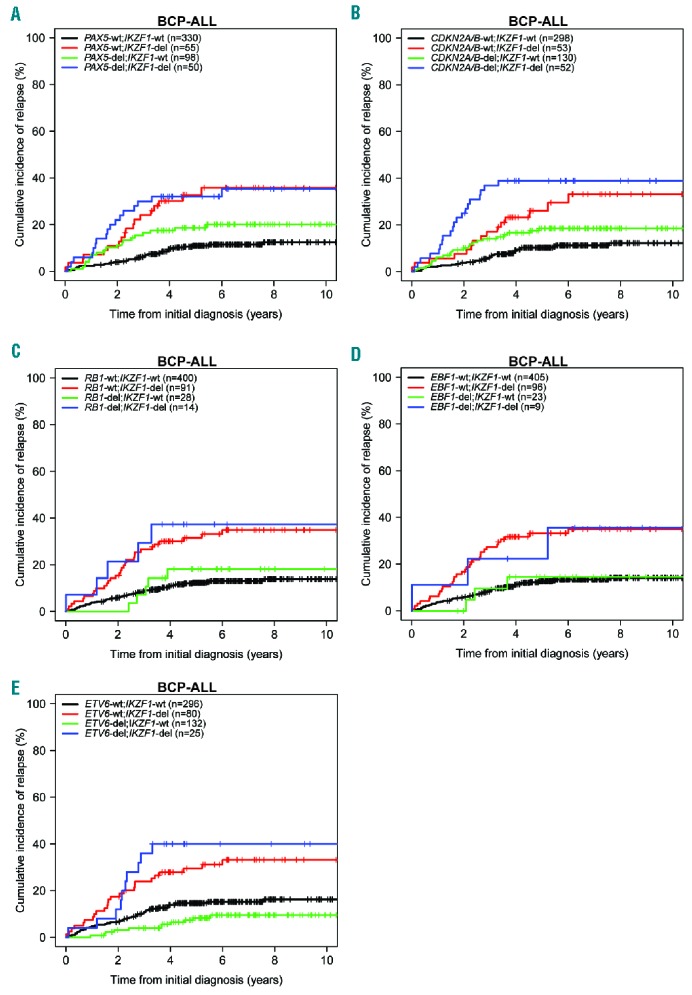
Cumulative incidence of relapse (CIR) curves for pediatric BCP-ALL cases with or without IKZF1 deletions in combination with other single common gene deletions. (A) IKZF1 plus PAX5, (B) IKZF1 plus CDKN2A/B, (C) IKZF1 plus RB1, (D) IKZF1 plus EBF1, (E) IKZF1 plus ETV6 (n=533). Colors: black, none of the indicated deletions; green, IKZF1 wild-type, indicated gene deleted; red, IKZF1-deleted, indicated gene wild-type; blue, both IKZF1 and indicated gene deleted. wt: wild-type; del: deletion; n: total number; BCP-ALL: B-cell precursor acute lymphoblastic leukemia.
Leukemia predisposition in Btg1 knockout mice
Although deletions affecting the BTG1 gene are a frequent event in BCP-ALL, a direct role for BTG1 in leukemia development has not been reported. Therefore, we first examined the tumor suppressive function of BTG1 using a constitutive Btg1 knockout line harboring a Neo-cassette in the first coding exon.28 We previously reported that these Btg1 knockout mice display defective B-cell development with a 25% reduction in the amount of progenitor B cells within the bone marrow compartment, mainly affecting the pre-B and immature B-cell stage.33 Furthermore, Btg1 is required for optimal proliferative expansion of early progenitor B cells in methylcellulose in response to interleukin-7. At the same time, there was no obvious defect in the development of myeloid and T-lymphoid cells in these Btg1-deficient animals.33 In the study herein, mice that carried either one or two copies of the Btg1 knockout allele were followed over a period of 18 months, along with control littermates. About 6% (n=3/49) of the wild-type C57BL6/J mice developed B-cell lymphomas between the age of 14 and 18 months (Table 4), which is consistent with previous observations.34 Within the same time period 6% of the Btg1+/− (n=2/34) and 18% of the Btg1−/− (n=6/33) mice developed T-cell leukemia exclusively (Table 4), characterized by enlarged primary lymphoid organs, such as the spleen and lymph nodes, and focal infiltration of leukemic T cells into peripheral organs, such as the lungs and liver. These Btg1-deficient T-cell leukemias expressed the T-cell surface marker CD3, and displayed clonal T-cell receptor (TR) rearrangements (Figure 3B). In addition, these CD3+ T-cell leukemias not only showed increased expression of the T-cell activation marker CD44, but also large numbers of B220+ cells within the infiltrated areas of tissues, such as the liver or lungs, and affected lymph nodes (Online Supplementary Figure S2). There was no evidence for clonal immunoglobulin gene rearrangements in these Btg1−/−T-cell leukemias (Online Supplementary Figure S2), suggesting the presence of a substantial number of non-malignant B lymphocytes in proximity to these leukemic T cells. These data show that, although somatic BTG1 deletions predominantly occur in BCP-ALL, Btg1-deficiency in the mouse germline predisposes exclusively to T-cell malignancies. This predilection for T-lineage leukemias is also observed in other knockout mouse models targeting genes commonly deleted in BCP-ALL, such as Ikzf1 mutant mice.35,36
Table 4.
Characteristics of lymphoid tumors derived from single and intercrossed Btg1 and Ikzf1 knockout lines.
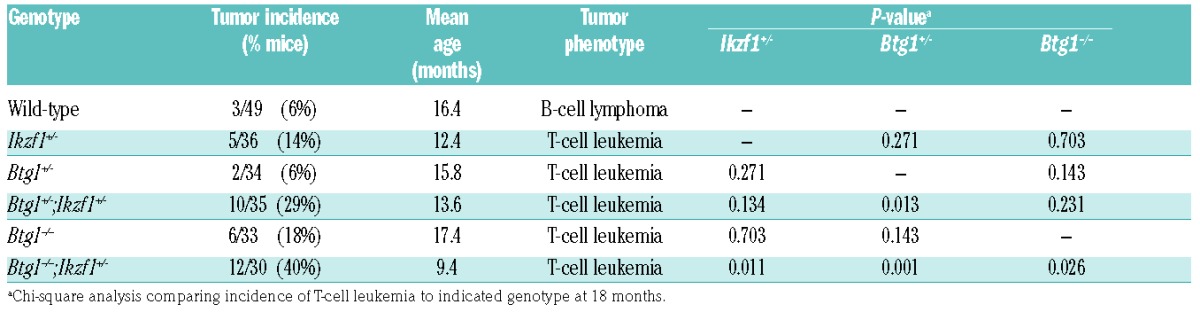
Figure 3.

Leukemia incidence and phenotype of Btg1 knockout mice intercrossed with haplodeficient Ikzf1 animals. (A) Kaplan-Meier survival curve indicates the leukemia event-free survival in wild-type, Ikzf1+/−, Btg1+/−, Btg1+/−;Ikzf1+/−, Btg1−/−, and Btg1−/−;Ikzf1+/− mice over a time period of 17 months. Leukemia incidence is significantly increased in Btg1−/−;Ikzf1+/− mice as compared to Ikzf1+/− mice (P=0.011). (B) Peripheral blood smear stained with Giemsa and immunohistochemistry for CD3 on the lung and liver tissues of diseased Ikzf1+/−, Btg1−/−, Btg1+/−;Ikzf1+/−, and Btg1−/−;Ikzf1+/− mice. (C) Quantification of blast counts in peripheral blood smear stained with Giemsa of diseased wild-type, Ikzf1+/−, Btg1−/−, Btg1+/−;Ikzf1+/− and Btg1−/−;Ikzf1+/− mice (n=3). The percentage of leukemic blasts is indicated. ***P<0.001. (D) Quantification of T-cell (CD3) infiltration into the liver and lungs of diseased wild-type, Ikzf1+/−, Btg1−/−, Btg1+/−;Ikzf1+/− and Btg1−/−;Ikzf1+/− mice (n=3) using FIJI software. Data are represented as the positively stained area divided by the total area measured, with standard errors of the mean P-values (two-sided t-test). *P<0.05, **P<0.01, and ***P<0.001. (E) T-cell receptor beta 2 (Trb2) gene rearrangement analysis by PCR on control tissue, B-cell lymphoma derived from wild-type mice (#3863), and T-cell leukemias derived from Ikzf1+/− (#13135, #350), Btg1−/− (#160), Btg1+/−;Ikzf1+/− (#5103 and #5102) and Btg1−/−;Ikzf1+/− mice (#9073, #7169, #7173). (F) Flow cytometry analyzing CD4 and CD8 expression on T-cell leukemia samples of two Ikzf1+/−, Btg1−/−, Btg1+/−;Ikzf1+/− and Btg1−/−;Ikzf1+/− mice. WT: wild-type; M: 1 kb DNA ladder marker; Ctrl: control DNA.
Loss of Btg1 increases leukemia incidence in Ikzf1+/− mice
To investigate cooperation between BTG1 and IKZF1 during leukemogenesis, we intercrossed Btg1-deficient mice with haplodeficient Ikzf1 mice using the IkNeo mouse line,29 which harbors a Neo-floxed knock-in allele combined with a Pax5-IRES-GFP complementary DNA (cDNA) at the first coding exon of Ikzf1, thereby creating an Ikzf1 null allele.29 These mice are only viable as a heterozygous knockout line (Ikzf1+/−). First, we analyzed the phenotype of young animals (age 6–12 weeks) to assess the effect of the Ikzf1+/− allele on Btg1-deficiency in B- and T-lymphoid development. Ikzf1+/−mice, like Btg1−/− mice, displayed a moderate reduction in the fraction of B220+ cells in bone marrow (BM) and the spleen (Online Supplementary Figure S3). This correlated with a partial block at the pre-pro-B-cell stage (Hardy fraction A) and the pre-B-cell stage (Hardy fraction D) in Ikzf1+/− mice (Online Supplementary Figure S3). Btg1−/−;Ikzf1+/− mice showed an even stronger reduction in B220+ cells, with additive effects at both B220+CD43+ and B220+CD43− differentiation stages in BM (Hardy fractions A to E) (Online Supplementary Figure S3). In contrast, Btg1−/−;Ikzf1+/− mice, similar to Btg1−/− and Ikzf1+/− single knockout animals, showed no major defects in postnatal thymic T-cell development (Online Supplementary Figure S4).
Next, we followed Ikzf1+/−, Btg1−/−;Ikzf1+/− and Btg1−/−;Ikzf1+/− mice for a period of 18 months. In our cohort, 14% (n=5/36) of the Ikzf1+/− mice developed T-cell leukemia between 3 and 18 months of age, with a median age of 12 months (Figure 3A; Table 4). Similar to Btg1-deficient leukemia, the Ikzf1+/− T-cell leukemias showed infiltration of CD3+ leukemic blasts into distant organs and clonal TR rearrangements (Figure 3). Interestingly, we observed an increased leukemia incidence upon combined loss of Btg1 and Ikzf1 (Figure 3A), which is consistent with our hypothesis that BTG1 deletions cooperate with IKZF1 aberration to induce human BCP-ALL. We found that 26% of the Btg1+/−;Ikzf1+/− mice (n=10/35) and 40% of the Btg1−/−;Ikzf1+/− (n=12/30) animals developed T-cell leukemia, while leukemias in Btg1−/−;Ikzf1+/− appeared with a slightly shorter latency (9.4 months) relative to Ikzf1+/− mice (12.4 months) (P=0.011) (Table 4). Tumors in the Btg1−/−;Ikzf1+/−compound mice were characterized by significantly higher leukocyte counts in peripheral blood compared to single knockout animals (Figure 3B,C), and strong infiltration of leukemic cells into the liver and lungs (Figure 3B,D), as well as clonal TR rearrangements (Figure 3E). Flow cytometric analysis of the different T-cell leukemias revealed that most of the Btg1−/−;Ikzf1+/− leukemias were CD4+CD8+ double positive T cells with ongoing differentiation towards CD4 or CD8 single positive stage (Figure 3F, Online Supplementary Table S5). Moreover, isolated leukemic T cells, derived from all the different genetic backgrounds included in our studies (Ikzf1+/−, Btg1−/− and Btg1−/−;Ikzf1+/−), could be serially transplanted into syngeneic C57BL6/J mice giving rise to similar (oligo)clonal T-cell leukemias (Online Supplementary Figure S5). Taken together, our data demonstrate that loss of Btg1 cooperates with haplodeficiency for Ikzf1 during mouse leukemia development in a dose-dependent manner.
BTG1 modifies glucocorticoid resistance mediated by loss of IKZF1
While these experiments confirm the genetic interaction between BTG1 deletions and IKZF1 aberrations during leukemogenesis, they do not explain the poor outcome observed in patients showing a combined loss of BTG1 and IKZF1. Recently, we established that inferior outcome related to IKZF1 deletions in BCP-ALL is correlated with an attenuated in vivo day 8 prednisolone response and increased GC resistance in IKZF1-deleted primary leukemic cells, as determined by in vitro MTT assays.6 These results could be recapitulated using primary splenic B cells isolated from Ikzf1+/− mice, which revealed that non-leukemic Ikzf1+/− B cells are also less sensitive towards GC-induced apoptosis.6 Based on our previous findings that BTG1 regulates glucocorticoid receptor activation,37 we investigated whether loss of BTG1 would impact the GC response in primary murine B cells. To this end, B cells isolated from WT, Btg1−/−, Ikzf1−/− and Btg1−/−;Ikzf1+/− mice, and obtained after lipopolysaccharide activation, were stimulated for 48 hours with increasing concentrations of prednisolone or dexamethasone and subjected to MTS assays to assess relative cell survival. While Btg1-deficiency alone had no effect on GC-induced apoptosis, Ikzf1-haplodeficient B cells showed enhanced cell survival as compared to WT (Figure 4A), similar to our previous findings.6 Importantly, Btg1−/−;Ikzf1+/− B cells showed an even stronger resistance to GC-induced apoptosis when compared to Ikzf1+/− B cells (P<0.001). These findings were confirmed by AnnexinV staining, demonstrating a significantly smaller apoptotic fraction in Btg1−/−;Ikzf1+/− B cells relative to Ikzf1+/− B cells (P<0.001) (Figure 4B). Analyses of primary Btg1−/−, Ikzf1+/− and Btg1−/−;Ikzf1+/− thymocytes revealed no differential sensitivity to GC-induced apoptosis as compared to WT (Online Supplementary Figure S6A). Next, we assessed whether loss of Btg1 and Ikzf1 would promote resistance in B cells to other chemotherapeutic drugs commonly used in the treatment of BCP-ALL patients, including 6-mercaptopurine, doxorubicin, vincristine and asparaginase. However, Btg1−/−;Ikzf1+/− and Btg1−/−;Ikzf1+/− B cells showed similar cell survival in comparison to control cells (Online Supplementary Figure S6B).
Figure 4.
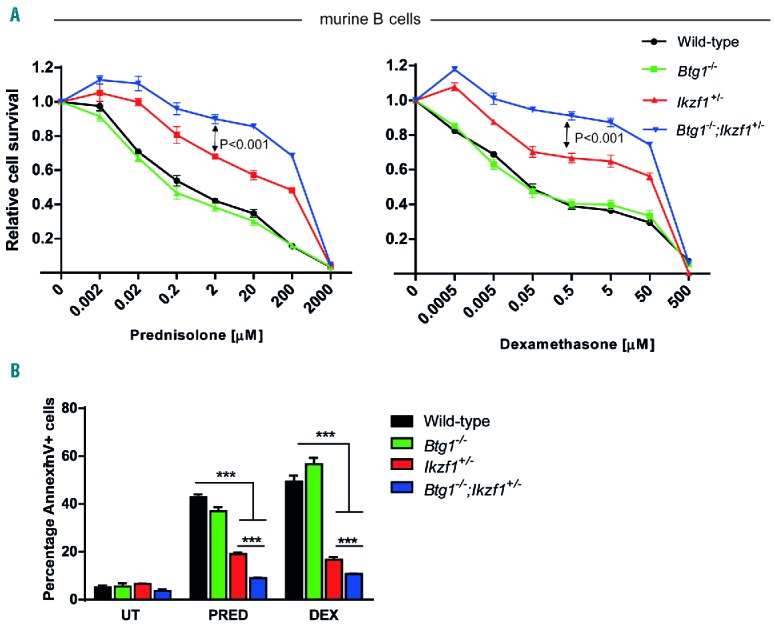
Glucocorticoid resistance of B cells isolated from Btg1 knockout mice intercrossed with haplodeficient Ikzf1 animals. (A) Splenic B cells isolated from wild-type, Ikzf1+/−, Btg1−/− and Btg1−/−;Ikzf1+/− mice were activated by LPS for 48 hours and subsequently treated in vitro for 48 hours with increasing concentrations of prednisolone (PRED, left panel) or dexamethasone (DEX, right panel) and analyzed by MTS assay (n=6). All values were normalized to untreated (UT) B cells. Error bars represent ± standard error of the mean (SEM). P-values were calculated based on the differences of the best-fit curve using two-way ANOVA. (B) AnnexinV/7-AAD staining of WT, Ikzf1+/−, Btg1−/− and Btg1−/−;Ikzf1+/− B-lymphocytes after 2 mM prednisolone or 5 mM dexamethasone treatment for 48 hours (n=4). The fraction of AnnexinV-positive cells was determined. Data represent means, and error bars represent SEM. P-values (two-sided t-test) are indicated. ***P<0.001.
Together, these data argue that loss of tumor suppressor BTG1 enhances GC resistance in the context of IKZF1 deletions, which may explain the inferior treatment outcome observed in patients showing combined loss of BTG1 and IKZF1.
Discussion
BCP-ALL is a heterogeneous disease characterized by recurrent deletions enriched in specific genetic subtypes.1 For instance, it is known that deletions affecting the transcriptional co-regulator BTG1 are unevenly distributed among cytogenetic subgroups, as we and others have shown that BTG1 deletions are strongly enriched in ETV6-RUNX1-positive leukemia as well as BCR-ABL1-positive ALL.30,31,38 The presence of these lesions in such distinct BCP-ALL subgroups may relate to the fact that deletions of IKZF1 and BTG1 appear to be the result of illegitimate RAG recombination,30 as is the case for several of the other commonly deleted genes, such as EBF1 and PAX5. In addition to these earlier observations, we find a specific co-occurrence of BTG1 and IKZF1 gene deletions across cytogenetic subtypes, suggesting that the combined loss of BTG1 and IKZF1 may actively contribute to leukemogenesis. Previous studies, mostly carried out in mouse models, revealed that Ikzf1 and Arf alterations in BCR-ABL1-positive ALL synergize to promote the development of leukemia by conferring stem cell-like properties.39 This leads us to hypothesize that the preponderance of BTG1;IKZF1 double-deletions in this particular subgroup may have similar consequences, although this remains to be assessed in well-established BCR-ABL1-positive mouse models. As BTG1 and IKZF1 deletions also (co)occur in lymphoid blast crises of chronic myeloid leukemia (CML),7,31 it will be interesting to study if and how the combined loss of BTG1 and IKZF1 drive the progression of this disease. Of all the common copy number losses analyzed in our study, including CDKN2A/B, PAX5, EBF1, and RB1, only loss of BTG1 appears to worsen the outcome of IKZF1-deleted ALL. Our data are consistent with the findings of Moorman et al. showing that specific combinations of different deletions impact the outcome in BCP-ALL.40
A number of different knockout mouse models have provided insight into the role of commonly deleted transcription factors in early hematopoiesis and spontaneous tumor incidence. It is evident that several of these transcriptional regulators play an important role as lymphoid specification factors and are essential for normal lymphopoiesis.41–44 However, in the mouse, loss of these early B-cell transcription factors affected in BCP-ALL, such as E2A45 and IKZF1,46 gives rise to T-cell malignancies. E2a-deficient tumors are characterized by a strong increase in c-Myc expression,45 an oncogene known to promote the development of T-cell lymphomas.47 Similarly, while IKZF1 deletions predominantly occur in human BCP-ALL, heterozygous Ikzf1 knockout and dominant-negative Ikzf1 mice develop T-cell malignancies exclusively, which has been attributed to activation of the Notch pathway.36,48 In our studies we observed a lower incidence of T-cell leukemia in mice as compared to what has been reported for some other genetically engineered Ikzf1 mouse models, where expression of dominant-negative isoforms or hypomorphic knockout alleles of Ikzf1 yielded a higher susceptibility to T-cell malignancies.35,36,48 Similar to mice heterozygous knockout for Ikzf1, Btg1 knockout mice develop T-cell leukemia, while BTG1 deletions are almost exclusively found in human BCP-ALL.30 However, consistent with our finding that monoallelic BTG1 deletions are enriched in human BCP-ALL cases with IKZF1 aberrations, Btg1+/−;Ikzf1+/− mice are more prone to develop leukemia relative to Btg1+/− single knockout mice (P=0.013). In addition, we observed a significant acceleration in the onset of disease in Btg1−/−;Ikzf1+/− mice as compared to Btg1−/− (P=0.026) or Ikzf1+/− mice (P=0.011), indicating that loss of these tumor suppressor genes cooperates during leukemogenesis. Genomic DNA analyses further indicate that both the wild-type Btg1 allele and Ikzf1 allele are maintained in the Btg1+/−;Ikzf1+/− leukemias (data not shown), arguing that Btg1 and Ikzf1 dosage contribute to leukemia development. These data confirm that BTG1 acts as a tumor suppressor gene that cooperates with IKZF1 loss during leukemia development.
Another important finding in this study is that BTG1 deletions define a high-risk group within the IKZF1-deleted subtype. Our finding that loss of BTG1 specifically enhances GC resistance mediated by Ikzf1-haplodeficiency implies that the prognostic value of BTG1 and IKZF1 deletions could be dependent on the upfront treatment and dose of synthetic glucocorticoids used. However, this remains to be established in future studies. The relation between BTG1 deletions and inferior outcome was recently confirmed with the analyses of a relapsed BCP-ALL cohort, showing that BTG1 and NR3C1 deletions were associated with a higher risk of disease progression.49 Collectively, our data demonstrate that BTG1 is a prognostic factor and regulator of the GC response, particularly in the context of IKZF1-deletions.
Supplementary Material
Acknowledgments
The authors would like to thank Marieke von Lindern and Meinrad Busslinger for providing the Btg1 and Ikzf1 knockout mice, respectively. We would like to thank Arian van der Veer for performing the MLPA experiments.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/3/541
Funding
This work was supported by grants of the Stichting Kinderen Kankervrij (KiKa 2009-55; KiKa 2010-77; KiKa 2014-132) and Stichting KOC Nijmegen. Work on the validation cohort was supported by NHMRC Australian APP1057746. MLdB has been supported by grants from the Dutch Cancer Society KWF (AMC 2008-4265), the Paediatric Oncology Foundation Rotterdam, the European Union’s Seventh Framework Program (FP7/2007-2013 ENCCA grant HEALTH-F2-2011-261474); RPK and PH have been supported by grants from the Dutch Cancer Society KWF (KUN 2009-4298); EW is supported by a fellowship from the Dutch Cancer Society KWF.
References
- 1.Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015;125(26): 3977–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuiper RP, Waanders E, van der Velden VH, et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24(7):1258–1264. [DOI] [PubMed] [Google Scholar]
- 4.Waanders E, van der Velden VH, van der Schoot CE, et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia. 2011;25(2):254–258. [DOI] [PubMed] [Google Scholar]
- 5.van der Veer A, Waanders E, Pieters R, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122(15):2622–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marke R, Havinga J, Cloos J, et al. Tumor suppressor IKZF1 mediates glucocorticoid resistance in B-cell precursor acute lymphoblastic leukemia. Leukemia. 2016;30(7): 1599–1603. [DOI] [PubMed] [Google Scholar]
- 7.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–114. [DOI] [PubMed] [Google Scholar]
- 8.van der Veer A, Zaliova M, Mottadelli F, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood. 2014;123(11):1691–1698. [DOI] [PubMed] [Google Scholar]
- 9.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009; 10(2):125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118(11): 3080–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts KG, Pei D, Campana D, et al. Outcomes of children with BCR-ABL1-like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol. 2014;32(27):3012–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dail M, Li Q, McDaniel A, et al. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci U S A. 2010;107(11):5106–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Virely C, Moulin S, Cobaleda C, et al. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia. Leukemia. 2010;24(6):1200–1204. [DOI] [PubMed] [Google Scholar]
- 15.Clappier E, Auclerc MF, Rapion J, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014;28(1):70–77. [DOI] [PubMed] [Google Scholar]
- 16.Zaliova M, Zimmermannova O, Dorge P, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. 2014;28(1): 182–185. [DOI] [PubMed] [Google Scholar]
- 17.Kuiper RP, Schoenmakers EF, van Reijmersdal SV, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21(6):1258–1266. [DOI] [PubMed] [Google Scholar]
- 18.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007. 12;446(7137):758–764. [DOI] [PubMed] [Google Scholar]
- 19.Tijchon E, Havinga J, van Leeuwen FN, Scheijen B. B-lineage transcription factors and cooperating gene lesions required for leukemia development. Leukemia. 2013;27(3):541–552. [DOI] [PubMed] [Google Scholar]
- 20.Hauer J, Mullighan C, Morillon E, et al. Loss of p19Arf in a Rag1(−/−) B-cell precursor population initiates acute B-lymphoblastic leukemia. Blood. 2011;118(3): 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel I, Murre C. Disruption of pre-TCR expression accelerates lymphomagenesis in E2A-deficient mice. Proc Natl Acad Sci U S A. 2002;99(17):11322–11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasad MA, Ungerback J, Ahsberg J, et al. Ebf1 heterozygosity results in increased DNA damage in pro-B cells and their synergistic transformation by Pax5 haploinsufficiency. Blood. 2015;125(26):4052–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshall GM, Dalla Pozza L, Sutton R, et al. High-risk childhood acute lymphoblastic leukemia in first remission treated with novel intensive chemotherapy and allogeneic transplantation. Leukemia. 2013;27(7):1497–1503. [DOI] [PubMed] [Google Scholar]
- 24.Sutton R, Shaw PJ, Venn NC, et al. Persistent MRD before and after allogeneic BMT predicts relapse in children with acute lymphoblastic leukaemia. Br J Haematol. 2015;168(3):395–404. [DOI] [PubMed] [Google Scholar]
- 25.Gray RJ. cmprsk: Subdistribution Analysis of Competing Risks. R package version 2.2-6 2013. [Available from: http://CRAN.R-project.org/package=cmprsk] Last accessed: 12th January 2017.
- 26.de Wreede LC, Fiocco M, Putter H. mstate: An R Package for the Analysis of Competing Risks and Multi-State Models. Stat Med. 2011;38(7):1–30. [Google Scholar]
- 27.Therneau T. A Package for Survival Analysis in S. R package version 2.36-12. 2012. [Google Scholar]
- 28.Farioli-Vecchioli S, Micheli L, Saraulli D, et al. Btg1 is Required to Maintain the Pool of Stem and Progenitor Cells of the Dentate Gyrus and Subventricular Zone. Front Neurosci. 2012;6:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Souabni A, Cobaleda C, Schebesta M, Busslinger M. Pax5 promotes B lymphopoiesis and blocks T cell development by repressing Notch1. Immunity. 2002; 17(6):781–793. [DOI] [PubMed] [Google Scholar]
- 30.Waanders E, Scheijen B, van der Meer LT, et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet. 2012;8(2):e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie J, Wang Q, Wang Q, et al. High frequency of BTG1 deletions in patients with BCR-ABL1-positive acute leukemia. Cancer Genet. 2014;207(5):226–230. [DOI] [PubMed] [Google Scholar]
- 32.Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14(1):18–24. [DOI] [PubMed] [Google Scholar]
- 33.Tijchon E, van Emst L, Yuniati L, et al. Tumor suppressors BTG1 and BTG2 regulate early mouse B-cell development. Haematologica. 2016;101(7):e272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haines DC, Chattopadhyay S, Ward JM. Pathology of aging B6;129 mice. Toxicol Pathol. 2001;29(6):653–61. [DOI] [PubMed] [Google Scholar]
- 35.Winandy S, Wu P, Georgopoulos K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell. 1995;83(2):289–99. [DOI] [PubMed] [Google Scholar]
- 36.Mantha S, Ward M, McCafferty J, et al. Activating Notch1 mutations are an early event in T-cell malignancy of Ikaros point mutant Plastic/+ mice. Leuk Res. 2007;31(3):321–327. [DOI] [PubMed] [Google Scholar]
- 37.van Galen JC, Kuiper RP, van Emst L, et al. BTG1 regulates glucocorticoid receptor autoinduction in acute lymphoblastic leukemia. Blood. 2010;115(23):4810–4819. [DOI] [PubMed] [Google Scholar]
- 38.Tsuzuki S, Karnan S, Horibe K, et al. Genetic abnormalities involved in t(12;21) TEL-AML1 acute lymphoblastic leukemia: analysis by means of array-based comparative genomic hybridization. Cancer Sci. 2007;98(5):698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Churchman ML, Low J, Qu C, et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell. 2015;28(3):343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moorman AV, Enshaei A, Schwab C, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124(9):1434–1444. [DOI] [PubMed] [Google Scholar]
- 41.Georgopoulos K, Bigby M, Wang JH, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79(1):143–156. [DOI] [PubMed] [Google Scholar]
- 42.Wang JH, Nichogiannopoulou A, Wu L, et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity. 1996;5(6):537–549. [DOI] [PubMed] [Google Scholar]
- 43.Engel I, Johns C, Bain G, Rivera RR, Murre C. Early thymocyte development is regulated by modulation of E2A protein activity. The Journal of experimental medicine. 2001;194(6):733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kee BL, Bain G, Murre C. IL-7Ralpha and E47: independent pathways required for development of multipotent lymphoid progenitors. The EMBO journal. 2002;21(1–2):103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bain G, Engel I, Robanus Maandag EC, et al. E2A deficiency leads to abnormalities in alphabeta T-cell development and to rapid development of T-cell lymphomas. Mol Cell Biol. 1997;17(8):4782–4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kastner P, Chan S. Role of Ikaros in T-cell acute lymphoblastic leukemia. World J Biol Chem. 2011;2(6):108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart M, Cameron E, Campbell M, et al. Conditional expression and oncogenicity of c-myc linked to a CD2 gene dominant control region. Int J Cancer. 1993;53(6):1023–1030. [DOI] [PubMed] [Google Scholar]
- 48.Dumortier A, Jeannet R, Kirstetter P, et al. Notch activation is an early and critical event during T-Cell leukemogenesis in Ikaros-deficient mice. Mol Cell Biol. 2006;26(1):209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irving JA, Enshaei A, Parker CA, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2016; 128(7):911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.