

Ibrutinib is an irreversible inhibitor of Bruton’s tyrosine kinase (BTK), which has emerged as a potent molecular therapy in the treatment of B-cell malignancies, and continues to be investigated for treatment in a variety of blood cancers. Despite an excellent safety profile and high tolerability, a large number of patients have bleeding as a side effect.1–3 These events are typically low grade, presenting as mucosal or skin bleeding;1,3 however, more severe bleeding has been reported, and is a concern when scheduling surgery or if anti-coagulants are needed, such as in patients with atrial fibrillation.2 Therefore, identifying the cause of ibrutinib-associated bleeding is critical for improved treatment management in these patients.
In platelets, BTK acts within the signalosome downstream of several receptors, including the collagen receptor GPVI and the podoplanin receptor CLEC-2,4 where it promotes phospholipase (PL) Cγ phosphorylation. Several labs have shown that ibrutinib can impair platelet activation in vitro,4,5 and platelets taken from ibrutinib-treated patients have an impaired response to collagen and reduced adhesion to von Willebrand Factor (vWF) ex vivo.6,7 It has therefore been suggested that ibrutinib-induced platelet dysfunction underlies bleeding in patients. In contrast, administration of ibrutinib analogs in non-human primates had no significant effect on bleeding time despite the ability to inhibit collagen-mediated platelet activation in vitro and ex vivo following treatment.8 This begs the question as to whether ibrutinib alone is responsible for causing bleeding in patients. In the present study, we examined the impact of ibrutinib on platelet function in the context of inflammatory hemorrhage and primary hemostasis in mice.
Platelets are responsible for maintaining vascular integrity even in the absence of overt vascular injury, such as during development and inflammation. In the setting of inflammation, thrombocytopenia leads to loss of vascular integrity and localized hemorrhaging.9 This protective effect of platelets was shown to be dependent on signaling through the (hem)ITAM receptors GPVI and CLEC-2.10 Infections and inflammation are common in cancer patients, so we hypothesized that since ibrutinib inhibits (hem)ITAM receptor activation in platelets, bleeding events such as petechiae may be due to inflammatory hemorrhage in ibrutinib-treated patients. We first established the ibrutinib dose required to inhibit murine platelet activation via the (hem)ITAM receptors GPVI (collagen) and CLEC-2 (podoplanin). While 0.5 μM ibrutinib inhibited aggregation in response to low dose collagen, a dose of 5 μM ibrutinib was required to completely inhibit aggregation in response to high dose collagen (Figure 1A). Similarly, podoplanin-induced aggregation was slightly delayed by pre-treatment with 0.5 μM ibrutinib, but was completely inhibited by 5 μM ibrutinib (Figure 1B). Interestingly, the inhibitory effect of ibrutinib on platelet aggregation was lessened in the presence of plasma, suggesting that residual integrin activation occurred even when cells were treated with 5 μM ibrutinib (Online Supplementary Figure S1). These findings were mirrored in flow cytometry experiments, where integrin activation and granule secretion in response to convulxin stimulation (GPVI-specific agonist) were slightly impaired in the presence of 0.5 μM ibrutinib but almost completely abolished in the presence of 5 μM ibrutinib (Online Supplementary Figure S2A). Importantly, the platelet response to PAR4 activating peptide or ADP was not affected with 5 μM ibrutinib treatment (Online Supplementary Figure S2 B, C).
Figure 1.

Ibrutinib impairs (hem)ITAM-mediated platelet activation but does not cause bleeding in mouse models of inflammatory hemorrhage. A, B) washed murine platelets were resuspended in modified Tyrode’s buffer with 1 mM Ca2+ in the absence of fibrinogen under stirring conditions. Platelets were pre-incubated with 0.5% DMSO or ibrutinib (0.5 or 5 μM) for 5 minutes, followed by the addition of collagen (A) or podoplanin-Fc (PDPN-Fc) and anti-mouse F(ab’)2 IgG (B). C–F) hIL-4Rα/GPIbα transgenic (Tg) mice were depleted of endogenous platelets by administration of an anti-hIL-4Rα antibody, and then transfused with wild-type platelets treated with vehicle or 5 μM ibrutinib. Non-transfused thrombocytopenic mice served as positive controls for hemorrhage. C) hIL-4Rα/GPIbα Tg mice were subjected to the intradermal reverse passive Arthus (rpA) inflammatory model. Anti-bovine serum albumin (BSA) antibody (dashed circles) was injected intradermally and buffer (closed circles) was injected as a control for bleeding from needle puncture, and inflammation was initiated by intravenous administration of BSA. Mice were sacrificed after 4 hours, and the dorsal skin was removed and reversed for imaging. D) Tissue hemoglobin was assessed in the rpA reaction. Skin biopsies were taken at sites of inflammation and homogenized in PBS. After centrifugation, hemoglobin in the supernatant was determined after addition of formic acid and measuring the optical denstiy at 405 nm. ** P<0.01 vs. no plts. E) Lung inflammation was induced by intranasal inoculation with P. aeruginosa LPS in hIL-4Rα/GPIbα-Tg mice which were platelet-depleted and then transfused with vehicle- or ibrutinib-treated platelets or not transfused for thrombocytopenic control. 24 hours later, mice were sacrificed and lungs were lavaged with PBS. Representative images of bronchoalveolar lavage (BAL) fluid are shown. F) Whole blood was collected from hIL-4Rα/GPIbα-Tg mice transfused with either vehicle- or ibrutinib-treated platelets and treated with convulxin (250 ng/ml) in the presence of JON/A-PE to determine levels of activated αIIbβ3 integrin. * P<0.05, ** P<0.01, *** P<0.001, n=4.
Next, we tested the impact of ibrutinib on the ability of platelets to secure vascular integrity during inflammation. We used two models of inflammation; the reverse passive Arthus (rpA) reaction in the skin, and LPS-induced lung inflammation. For these studies, we took advantage of a model for adoptive platelet transfer where vehicle- or ibrutinib-treated platelets can be transfused into platelet-depleted mice (Online Supplementary Figure S3A). This bypasses the need to administer ibrutinib, which can impair neutrophil function and suppress inflammation in mice. Platelet counts of recipient hIL-4Rα/GPIbα-Tg mice before and after platelet depletion/transfusion are shown in Online Supplementary Figure S3B. As expected, thrombocytopenic mice had robust hemorrhage at sites of inflammation in the skin (Figure 1C, left panel). Transfusion of vehicle-treated platelets significantly protected against hemorrhage (Figure 1C, middle panel). Surprisingly, however, transfusion of ibrutinib-treated platelets was also able to prevent inflammatory hemorrhage in the skin (Figure 1C, right panel). Tissue hemoglobin analysis of skin biopsies confirmed visual observations in the rpA model (Figure 1D). We observed similar results in the LPS-induced lung inflammation model, where both vehicle- and ibrutinib-treated platelets could prevent bleeding into the bronchoalveolar lavage (BAL) fluid (Figure 1E). Platelet activation was tested at the end of each experiment, and ibrutinib-treated platelets had markedly reduced αIIbβ3 integrin activation in response to convulxin stimulation (Figure 1F). Importantly, no change in surface expression of important adhesion receptors such as αIIbβ3, GPIbα, GPVI or β1 integrin subunit was observed in ibrutinib-treated platelets (not shown). Together, these findings suggest that while ibrutinib can almost completely inhibit (hem)ITAM-induced platelet activation and aggregation in vitro, platelets treated with ibrutinib retained the capacity to secure vascular integrity during inflammation.
Finally, we assessed the effect of ibrutinib on platelet function during hemostatic plug formation, both in the presence and absence of an additional platelet defect. To visualize platelet plug formation, we performed intravital microscopy on small lesions induced by laser ablation in the saphenous vein.11 In wild-type mice, ibrutinib administration did not significantly increase the bleeding time after laser injury (Figure 2A). We also observed these findings in a small cohort of mice using the saphenous vein needle injury model, where the saphenous vein is fully transected using a 23G needle (Online Supplementary Figure S4). We next investigated whether ibrutinib impairs hemostatic plug formation when given to mice lacking the P2Y12 receptor (P2ry12−/−), i.e., mice with an additional platelet defect.12 In our laser injury model, P2ry12−/− mice exhibited reduced platelet adhesion (anti-GPIX-488 intensity) at the site of injury (Figure 2C, left panel vs. middle panel) but no significant increase in the bleeding time (Figure 2A). However, administration of ibrutinib significantly increased the bleeding time in P2ry12−/− mice (Figure 2A). A significant increase in the number of platelet plug disruptions and re-bleeding was also observed (Figure 2B; Figure 2C, white arrow). These results suggest that ibrutinib can compromise hemostasis when additional platelet defects are present, but has minimal effects in healthy mice.
Figure 2.
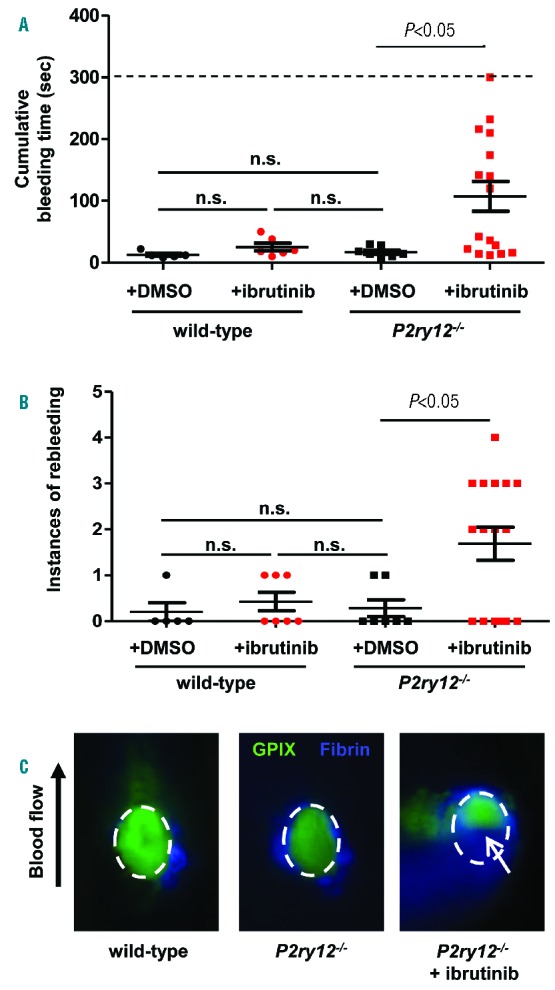
Ibrutinib impairs primary hemostasis in the presence of additional platelet defects. To assess hemostasis, bleeding time was determined using the saphenous vein laser injury model developed in our lab.11 Mice were anesthetized and the saphenous vein exposed. Before injury, mice received an injection of anti-GPIX-AlexaFluor488 and anti-fibrin-AlexaFluor647 antibodies. The saphenous vein was then injured and the injury site was recorded for 5 minutes. A) Cumulative bleeding time was determined by the amount of time blood could be seen flowing from the injury site, including both the bleeding time following initial injury and during injury rebleeding. The dashed line indicates the maximum bleeding time, which was only achieved in one injury site in a P2ry12−/− mouse treated with ibrutinib. n.s. = not statistically significant. B) The number of rebleeds, or injury re-openings, per injury site following occlusion of the initial injury. n.s. = not statistically significant. C) Platelet and fibrin accumulation was observed at the site of laser injury. Wild-type mice quickly occluded the injury site and robust platelet accumulation was observed within the first minute, after which loosely-adhered platelets began to embolize and the platelet plug stabilized. In P2ry12−/− mice, the vascular lesion occluded in a time frame comparable to wild-type mice. However, the peak of platelet accumulation was lost, and rebleeding occurred in several mice. In P2ry12−/− mice treated with ibrutinib, the cumulative bleeding time was significantly increased (Fig 2A), and a significant number of rebleedings occurred (Fig 2B), which were visually observed by blood flowing from the injury site (white arrow). In this study, P2ry12−/− mice were used as their own control; mice were injured at 1–2 sites, followed by ibrutinib administration, and then subjected to 2–3 more injuries at upstream sites along the saphenous vein.
We were surprised to find that ibrutinib-treated platelets could prevent hemorrhage during inflammation. The (hem)ITAM receptors GPVI and CLEC-2 have been shown to be necessary for platelets to secure vascular integrity during inflammation,10,13 and we expected the in vitro inhibitory effects of ibrutinib to manifest in a mouse model of inflammatory hemorrhage. However, when we tested platelet activation at the conclusion of the rpA reaction, we observed residual activation in ibrutinib-treated platelets activated via GPVI or CLEC-2 even at very high doses of the inhibitor. Considering that very few platelets are sufficient to prevent inflammatory hemorrhage,9 this residual platelet activation response may explain the lack of inflammatory hemorrhage in the presence of ibrutinib. At sites of more severe vascular lesions, such as after laser ablation injury, ITAM receptors play a minor role for platelet activation and plug formation when compared to GPCRs.14 Consistently, ibrutinib treatment reduced platelet accumulation at sites of injury, but did not cause prolonged bleeding times in mice. A significant increase in the bleeding time, however, was observed when ibrutinib was given to P2ry12−/− mice, i.e., mice with a partial defect in platelet GPCR signaling. Thus, our studies in mice suggest that ibrutinib alone does not cause bleeding after mechanical injury or at sites of inflammation, but that it can exacerbate disease/therapy-related defects in platelet function and hemostasis.
We believe that these results have important implications for our understanding of the cause of bleeding in humans taking ibrutinib. Patients on ibrutinib are typically elderly and may be on various medications, reported or unreported. The interaction of ibrutinib and aspirin or fish oils leading to increased bleeding risk has recently been suggested.1 Additionally, non-prescription NSAIDS such as ibuprofen can affect platelet function and may have important interactions with ibrutinib.15 Not all patients are on platelet inhibitors, and this interaction cannot explain all bleeding events in patients; however, the concept of multiple “hits” being required to impair hemostasis may be pertinent to these patients. Interestingly, in patients with CLL, it was also shown that the disease itself was a risk factor for impaired platelet function, independent of platelet count.1 A critical limitation of our study is that it was performed in young, healthy mice. In future studies, the effects of ibrutinib on hemostasis should also be tested in mouse models of B-cell mediated malignancies, and the whole of these complicating factors should be considered when examining the impact of ibrutinib on platelet function in vivo. Overall we conclude that, based on our results, caution should be taken in over-interpretation of the effects of ibrutinib from in vitro platelet function assays when discussing the in vivo situation, and further studies on the interaction of ibrutinib with other factors that inhibit platelet function are warranted.
Supplementary Material
Acknowledgments
The authors would like to thank Drs. Yacine Boulaftali and David Paul for their technical expertise in inflammatory mouse models, Kathryn Poe for her assistance with mouse husbandry and genotyping of mice used in this study, and Dr. Brandi Reeves for helpful discussions.
Footnotes
The online version of this paper contains a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Lipsky AH, Farooqui MZ, Tian X, et al. Incidence and risk factors of bleeding-related adverse events in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica. 2015;100(12):1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N Engl J Med. 2014;371(3):213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winqvist M, Asklid A, Andersson PO, et al. Real–world results of ibrutinib in patients with relapsed or refractory chronic lymphocytic leukemia: Data from 95 consecutive patients treated in a compassionate use program. Haematologica. 2016;101(12):1573–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manne BK, Badolia R, Dangelmaier C, et al. Distinct pathways regulate Syk protein activation downstream of immune tyrosine activation motif (ITAM) and hemITAM receptors in platelets. J Biol Chem. 2015;290(18):11557–11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bye AP, Unsworth AJ, Vaiyapuri S, Stainer AR, Fry MJ, Gibbins JM. Ibrutinib Inhibits Platelet Integrin alphaIIbbeta3 Outside-In Signaling and Thrombus Stability But Not Adhesion to Collagen. Arterioscler Thromb Vasc Biol. 2015;35(11):2326–2335. [DOI] [PubMed] [Google Scholar]
- 6.Levade M, David E, Garcia C, et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2014;124(26):3991–3995. [DOI] [PubMed] [Google Scholar]
- 7.Kamel S, Horton L, Ysebaert L, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29(4):783–787. [DOI] [PubMed] [Google Scholar]
- 8.Rigg RA, Aslan JE, Healy LD, et al. Oral administration of Bruton’s tyrosine kinase inhibitors impairs GPVI-mediated platelet function. Am J Physiol Cell Physiol. 2016;310(5):C373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goerge T, Ho-Tin-Noe B, Carbo C, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111(10):4958–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulaftali Y, Hess PR, Getz TM, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123(2):908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Getz TM, Piatt R, Petrich BG, Monroe D, Mackman N, Bergmeier W. Novel mouse hemostasis model for real-time determination of bleeding time and hemostatic plug composition. J Thromb Haemost. 2014;13(3):417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andre P, Delaney SM, LaRocca T, et al. P2Y12 regulates platelet adhesion/activation, thrombus growth, and thrombus stability in injured arteries. J Clin Invest. 2003;112(3):398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gros A, Syvannarath V, Lamrani L, et al. Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex-mediated inflammation in mice. Blood. 2015;126(8):1017–1026. [DOI] [PubMed] [Google Scholar]
- 14.Nonne C, Lenain N, Hechler B, et al. Importance of platelet phospholipase Cgamma2 signaling in arterial thrombosis as a function of lesion severity. Arterioscler Thromb Vasc Biol. 2005;25(6):1293–1298. [DOI] [PubMed] [Google Scholar]
- 15.Schafer AI. Effects of nonsteroidal antiinflammatory drugs on platelet function and systemic hemostasis. J Clin Pharmacol. 1995;35(3):209–219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.