

Abstract
A vailable tyrosine kinase inhibitors for chronic myeloid leukemia bind in an adenosine 5′-triphosphate-binding pocket and are affected by evolving mutations that confer resistance. Rebastinib was identified as a switch control inhibitor of BCR-ABL1 and FLT3 and may be active against resistant mutations. A Phase 1, first-in-human, single-agent study investigated rebastinib in relapsed or refractory chronic or acute myeloid leukemia. The primary objectives were to investigate the safety of rebastinib and establish the maximum tolerated dose and recommended Phase 2 dose. Fifty-seven patients received treatment with rebastinib. Sixteen patients were treated using powder-in-capsule preparations at doses from 57 mg to 1200 mg daily, and 41 received tablet preparations at doses of 100 mg to 400 mg daily. Dose-limiting toxicities were dysarthria, muscle weakness, and peripheral neuropathy. The maximum tolerated dose was 150 mg tablets administered twice daily. Rebastinib was rapidly absorbed. Bioavailability was 3- to 4-fold greater with formulated tablets compared to unformulated capsules. Eight complete hematologic responses were achieved in 40 evaluable chronic myeloid leukemia patients, 4 of which had a T315I mutation. None of the 5 patients with acute myeloid leukemia responded. Pharmacodynamic analysis showed inhibition of phosphorylation of substrates of BCR-ABL1 or FLT3 by rebastinib. Although clinical activity was observed, clinical benefit was insufficient to justify continued development in chronic or acute myeloid leukemia. Pharmacodynamic analyses suggest that other kinases inhibited by rebastinib, such as TIE2, may be more relevant targets for the clinical development of rebastinib (clinicaltrials.gov Identifier:00827138).
Introduction
Genetic changes that alter tyrosine kinase function are found in multiple malignancies, including chronic myeloid leukemia (CML) and acute myeloid leukemia (AML). In CML, the BCR-ABL1 translocation is the hallmark of the disease, leading to constitutive kinase signaling.1 Tyrosine kinase inhibitors (TKIs) have altered the natural history of CML.2,3 In AML, FLT3 internal tandem duplication (ITD) also leads to constitutive activation of FLT3, and likewise, FLT3 inhibitors have shown clinical efficacy in the treatment of FLT3-mutated AML.4,5–8
Although TKIs are very effective treatments for CML, resistance and disease progression eventually occur in some patients. Second generation ABL1 TKIs (dasatinib, nilotinib, and bosutinib) are not effective against the T315I BCR-ABL1 mutation, which is the most prevalent mutation found in patients progressing on these agents.9,10 The recent approval of ponatinib has added a treatment effective against the T315I mutation to the therapeutic armamentarium.11 However, resistance to ponatinib has been observed in some patients, making the development of new T315I-targeted inhibitors a necessity.
All TKIs currently approved by the United States Food and Drug Administration inhibit their target kinases by competing with adenosine 5′-triphosphate (ATP) for binding. Rebastinib (DCC 2036) is a “switch control” inhibitor of several tyrosine kinases, including ABL1, FLT3, and TIE2.12 Based on crystallographic analyses, rebastinib binds as a type II inhibitor that additionally penetrates into and binds the switch control pocket of the ABL1 kinase domain, key amino acid residues that mediate the change from inactive to active conformation. Through occupancy of this switch control pocket, rebastinib blocks the conformational process required for kinase activation. This unique binding mode enables rebastinib to maintain the ABL1 kinase domain in the inactive conformation, independent of the state of phosphorylation of the regulatory Tyr 393. Additionally, for certain TKI resistance mutations that also dysregulate and activate c-ABL1 (e.g., T315I and Y253F), rebastinib binding forces these mutant variants of BCR-ABL1 to adopt inactive conformations, durably blocking downstream cellular signaling pathways.12–16 Rebastinib inhibits the BCR-ABL1 T315I mutant kinase in an ATP noncompetitive manner, even at the high millimolar concentrations of ATP found intracellularly. In vivo, rebastinib is effective in a mouse model of CML-like myeloproliferative neoplasia induced by BCR-ABL1 T315I.12 Potential advantages of switch pocket inhibitors compared to ATP-competitive inhibitors include longer off-rates compared to ATP-binding site inhibitors due to their binding into the deeply embedded switch pocket, and the fact that switch pockets are more varied in sequence from kinase to kinase than are ATP-binding pockets. The latter could potentially lead to inhibition of fewer off-target kinases.
This first-in-human study with rebastinib was performed in order to determine the maximum tolerated dose (MTD) when administered daily, and to evaluate preliminary efficacy in patients with relapsed CML or AML.
Methods
Trial design
This first-in-human, multi-center, single arm study evaluated the safety, tolerability, pharmacokinetics (PK), efficacy and pharmacodynamic (PD) effects of rebastinib in patients with CML or AML. The study was conducted in accordance with the Declaration of Helsinki and approved by the institutional review boards at each participating institution.
Patients
Patients ≥18 years old were eligible if they had a diagnosis of CML in chronic, accelerated or blast phase resistant to ≥2 TKIs or with T315I mutation, or had resistant or refractory AML with FLT3 ITD and had not received prior FLT3 inhibitors, or were ≥60 years old and not candidates for standard chemotherapy. An Eastern Cooperative Oncology Group performance status of ≤2 for patients with CML and ≤1 for those with AML was required. After the MTD was established, only patients with chronic phase chronic myeloid leukemia (CP CML) and accelerated phase chronic myeloid leukemia (AP CML) were enrolled into a CML expansion portion. After treating 13 patients in the CML expansion portion, enrollment was further limited to patients with CP-CML with the T315I mutation. Detailed inclusion and exclusion criteria are available in the Online Supplementary Information.
Rebastinib was administered orally daily in continuous 28 day cycles. Dosing started with powder-in-capsules (PIC) at 57 mg once daily (QD), escalating in 11 consecutive cohorts up to 1200 mg QD. Doses between the planned initial single-patient cohorts were increased by 100%. Subsequent cohorts enrolled at least 3 patients each in an accelerated 3+3 dose-escalation design, whereupon occurrence of grade ≥2 adverse events (AEs) accelerated titration stopped and a standard 3+3 design came into effect. During the trial, formulated tablets were introduced to seek more predictable pharmacokinetics. These were administered to patients in 3 cohorts from 100 mg to 200 mg. Patients receiving PIC were transferred to tablets after the safety of tablets had been confirmed. Intra-patient dose-escalation was permitted to a higher dose that had been evaluated in other patients for at least 28 days.
Safety and efficacy assessments
Safety was assessed using the National Cancer Institiute Common Toxicity Criteria (NCI CTC), Version 3.0.17 During the course of the study, serial ophthalmologic examinations, echocardiograms, and measurements of N-terminal pro-hormone brain natriuretic peptide (NT-proBNP) were added. Disease response was evaluated locally every 3 cycles using standard criteria.18,19
Pharmacokinetic and pharmacodynamic analyses
Plasma samples were collected on Days 1, 8, 15, and 22 of Cycle 1 and analyzed by liquid chromatography–mass spectrometry (LC MS)/mass spectrometry (MS) after single and multiple doses of either PIC or tablets. Pharmacokinetic samples were collected at predose, and up to 24 hr postdose on Days 1 and 8, and single predose samples were collected on Days 15 and 22. Pharmacokinetic parameters were derived using non-compartmental methods and based on actual blood sampling and dosing times for each individual. Whole blood samples for pharmacodynamic evaluation were collected at baseline and Days 1 and 8 of Cycle 1. In whole blood assays, mononuclear cells were fixed and permeabilized after incubation. pCRKL was analyzed by flow cytometry. For assays using isolated white blood cells, phospho-protein levels were determined by immunoblot. Target inhibition and rebastinib plasma concentrations were used for determination of pharmacokinetic/pharmacodynamic relationships.
Results
Patient population
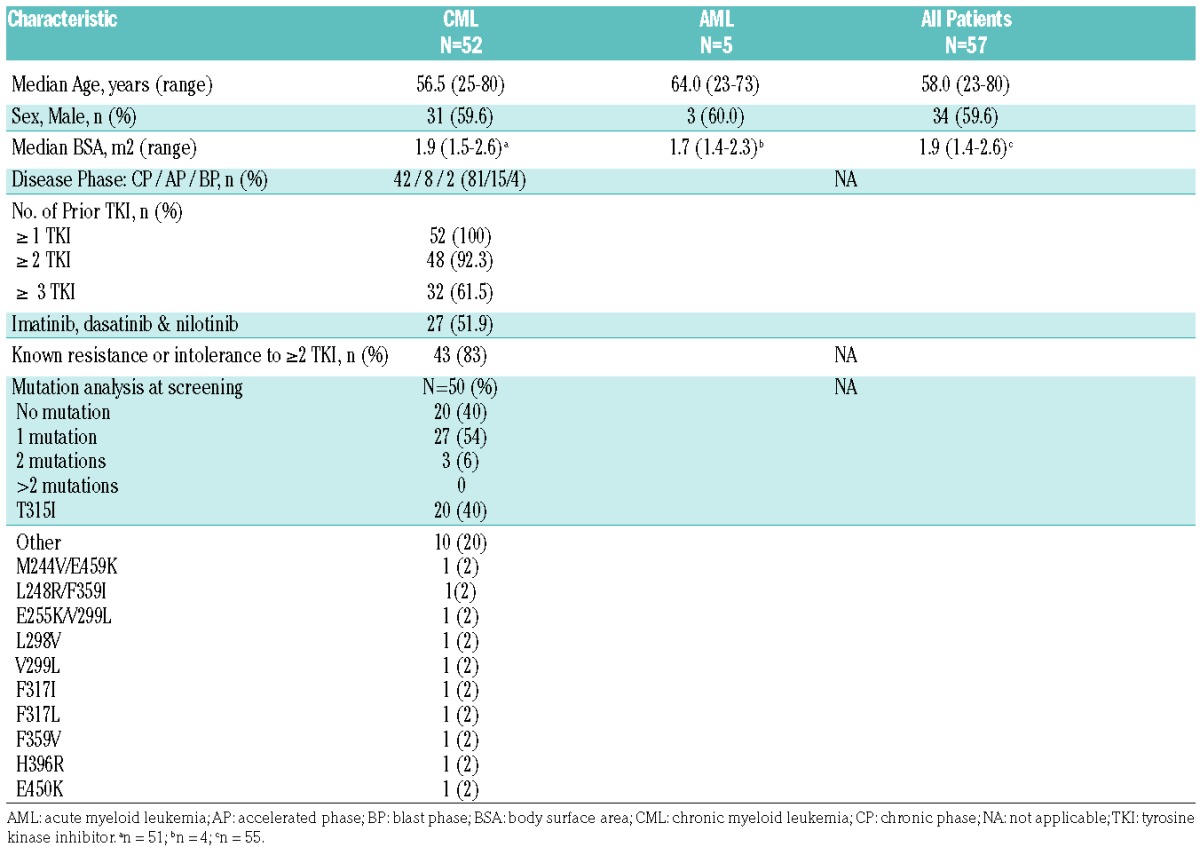
Sixty-nine patients were enrolled between March 2009 and February 2012, and 57 patients received rebastinib (CML:52 and AML:5). At data cutoff on 17 January 2013, all patients were off study (Online Supplementary Figure S1). The median age was 58.0 years (range: 25 to 80) and 59.6% of patients were male. All of the 52 patients with CML had received at least 1 prior TKI, 92% received 2 prior TKIs, and 62% received 3 prior TKIs. More than half (52%) of the CML patients had previously received imatinib, dasatinib, and nilotinib. Twenty (40%) of the CML patients harbored a T315I mutation (Table 1).
Table 1.
Baseline Patient Characteristics.
Maximum tolerated dose and safety
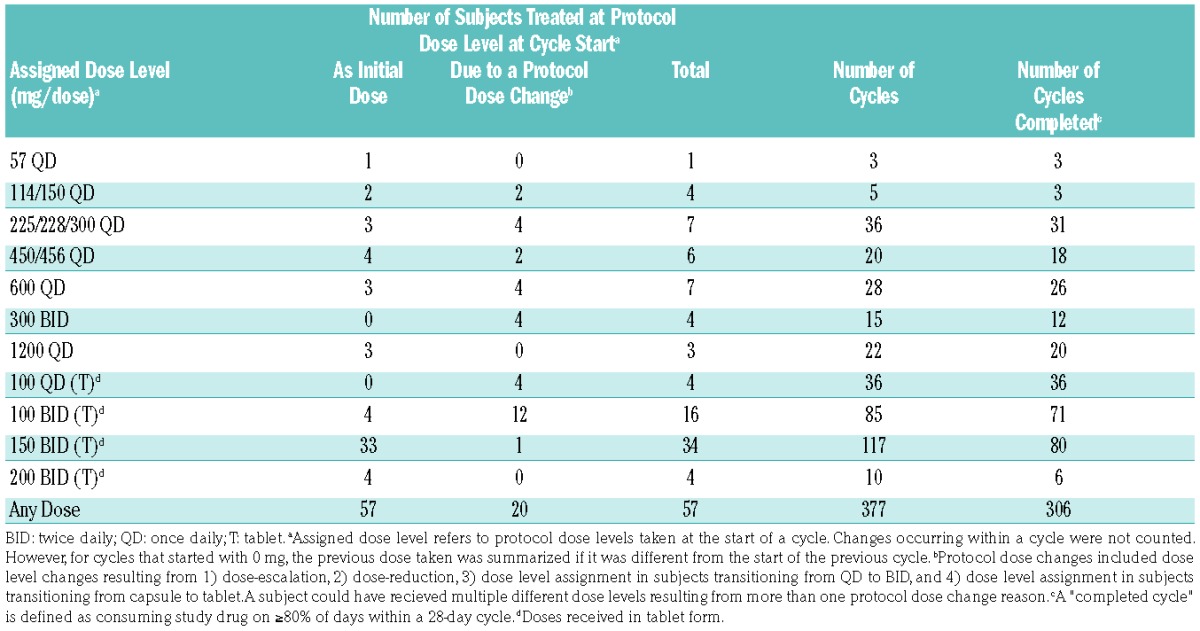
Fifty-seven patients received rebastinib at a total of 15 dosing regimens using the two formulations, PIC and tablet, and were analyzed in 11 dose groups (Table 2). A group represented different formulations with similar amounts of active ingredients. A total of 306 completed cycles were administered to 57 patients. Intra-patient dose-escalation was permitted and 20 patients received one or more dose levels in addition to their initial level.
Table 2.
Exposure to Rebastinib by Dose Level.
Dose-limiting toxicities (DLTs) were defined as the occurrence of one of the following AEs, which were considered to be at least possibly related to rebastinib during the first treatment cycle of the dose-escalation portion: 1) Grade 4 hematologic toxicity sustained for ≥4 weeks (≥6 weeks in patients with prior bone marrow transplantation), 2) ≥Grade 3 non-hematologic toxicity (except for nausea, vomiting or diarrhea that could be managed to ≤Grade 2 severity), and 3) any rebastinib-attributed AE that resulted in a treatment delay ≥28 days. Twenty seven patients were treated in the dose-escalation portion. Three patients experienced a DLT in the first cycle of treatment (Table 3). After exceeding the MTD at 200 mg tablets twice daily (BID), an additional 30 patients were enrolled in the expansion portion at 150 mg tablets BID. In the dose-escalation portion, three patients experienced a DLT in the first cycle of treatment at doses of 1200 mg QD (PIC, n=1) or 200 mg BID (tablet, n=2) (Table 3).
Table 3.
Summary of Adverse Events.
One patient in the 1200 mg QD dose level (PIC) experienced 2 DLTs (Grade 3 dysarthria and muscular weakness). This dose level was not expanded to 6 patients because of a planned change to tablet formulation. Two patients experienced a DLT in the first cycle of the 200 mg BID dose level with tablets; one experienced Grade 3 muscle weakness of their upper and lower extremities and the other experienced Grade 3 peripheral neuropathy. Consequently, the tablet dose of 150 mg BID was determined to be the MTD, and this dose cohort was expanded with an additional 27 patients to further characterize the safety profile. Of these 27 patients, 18 experienced a Grade 3 adverse event of special interest (AESI) considered possibly or probably related.
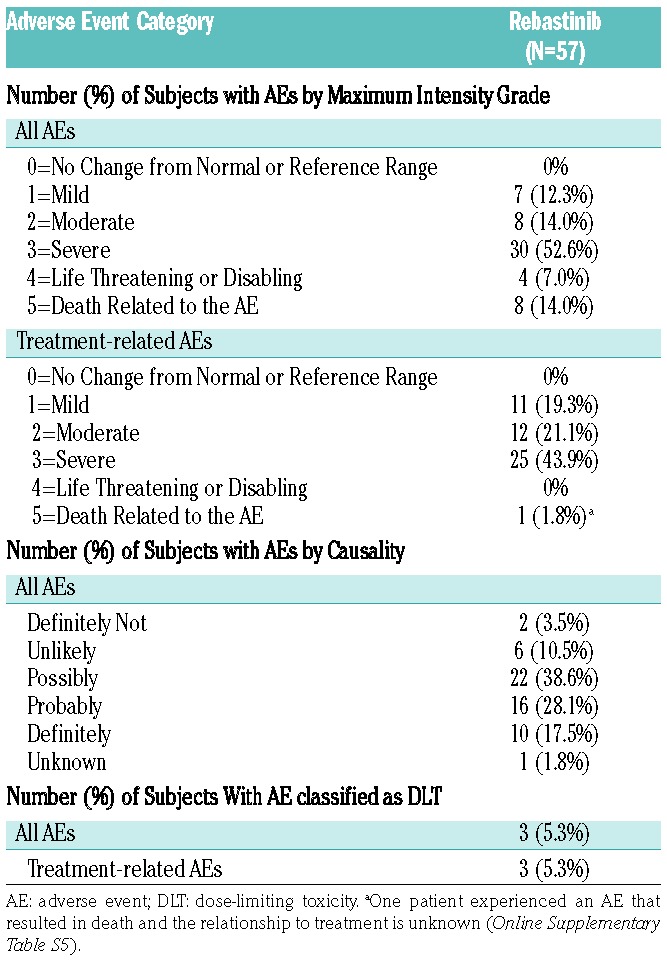
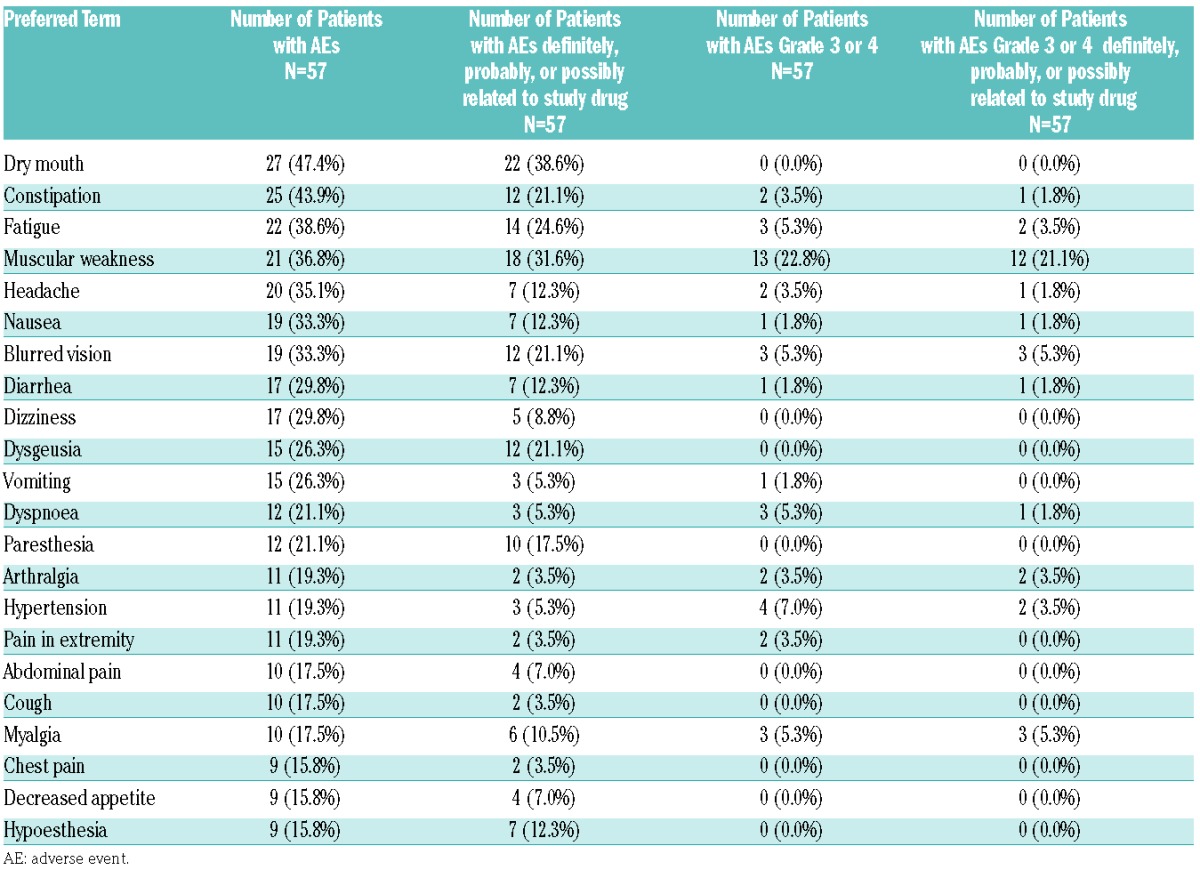
Analyses of all 57 (100%) patients showed that all experienced at least one AE of any grade during the entire treatment. Fifteen patients (26%) reported an AE with a maximum intensity of Grade 1 or 2, 34 patients (60%) reported a Grade 3 or 4 AE and 8 patients experienced an AE that resulted in death. Forty-nine patients (86%) reported AEs that were considered study drug related; related Grade 1 or 2 AEs by 23 patients (40%), related Grade 3 AEs by 25 patients (44%), and 1 patient experienced an AE that resulted in death, and the relationship to treatment is unknown; no related Grade 4 AE was reported (Table 3). Adverse events of any grade that were reported with an incidence of ≥30% were: dry mouth (27 patients, 47%), constipation (25 patients, 44%), fatigue (22 patients, 39%), muscular weakness (21 patients, 37%), headache (20 patients, 35%), and nausea and blurred vision (19 patients each, 33%) (Table 4). An overview of AEs occurring by dose level is shown in the Online Supplementary Table S1.
Table 4.
Frequency of patients with adverse events of any grade and grade 3 or 4 adverse events observed in ≥15% of patients by causality.
A total of 42 patients (74%) experienced one or more AE of ≥Grade 3 intensity: 30 patients (53%) experienced Grade 3 events, 4 patients (7%) experienced AEs of Grade 4, and 8 patients (14%) experienced AEs of Grade 5. The most common Grade 3 and 4 AEs (incidence >5%) were: muscular weakness (13 patients, 23%), hypertension (4 patients, 7.0%), and dyspnea, fatigue, myalgia, and blurred vision (3 patients each, 5%) (Table 4). Twenty six (46%) of the ≥Grade 3 AEs were considered related (Grade 3/4/5: 25 patients [44%]/0 patients/1 patient [2%], respectively). The most common Grade 3 and 4 related AEs (incidence >5%) were muscular weakness (12 patients, 21%), and myalgia and blurred vision (3 patients each, 5%). A total of 34 patients (60%) experienced serious adverse events (SAEs) (as reported by the investigator) during the study. The most common SAEs included muscular weakness (4 patients, 7%), disease progression, and pneumonia (3 patients each, 5%). In 18 patients (32%), these SAEs were considered related to study treatment. The most common study drug related SAE was muscular weakness (4 patients, 7%). Related SAEs occurred at dose levels of 300 mg BID and 1200 mg QD with PIC; and 100 mg, 150 mg, and 200 mg BID with tablets (Online Supplementary Table S2). No treatment-related SAE was reported for patients treated at dose levels below 300 mg BID (PIC) and 100 mg QD (tablet).
Events suggestive of musculoskeletal disorders (e.g., muscular weakness and myopathy), nervous system disorders (e.g., peripheral neuropathy, paresthesia, and peripheral sensory neuropathy), as well as visual abnormalities (e.g., blurred vision) were identified as AESIs (Online Supplementary Table S3 and Table S4). Musculoskeletal disorders of all grades was reported in 22 patients who experienced 34 events. In 14 of the 22 patients, musculoskeletal disorders occurred during Cycle 1. Among the 22 patients, 13 experienced Grade 3 events, and no Grade 4 event occurred. Recovery was documented in 11 of the patients within several days of halting rebastinib administration, and 2 patients discontinued study treatment due to musculoskeletal disorders. In the majority of cases, these events were reported during the first cycle of therapy. Nineteen patients (33%) experienced AEs of any grade collectively suggestive of nervous system disorders. Two patients with Grade 3 nervous system disorders continued on decreased rebastinib dosages, and the nervous system disorder resolved. Twenty-four AEs of visual abnormalities were reported in 19 patients (33%). Four out of 5 patients with 2 reported AEs recovered from at least 1 event. Thirteen out of 14 patients with Grade 1 as the highest severity, 2/2 with Grade 2, and 3/3 with Grade 3 reported recovery from visual abnormalities. Two patients with Grade 3 visual abnormalities discontinued from the study due to the AE. In 10 of the 19 patients, visual abnormalities were first reported during Cycle 1 (Grade 1/2/3: 6/1/3 patients, respectively). Treatment interruption was required in 3 patients; 1 patient with Grade 1, Grade 2, and Grade 3 visual abnormalities each and recovery from the event was documented (data not shown).
In addition, 4 CML patients (7%) experienced AEs suggestive of a potential ≥Grade 3 cardiomyopathy (congestive cardiac failure in 2 patients, viral cardiomyopathy in 1 patient, and cardiomyopathy in 1 patient). The patient who was diagnosed with viral cardiomyopathy on Day 1 had a medical history of allogeneic bone marrow transplant. He was immediately withdrawn from the study due to this event. Grade 3 congestive cardiac failure in another patient occurred on Day 4 and led to death on Day 7. Since 1998 this patient had received, among other therapies, interferon, imatinib, nilotinib, and dasatinib. The patient had a history of previous pleural effusion on dasatinib, hypertension, coronary artery disease, chronic dyspnea, and tachycardia. The third patient presented on Day 7 with Grade 3 cardiomyopathy; his symptoms included Grade 3 elevated cardiac enzymes and Grade 2 atrial fibrillation. He had a history of hypertension and received imatinib, nilotinib, and dasatinib as prior CML treatment. This patient recovered after cardiac conversion to a normal sinus rhythm. The fourth patient presented with Grade 3 bronchitis on Day 211, followed by Grade 3 congestive cardiac failure on Day 213 and died due to cardiac failure 2 days later. This patients prior therapies for CML included imatinib and dasatinib. His medical history included chronic obstructive pulmonary disease, hypertension, and bicuspid aortic stenosis with valve replacement. A summary of all deaths of study patients (n=8) and the attributed causality is provided in the Online Supplementary Table S5; none of the deaths were deemed related to rebastinib.
Pharmacokinetics
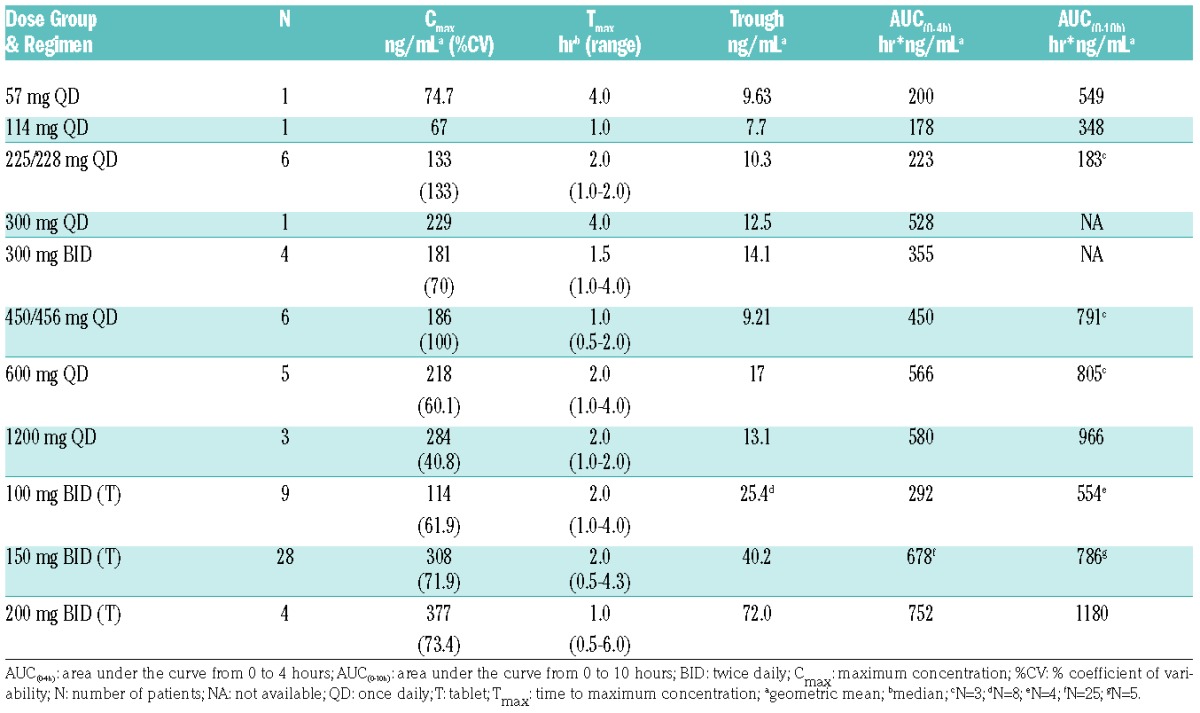
Rebastinib absorption was rapid with both tablet and PIC. Mean peak plasma concentrations occurred within a median time to maximum concentration (Tmax) of 1 to 1.5 hours using tablets (Figure 1) and 2 hours using PIC (Online Supplementary Figure S2). Exposure, including mean maximum concentration (Cmax), area under the curve from 0 to 4 hours (AUC(0–4h)), area under the curve from 0 to 10 hours (AUC(0–10h)), and trough values, increased with dose (Table 5), but in a less than dose-proportional manner. The elimination of rebastinib appeared to be biphasic, with an initial distribution phase from Tmax of 6–8 hours postdose, followed by a terminal elimination with a half-life (T1/2) of 12–15 hr (data not shown). Terminal elimination rates appeared to be similar across dose levels. Exposure was approximately 3-fold greater for tablets compared to PIC (Table 5).
Figure 1.
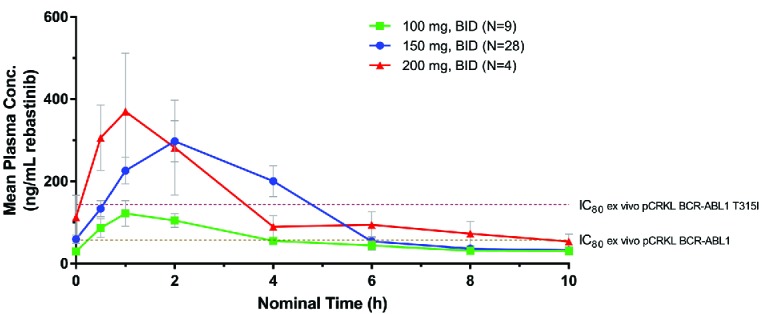
Mean plasma rebastinib concentration-time profile following multiple-dose administration of rebastinib tablets to steady state. Peripheral blood samples were obtained at the indicated times following administration of an oral dose of rebastinib (tablet formulation at doses of 100 mg BID, 150 mg BID, and 200 mg BID) in patients who had received at least 1 week of continuous dosing. Plasma concentrations of rebastinib were determined by LC-MS/MS as described in Methods. IC80 values for inhibition of BCR-ABL1 phosphorylation of CRKL in ex vivo assays using whole blood from CML patients with BCR-ABL1 or BCR-ABL1 T315I are indicated. BID: twice daily; Conc: concentration; pCRKL: phosphorylated CT10 Regulator of Kinase-like.
Table 5.
Summary of pharmacokinetic parameters following multiple doses of oral rebastinib as powder in capsule or tablet taken once or twice daily.
Safety Exposure Analyses
Safety exposure analyses investigated the relationship between rebastinib exposure, Cmax and AUC(0–4h), and the occurrence of AESIs in the medical categories of muscle weakness, peripheral neuropathy, cardiomyopathy and visual abnormalities (see Online Supplementary Table S3 for AE terms included in each system organ class). The analysis for Cmax included a total of 50 patients, and 47 patients for AUC(0–4h), who had pharmacokinetics data available for Cycle 1 (Day 8 or Day 15). The values for either Cmax or AUC(0–4h) of evaluable patients were divided into low, middle, and high tertiles. Rebastinib doses were identified that patients received at the time of the onset of a particular AESI, and patients were then analyzed based on the highest toxicity grade for a particular AE category within each Cmax or AUC(0–4h) value.
This analysis showed a trend towards the occurrence of more AESIs or AESIs of higher severity with increasing Cmax and AUC(0–4h) tertiles (Online Supplementary Table S6). For example, in the AESI analyses by Cmax, 23, 24, and 24 patients were categorized in the low/middle/high Cmax subgroup. A total of 7 patients (30%), 5 patients (22%), and 13 patients (54%) categorized in the low/middle/high Cmax subgroup experienced nervous system disorders. Whereas Grade 1 and 2 AESIs occurred in all Cmax subgroups, in the medium and high Cmax subgroups, one Grade 3 nervous system disorder each was also observed.
Clinical efficacy
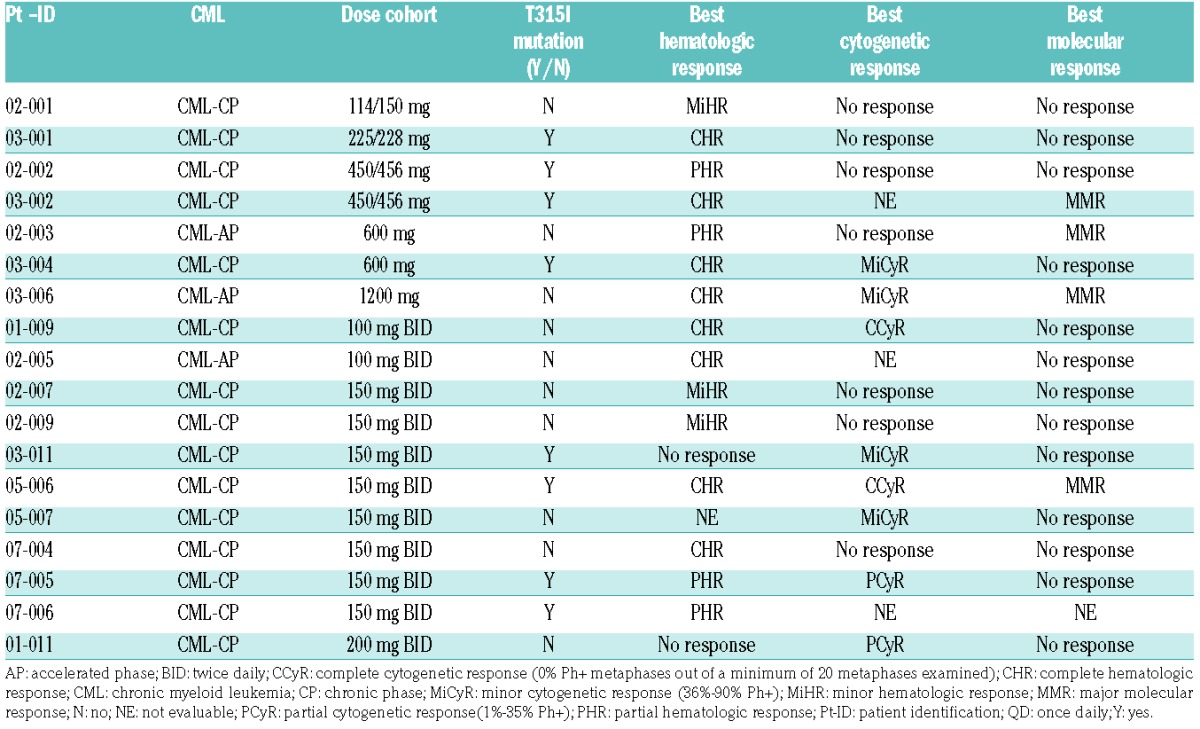
Response assessment was performed locally. All treated CML and AML patients were evaluable for response. Eighteen of the 52 CML patients (35%) responded. Fifty patients were evaluable for hematologic response. The best hematologic responses were 8 patients with complete hematologic response (T315I: 4 patients), 4 patients with partial hematologic response (T315I: 3), and 3 patients with minor hematologic response (T315I: 0), including 15 patients with chronic phase (CP) CML, and 3 patients with acute phase (AP) CML. Of the 37 patients evaluable for cytogenetic response, 8 patients showed a response (2 with complete cytogenetic response [T315I: 1], 2 with partial cytogenetic response [T315I: 1], and 4 with minor cytogenetic response [T315I: 3]). Four of the 40 evaluable patients achieved a molecular response (all had major molecular response; T315I: 2) (Table 6). Two of these occurred between Cycles 17–19 and were maintained for the duration of the study, while the other two were not durable (maintained for 3–6 cycles). All AML patients were evaluable for hematological and morphological responses. None of the 5 patients with AML showed a response to rebastinib.
Table 6.
Chronic myeloid leukemia subgroup: individual responses.
Pharmacokinetic/Pharmacodynamic evaluation
Screening (predose) whole blood samples were collected from 31 patients. To determine if rebastinib could inhibit BCR-ABL1 or FLT3 in samples from TKI-resistant patients, blood samples were incubated with rebastinib ex vivo and the phosphorylation of CRKL or FLT3 was measured. Of the 28 samples that were evaluable, in all cases the IC50 for inhibition of phosphorylation was less than 200 nM, including in samples from patients with ≥1 mutation in BCR-ABL1, such as T315I. To determine if BCR-ABL1 or FLT3 was inhibited in patients dosed with rebastinib, predose and postdose blood samples from Cycle 1 Day 1 and Cycle 1 Day 8 were analyzed from 48 patients. Of the 45 patients with evaluable samples, significant (>50%) inhibition of phosphorylation of CRKL and/or STAT5 was observed for 20 patients (Figure 2). Clinical response did not appear to be correlated closely to inhibition of phosphorylation of CRKL and/or STAT5 as measured in this assay, as only 8 of the 20 patients with significant inhibition of CRKL or STAT5 had a clinical response. Conversely, 9 patients who had a clinical response exhibited <50% inhibition of phosphorylation of CRKL and STAT5 at the time points tested. Samples were not collected for 1 patient who had a clinical response.
Figure 2.
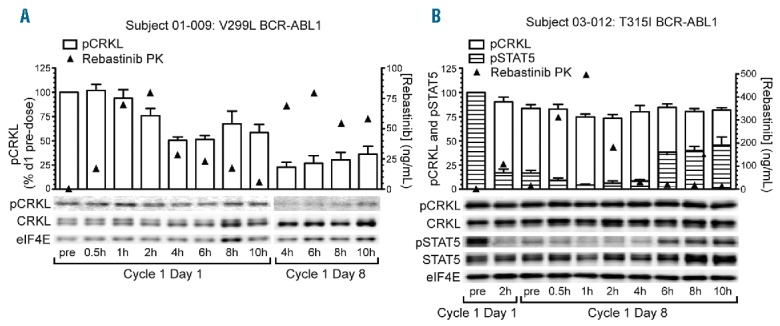
Pharmacodynamic analyses of pCRKL and pSTAT5 inhibition pre- and post-rebastinib dose. Serial peripheral blood samples were obtained from patients following the first dose of rebastinib (Cycle 1 Day 1) and on Day 8 of continuous twice daily dosing (Cycle 1 Day 8). Mononuclear cells were isolated, lysed, and the indicated proteins and phosphoproteins analyzed by immunoblotting as described in Methods. (A) Patient with relapsed chronic phase CML BCR-ABL1 V299L mutation who demonstrated >75% inhibition of pCRKL 4h following the Day 8 morning dose. (B) Patient with relapsed chronic phase CML and BCR-ABL1 T315I mutation who demonstrated >90% inhibition of pSTAT5 at 1h following the Day 8 morning dose. In this patient, the degree of inhibition of pCRKL was less pronounced (~25%). PK: pharmacokinetics; pCRKL: phosphorylated CT10 regulator of kinase-like; CRKL: CT10 regulator of kinase-like; eIF4E: eukaryotic translation initiation factor 4E; pSTAT5: phosphorylated signal transducer and activator of transcription 5; STAT5: signal transducer and activator of transcription 5.
In addition to BCR-ABL1 and FLT3, rebastinib is a potent TIE2 inhibitor (unpublished data). In selected patients, elevated plasma levels of the TIE2 ligand angiopoietin 2 (ANG2) were observed on Day 22 compared to baseline levels. ANG2 increases were observed in 19/20 of the Day 22 patient samples, with no change observed in 1/20 samples (data not shown). Both Cmax and trough exposures of rebastinib on Day 8 significantly correlated with increased levels of plasma ANG2. Increased plasma ANG2 levels also correlated with trough rebastinib exposures on Day 22 (data not shown).
Discussion
This Phase 1 study of the switch-control inhibitor, rebastinib, was conducted in patients with relapsed CML and AML based on its potency against ABL1 and FLT3. In vitro studies had established that rebastinib is an effective inhibitor of BCR-ABL1 and other kinases.12 The MTD of continuously administered rebastinib was determined to be 150 mg BID, with dysarthria, muscular weakness, and peripheral neuropathy representing the DLTs at a dose of 200 mg BID. Expansion of the 150 mg BID dose cohort consolidated the safety findings of the dose-escalation. The most common AEs included muscular weakness, peripheral neuropathy, and visual abnormalities. In addition, 4 patients experienced severe cardiac AEs during the study.
Visual abnormalities have been recently described with other TKIs.20,21 Such events observed with rebastinib were mostly of mild severity, and many of the events resolved with no change to rebastinib dosing. Since there were no significant ocular findings in preclinical animal studies, detailed baseline ophthalmologic examinations were not performed at baseline in this study, and a potential contribution of pre-existing conditions or the underlying disease could not be ruled out. Baseline and serial ophthalmologic examinations will be included in forthcoming studies in order to gain insight into the relatedness of the blurred vision seen with rebastinib in CML and AML patients. The mechanism by which these events are mediated will also need to be investigated.
A number of TKIs have been associated with a low incidence of cardiomyopathy occurring after months of treatment.22 The rebastinib study herein documented 3 cases of cardiomyopathy occurring during the first 2 weeks of treatment. Since these patients had alternative potential causes of cardiomyopathy, and there was no screening of cardiac function at baseline in this study, the relationship seen between cardiomyopathy events and rebastinib exposure is not clear. However, preclinical studies have suggested that ABL inhibition may lead to myocardial toxicity.23 Future rebastinib studies will include baseline and serial echocardiography studies to carefully investigate cardiac function. The pharmacokinetics parameters of rebastinib indicated rapid absorption of the drug, and an increase, though less than dose-proportional, in exposure with dosage. The T1/2 was 12–15 hours; however, due to the biphasic elimination of rebastinib resulting in trough concentrations of generally 10–20% of Cmax, some accumulation should be expected with a BID dosing regimen. Average plasma levels of 200 ng/mL (360 nM) were achieved transiently from ~1–4 hours postdose at the MTD of 150 mg BID. At this exposure level, the amount of free drug in circulation is expected to be ~1.3 nM (based on determined human plasma protein binding of 99.65%). With regards to potential off-target effects of rebastinib, the only kinases that would be inhibited by ≥50% at concentrations of <1.3 nM include c-ABL1 (IC50 0.7 nM), TIE2 (IC50 0.058 nM), and TRKA and TRKB (IC50 0.17 nM and 0.42 nM, respectively).12 Further monitoring of AESIs will help to understand the off-target effects or collateral “target” effects of rebastinib.
A safety exposure analysis that investigated the relationship of AESIs of cardiac, eye, musculoskeletal, and peripheral neuropathy/nervous systems disorders to rebastinib exposure measured as Cmax or AUC(0–4h) suggested that AESIs of musculoskeletal and peripheral neuropathy/nervous systems disorders may be related to rebastinib exposure. Regarding the severity of cardiac and ocular disorders, an increase in these disorders with an increase in rebastinib exposure cannot be ruled out. The understanding of safety exposure outcomes for cardiac and ocular disorders will improve from longitudinal monitoring in future studies.
Pharmacodynamic data show that the rebastinib plasma concentrations achieved in patients inhibited BCR-ABL1 in patient samples, including mutant alleles of BCR-ABL1, like T315I. Patients with CML who entered this trial were resistant or intolerant to > 1 of other BCR-ABL1 TKIs. In approximately half of the cases, no mutations were present in BCR-ABL1, possibly indicating activation of other pathways for cell survival. Inhibition of BCR-ABL1 in these patients, as monitored by pharmacodynamic analysis, may not necessarily correlate with clinical response. In addition, postdose levels of rebastinib in certain patients may not have inhibited phosphorylation of targets to the level needed to irreversibly commit leukemic progenitors to apoptosis, and thereby achieve a response.24 While rebastinib exposures at 150 mg BID on average were above or near the level required for 80% inhibition of BCR-ABL1 for the entire dosing period, exposures were only above the IC80 for inhibition of T315I mutant BCR-ABL1 for ~3 hours after each dose (Figure 2). Based on experience with other BCR-ABL1 inhibitors, a higher dose, e.g., 175 mg BID, if it would have been tested and tolerated, would probably not have led to an improved efficacy outcome in the case of BCR-ABL1 T315I resistance mutations.
When this study was designed, the only drug with activity against the BCR-ABL1 T315I mutation was omacetaxine mepesuccinate, which yields cytogenetic responses in ~20% of CP-CML patients.25 Although the overall cytogenetic response rate of CML patients to rebastinib (21%) was comparable to that of omacetaxine, the response rate was considerably lower than that which has subsequently been reported with ponatinib in a comparable patient population.11 Thus, continued development of rebastinib in CML may not be justified. Pharmacodynamic analysis suggested the inhibition of other rebastinib-targeted kinases in patients receiving rebastinib, such as TIE2 in particular. Compensatory elevations in circulating ligands upon the inhibition of their cognate receptor kinases have been frequently observed clinically. For example, the inhibition of VEGF receptors 1, 2, and 3 by sunitinib leads to increases in the plasma levels of VEGF and PlGF in patients.26 Thus, increased plasma ANG2 levels observed in patients on the trial herein provide evidence of TIE2 inhibition by rebastinib in humans. Future clinical development of rebastinib will focus more on diseases involving TIE2 or TIE2-expressing macrophages and the tumor microenvironment. The ANG/TIE2 kinase signaling pathway is a pivotal signaling axis in the tumor microenvironment, mediating tumor angiogenesis, tumor cell intravasation, extravasation, therapy resistance, and immunosuppression.27–30 TIE2 is expressed in endothelial cells and a subset of highly protumoral macrophages, referred to as TIE2-expressing macrophages (TEMs), both of which contribute to tumor angiogenesis.31 It has been demonstrated that TEMs expand an immunosuppressive population of Treg cells in the tumor microenvironment.29,32 In addition, it was reported that TEMs mediate tumor cell vascular intravasation and extravasation, and ultimately the formation of metastases.33–35 Data from in vitro assays indicate that the testing of a daily dose lower than the MTD defined in the study herein is warranted for TIE2 inhibition in future studies. This hypothesis is being tested in an ongoing clinical trial (clinicaltrials.gov Identifier:02824575).
Supplementary Material
Acknowledgments
The conduct of the study at MD Anderson Cancer Center was supported in part by MD Anderson Cancer Center Support Grant P30 CA016672 (PI: Dr. Ronald DePinho) and P01 CA049639 (PI: Dr. Richard Champlin). Studies at Tufts Medical Center were supported in part by R01 CA090576 (PI: Dr. Richard Van Etten). The authors thank Molly Hood and Cynthia Leary for performing ex vivo analyses on patient-derived plasma samples, Linda Martin of Deciphera Pharmaceuticals LLC for her assistance in the compilation and verification of key data to support this manuscript, and thank Korinna Pilz for medical writing support.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/3/519
References
- 1.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247(4946):1079–1082. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian HM, Talpaz M, O’Brien S, et al. Imatinib mesylate for Philadelphia Chromosome-positive, chronic-phase myeloid leukemia after failure of interferon: follow-up results. Clin Cancer Res. 2002;8(7):2177–2187. [PubMed] [Google Scholar]
- 3.Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–2551. [DOI] [PubMed] [Google Scholar]
- 4.Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10(12):1911–1918. [PubMed] [Google Scholar]
- 5.Schiller GJ, Tallman MS, Goldberg SL, et al. Final results of a randomized phase 2 study showing the clinical benefit of quizartinib (AC220) in patients with FLT3-ITD positive relapsed or refractory acute myeloid leukemia. J Clin Oncol. 2014;32:5s, (suppl;abstr 7100). [Google Scholar]
- 6.Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5(3):65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Röllig C, Müller-Tidow C, Hüttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–1699. [DOI] [PubMed] [Google Scholar]
- 8.Stone RM, Mandrekar S, Sanford BL, et al. The multi-kinase inhibitor midostaurin (M) prolongs survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine(C) induction (Ind), high-dose C consolidation (consol), and as main tenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18–60 with Flt3 mutations (muts): An international prospective randomized (rand) p-controlled double-blind trial (CALGB10603/RATIFY [Alliance]. Blood 2015;126(23):abstract 6. [Google Scholar]
- 9.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–880. [DOI] [PubMed] [Google Scholar]
- 10.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117 125. [DOI] [PubMed] [Google Scholar]
- 11.Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A Phase 2 trial of ponatinib in Philadelphia chromosome–positive leukemias. N Engl J Med. 2013;369(19):1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan WW, Wise SC, Kaufman MD, et al. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch control inhibitor DCC-2036. Cancer Cell. 2011;19(4):556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15(10):1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roumiantsev S, Shah NP, Gorre ME, et al. Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr-253 in the Abl kinase domain P-loop. Proc Natl Acad Sci USA. 2002;99(16):10700–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eide CA, Adrian LT, Tyner JW, et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Research. 2011;71(9):3189–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redaelli S, Mologni L, Rostagno R, et al. Three novel patient-derived BCR/ABL mutants show different sensitivity to second and third generation tyrosine kinase inhibitors. Am J Hematol. 2012;87(11): E125–128. [DOI] [PubMed] [Google Scholar]
- 17.National Cancer Institute Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events v3.0. Available from: http://ctep.cancer.gov/reporting/ctc.html Last accessed: 8th October 2008.
- 18.NCCN Practice Guidelines in Oncology. Available from: http://www.nccn.org/ Last accessed: 8th October 2008.
- 19.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2003;21(24):4642–4649. [DOI] [PubMed] [Google Scholar]
- 20.Renouf DJ, Velazquez-Martin JP, Simpson R, Siu LL, Bedard PL. Ocular toxicity of targeted therapies. J Clin Oncol. 2012; 30(26):3277–3286. [DOI] [PubMed] [Google Scholar]
- 21.Stjepanovic N, Velazquez-Martin JP, Bedard PL. Ocular toxicities of MEK inhibitors and other targeted therapies. Ann Oncol. 2016; 27(6):998–1005. [DOI] [PubMed] [Google Scholar]
- 22.Mellor HR, Bell AR, Valentin JP, Roberts RR. Cardiotoxicity associated with targeting kinase pathways in cancer. Toxicol Sci. 2011;120(1):14–32. [DOI] [PubMed] [Google Scholar]
- 23.Kerkala R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006; 12(8):908–916. [DOI] [PubMed] [Google Scholar]
- 24.Shah NP, Kasap C, Weier C, et al. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell. 2008; 14(6):485–493. [DOI] [PubMed] [Google Scholar]
- 25.Cortes JE, Kantarjian HM, Rea D, et al. Final analysis of the efficacy and safety of omacetaxine mepesuccinate in patients with chronic- or accelerated-phase chronic myeloid leukemia: Results with 24 months of follow-up. Cancer. 2015; 15;121(10): 1637–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DePrimo SE, Bello CL, Smeraglia J, et al. Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF-related proteins. J Transl Med. 2007;5:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis CE, Ferrara N. Multiple effects of angiopoietin-2 blockade on tumors. Cancer Cell. 2011;19(4):431–433. [DOI] [PubMed] [Google Scholar]
- 28.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012. March 20;21(3):309–322. [DOI] [PubMed] [Google Scholar]
- 29.Coffelt SB, Chen YY, Muthana M, et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol. 2011;186(7):4183–4190. [DOI] [PubMed] [Google Scholar]
- 30.Hughes R, Qian B-Z, Rowan C, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015;75(17):3479–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazzieri R, Pucci F, Moi D, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19(4):512–526. [DOI] [PubMed] [Google Scholar]
- 32.Ibberson M, Bron S, Guex N, et al. TIE2 and VEGFR kinase activities drive immunosuppressive function of TIE2-expressing monocytes in human breast cancer. Clin Cancer Res. 2013;19(13):3439–3449. [DOI] [PubMed] [Google Scholar]
- 33.Roussos ET, Goswami S, Balsamo M, et al. Mena invasive (Mena(INV)) and Mena11a isoforms play distinct roles in breast cancer cell cohesion and association with TMEM. Clin Exp Metastasis. 2011;28(6):515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dovas A, Gligorijevic B, Chen X, Entenberg D, Condeelis J, Cox D. Visualization of actin polymerization in invasive structures of macrophages and carcinoma cells using photoconvertible beta-actin-Dendra2 fusion proteins. PLoS One. 2011;6(2): e16485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian B-Z, et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage–derived VEGFA. Cancer Discov. 2015;5(9):932 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.