

Abstract
High mobility group AT-hook 2 (HMGA2) is an architectural transcription factor that is negatively regulated by let-7 microRNA through binding to it’s 3′-untranslated region. Transgenic mice expressing Hmga2 with a truncation of its 3′-untranslated region has been shown to exhibit a myeloproliferative phenotype. To decipher the let-7-HMGA2 axis in myeloproliferative neoplasms, we employed an in vitro model supplemented with clinical correlation. Ba/F3 cells with inducible JAK2V617F expression (Ton.JAK2.V617F cells) showed upregulation of HMGA2 with concurrent let-7a repression. Ton.JAK2.V617F cells treated with a let-7a inhibitor exhibited further escalation of Hmga2 expression, while a let-7a mimic diminished the Hmga2 transcript level. Hmga2 overexpression conferred JAK2-mutated cells with a survival advantage through inhibited apoptosis. A pan-JAK inhibitor, INC424, increased the expression of let-7a, downregulated the level of Hmga2, and led to increased apoptosis in Ton.JAK2.V617F cells in a dose-dependent manner. In samples from 151 patients with myeloproliferative neoplasms, there was a modest inverse correlation between the expression levels of let-7a and HMGA2. Overexpression of HMGA2 was detected in 29 (19.2%) of the cases, and it was more commonly seen in patients with essential thrombocythemia than in those with polycythemia vera (26.9% vs. 12.7%, P=0.044). Patients with upregulated HMGA2 showed an increased propensity for developing major thrombotic events, and they were more likely to harbor one of the 3 driver myeloproliferative neoplasm mutations in JAK2, MPL and CALR. Our findings suggest that, in a subset of myeloproliferative neoplasm patients, the let-7-HMGA2 axis plays a prominent role in the pathogenesis of the disease that leads to unique clinical phenotypes.
Introduction
Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs), which include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are a group of clonal disorders of the hematopoietic system characterized by excessive production of differentiated myeloid cells. With the discoveries of underlying driver mutations in JAK2, MPL, and calreticulin (CALR) that together account for 90% of patients with myeloproliferative neoplasm (MPN), it is now clear these conditions are characterized by dysregulated JAK-STAT (signal transducer and activator of transcription) signaling pathways.1 However, none of these mutations have been proven to be specific to disease subtype. As a result, they cannot be used in the molecular classification of MPNs. In addition, it remains unclear why the same acquired mutation in one of these genes causes similar clinical entities with distinct phenotypes.
Some studies allude to the fact that JAK2V617F confers only a weak growth advantage to hematopoietic stem cells.2,3 An emerging hypothesis suggests that several cooperating genetic hits might be required to induce disease and allow progression, as mutations in signaling molecules are not sufficient for disease development in humans.4 There are quite a few other genetic alterations identified in MPN;5 among them, several are implicated in epigenetic regulation, either in histone modifications or in DNA methylation control.6 Studies have shown that these epigenetic regulator genes and JAK2 mutations are synergistic by combining an early and late amplification, with mutation of the former mainly expanding the hematopoietic progenitor cells, whereas JAK2V617F mainly expands the mature fraction.4 Therefore, it is widely postulated that epigenetic regulation might also play an important role in the pathogenesis of MPN.
The high mobility group A2 (HMGA2) gene codes for a non-histone protein that has no intrinsic transcriptional activity, but can modulate transcription by altering the chromatin architecture.7,8 The HMGA2 protein, highly expressed during embryogenesis but diminished in normal adult tissues, is thought to play an essential role in self-renewal and the control of differentiation of embryonic stem cells.9 However, high levels of HMGA2 are found in various tumors, especially those of mesenchymal origin.10 The 3′-untranslated region (3′-UTR) of HMGA2 contains 7 sequences complementary to the let-7 microRNA (miRNA), which negatively regulates HMGA2 expression.11 In some tumors, rearrangement around the region of chromosome 12q14–15, the location of the HMGA2 gene, can lead to a deletion of the HMGA2 3′-UTR and loss of let-7 binding sites. This results in overexpression of a full-length or truncated HMGA2 protein which promotes tumor formation.2
Guglielmelli et al. first reported the association between HMGA2 upregulation and MPNs. In their seminal work studying the molecular profiling of CD34+ cells in PMF, they found that abnormal expression of HMGA2 was dependent on the presence of JAK2V617F mutation.12 Subsequently, Ikeda and colleagues demonstrated that transgenic mice expressing 3’UTR-truncated Hmga2 (ΔHmga2) cDNA exhibited a myeloproliferative phenotype.13 Enforced expression of ΔHmga2 led to a proliferative advantage in hematopoietic stem and progenitor cells. However, in spite of these studies, there are only scarce data available on the frequencies of dysregulated HMGA2 signaling activity in MPN patients, which severely limits the kinds of conclusions one can draw. Moreover, it remains unclear how HMGA2 and JAK2V617F interact with each other, and whether upregulation of HMGA2 plays specific roles in the pathogenesis of JAK2-mutated MPNs.
In the study herein, we aimed to address some of these issues. An in vitro model was employed to elucidate the correlation between JAK2V617F and HMGA2 expression. Furthermore, the phenotypic influences of HMGA2 overexpression on JAK2-mutated MPNs and the cause of HMGA2 upregulation were also explored.
Methods
Study population and mutational analysis
Relevant information on the patient enrollment, diagnosis,14 treatment,15 definition of events,16,17 and measurement of survival are listed in the Online Supplementary File. All participants provided informed consent in accordance with the Declaration of Helsinki. The study was approved by our Institutional Review Board. The detection of JAK2V617F, JAK2 Exon 12, CALR, and MPL mutations in clinical samples was performed as previously described.18
Cell lines and doxycycline induction
Interleukin-3 (IL-3)-dependent Ba/F3 cells with inducible expression of JAK2V617F (Ton.JAK2.V617F) or wild-type (WT) JAK2 (Ton.JAK2.WT) were kindly provided by Professor Gregor Hoermann and Professor Matthias Mayerhofer (Medical University of Vienna, Austria). The expression of JAK2 was induced by the addition of doxycycline (1μg/ml). The cells were maintained in IL-3 throughout the experiments until 3 hours before they were subjected to real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and western blot analysis. Sources of other cells used are listed in the Online Supplementary File.
Gene silencing by small interfering RNA transfection, miRNA ectopic expression and inhibition
The small interfering RNA (siRNA) oligos that directed against the mouse Hmga2 messenger RNA (mRNA, siHmga2) or a scrambled sequence (siScr) were purchased from Sigma-Aldrich. Small oligos that either mimic or inhibit endogenous let-7a were purchased from ABI (mirVana, Thermo Fisher Scientific Inc.). All the transfection was performed using X-tremeGENE siRNA Transfection Reagent (Roche) according to the manufacturer’s specifications. The efficiency of various siRNA oligos is demonstrated in the Online Supplementary Figure S1. The si-1 Hmga2 siRNA (siHmga2) was considered an ideal option and chosen for further experiments. The oligo concentrations used for let-7a inhibition were 0.2 and 0.5 nM according to the manufacturer’s suggestion, whereas 0.5 nM was used for the ectopic expression of let-7a. All the cells were harvested and validated by qRT-PCR or western blotting after 48 hours of transfection.
Cell viability and apoptosis assays
Cell survival was measured by the XTT assay according to the manufacturer’s instructions (Biological Industries). The percentages of apoptotic cells were determined using a FACS Canto II flow cytometer (Becton, Dickinson and Company) after the cells were treated with a 7-AAD and PE-Annexin V Apoptosis Detection Kit (BD Pharmingen). Each data result and its respective error bar were measured by 3 independent experiments run in triplicate.
Growth inhibition assay
The pan-JAK inhibitor INC424 (ruxolitinib) was kindly provided by Novartis Pharmaceuticals. INC424 was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM as the stock solution. 0.5% DMSO was added to the culture medium as control. Candidate cells were exposed to various concentrations of hydroxyurea (Bristol-Myers Squibb) or INC424 for 72 h. The drug concentrations that inhibited cell growth by 50% (IC50) and 90% (IC90) were determined by the XTT assay.
Others
Protocols on western blotting, qRT-PCR, and fluorescence in situ hybridization (FISH) are all listed in the Online Supplementary File.19
Statistical analysis
All calculations were performed using the Statistical Package for Social Sciences software (version 17.0; SPSS, Inc.). The level of statistical significance was set at 0.05 for all tests.
Results
Mutated JAK2 activates JAK-STAT pathway and up-regulates HMGA2 expression
We hypothesized that HMGA2 upregulation could be seen in cells with JAK-STAT signaling pathway activation, and chose to check its expressional status in MPN cells harboring either one of the two most common driver mutations (JAK2 and CALR). As demonstrated in Figure 1A, there was an 8-fold increase in Hmga2 levels in Ton.JAK2.V617F cells. The increment, however, was only around 2-fold in both Ba/F3 cells co-transduced with wild-type MPL and either type I (deletion) or type II (insertion) CALR mutants. Knowing that both mutated JAK2 and CALR activated JAK-STAT signaling,20–22 and considering the fact that a rise in Hmga2 expression was more prominent in JAK2-mutated cells, we used mutated JAK2 as our model of current investigation, but did not further explore CALR-mutated cells. To further validate the rise of HMGA2 expression in JAK2-mutated cells, we screened four different human acute myeloid leukemia (AML) cell lines. Western blotting showed that JAK2V617F-carrying HEL and UKE-1 cells exhibited increased JAK2 phosphorylation and enhanced HMGA2 expression (Figure 1B). On the contrary, HMGA2 expression was not increased in either JAK2-unmutated KG-1a or HL-60 cells. We next examined the signaling activity in cells with induced expression of JAK2. As demonstrated in Figure 1C, upon addition of doxycycline, Ton.JAK2.V617F cells exhibited enhanced JAK-STAT signaling as shown by increased phosphorylation of JAK2, STAT3, and STAT5. The level of HMGA2 protein was also increased significantly. On the other hand, the phosphorylation of JAK2 protein was rather weak in Ton.JAK2.WT cells. Although there was HMGA2 upregulation in these wild-type cells, the increment in HMGA2 expression was not as substantial as that seen in Ton.JAK2.V617F cells. Increased Hmga2 transcripts could be observed at 2 days after induction of JAK2V617F expression (Figure 1D), and the levels of transcripts continued to rise after extended exposure to doxycycline in Ton.JAK2.V617F cells. These data demonstrate that mutated JAK2 could activate the JAK-STAT pathway and upregulate Hmga2 expression in Ba/F3 cells.
Figure 1.
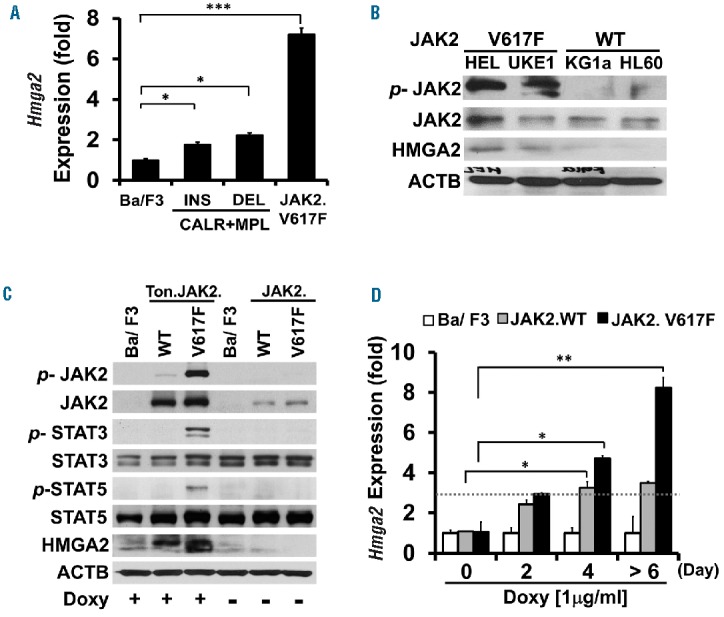
The levels of HMGA2 expression in cells with various JAK-STAT signaling activity. (A) Quantitative RT-PCR analysis of Hmga2 transcript levels in parental Ba/F3 cells, stable Ba/F3 cells co-transfected with MPL and either type 1 (deletion; DEL) or type 2 (insertion; INS) CALR mutant, and stable, inducible Ton.JAK2.V617F cells. The Ton.JAK2.V617F cells were treated with doxycycline (1 μg/ml) for at least 6 days before being subjected to analysis. Representative data from three independent experiments are presented. The error bars show the standard deviation (± SD) of three independent experiments. Asterisk indicates statistical significance (t-test; *P<0.01; ***P<0.0001). (B) Western blot analysis of JAK2, phosphorylated JAK2 (p-JAK2), and HMGA2 levels in 4 human acute myeloid leukemia cell lines: JAK2V61F-carrying HEL and UKE-1 cells, and JAK2-unmutated KG-1a and HL-60 cells. (C) The expression levels of JAK2, p-JAK2, STAT3, p-STAT3, STAT5, p-STAT5, and HMGA2 proteins in stable, inducible Ton.JAK2.WT and Ton.JAK2.V617F cells. Cells were treated with doxycycline (1 μg/ml; for at least 6 days) before being harvested concurrently with untreated controls for subsequent analysis. (D) Quantitative RT-PCR analysis of Hmga2 transcripts in parental Ba/F3, Ton.JAK2.WT and Ton.JAK2.V617F cells at baseline as well as 2, 4, and 6 days after addition of doxycycline (1 μg/ml), respectively. The error bars show the standard deviation of three independent experiments. Asterisk indicates statistical significance (t-test; *P<0.01; **P<0.001). WT: wild-type.
HMGA2-overexpressing MPN patients exhibit unique clinical characteristics
To assess whether increased expression of HMGA2 could be detected in clinical samples, we checked the levels of HMGA2 transcripts in the granulocytes of 151 patients with MPN and 27 adult healthy controls. The cycle numbers (Ct) in all healthy individuals were greater than 40, indicating the expression of HMGA2 was rather low in this population. We used the 2−ΔΔCT method to calculate the relative HMGA2 expression in our patients. Among the healthy individuals, the distribution of HMGA2 transcript levels ranged between 0.02–3.0 times the mean level of the whole control population. Therefore, we defined the patients with a 2−ΔΔCT value of HMGA2 transcript greater than 3 as HMGA2(+), whereas the remainders were designated as HMGA2(−). Overall, 29 (19.2%) patients exhibited overexpression of HMGA2 in their peripheral blood (PB) granulocytes. Table 1 demonstrates the clinical and laboratory characteristics of the 151 MPN patients subcategorized into the two groups. Patients whose granulocytes harbored increased HMGA2 expression had a higher probability of carrying a driver mutation than those without, though the comparison was of borderline significance (93.1% vs. 77.0%, P=0.051). When taking only JAK2-mutated and triple negative (TN) patients into consideration, we had 22 (91.7%) JAK2-mutated and 2 (8.3%) TN patients in the HMGA2(+) group, and 78 (73.6%) and 28 (26.4%) in the HMGA2(−) group, respectively. The comparison regarding their difference was more significant (P=0.040, Table 1). Based on our in vitro data, it’s plausible that JAK2V617F induced a higher degree of Hmga2 upregulation (Figure 1A). Therefore, the difference in the frequencies of JAK2 mutation between the HMGA2(+) and (−) groups became more prominent when we included only JAK2 -mutated and TN patients as the denominator. Female patients constituted the main population of HMGA2(+) patients (65.5%), in contrast with that seen in the HMGA2(−) group (41.0%, P=0.017). Strikingly, compared to those without HMGA2 overexpression, HMGA2-upregulated patients presented with a higher platelet count (789 ± 445 × 109/L vs. 576 ± 414 × 109L, P=0.016). This could be the result of a higher prevalent rate of HMGA2 overexpression among ET patients (26.7%) as compared to that seen in PV patients (12.7%, P=0.044). Overall, HMGA2(+) MPN patients were more likely to suffer from major thrombotic events (34.5% vs. 15.6%, P=0.021). Consistently, Kaplan-Meier estimates showed that HMGA2-overexpressing MPN patients had a significantly inferior thrombosis-free survival (P=0.049, Figure 2). The cumulative incidences of thrombosis at 10 years were 31.0% and 13.9% in HMGA2(+) and HMGA2(−) patients, respectively. Coupled with the finding that HMGA2-overexpressing MPN patients were slightly older (65.7 ± 14.4 years old, vs. 59.5 ± 16.1, P=0.058), it’s not surprising that HMGA2(+) ET/PV patients were more likely to carry high-risk disease (21/26; 80.8%), as defined by the European LeukemiaNet criteria,15 than their HMGA2(−) counterparts (59/104; 56.7%; P=0.026). These data suggest that upregulation of HMGA2 confers unique characteristics in patients with MPN, including a heightened probability of harboring either one of the 3 driver mutations, an increased propensity for the ET phenotype, and a higher likelihood of major thrombotic events.
Table 1.
Clinical and laboratory features of 151 patients with myeloproliferative neoplasm, stratified by expressional status of the HMGA2 gene.
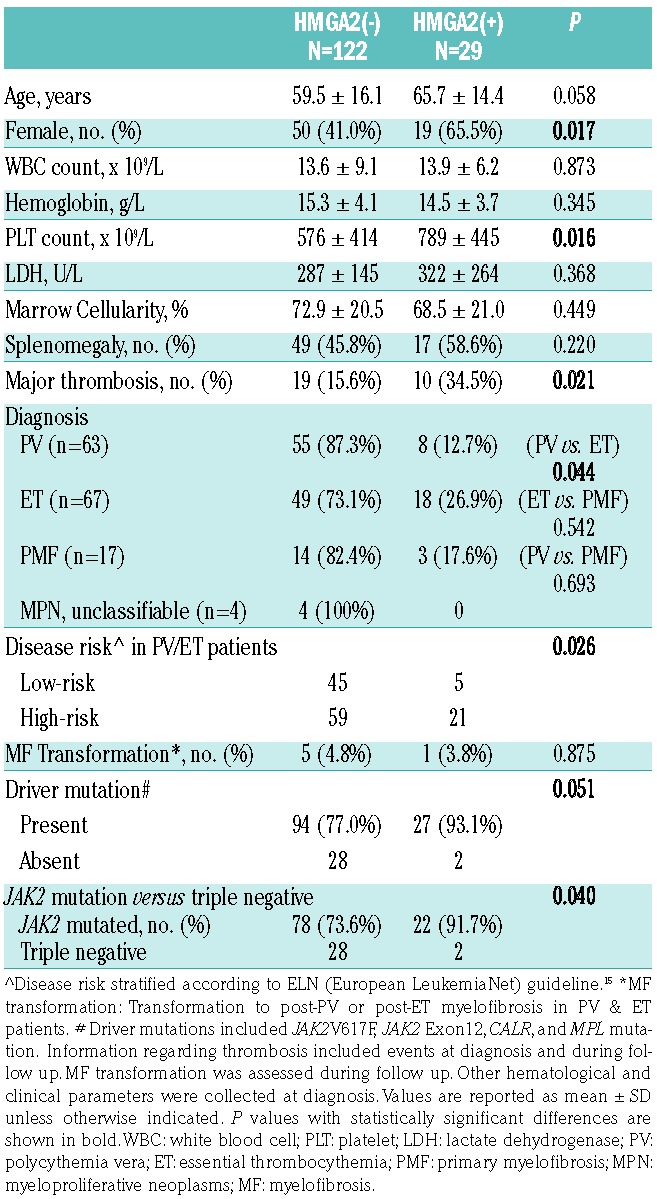
Figure 2.
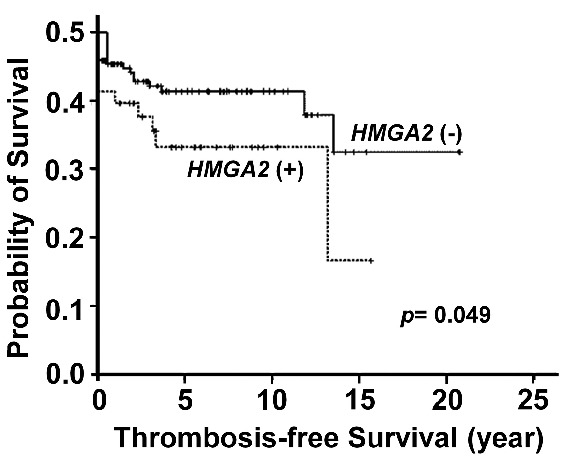
Impacts of HMGA2 overexpression on clinical outcome of patients with MPN. Kaplan–Meier estimates of thrombosis-free survival (TFS) in 151 MPN patients stratified by the expression status of HMGA2 showed those with upregulated HMGA2 had significantly inferior TFS than those with lower expression (P=0.049, log-rank test).
Hmga2 overexpression confers JAK2V617F-mutated cells a survival advantage
To delineate if Hmga2 overexpression altered cellular phenotypes, candidate cells were treated with either Hmga2 siRNA or a scramble control to assess their respective characteristics (Figure 3). Figure 3A demonstrates the efficacy of Hmga2 downregulation in both Ton.JAK2.WT and Ton.JAK2.V617F cells. Compared with a mock control or scrambled siRNA-treated control, Ton.JAK2.V617F cells treated with Hmga2 siRNA exhibited decreased viability by 50–60% (Figure 3B). The apoptosis assay revealed that the siHmga2-treated Ton.JAK2.V617F population had more cells in apoptosis (Figure 3C,D), a finding further confirmed by increased cleaved poly (ADP-ribose) polymerase (PARP) on western blotting (Figure 3E). Noticeably, the Hmga2 knockdown efficiency was also confirmed by western blot analysis (Figure 3E). The data illustrate that Hmga2 overexpression confers JAK2V617F-mutated cells with a survival advantage by rendering them more resistant to apoptotic death.
Figure 3.

Effects of Hmga2 downregulation in JAK2-mutated cells. Stable, inducible Ton.JAK2.WT and Ton.JAK2.V61F cells were pretreated with doxycycline (1 μg/mL) for 4 days and then transfected with either scrambled (siScr) or Hmga2 siRNA (siHmga2). (A) Quantitative RT-PCR confirmed the knockdown efficiency. (t-test; **P<0.001; ***P<0.0001). (B) Viability assay was assessed at 48 hours posttreatment with mock, siScr or siHmga2 in Ton.JAK2.WT and Ton.JAK2.V61F cells. The error bars show the standard deviation of 3 independent experiments run in triplicate. Asterisks indicate t-test statistical significance (*P<0.01; ***P<0.0001). (C) Cell apoptotic rates were detected by flow cytometric analysis using PE conjugated-Annexin V and 7-AAD double staining. Cells positively stained with Annexin V only were defined as being in early apoptosis, whereas those with positive stains for both Annexin V and 7-AAD were considered as being late apoptotic cells. (D) Histogram of apoptotic percentages (*P<0.01; **P<0.001; ***P<0.0001, statistical significance by t-test). The data were collected from three individual experiments. (E) To detect the discrepancy of apoptotic rates between siHmga2- and siScr-treated cells, western blotting was performed to assess the amount of full and cleavage form of poly (ADP-ribose) polymerase (PARP). Arrowhead indicates the full-length of PARP protein (116kDa) and the asterisk represents the cleavage form of PARP (89kDa). WT: wild-type.
HMGA2 overexpression is associated with let-7a downregulation instead of chromosomal rearrangement around the 12q15 region
To clarify the mechanism that leads to abnormal HMGA2 expression, we employed interphase FISH to detect potential cytogenetic abnormalities around this region, but found no abnormal rearrangement in all the cases evaluated (data not shown). We next assessed whether let-7a microRNA might play a role in the upregulation of HMGA2 in MPN patients. As illustrated in Figure 4A, there was a modest inverse correlation between the expression levels of let-7a and HMGA2 transcripts in the granulocytes of our patients (Pearson’s correlation coefficient r=−0.291, P=0.0002). In vitro experiments disclosed that the gradual decline of the let-7a expression could be observed in Ton.JAK2.V617F cells upon the addition of doxycycline in a time-dependent manner (Figure 4B), as opposed to the progressive increment of Hmga2 expression seen in similar assays (Figure 1D). To further validate whether the expression of HMGA2 could be regulated by let-7a, we first treated parental Ba/F3 cells with two different concentrations of a let-7a inhibitor. The result demonstrated that upon let-7a suppression, the expression of HMGA2 was increased in a dose-dependent manner at the protein (Figure 4C, upper panel) and mRNA (Figure 4C, lower panel) levels. Ectopic expression of let-7a in Ton.JAK2.V617F cells resulted in the downregulation of Hmga2 mRNA (Figure 4D, right panel) and protein (Figure 4D, left panel), whereas cells treated with a let-7a inhibitor had increased HMGA2 expression. There was an inverse correlation between the expression levels of Hmga2 and let-7a in those treated Ton.JAK2.V617F cells (r=−0.9281, P=0.002; Pearson’s correlation). Overexpression of let-7a in Ton.JAK2.V617F cells also led to increased apoptosis, as shown by increased cleaved PARP and decreased phosphorylation of the pro-apoptotic protein Bcl-2-associated death promoter (BAD) (Figure 4D, left panel). Conversely, treatment with the let-7a inhibitor endued the Ton.JAK2.V617F cells with more resistance to apoptosis, as the cleaved PARP was reduced but the phosphorylated BAD was increased (Figure 4D). Importantly, the let-7a inhibitor significantly increased the cellular viability in both parental Ba/F3 cells and inducible Ton.JAK2.V617F cells, whereas overexpressed let-7a suppressed these JAK2-mutated cells’ viability (Figure 4E). It was noteworthy that the levels of p-JAK2 were not significantly affected by either ectopically expressed let-7a or let-7a inhibition (Figure 4D), indicating that alteration in the let-7a-HMGA2 axis would not lead to a change in JAK-STAT signaling activity. These results imply that in MPN, HMGA2 overexpression is mediated through let-7a downregulation, whereas chromosomal rearrangement around the 12q15 region might not play a potential role in this aspect. The data also further complement our earlier experiments showing that HMGA2 overexpression confers JAK2-mutated cells with a survival advantage (Figure 3).
Figure 4.
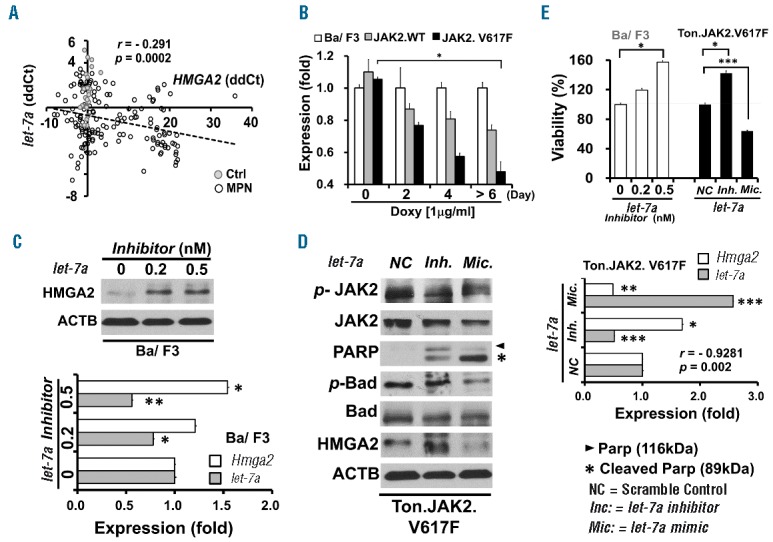
Correlation between the expression levels of let-7a and HMGA2 transcripts in MPN cells. (A) Scatter plot illustrating the correlation between the expression levels of let-7a and HMGA2 transcripts in the granulocytes of 151 patients with MPN (hollow circles; correlation coefficient r=−0.291, Pearson’s correlation, P=0.0002). Note that data from healthy individuals (solid gray circles) mostly fell on the y-axis, and many of them were very close to the origin. Data are shown in ΔΔCT. (B) Quantitative RT-PCR was performed to detect the levels of let-7a miRNA transcripts in parental Ba/F3, Ton.JAK2.WT and Ton.JAK2.V61F cells after doxycycline treatment (1 μg/ml) for indicated periods of time. (C) Effects on a let-7a inhibitory oligo on the expression of let-7a and Hmga2 in parental Ba/F3 cells. Two oligo concentrations (0.2 and 0.5 nM) were used for let-7a inhibition. All cells were harvested after 48 hours of transfection. Upper panel: western blot analysis of HMGA2; Lower panel: qRT-PCR analysis of let-7a and Hmga2 transcript levels. (t-test; *P<0.01; **P<0.001). (D) Effects of let-7a inhibitory and mimic oligos on Ton.JAK2.V617F cells. The working concentration was 0.5 nM for both oligos. Right panel: qRT-PCR analysis of let-7a and Hmga2 transcript levels. Note the inverse correlation between the expression levels of let-7a and Hmga2 (correlation coefficient r=−0.9281, Pearson’s correlation, P=0.002). Left panel: western blot analysis on the expression levels of JAK2, p-JAK2, PARP, cleaved PARP, BAD (Bcl-2-associated death promoter protein), p-BAD, and HMGA2. NC: scramble control; “−”: treatment with a let-7a inhibitor; “+”: treatment with a let-7a mimic. (E) The viability of parental Ba/F3 and inducible Ton.JAK2.V617F cells treated with indicated let-7a oligos. The error bars show the standard deviation of three independent experiments. Asterisks indicate statistical significance (t-test; *P<0.01; **P<0.001; ***P<0.0001).
Previous reports have demonstrated that LIN28A expression hinders the maturation of let-7.23 Therefore, we also explored the potential roles of LIN28A in the JAK2V617F-mediated upregulation of Hmga2. We found that the expression levels of LIN28A in Ton.JAK2.V617F cells were also increased (Online Supplementary Figure S2). Nevertheless, when clinical samples were tested, we could not identify any association between the expression levels of LIN28A and let-7a (Online Supplementary Figure S2C).
Both hydroxyurea and pan-JAK inhibitor INC424 ameliorate JAK-STAT activation and alleviate HMGA2 upregulation
Hydroxyurea (HU) is considered an integral part in the treatment of patients with MPN.15 To appraise the potential effects of HU on JAK-STAT signaling activity and HMGA2 expressional status, we first identified its estimated IC50 concentration (5.5μg/ml) in Ton.JAK2.V617F cells in a growth inhibition assay (Figure 5A). Subsequently, the cells were treated with hydroxyurea at the concentration for 3 days and then subjected to western blot analysis. As shown in Figure 5B, hydroxyurea abolished JAK2 phosphorylation almost completely, which in turn ameliorated downstream STAT3/STAT5 activity and significantly suppressed HMGA2 expression. To be more specific, we also selected INC424 (ruxolitinib, a pan-JAK inhibitor used in the treatment of MPN24,25) to assess the influence of JAK-STAT activity on the expression of HMGA2. Upon obtaining the IC50 and IC90 concentrations of INC424 in Ton.JAK2.WT and Ton.JAK2.V617F cells (Figure 5C), we treated the cells with the indicated drug concentrations for 3 days. Both sets of cells showed decreased JAK/STAT activity, abolished HMGA2 expression, and enhanced apoptosis in a dose-dependent manner (Figure 5D). The decreased Hmga2 expression, as well as upregulated let-7a in INC424-treated cells, was also confirmed by qRT-PCR (Figure 5E). The results provide complementary evidence supporting a non-redundant role of JAK/STAT activation in let-7a inhibition and HMGA2 upregulation in MPN cells.
Figure 5.

Influence of hydroxyurea and pan-JAK inhibitor INC424 on the phenotypes of MPN cells. Stable, inducible Ton.JAK2.V617F and Ton.JAK2.WT cells were treated with 1 μg/mL doxycycline for 6 days and then exposed to various concentrations of hydroxyurea or INC424 for 72 h. The drug concentrations that inhibited cell growth by 50% (IC50) and 90% (IC90) were determined by the XTT assay. (A) The viability of Ton.JAK2.V617F cells plotted against various concentrations of hydroxyurea. (B) Western blot analysis on various proteins of the Ton.JAK2.V617F cells treated with or without hydroxyurea at the concentration of 5.55 μg/ml (the IC50 concentration for HU in these cells) for 3 days. (C) The viability of Ton.JAK2.WT (left panel) and Ton.JAK2.V617F (right panel) cells plotted against various concentrations of INC424. (D) Western blot analysis on various proteins of the Ton.JAK2.WT and Ton.JAK2.V617F cells treated with or without INC424 at the concentration of either IC50 or IC90 for 3 days. (E) Effects of various concentrations of INC424 on the expression of let-7a and Hmga2 in Ton.JAK2.WT and Ton.JAK2.V617F cells. Cells were harvested for qRT-PCR analysis at 3 days after treatment with the drug at indicated concentrations. The error bars show the standard deviation of three independent experiments. Asterisks indicate statistical significance (t-test; *P<0.01; **P<0.001; ***P<0.0001). WT: wild-type; HU: hydroxyurea; Conc: concentration.
Discussion
In the study, we used Ba/F3 cells with conditional expression of JAK2V617F or WT JAK2 as a working model to recapitulate the phenotypic comparison between JAK2-mutated and -unmutated MPN cells. We demonstrate that JAK2-mutated cells exhibit upregulation of HMGA2, and the expression of HMGA2 could be affected by the level of let-7a miRNA. HMGA2 overexpression confers JAK2-mutated cells with a survival advantage through inhibited apoptosis, and MPN patients harboring upregulated HMGA2 show an increased propensity for ET phenotype as well as a higher likelihood of developing thrombosis.
The finding, in the study herein, that HMGA2-overex-pressing MPN patients are more likely to belong to the ET subtype and have higher platelet counts is actually supported by several in vivo studies. Oguro et al. disclosed that overexpression of Hmga2 in hematopoietic stem cells induced a myeloproliferative state with enhanced megakaryopoiesis in mice,26 whereas Yang et al. similarly demonstrated that Hmga2 significantly increased megakaryocytic colonies in the bone marrow of JAK2V617F mice.27 Interestingly, Ikeda et al. showed that overexpression of Hmga2 could lead to an increased level of Jak2 transcripts and a rise in STAT3 phosphorylation.13 As a result, the proliferation of hematopoietic stem cells was expanded. In the study herein, we further illustrated that HMGA2 knockdown in JAK2-mutated cells resulted in growth inhibition and a significant increase in apoptosis, a finding consistently seen in other cancer types.28–30 Coupled with the fact that Hmga2-overexpressing BM cells have a growth advantage over control cells in mice competitive repopulation and serial BM transplantation models,13 it is conceivable that the upregulated HMGA2 “turns on” a state of proliferative hematopoiesis as well as inhibited apoptosis, and the predilection for megakaryocytic expansion26,27 may account for a higher platelet count and ET phenotype in HMGA2-overexpressing patients.
However, the stronger association between HMGA2 overexpression and ET subtype in the work herein contrasted with previous reports showing a higher prevalence of upregulated HMGA2 in patients with PMF.31,32 There are several possibilities that lead to such a discrepancy. Most likely, HMGA2-overexpressing ET and PMF might represent a continuum of disease entities at different stages, in which the overexpressed HMGA2 collaborates with coexisting driver mutations or other pathognomonic mutations to cultivate the phenotypic expression. The argument can be supported by the fact that overexpression of Hmga2 leads to both increased megakaryopoiesis and accelerated progression of myelofibrosis in animal models.26,27 Secondly, patients from various studies have different ethnical and environmental backgrounds, which may contribute to the discrepancy in the HMGA2-overexpression rates in these reports, including ours. Lastly, the composition of the enrolled patients and the sample sizes across studies are different. Although currently available data suggest that overexpressed HMGA2 might play a critical role in the pathogenesis of PMF,12,13,26,27,31–34 in some studies the reproducibility of clinical data is hampered by either the low number of PMF patients evaluated or insufficient inclusion of PV and ET patients.31–33 The Italian study12 and ours enrolled the largest cohorts of patients, with the PMF population being the largest representative subgroup [n=88 (out of 158); 55.7%] in the former study. Supplemented by other smaller series, investigators from that study convincingly showed a high frequency of HMGA2 overexpression in patients with PMF.12 It is very likely that we had too few PMF patients (17 cases only) to make our estimation justifiable. On the other hand, our study enrolled by far the largest number of ET (n=67) and PV (n=63) patients for the assessment of HMGA2 expression. We revealed that about one-fourth of ET patients harbored upregulated HMGA2, a finding mirrored by Harada-Shirado et al.34 Although PMF patients comprised the major subgroup that showed upregulated HMGA2 in the Italian study (as compared to ET and PV patients as well as normal controls), there was still a specific portion of ET patients who harbored overexpressed HMGA2.12 Supported by the findings from these two studies,12,34 our work provides further proof of evidence endorsing the notion that HMGA2 overexpression not only plays an essential role in the pathogenesis of PMF, but also exerts specific effects on the phenotypic presentation of ET.
Although case reports suggested that chromosomal translocation involving the 12q14–15 region led to overexpression of HMGA2 in patients with MPN,35–38 our data nonetheless disclosed an essential role of let-7a miRNA regulation on the expression of HMGA2 in patients with MPN. These findings are in line with a recent report demonstrating that decreased let-7 miRNA expression, instead of generating the loss of 3′-UTR of the HMGA2 gene, is the major cause of dysregulated HMGA2 mRNA expression in MPN.34 The aberrancy in microRNA expression that leads to the genetic complexity of MPN is also supported by some other studies, as the reduction of a wide variety of miRNAs, including let-7a,31 miR-149,32 miR-150,32 let-7f,39 and let-7g39 has been reported, and the former three are also associated with upregulated HMGA2 mRNA in these reports.31,32
The correlation between aberrant let-7a-HMGA2 activity and JAK2V617F mutation has been partially dissected by several investigators. Through transcriptome comparative microarray analysis, Guglielmelli et al. revealed that abnormal HMGA2 expression in the granulocytes of patients with PMF was dependent on the presence of JAK2V617F mutation.12 Potential alteration in the expression of microRNAs, however, was not assessed in this study. Bruchova et al. performed gene expression profiling and found that JAK2V617F frequency was inversely correlated with let-7a expression in PV granulocytes.31 Nevertheless, upregulated HMGA2 was detected in PMF (but not in PV) patients, and no correlation between the expression levels of let-7a and HMGA2 were identified.31 With only 35 patients included, the study was probably hampered by small sample size. Although an inverse correlation between the expression levels of let-7a and HMGA2 was found, Harada-Shirado et al. could not identify any potential relationship between HMGA2 expressional status and JAK2V617F mutation.34 Our study, enrolling significantly more patients, clearly demonstrates that HMGA2-overexpressing MPN patients are more likely to carry either one of the 3 driver mutations (Table 1). Coupled with our in vitro findings showing an apparent correlation between JAK-STAT activation and the let-7a-HMGA2 axis, we comprehensively elaborated the dependence of aberrant let-7a-HMGA2 axis activity on the presence of JAK2V617F mutation. A major flaw of our study might lie in the fact that we were unable to identify how mutant JAK2 regulates the let-7a-HMGA2 axis activity. Further exploratory work would be important to delineate the missing link between them.
In conclusion, we have demonstrated that enhanced Hmga2 expression could be seen in Ba/F3 cells with enforced JAK2V617F expression. HMGA2-overexpressing patients exhibit a trend of a higher likelihood of carrying one of the 3 MPN-relevant driver mutations. In vitro data confirm that downregulation of let-7 miRNA plays an essential role in the dysregulated expression of Hmga2; a result supplemented by the inverse correlation between the expression levels of let-7 and HMGA2 in clinical samples. Strikingly, expression of HMGA2 confers JAK2-mutated cells with a survival advantage and endues MPN patients with a unique clinical phenotype. Our findings suggest that, in a subset of MPN patients, the let-7-HMGA2 axis plays a prominent role in the pathogenesis of the disease that leads to unique clinical phenotypes.
Supplementary Material
Acknowledgments
The authors thank Professor Gregor Hoermann and Professor Matthias Mayerhofer (both of the Medical University of Vienna, Austria) for kindly providing Ba/F3 cells with inducible expression of JAK2V617F (Ton.JAK2.V617F) or wild-type (WT) JAK2 (Ton.JAK2.WT). We also thank Miss Hsing-Yi Tsou and Miss I-Shan Chen for their assistance with data collection.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/3/509
Funding
The study was supported by the Medical Ministry of Science and Technology (Taiwan, R.O.C) grant to CC Chen (MOST 103-2314-B-182-051-MY3) and Chang Gung Memorial Hospital grants to CC Chen (CMRPG6B0223, CORPG6B0373 and CORPG6F0031).
References
- 1.Nangalia J, Green TR. The evolving genomic landscape of myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2014;2014(1):287–296. [DOI] [PubMed] [Google Scholar]
- 2.James C, Mazurier F, Dupont S, et al. The hematopoietic stem cell compartment of JAK2V617F-positive myeloproliferative disorders is a reflection of disease heterogeneity. Blood. 2008;112(6):2429–2438. [DOI] [PubMed] [Google Scholar]
- 3.Stein BL, Williams DM, Rogers O, Isaacs MA, Spivak JL, Moliterno AR. Disease burden at the progenitor level is a feature of primary myelofibrosis: a multivariable analysis of 164 JAK2 V617F-positive myeloproliferative neoplasm patients. Exp Hematol. 2011;39(1):95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011; 118(7): 1723–1735. [DOI] [PubMed] [Google Scholar]
- 5.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24(6):1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nature Rev Cancer. 2012;12(9):599–612. [DOI] [PubMed] [Google Scholar]
- 7.Sgarra R, Rustighi A, Tessari MA, et al. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS letters. 2004;574(1–3):1–8. [DOI] [PubMed] [Google Scholar]
- 8.Fedele M, Battista S, Kenyon L, et al. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene. 2002;21(20): 3190–3198. [DOI] [PubMed] [Google Scholar]
- 9.Zhou X, Benson KF, Przybysz K, et al. Genomic structure and expression of the murine Hmgi-c gene. Nucleic Acids Res. 1996;24(20):4071–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nature Rev Cancer. 2007;7(12):899–910. [DOI] [PubMed] [Google Scholar]
- 11.Young AR, Narita M. Oncogenic HMGA2: short or small? Genes Dev. 2007;21(9):1005–1009. [DOI] [PubMed] [Google Scholar]
- 12.Guglielmelli P, Zini R, Bogani C, et al. Molecular profiling of CD34+ cells in idiopathic myelofibrosis identifies a set of disease-associated genes and reveals the clinical significance of Wilms’ tumor gene 1 (WT1). Stem Cells. 2007;25(1):165–173. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda K, Mason PJ, Bessler M. 3′UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood. 2011;117(22):5860–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swerdlow SH, C E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer, 2008. [Google Scholar]
- 15.Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2014;123(10):1552–1555. [DOI] [PubMed] [Google Scholar]
- 17.Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110(3):840–846. [DOI] [PubMed] [Google Scholar]
- 18.Chen CC, Gau JP, Chou HJ, et al. Frequencies, clinical characteristics, and outcome of somatic CALR mutations in JAK2-unmutated essential thrombocythemia. Ann Hematol. 2014;93(12):2029–2036. [DOI] [PubMed] [Google Scholar]
- 19.Chen CC, You JY, Gau JP, et al. Favorable clinical outcome and unique characteristics in association with Twist1 overexpression in de novo acute myeloid leukemia. Blood Cancer J. 2015;5:e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. [DOI] [PubMed] [Google Scholar]
- 21.Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016; 127(10):1325–1335. [DOI] [PubMed] [Google Scholar]
- 22.Marty C, Pecquet C, Nivarthi H, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127(10):1317–1324. [DOI] [PubMed] [Google Scholar]
- 23.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320(5872):97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, place-bocontrolled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oguro H, Yuan J, Tanaka S, et al. Lethal myelofibrosis induced by Bmi1-deficient hematopoietic cells unveils a tumor suppressor function of the polycomb group genes. J Exp Med. 2012;209(3):445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y, Akada H, Nath D, Hutchison RE, Mohi G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood. 2016;127(26):3410–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pentimalli F, Dentice M, Fedele M, et al. Suppression of HMGA2 protein synthesis could be a tool for the therapy of well differentiated liposarcomas overexpressing HMGA2. Cancer Res. 2003;63(21):7423–7427. [PubMed] [Google Scholar]
- 29.Malek A, Bakhidze E, Noske A, et al. HMGA2 gene is a promising target for ovarian cancer silencing therapy. Int J Cancer. 2008;123(2):348–356. [DOI] [PubMed] [Google Scholar]
- 30.Cai J, Shen G, Liu S, Meng Q. Downregulation of HMGA2 inhibits cellular proliferation and invasion, improves cellular apoptosis in prostate cancer. Tumour Biol. 2016;37(1):699–707. [DOI] [PubMed] [Google Scholar]
- 31.Bruchova H, Merkerova M, Prchal JT. Aberrant expression of microRNA in polycythemia vera. Haematologica. 2008; 93(7):1009–1016. [DOI] [PubMed] [Google Scholar]
- 32.Guglielmelli P, Tozzi L, Pancrazzi A, et al. MicroRNA expression profile in granulocytes from primary myelofibrosis patients. Exp Hematol. 2007;35(11):1708–1718. [DOI] [PubMed] [Google Scholar]
- 33.Andrieux J, Bilhou-Nabera C, Lippert E, et al. Expression of HMGA2 in PB leukocytes and purified CD34+ cells from controls and patients with Myelofibrosis and myeloid metaplasia. Leuk Lymphoma. 2006; 47(9):1956–1959. [DOI] [PubMed] [Google Scholar]
- 34.Harada-Shirado K, Ikeda K, Ogawa K, et al. Dysregulation of the MIRLET7/HMGA2 axis with methylation of the CDKN2A promoter in myeloproliferative neoplasms. Br J Haematol. 2015;168(3):338–349. [DOI] [PubMed] [Google Scholar]
- 35.Martin SE, Sausen M, Joseph A, Kingham BF, Martin ES. Identification of a HMGA2-EFCAB6 gene rearrangement following next-generation sequencing in a patient with a t(12;22)(q14.3;q13.2) and JAK2V617F-positive myeloproliferative neoplasm. Cancer Genet. 2012;205(6):295–303. [DOI] [PubMed] [Google Scholar]
- 36.Aliano S, Cirmena G, Garuti A, et al. HMGA2 overexpression in polycythemia vera with t(12;21)(q14;q22). Cancer Genet Cytogenet. 2007;177(2):115–119. [DOI] [PubMed] [Google Scholar]
- 37.Andrieux J, Demory JL, Dupriez B, et al. Dysregulation and overexpression of HMGA2 in myelofibrosis with myeloid metaplasia. Genes Chromosomes Cancer. 2004;39(1):82–87. [DOI] [PubMed] [Google Scholar]
- 38.Storlazzi CT, Albano F, Locunsolo C, et al. t(3;12)(q26;q14) in polycythemia vera is associated with upregulation of the HMGA2 gene. Leukemia. 2006;20(12): 2190–2192. [DOI] [PubMed] [Google Scholar]
- 39.Zhan H, Cardozo C, Yu W, et al. MicroRNA deregulation in polycythemia vera and essential thrombocythemia patients. Blood Cells Mol Dis. 2013; 50(3):190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.